

Abstract
Objectives
The purpose was to clarify the characteristics of interstitial lung disease (ILD) in immune-mediated necrotizing myopathy (IMNM) patients with anti-signal recognition particle (SRP) antibodies.
Methods
Medical records of IMNM patients with anti-SRP antibodies were reviewed retrospectively.
Results
A total of 60 patients were identified. Twenty-seven (45.0%) patients were diagnosed with ILD based on lung imaging: nonspecific interstitial pneumonia (NSIP) in 17 patients (63.0%) and organizing pneumonia in 9 patients (33.3%). Reticulation pattern was identified in 17 patients (63.0%) whereas 10 cases (37.0%) showed ground glass opacity and patchy shadows by high-resolution computed tomography (HRCT). Pulmonary function tests (PFTs) were available in 18 patients, 6 (33.3%) and 10 (55.6%) patients were included in the mild and moderate group, respectively. The average age at the time of ILD onset was significantly older than those without ILD (48.6 ± 14.4 years vs. 41.2 ± 15.4 years, p < 0.05), and the frequency of dysphagia in the ILD group was higher than the group without ILD (p < 0.05). Long-term follow-up was available on 9 patients. PFTs were stable in 8 (88.9%), and the HRCT remained stable in 6 (66.7%) patients.
Conclusions
ILD is not rare in IMNM patients with anti-SRP antibodies, most being characterized as mild to moderate in severity. NSIP is the principal radiologic pattern, and ILD typically remains stable following treatment.
Keywords: Immune-mediated necrotizing myopathy, Interstitial lung disease, Anti-signal recognition particle antibodies, Nonspecific interstitial pneumonia
Introduction
Idiopathic inflammatory myopathies (IIMs) are a group of clinically heterogeneous, autoimmune inflammatory disorders characterized by muscular weakness and multi-system involvement such as lung and skin. Dermatomyositis (DM), antisynthetase syndrome (ASS) and inclusion body myositis (IBM) have long been recognized as distinct subtypes of IIMs. However, in recent years, it has been recognized that muscle biopsies from some patients with myositis have significant myofiber necrosis yet minimal, if any, lymphocytic infiltrates. These patients are now widely recognized to have immune-mediated necrotizing myopathy (IMNM).
The presence of myositis specific autoantibodies (MSAs) and myositis associated autoantibodies (MAAs) have become key features for the classification and diagnosis of IIMs. To date, two different autoantibodies have been described in association with IMNM, those recognizing the signal recognition particle (SRP) and those targeting hydroxy-3-methylglutaryl-CoA reductase (HMGCR).
Autoantibodies recognizing the SRP were first identified in the 1980s [1–3]. Recently, it has been shown that the anti-SRP antibody is significantly associated with necrotizing myopathy, and the antibody is now recognized as a marker of IMNM [4].
Interstitial lung disease (ILD) is frequently associated with IIMs, especially in ASS and anti-melanoma differentiation-associated gene 5 (anti-MDA5) antibody-positive DM [5, 6]. Because ILD significantly contributes to the morbidity and mortality in patients with IIMs, it is important to clarify the clinical characteristics of ILD in such patients. Previous studies suggested that ILD was rare in IMNM patients with anti-SRP antibodies (SRP-IMNM), yet in clinical practice, ILD often occurred in conjunction with SRP-IMNM. Moreover, ILD complicating SRP-IMNM has been further substantiated in more recent reports [7–10]. However, little is known regarding the frequency, clinical characteristics, or the pattern of onset of ILD in SRP-IMNM patients. To address these questions, we reviewed the clinical data of SRP-IMNM patients intending to elucidate risk factors of ILD in these patients. Additionally, with appropriate clinical follow-up, we aimed to investigate the response to therapy of the patient subgroup, ILD in SRP-IMNM.
Methods and materials
Patients
All initially screened patients met either the Bohan and Peter criteria or the European Neuromuscular Centre criteria (ENMC) for IIMs between March 2010 and February 2020. All patients with IMNM were confirmed by muscle pathology. IMNM patients whose sera carried anti-SRP antibodies were selected for study. The anti-SRP antibody in the serum of patients was detected by the Euroline myositis line-blot assay of Euroimmun (Lübeck, Germany) according to the instructions of the manufacturer. Clinical and laboratory data were retrieved from their medical records, such as age, skin manifestations, muscle strength, muscle pathology, pulmonary function tests (PFTs), high-resolution computed tomography (HRCT), lung biopsy, presence of other MAAs, treatment and follow-up. If anti-SRP-positive IMNM patients were combined with other MSAs or other connective tissue diseases (CTDs), they were excluded.
This study was approved by the Ethics Institutional Review Board of the China-Japan Friendship Hospital.
Radiologic analysis of SRP-IMNM patients
All pulmonary HRCT for the diagnosis of ILD were reviewed independently by a single experienced radiologist and a single rheumatologist. In accordance with previous reports, the HRCT patterns were classified as usual interstitial pneumonia (UIP), organizing pneumonia (OP), nonspecific interstitial pneumonia (NSIP) and lymphocytic interstitial pneumonia (LIP) [11]. The HRCT findings and disease distribution were also evaluated in detail.
Pulmonary function tests in SRP-IMNM patients
Patients were initially divided into three groups according to the initial PFT values as referenced in previous studies: mild group (% predicted forced vital capacity [FVC] > 75% and % predicted diffusing capacity of the lung for carbon monoxide [DLCO] > 55%), moderate group (% predicted FVC from 50 to 75% or % predicted DLCO from > 35 to 55%), and severe group (% predicted FVC < 50% and % predicted DLCO < 35%) [12, 13]. Rapidly progressive ILD (RP-ILD) was defined as progressive dyspnea, progressive hypoxemia, and worsening of interstitial changes on chest radiography within 3 months from the onset of respiratory symptoms [14].
According to the consensus statement of the American Thoracic Society (ATS) on idiopathic pulmonary fibrosis, a decrease or increase of ≥ 10% in FVC was considered deterioration or improvement, respectively; either a decrease or increase of < 10% in FVC was considered stable [15].
Statistical analysis
All analyses were completed using SPSS 21.0 and p < 0.05 were considered significant. Quantitative variables are reported as means and were compared using a nonparametric test. Categorical variables are reported as numbers and/or percentages and were compared using the chi-square or, when appropriate, Fisher exact test.
Results
Characteristics of SRP-IMNM patients
A total of 138 patients with IMNM were screened initially, included 78 patients with anti-SRP antibody. After excluding patients with other MSAs or combined with other CTDs (see Table 1), 60 IMNM patients carrying the anti-SRP antibody were identified in the study, including 45 (75.0%) females and 15 (25.0%) males. The selection process was shown in Fig. 1. The age of onset ranged from 10 to 84 years with a mean age of 44.5 ± 15.3 years. The age at onset was younger than 18 years old in 2 patients: a 12-year-old female IMNM patient with ILD and atypical skin rash; the other a 10-year-old male with IMNM.
Table 1.
Differences between ILD and those without ILD in SRP-IMNM
| Characteristic | ILD patients (n = 27) | Patients without ILD (n = 33) | p value |
|---|---|---|---|
| Female/male | 22/5 | 23/10 | > 0.05 |
| Smoking history | 3/27 (11.1%) | 2/33 (6.1%) | > 0.05 |
| Age (years) | 48.6 ± 14.4 | 41.2 ± 15.4 | < 0.05 |
| Arthritis | 1/27 (3.7%) | 3/33 (9.1%) | > 0.05 |
| Fever | 3/27 (11.1%) | 1/33 (3.0%) | > 0.05 |
| Myalgia | 11/27 (40.7%) | 10/33 (30.3%) | > 0.05 |
| Dysphagia | 14/27 (51.9%) | 7/33 (21.2%) | < 0.05 |
| AST (U/L) | 101.2 ± 89.5 | 130.8 ± 113.0 | > 0.05 |
| ALT (U/L) | 126.5 ± 102.9 | 158.7 ± 105.8 | > 0.05 |
| CK (U/L) | 2520.4 ± 2409.1 | 3493.4 ± 3161.7 | > 0.05 |
| LDH (U/L) | 564.8 ± 316.7 | 705.1 ± 390.6 | > 0.05 |
| Serum ferrtin (ng/L) | 162.9 ± 142.5 | 184.5 ± 178.4 | > 0.05 |
| ANA positive | 24/27 (88.9%) | 27/33 (81.8%) | > 0.05 |
| Ro-52 | 13/27 (48.1%) | 11/33 (33.3%) | > 0.05 |
| SSA | 4/27 (14.8%) | 4/33 (12.1%) | > 0.05 |
LDH, lactate dehydrogenase; ALT, alanine transaminase; AST, aspartate transaminase; CK, creatine kinase
Fig. 1.

Selection flowchart
Five patients had a history of smoking, but none of them with history of dust exposures. Twenty-one (35.0%) patients suffered myalgia and 4 (6.7%) had arthralgia. Mechanic’s hands in 4/50 (8%), atypical rash in 5/60 (8.3%) and Raynaud's phenomenon occurred in 2/50 patients (4%).
Cardiac involvement was observed in 6/60 patients (10.0%), presenting as cardiac troponin I (cTnI) elevation, myocardial ischemia, arrhythmia, or myocardial MRI that demonstrated myocardial fibrosis. Five (9.1%) patients developed mild pulmonary arterial hypertension (PAH). Cancers were observed in 5 patients (8.3%).
Fifty-one (85.0%) patients were ANA-positive, with a titer ranging from 1:80 to 1:640, and the most common pattern of ANA was cytoplasmic granules. Patients testing positive for other MAAs included 24 (40.0%) patients with Ro-52, 8 (13.3%) patients carried the SSA antibody.
Radiologic analysis of ILD in SRP-IMNM
Fifty patients were evaluated with HRCT and and 10 patients by X-ray. In total, 27 (45.0%) patients were diagnosed with ILD on the basis of lung images or pulmonary histopathology. The radiologic patterns of ILD included NSIP in 17 patients (63.0%), OP in 9 (33.3%), and one patient with LIP (3.7%).
In most cases, abnormal opacities were predominantly distributed in the lower lobes and peribronchovascular sites. Ground glass opacity (GGO) was identified in 10 (37.0%) patients, 17 (63.0%) showed reticulation, 10 (37.0%) had patchy shadows, 6 (22.2%) had interlobular septal thickening (IST), there were 5 (18.5%) with pleural thickening, and consolidation was found in 9 (33.3%) cases. In contrast, traction bronchiectasis (TB, 7.4%) and cyst (3.7%) were infrequently seen. Representative HRCT images are shown in Fig. 2.
Fig. 2.
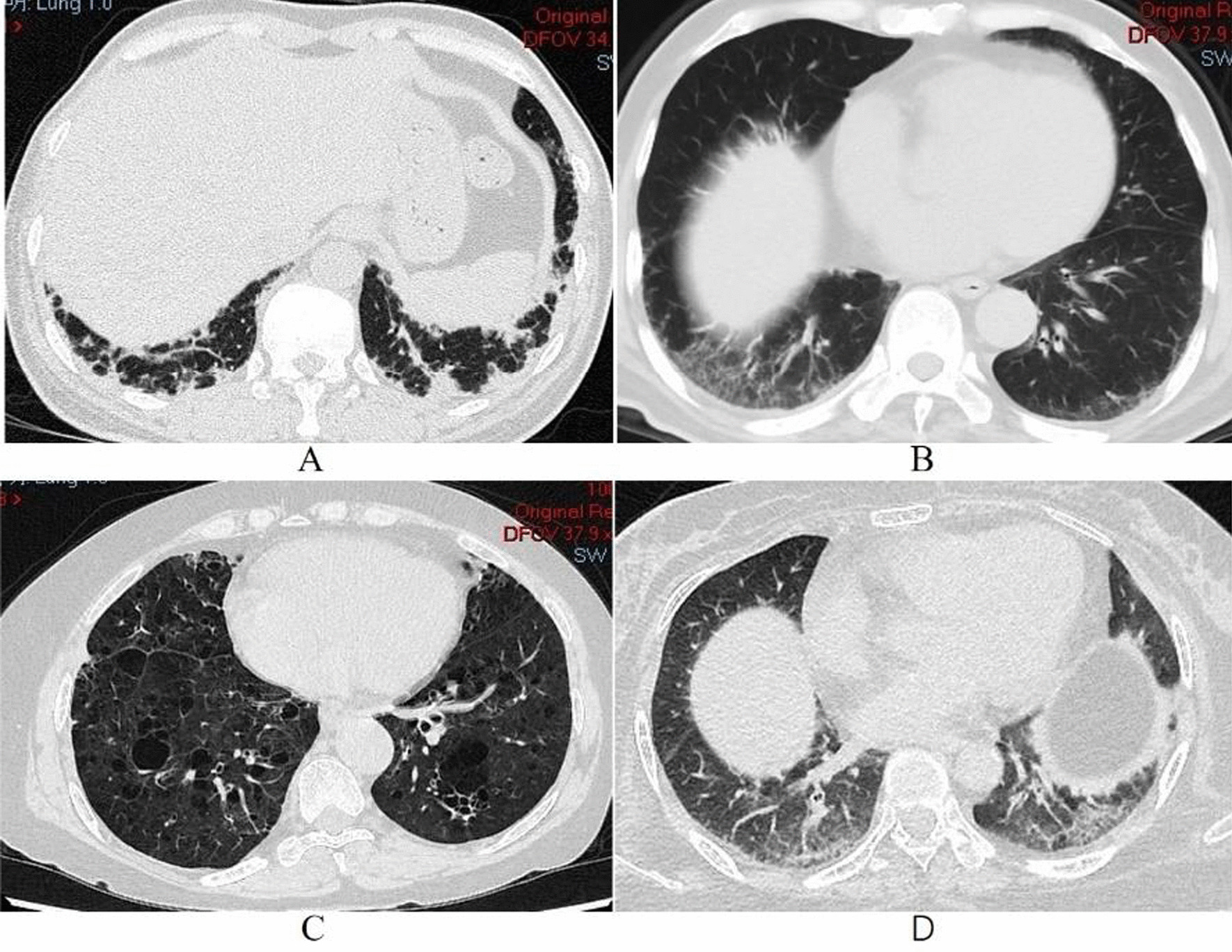
Abnormal opacities of HRCT from different SRP-IMNM patients. A 45-year-old woman with OP; B 54-year-old man with NSIP; C 55-year-old woman with LIP; D 45-year-old woman with NSIP
Severity of ILD in SRP-IMNM
Of the 27 SRP-IMNM patients with ILD, most were asymptomatic at the time of disease onset with slow development without respiratory symptoms; only 12 (44.4%) patients experienced dry cough and breathing difficulty with activity.
The PFTs were available for 18 patients: 6 (33.3%) met criteria for inclusion in the mild group, 10 (55.6%) comprised the moderate group, and two (11.1%) was in the severe group based on PFTs. None of the patients progressed to RPILD. In addition, all patients had FEV1/FVC ≥ 70%.
Comparison of SRP-IMNM patients with and without ILD
Comparing the two groups, the average age of onset among SRP-IMNM patients with ILD was significantly older than those without ILD (48.6 ± 14.4 years vs. 41.2 ± 15.4 years, p < 0.05) (see Table 1). The frequency of dysphagia in the ILD group was higher than the group without ILD (p < 0.05). The frequency of smoking history, skin rashes, arthritis, fever, myalgia and mechanic’s hands in the ILD group was not significantly different from the group without ILD (p > 0.05). Serum creatine kinase (CK), lactate dehydrogenase (LDH), as well as ALT and AST levels in ILD patients were not significantly different from those without ILD (p > 0.05). Regarding MAAs, the frequency of ANA, SSA and Ro-52 antibody were somewhat higher in the ILD group than patients without ILD, but the differences did not reach statistical significance (all p values > 0.05).
Follow-up of ILD in SRP-IMNM patients
Of the 60 patients, three patients with ILD died during follow-up; one patient died of pulmonary embolism and infection, one patient died of severe pulmonary infection, and another patient died due to the deterioration of ILD. Nine of the ILD patients were followed with HRCT and PFTs for a median of 23 months (range, 6–78 months) (Table 2). One patient received only glucocorticoids whereas the remaining 8 patients received a combination of immunosuppressive agents such as azathioprine (AZA), tacrolimus (TAC), cyclophosphamide (CYC), intravenous immunoglobulin (IVIG), and Tocilizumab (TCZ). Eight patients responded well to glucocorticoid and/or immunosuppressive agents with improvement in myositis. Moreover, patients 2 and 8 experienced restoration of normal muscle strength and CK levels. In accordance with performance standards for PFTs, 8 (88.9%) patients remained stable, and worsened in one. On assessment of HRCT, 6 (66.7%) patients remained stable (see Fig. 3), but progression of ILD occurred in 3 patients with the appearance of a new abnormal shadow. Kaplan–Meier showed that the survival rate of ILD group was lower than that of non ILD group, but the results were not statistically different (Fig. 4).
Table 2.
Changes in ILD patients at admission and the last follow-up
| Patients, gender, age | Admission | Last visit | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MMT | CK | FVC (%) | FEV1/FVC (%) | DLCO (%) | HRCT | MMT | CK | FVC (%) | FEV1/FVC (%) | DLCO (%) | HRCT | Fellow-up (months) | |
| P1, F, 48 | 65 | 7203 | 71 | 81 | 63 | GGO | 78 | 1180 | 59 | 82 | 58 | GGO, reticulation | 43 |
| P2, F, 66 | 72 | 1436 | 99 | 71 | 88 | Reticulation | 80 | 25 | 114 | 72 | 74 | Reticulation | 6 |
| P3, F, 41 | 68 | 4113 | 82 | 85 | 82 | IST | 70 | 51 | 84 | 100 | 66 | IST | 40 |
| P4, F, 51 | 70 | 2748 | 71 | 86 | 58 | Reticulation | 72 | 171 | 69 | 88 | 68 | Reticulation | 78 |
| P5, F, 24 | 56 | 6367 | 69 | 83 | 70 | Reticulation, IST | 75 | 43 | 77 | 100 | 49 | Reticulation, IST | 23 |
| P6, M, 55 | 55 | 1620 | 83 | 90 | 64 | GGO, consolidation | 63 | 661 | 83 | 93 | 65 | GGO, consolidation | 9 |
| P7, F, 50 | 80 | 1176 | 90 | 85 | 50 | Reticulation | 80 | 1702 | 97 | 87 | 56 | Reticulation, IST, TB, | 48 |
| P8, F, 52 | 75 | 535 | 70 | 83 | 58 | Reticulation, IST | 80 | 34 | 65 | 83 | 55 | Consolidation, Patchy shadows | 6 |
| P9, M, 54 | 45 | 3196 | 57 | 96 | 71 | GGO, patchy shadows, PT | 44 | 2459 | 59 | 81 | 60 | GGO, PT, patchy shadows | 7 |
GGO, Ground glass opacity; IST, interlobular septal thickening; TB, traction bronchiectasis; MMT, manual muscle test; PT, pleural thickening
Fig. 3.
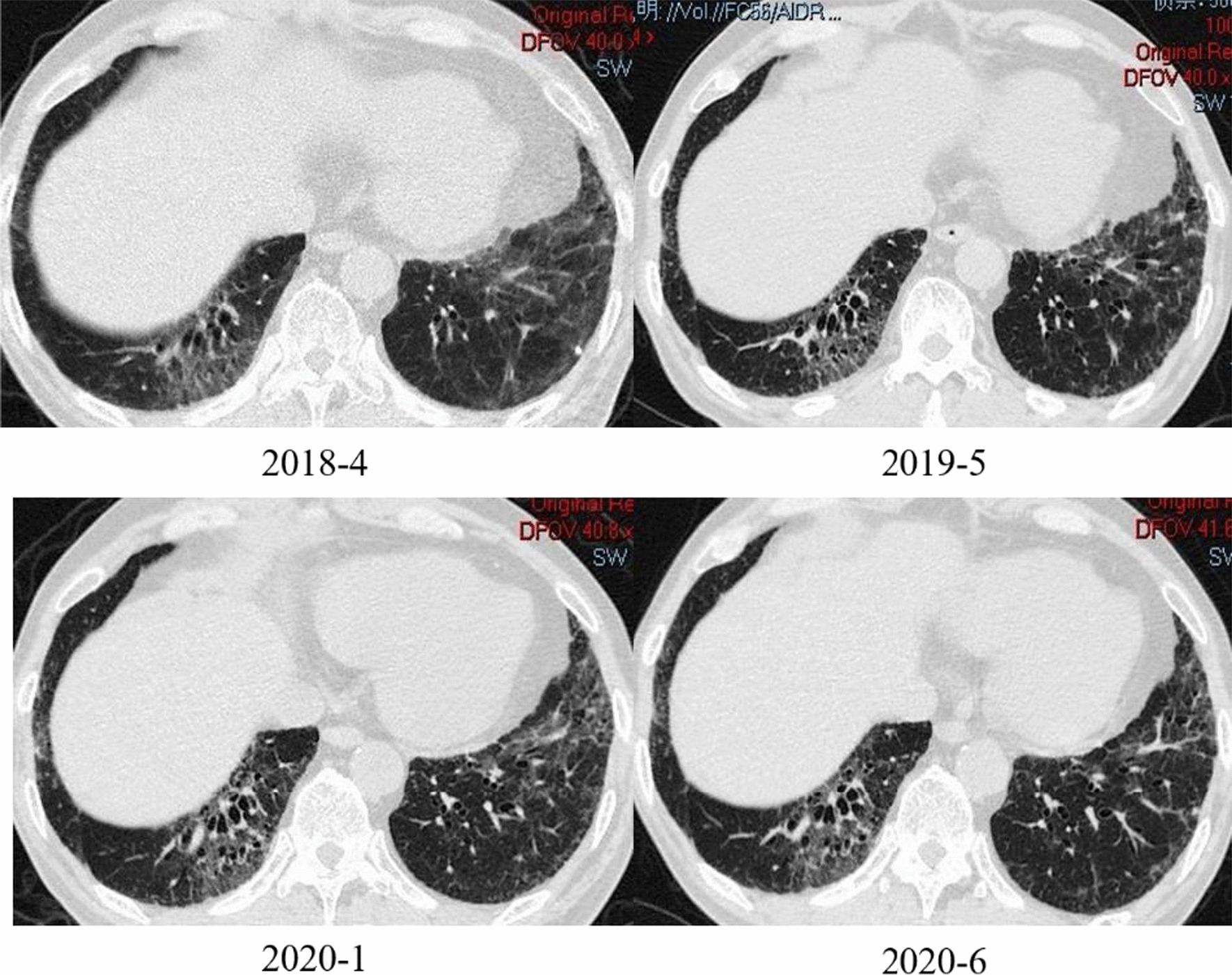
A 55-year-old male patient's lung CT, almost unchanged in 26 months
Fig. 4.
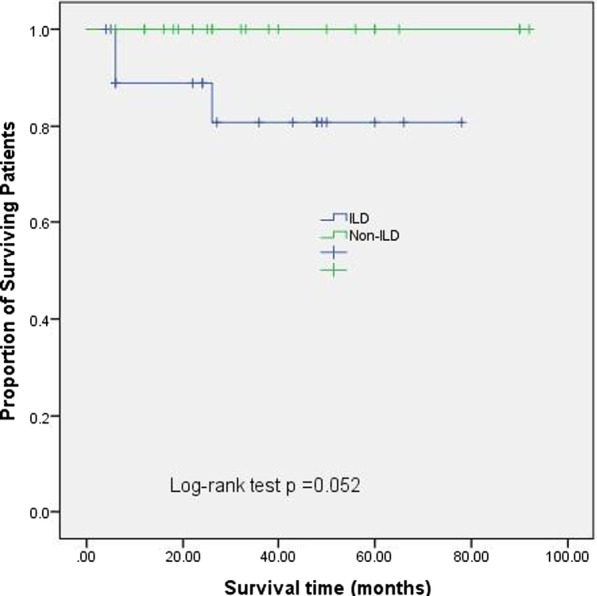
Kaplan–Meier survival curves for patients with and without ILD
Discussion
The present study investigated the clinical and radiological features and prognosis in SRP-IMNM patients with ILD. About half of SRP-IMNM patients were diagnosed with ILD. On HRCT, greater than half of the patients showed reticulation, followed by a pattern of GGO and patchy shadows. NSIP was the principal radiological pattern in ILD of SRP-IMNM patients. The severity of ILD in most patients was classified as mild to moderate as judged by PFTs and remained stable in most patients.
SRP is a complex consisting of six polypeptides and a 7S RNA molecule that is important for the translocation of newly formed proteins to the endoplasmic reticulum [2]. The hallmark feature of patients with anti-SRP antibodies is the presence of proximal muscle weakness, myofiber necrosis with minimal inflammatory cell infiltrate on muscle biopsy. The clinical picture of muscle weakness and characteristic pathologic changes of IMNM were present in all patients in our cohort.
Extra-muscular manifestations such as skin, lung and heart involvement may also be seen in SRP-IMNM patients. Pinal-Fernandez et al. reviewed the characteristics of patients with IMNM, and ILD occurred in only 10–20% of SRP-IMNM patients and fewer than 5% of those with HMGCR-IMNM [4]. Similarly, in a study of 100 Japanese IIM patients with anti-SRP antibodies, 13 (13%) patients had ILD [7]. In a literature review of SRP-IMNM, including 9 case series with a total of 239 patients, only 41 (17%) patients had ILD [8]. In sharp contrast, nearly half of our SRP-IMNM patients had documented ILD. The prevalence of ILD was higher in our study, but it still needed to be confirmed by a larger cohort in the future.
In our cohort, analysis of HRCT revealed that the abnormalities were predominantly distributed in the lung bases, and greater than half of the SRP-IMNM patients had reticulation. Additionally, GGO, patchy shadows, and IST were commonly identified by HRCT. Furthermore, the HRCT patterns of ILD in SRP-IMNM patients were most commonly NSIP, followed by OP; LIP and UIP were rarely identified. In 2010, Wantke et al. reported a 46-year-old female SRP-IMNM patient with OP, yet the patient was completely stable and lung function even improved. The HRCT showed no sign of active OP but rather, diffuse fibrosis especially in the middle lobe [9]. In 2015, Suzuki described the clinical characteristics of 100 patients with anti-SRP antibodies. Chest CT revealed ILD in 13 patients, all of whom had NSIP, and their respiratory symptoms were generally mild [7]. Kusumoto et al. described NSIP in a 72-year-old male SRP-IMNM patient: chest CT revealed GGO and reticular shadows with a peripheral, predominantly lower lobe distribution. Treatment with prednisolone and immunosuppressive agents resulted in normalization of CK, mild muscle weakness, and CT and PFT showed no further progression of ILD [8].
Because the vast majority of our patients did not undergo bronchoscopy or surgical lung biopsy, confirmation of HRCT-based subtyping of ILD was lacking and may not have been entirely accurate. In our cohort, only a single female child with ILD was found, an extremely rare occurrence. Standard chest X-ray or plain CT scan may not be adequate to diagnose ILD, so screening for it should include HRCT.
Although ILD is frequently present in SRP-IMNM patients, it is typically not severe, as only a small number of patients in our series noted dyspnea with exercise. With objective PFT measurements, about 90% of patients with ILD were classified as mild or moderate. In contradistinction, for patients with ASS and anti-MDA5 antibody positive DM, the severity of ILD is often moderate to severe, and some progress to RP-ILD. Another study observed that subclinical lung involvement was not rare in patients with IMM associated with positive anti-NXP2 antibody, with various radiological patterns and a significant lung function defect [16]. Patients with SRP-IMNM may not only develop proximal muscle weakness, but diaphragmatic muscles can also be involved, resulting in decreased lung function. This may lead to restrictive ventilation dysfunction. In addition, our study showed that ILD group had a higher frequency of dysphagia, which was easy to lead to gastroesophageal reflux. These factors may be involved in the occurrence and development of ILD [17].
The patients with ILD in our cohort were middle aged, which suggests that screening for ILD with HRCT should be performed regularly in this age group of SRP-IMNM patients.
The prognosis for SRP-IMNM patients is generally good, as most patients can anticipate long term survival. Comparing the patients with and without ILD, the ILD group had a relatively higher death rate. However, only one patient with ILD died as a direct consequence of this complication whereas the 2 other deaths in this group resulted from a lung infection and a pulmonary embolism.
Despite the severity of the ILD being classified as mild to moderate, it did not improve with the recovery of muscle strength, ILD remained relatively stable. This outcome may be a function of relatively short follow-up.
Two of our patients did have severe ILD. Similarly, Qureshi et al. presented a 40-year-old female who presented with acute hypoxic respiratory failure secondary to SRP-IMNM associated ILD. After struggling through a complicated 6-month hospitalization, the patient underwent successful double lung transplantation and was eventually discharged on room air [10].
The present study has several limitations. First, it was a retrospective, single-center study with a relatively small sample size. Second, by the nature of a retrospective, single center (specializing in respiratory diseases) study, selection bias might have played a role. Third, the gold standard assay for the detection of MSAs is the immunoprecipitation assay (IP), but MSAs were detected by line blot assay in our study, this may lead to false positive or false negative.
In conclusion, the present study demonstrated that the complication of ILD in SRP-IMNM patients occurred appreciably more frequently than in previously published reports, especially in older middle-aged patients. However, the severity was limited to only mild or moderate in most patients and remained stable throughout the follow-up period.
Acknowledgements
The authors would like to express their gratitude to EditSprings (https://www.editsprings.com/) for the expert linguistic services provided.
Authors' contributions
G.W. and Y.G. took responsibility for the content of this manuscript, including the data and analysis. H.Y., X. X. and L.L. designed the study, analyzed the result, and revised the manuscript. X.L. worked in study abstraction, data analysis, and the main text composition. All authors read and approved the final manuscript.
Funding
This work was supported by the Academy Level Project of China-Japan Friendship Hospital, China (2019-1-QN-8).
Data availability
The original contributions generated for the study are included in the article, further inquiries can be directed to the corresponding author.
Declarations
Ethics approval and consent to participate
This study was approved by the Research Review Committee (RRC) and the Ethical Review Committee (ERC) of the China-Japan Friendship Hospital (IRB number is 2016-117). In the retrospective non-interventional study, all patients’ data was anonymously used.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Nakao Y, Mukai R, Kabashima T, et al. A novel antibody which precipitates 7.5S RNA is isolated from a patient with autoimmune disease. Biochem Biophys Res Commun. 1982;109:1332–1338. doi: 10.1016/0006-291X(82)91923-4. [DOI] [PubMed] [Google Scholar]
- 2.Okada N, Mimori T, Mukai R, et al. Characterization of human autoantibodies that selectively precipitate the 7SL RNA component of the signal recognition particle. J Immunol. 1987;138:3219–3223. [PubMed] [Google Scholar]
- 3.Reeves WH, Nigam SK, Blobel G. Human autoantibodies reactive with the signal-recognition particle. Proc Natl Acad Sci U S A. 1986;83:9507–9511. doi: 10.1073/pnas.83.24.9507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pinal-Fernandez I, Casal-Dominguez M, Mammen AL. Immune-mediated necrotizing myopathy. Curr Rheumatol Rep. 2018;20:21. doi: 10.1007/s11926-018-0732-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shi J, Li S, Yang H, et al. Clinical profiles and prognosis of patients with distinct antisynthetase autoantibodies. J Rheumatol. 2017;44:1051–1057. doi: 10.3899/jrheum.161480. [DOI] [PubMed] [Google Scholar]
- 6.Moghadam-Kia S, Oddis CV, Aggarwal R. Anti-MDA5 antibody spectrum in western world. Curr Rheumatol Rep. 2018;20:78. doi: 10.1007/s11926-018-0798-1. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki S, Nishikawa A, Kuwana M, et al. Inflammatory myopathy with anti-signal recognition particle antibodies: case series of 100 patients. Orphanet J Rare Dis. 2015;10:61. doi: 10.1186/s13023-015-0277-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kusumoto T, Okamori S, Masuzawa K, et al. Development of necrotizing myopathy following interstitial lung disease with anti-signal recognition particle antibody. Intern Med. 2018;57:2045–2049. doi: 10.2169/internalmedicine.0303-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wantke F, Kneussl M, Hubner M, et al. Signal recognition particle (SRP) positive myositis in a patient with cryptogenic organizing pneumonia (COP) Rheumatol Int. 2010;30:1361–1365. doi: 10.1007/s00296-009-1059-4. [DOI] [PubMed] [Google Scholar]
- 10.Qureshi A, Brown D, Brent L. Anti-signal recognition particle antibody-associated severe interstitial lung disease requiring lung transplantation. Cureus. 2020;12:e7962. doi: 10.7759/cureus.7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188:733–748. doi: 10.1164/rccm.201308-1483ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aiko N, Yamakawa H, Iwasawa T, et al. Clinical, radiological, and pathological features of anti-asparaginyl tRNA synthetase antibody-related interstitial lung disease. Respir Investig. 2020;58:196–203. doi: 10.1016/j.resinv.2019.12.003. [DOI] [PubMed] [Google Scholar]
- 13.Ley B, Ryerson CJ, Vittinghoff E, et al. A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann Intern Med. 2012;156:684–691. doi: 10.7326/0003-4819-156-10-201205150-00004. [DOI] [PubMed] [Google Scholar]
- 14.Moghadam-Kia S, Oddis CV, Sato S, et al. Antimelanoma diferentiation-associated gene 5 antibody: expanding the clinical spectrum in North American patients with dermatomyositis. J Rheumatol. 2017;44:319–325. doi: 10.3899/jrheum.160682. [DOI] [PubMed] [Google Scholar]
- 15.American Thoracic Society Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS) Am J Respir Crit Care Med. 2000;161:646–664. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- 16.Bermudez J, Heim X, Bertin D, et al. Lung involvement associated with anti-NXP2 autoantibodies in inflammatory myopathies: a French monocenter series. Expert Rev Respir Med. 2020;14:845–850. doi: 10.1080/17476348.2020.1767598. [DOI] [PubMed] [Google Scholar]
- 17.Baqir M, Vasirreddy A, Vu AN, et al. Idiopathic pulmonary fibrosis and gastroesophageal reflux disease: a population-based, case–control study. Respir Med. 2021;178:106309. doi: 10.1016/j.rmed.2021.106309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The original contributions generated for the study are included in the article, further inquiries can be directed to the corresponding author.