

Abstract
INTRODUCTION:
The ribosome catalyzes protein synthesis in all cells by coupling the decoding of messenger RNA by the small ribosomal subunit with peptide bond formation by the large ribosomal subunit. As both subunits are composed of ribosomal RNA and ribosomal proteins, the assembly of these molecular machines is tightly controlled. In human cells, more than 200 ribosome assembly factors catalyze the maturation of both ribosomal subunits. Ribosome assembly is initiated in the nucleolus, a biomolecular condensate formed through multivalent protein–nucleic acid interactions. Inside this biomolecular condensate, the first stable eukaryotic ribosome assembly intermediate of the small subunit, the small subunit (SSU) processome, is formed. Within the SSU processome, more than 70 proteins and an RNA chaperone, the small nucleolar RNA (snoRNA) U3, work in concert to bring about RNA folding, modifications, rearrangements, and cleavage as well as targeted degradation of preribosomal RNA by the RNA exosome. The essential nature of this process is highlighted by mutations in ribosomal proteins and ribosome assembly factors that have been associated with human diseases.
RATIONALE:
Little is known about the structure, function, and maturation of the human SSU processome in the nucleolus as it transitions through different states to assemble the small ribosomal subunit. The retention of preribosomal particles within the biomolecular condensate of the nucleolus has thus far prevented their biochemical and structural characterization. To elucidate the nucleolar maturation of the human SSU processome, the isolation of these preribosomal particles from the nucleolus was therefore a key hurdle. To this end, we have developed a human genome editing platform to biallelically affinity tag the endogenous SSU processome and have established biochemical procedures for its isolation from the nucleolus for structural studies.
RESULTS:
The cryo–electron microscopy (cryo-EM) structures of three distinct states of the human SSU processome (denoted pre-A1, pre-A1*, and post-A1) were obtained at resolutions of 3.6, 3.9, and 2.7 Å, respectively. Together these structures reveal the molecular mechanisms responsible for the progression during nucleolar SSU processome maturation. Intricate layers of regulation are exerted through the concerted interplay of ribosome assembly factors that control irreversible steps of ribosome assembly, such as RNA processing by the RNA exosome, RNA unwinding by the DEAH-box helicase DHX37 (Dhr1 in yeast), and RNA cleavage by the endonuclease UTP24. The earliest state, pre-A1, highlights how only a small portion of a large preribosomal RNA segment, the 5′ external transcribed spacer (5′ ETS) is recognized by the SSU processome. We have generated a synthetic human ribosomal DNA (rDNA) locus that enables the production of recombinant ribosomes in human cells. By derivatizing this rDNA locus, we have identified a minimal 5′ ETS that can function to generate mature small ribosomal subunits in human cells. As the SSU processome transitions from states pre-A1 and pre-A1* to post-A1, a specific binding site for the RNA exosome-associated exoribonuclease EXOSC10 (Rrp6 in yeast) is generated. In the context of the post-A1 state, the DEAH-box helicase DHX37 is recruited, and its helicase activity must be tightly regulated to prevent the premature unwinding of preribosomal RNA from the RNA chaperone U3. By combining x-ray crystallography with cryo-EM, we show that DHX37 is initially autoinhibited and tightly controlled on the SSU processome through a precisely choreographed interplay that dictates the availability of binding sites and the accessibility of an activating peptide of the ribosome assembly factor UTP14. Within the core of the SSU processome, we have identified a network of proteins that guide cleavage at site A1, thereby generating the mature end of the small ribosomal subunit RNA. Lastly, the structure of the human SSU processome in state post-A1 provides further insight into human diseases, showing how the mammalian-specific ribosome assembly factor AROS stabilizes ribosomal protein eS19, which is most abundantly mutated in Diamond-Blackfan anemia.
CONCLUSION:
The cryo-EM structures of three human SSU processome complexes presented here reveal the structural plasticity of the human SSU processome, which endows this particle with the ability to coordinate conformational changes with distinct stages of ribosome assembly. The combined cryo-EM, x-ray crystallography, and functional data serve as a framework for the mechanistic understanding of the human SSU processome.
Graphical Abstract
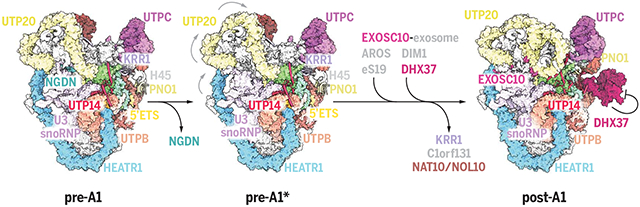
Schematic model of nucleolar maturation of the human SSU processome. The dissociation of neuroguidin (NGDN) signifies the transition from state pre-A1 to state pre-A1*, which is followed by large-scale conformational changes within the SSU processome (gray arrows) as well as the dissociation of pre-A1–specific factors and the association of the exosome and post-A1–specific factors. The RNA helicase DHX37 is autoinhibited in state post-A1. Protein complexes are color coded, and gray labels indicate proteins that are not shown.
The human small subunit processome mediates early maturation of the small ribosomal subunit by coupling RNA folding to subsequent RNA cleavage and processing steps. We report the high-resolution cryo–electron microscopy structures of maturing human small subunit (SSU) processomes at resolutions of 2.7 to 3.9 angstroms. These structures reveal the molecular mechanisms that enable crucial progressions during SSU processome maturation. RNA folding states within these particles are communicated to and coordinated with key enzymes that drive irreversible steps such as targeted exosome-mediated RNA degradation, protein-guided site-specific endonucleolytic RNA cleavage, and tightly controlled RNA unwinding. These conserved mechanisms highlight the SSU processome’s impressive structural plasticity, which endows this 4.5-megadalton nucleolar assembly with the distinctive ability to mature the small ribosomal subunit from within.
The biogenesis of human ribosomes requires more than 200 ribosome assembly factors, which facilitate transcription, RNA folding, and processing and modification of preribosomal RNA as well as the incorporation of 80 ribosomal proteins to form the small (40S) and large (60S) ribosomal subunits (1). This process starts in the human nucleolus, a tripartite biomolecular condensate formed through multivalent protein–nucleic acid interactions (2). Here, a 47S precursor transcript is formed, which contains coding segments for the small ribosomal subunit (18S) and large ribosomal subunit (28S and 5.8S) as well as key regulatory regions including the 5′ external transcribed spacer (5′ ETS) (3). The 5′ ETS plays a pivotal role during the initial stages of ribosome assembly, where it coordinates the recruitment of assembly factors and provides a structural blueprint for early assembly intermediates of the small subunit (SSU) (4). In contrast to lower eukaryotes such as Saccharomyces cerevisiae, human preribosomal RNA contains drastically larger spacer regions, so that the 5′ ETS is almost twice the size of the entire 18S rRNA. The trend toward larger 5′ ETS segments in mammals suggests that in addition to an immediate function in ribosome assembly, these 5′ ETS segments may have acquired additional macroscale functions, such as participation in biomolecular condensate formation in mammalian nucleoli (5).
After the transcription of preribosomal RNA, the first stable nucleolar ribosome assembly intermediate is the SSU processome, a giant precursor of the small ribosomal subunit (6). In yeast, the temporal order in which more than 50 ribosome assembly factors form the SSU processome has been elucidated (7, 8). Cryo–electron microscopy (cryo-EM) structures of yeast SSU processomes have been determined before cleavage at site A1, which separates the 5′ ETS and 18S (9-12), and during transition states toward a pre-40S particle that involve the RNA degradation machinery, the RNA exosome (13-15). However, despite these advances, limited resolution near 4 Å has so far only allowed a partial interpretation of these molecular snapshots. As a result, the molecular logic that drives SSU processome maturation at a mechanistic level has remained obscure, and the mechanisms and control of key enzymes, including the yeast RNA helicase Dhr1 (DHX37 in humans) within the SSU processome, or substrate recognition by the RNA exosome are still poorly understood.
Insights into human small ribosomal subunit assembly are even more modest and currently limited to late nuclear and cytoplasmic stages (16, 17), as the human nucleolus exhibits distinctive biochemical properties in the form of a biomolecular condensate whose contents have thus far proved impenetrable for biochemical and structural studies. An inability to purify intact human nucleolar ribosome assembly intermediates such as the human SSU processome has therefore limited our knowledge of their composition to known homologs of yeast ribosome assembly factors and large-scale genetic screens (18-20). The lack of structural information has further hindered our understanding of the roles and functions of about one-quarter of human ribosome assembly factors, precluding a mechanistic understanding of severe human diseases in which mutations of ribosomal proteins result in dysfunctional ribosome assembly at the level of the SSU processome (21).
In this study, we developed a human genome editing platform and biochemical methodologies to permeabilize the biomolecular condensate of the human nucleolus and determined the high-resolution cryo-EM structures of maturing human SSU processomes at resolutions of 2.7 to 3.9 Å. These structures reveal the exquisite levels of control that the SSU processome provides by enforcing a strict chronology and molecular choreography for irreversible steps of ribosome assembly, such as RNA degradation, site-specific RNA cleavage at site A1, and RNA unwinding.
Cryo-EM structures of maturing human SSU processomes
A critical hurdle for the study of endogenous human ribosome assembly intermediates is the ability to access all endogenous components. To overcome this challenge, we developed a human genome editing platform that couples the genome editing activity of the CRISPR RNA-guided endonuclease Cas9 with cell surface display to rapidly isolate biallelically edited cells by flow cytometry. In this platform, two DNA repair templates are used, one for each allele of the same gene. While both DNA repair templates code for an identical C-terminal tag of the gene of interest, they contain two different cell surface epitopes, which are produced as separate polypeptides. Thus, cells in which two alleles of a given gene have been repaired with two different DNA repair templates can be selected using flow cytometry (fig. S1, A to E). Using this editing tool, the endogenous human ribosome assembly factor NOC4L, which is present in human SSU processomes, was biallelically tagged and used as bait for subsequent purifications (fig. S1, F to H). To permeabilize the human nucleolus, we developed a sequential extraction procedure in which human nucleolar assembly intermediates containing tagged NOC4L are released in the last step of the extractions, thereby providing the starting material for subsequent nanobody-based purification and structural analysis by cryo-EM. The purified particles include pre-rRNA species that are uncut (30S and 26S) or cut (21S) at site A1 (fig. S2, B and C). A total of 84,904 cryo–electron micrographs were collected on a Titan Krios microscope equipped with a K3 detector, yielding 9.3 million autopicked particles (fig. S3). Extensive three-dimensional (3D) classification revealed three classes that represent distinct maturation states of the human SSU processome, here referred to as pre-A1, pre-A1*, and post-A1, which were resolved to resolutions of 3.5, 3.9, and 2.7 Å, respectively (Fig. 1A and fig. S3). As was done for the first complete yeast SSU processome structure (11), the use of focused classifications and refinements of subregions of particles belonging to states pre-A1 and post-A1 facilitated atomic model building and yielded high-resolution composite maps (figs. S4 to S9 and table S1). These reconstructions enabled the assignment and precise model building of human preribosomal RNA (5′ ETS and 18S rRNA), U3 small nucleolar RNA (snoRNA), 21 ribosomal proteins, and 50 human ribosome assembly factors (Fig. 1B, figs. S10 and S11, tables S2 to S4, and movies S1 to S4). For an in-depth view of these states at the atomic coordinate level, PyMOL sessions and morphs highlighting the differences between states pre-A1, pre-A1*, and post-A1 are available (data S1 and movies S5 and S6). For the post-A1 state, posttranscriptionally modified nucleotides could be built that are in agreement with prior mass spectrometry and structural data (22, 23). The three states are characterized by the maturation of preribosomal RNA as well as state-specific protein components. Whereas states pre-A1 and pre-A1* are characterized by the presence of the 5′ ETS, the post-A1 state only contains a minimal ordered segment of the 5′ ETS near the interacting UtpA and UtpB complexes (Fig. 1C and fig. S12). In addition to vertebrate-specific extension of the UtpA and UtpB complexes, the human UtpA complex lacks a Utp9 homolog and instead contains two copies of WDR43 (Utp5 in yeast), like what was previously observed in Chaetomium thermophilum (24). Similarly, we do not observe a human homolog of Utp30, a ribosome assembly factor that is replaced by the small ribosomal subunit protein eS19 during the assembly of the yeast SSU processome. The loss of neuroguidin (NGDN) signifies the transition from pre-A1 to pre-A1*, whereas a range of pre-A1–specific factors (NAT10, NOL10, AATF, C1orf131, and KRR1) are removed during the subsequent transition toward post-A1 in which DHX37, DIM1, the RNA exosome subunit EXOSC10, and eS19 and its chaperone AROS are recruited.
Fig. 1. Nucleolar maturation of human SSU processome.

(A) Cryo-EM density maps of nucleolar states pre-A1, pre-A1*, and post-A1 at 3.6, 3.9, and 2.7 Å, respectively. Modules, assembly factors, and RNA are labeled, and compositional changes are indicated at the bottom. (B) Atomic models of the three states displayed and labeled as in (A). (C) Structures of RNA elements in each state. 5′ domain, green; central domain, beige; 3′ major domain, gray; 3′ minor domain, light pink; 5′ ETS, yellow; U3 snoRNA, pink.
Recognition and processing of the human 5′ ETS
A distinctive feature of mammalian preribosomal RNAs is the drastically enlarged 5′ ETS (fig. S13), for which both structure and function have remained obscure. The critical structured elements of the human 5′ ETS can be visualized in states pre-A1 and pre-A1* (Fig. 2, A to D). Although the structured parts of the human 5′ ETS occupy a similar region as first reported in yeast (10), the overall topology of the human 5′ ETS differs from that observed in yeast (fig. S14). Recognition of the human 5′ ETS is mediated by extensive protein–RNA interactions with nearby assembly factors as well as base pairing with U3 snoRNA. Downstream of the cotranscriptional A′ cleavage site, the evolutionarily conserved motif (ECM) within the 5′ ETS (25) is specifically recognized by a composite binding site formed by UTP4 and WDR75 (Fig. 2C). Following a set of seven ordered helical segments, the 5′ ETS is first recognized by the U3 snoRNA via its 3′ hinge. Notably, the 5′ ETS segment paired with the 3′ hinge is located several hundred nucleotides away from the previously postulated position (26). After a largely single-stranded segment, the 5′ ETS protrudes into the solvent with a long disordered helical segment, which is followed by an ordered sequence that pairs with the U3 5′ hinge before once again extending into the solvent. As in yeast (11, 27), U3 snoRNA forms a central architectural anchor point to coordinate both the 5′ ETS via 3′ and 5′ hinges and the 18S via boxes A and A′ (Fig. 2B and figs. S10 and S14).
Fig. 2. Recognition and processing of the human 5′ ETS.

(A) Architectural view of state pre-A1 with pre-18S (white), 5′ ETS (yellow), and U3 snoRNA (pink). The ECM (orange) and U3 hinges are indicated. (B) Detailed view of U3 snoRNA and its interactions with pre-18S and 5′ ETS RNAs. (C) Recognition of the ECM by UTP4 (dark blue) and WDR75 (light blue). (D) Secondary structure of the observed 5′ ETS in state pre-A1. (E) Schematic representation of a synthetic human rDNA locus containing distinct sequences for the small and large ribosomal subunits (18S and 28S probe, respectively). Ordered regions of the 5′ ETS are indicated in dark yellow, and segments for truncations are indicated with Δ1, Δ2, and Δ3. (F) Northern blot analysis of cells transfected without plasmid (control, ctrl), wild-type (wt), and truncated synthetic human rDNA loci (Δ1; Δ3; Δ2,3; and Δ1,2,3). Precursors and mature 18S rRNA originating from the synthetic template are indicated. (G) Schematic illustrating the ratios between 5′ ETS and 18S of wild-type and truncated rDNAs. Ratios between 5′ ETS and 18S are indicated at the end of each bar, with yeast (bottom bar) serving as reference. nt, nucleotides.
The small fraction of the human 5′ ETS that we could visualize within the human SSU processome prompted us to analyze the functional significance of the other elements that make up this 3.6-kb segment of human preribosomal RNA. We generated a synthetic human ribosomal DNA (rDNA) locus under a native promoter on a plasmid for transfection into human cells. This synthetic rDNA contains distinctive sequences within the 18S and 28S sequences that allow mature ribosomal subunits originating specifically from this recombinant rDNA locus to be detected during Northern blotting analysis. On the basis of our secondary structure model and the built residues of the human 5′ ETS observed in states pre-A1 and pre-A1*, this system was derivatized to generate internal deletions of 5′ ETS regions that were not visualized in our cryo-EM reconstructions. These deletions included sequences upstream of the A′ cleavage site (Δ1), the long disordered helical element upstream of the 5′ hinge (Δ2), or a large helical segment between sites A0 and A1 (Δ3) (Fig. 2E and fig. S14).
Unexpectedly, single, double, and triple truncations of these sequences within the human 5′ ETS gave rise to mature small ribosomal subunits, as evidenced by the presence of a specific 18S band (Fig. 2F and fig. S15). Just as combined truncations in the 5′ ETS reduce the size of the transcript, a corresponding reduction in the size of the precursor species was observed, further supporting the specific assembly of mature 18S particles from the synthetic truncated rDNA locus. These data highlight that the human 5′ ETS can be separated into two functional units: the minimal functional core of the 5′ ETS that is required for SSU processome assembly (~25% of the 5′ ETS) and a large fraction of the 5′ ETS that is not required for SSU processome formation and may therefore function in biomolecular condensate formation (~75% of the 5′ ETS). The 75% reduction in size of the minimal 5′ ETS more closely matches the 5′ ETS:18S ratio observed in yeast (Fig. 2G).
Licensing for exosome-mediated remodeling
A critical function of the SSU processome is to signal the stage of assembly of the small ribosomal subunit that triggers subsequent maturation events of preribosomal RNA One of these key events is the exosome-mediated remodeling of the 5′ ETS. In yeast SSU processomes, large conformational changes within Utp20 have been observed that correlate with the dissociation of the assembly factor Lcp5 (the yeast homolog of NGDN) as well as the dissociation of the Kre33 module (yeast homolog of NAT10, NOL10, and AATF) (13). However, owing to limited resolution, the molecular significance and consequences of these events for eukaryotic ribosome assembly have remained elusive. The structures of the human SSU processomes reveal how conformational changes within the particles are used as a direct molecular readout for the recruitment of the RNA exosome component EXOSC10 (yeast Rrp6) (Fig. 3). In the pre-A1 state, the helical repeat proteins HEATR1 and UTP20 wrap around most of the SSU processome, with UTP20 lining both sides of NGDN, which is sandwiched between eS4 and uS4 (Fig. 3A).
Fig. 3. Structural remodeling facilitates licensing for exosome-mediated maturation.

(A) (Top) Architecture of state pre-A1. (Bottom) Zoomed view highlighting the NGDN (cyan, transparent surface) binding site near UTP20 (yellow), U3IP2 (purple), and ribosomal proteins eS4, eS24, and uS4. (B) (Top) Architecture of state pre-A1*. (Bottom) Zoomed view highlighting the loss of NGDN (cyan) and rearrangement of the U3IP2 N terminus. (C) (Top) Architecture of state post-A1. (Bottom) Zoomed view highlighting the presence of the EXOSC10 (pink, transparent surface) lasso with four peptide epitopes (I to IV) near the moved eS4.
The transition from states pre-A1 to pre-A1* is associated with the dissociation of NGDN and the conformational switch of the U3IP2 N terminus to a post-A1 conformation, thereby creating a largely vacant space in which helix 21 of the 18S rRNA is disordered (Fig. 3B). A key event occurring during the transition between pre-A1* and post-A1 states is the dissociation of the NAT10-NOL10-AATF module upon which large-scale movements of the 5′ domain of the 18S rRNA are transmitted via UTP20 and HEATR1. While HEATR1 moves upward and approaches the U3 snoRNP, UTP20 adopts a drastically different conformation, with its C-terminal region looping back toward the middle of the protein. In concert with these large conformational changes, eS4 adopts a different conformation to stabilize helix 21 in a new environment (movie S6). Unexpectedly, we observe the so-called “lasso” region of the exosome-associated nuclease EXOSC10 (28) in the post-A1 state, which is sensitive to these conformational changes and thus only binds to state post-A1 of the human SSU processome (Fig. 3C). Specifically, four peptide motifs of EXOSC10 probe this area for UTP20 (motif I), the formation of a UTP20-eS4 interface (motif II), the availability of eS24 for β sheet formation (motif III), and the proximity of HEATR1 (motif IV). Multiple sequence alignments of these motifs show that motifs I to III are present from yeast to humans whereas motif IV is a mammalian-specific extension (fig. S16). This mechanism of substrate recognition by EXOSC10 (Rrp6) upon conformational changes within the SSU processome is universal, as we have identified corresponding elements in exosome-associated SSU processomes from yeast in which these regions could not be interpreted previously (fig. S16, A to D) (13-15). These analyses highlight that there are two links between the SSU processome and the exosome: The exosome recognizes the SSU processome by way of the C terminus of EXOSC10, and the SSU processome recruits Mtr4 by way of arch-interacting motifs (AIMs) present in either Utp18 [in yeast (29)] or presumably TDIF2 (human), which contains a similar sequence motif in the solvent-exposed region (fig. S16E).
Regulation of the DEAH-box helicase DHX37
A key RNA helicase involved in the progression of later SSU processome intermediates is the RNA helicase DHX37, an enzyme of the DEAH-box family whose conformational state in the context of maturing SSU processomes remains the subject of controversy. Indeed, although high-resolution crystal structures of substrate-bound DHX37 (30) and a partially proteolyzed apo structure of the yeast homolog Dhr1 have been determined (31), a radically different configuration of the subdomains of Dhr1 was observed in yeast SSU processomes at lower resolution, a discrepancy originally attributed to a different nucleotide state (13). To elucidate the precise atomic basis of this altered state, we determined the 2.2-Å crystal structure of the yeast Dhr1 core enzyme (Fig. 4A and table S5). This structure reveals the presence of a loop within the RecA2 domain blocking the substrate channel. A comparison with the RNA-bound DHX37 shows that this RecA2 loop occupies the same region as the RNA substrate, suggesting that autoinhibition modulates enzymatic activity (Fig. 4, B and C, and fig. S17). This autoinhibited state of the enzyme can be docked into all yeast SSU processome states in which Dhr1 is not engaging its substrate, confirming the physiologically relevant role of the proposed mechanism (fig. S18, A to I) (13, 15). Moreover, the principle of DHX37 autoinhibition appears to be conserved in the human pathway as well, because the domain orientations observed at relatively low resolution in the human post-A1 state display a related overall topology (fig. S18, J to M).
Fig. 4. Regulation of the DEAH-box helicase DHX37.

(A) Crystal structure of the yeast Dhr1 core enzyme with the RecA2-associated autoinhibitory loop. A schematic of the yeast Dhr1 domain organization is shown at the bottom. ADP, adenosine diphosphate; WH, winged helix domain; OB, oligonucleotide/oligosaccharide binding-fold domain. (B and C) Detailed views of the Dhr1 autoinhibitory loop in the substrate channel (B) compared with the substrate RNA in the mouse Dhx37 structure (PDB ID 6O16) (C). (D) (Top) Structure of human pre-A1 state highlighting UTP14 proximal proteins. (Bottom) Detailed view of UTP14 and the recruitment domain (UTP14R). Black arrows depict subsequent movements of UTP14R, WDR46, and PNO1 from pre-A1 to post-A1. (E) (Top) Structure of human post-A1 state highlighting UTP14 proximal proteins. (Bottom) Detailed view of UTP14 and its recruitment, activation, and stabilizing domains (UTP14R, UTP14Act, and UTP14S, respectively). Positioning of these domains depends on movement of UTP20 (UTP14R), processing of the 5′ ETS (UTP14Act), and movement of Pno1 (UTP14S). The black arrow depicts the putative path of UTP14R toward its binding site, as seen in the yeast state Dis-C. The putative AIM of TDIF2 is indicated.
The autoinhibited DHX37 is recruited to the human SSU processome in the post-A1 state via its C-terminal domain, which is structurally distinct from the yeast homolog but occupies the same location between PWP2 and WDR36 of the UtpB complex (Fig. 4E). A prerequisite for this localization is the transition of the assembly factor PNO1 with helix 45 of the 18S rRNA into the interior of the particle to replace a site that was previously occupied by the assembly factor KRR1 (Fig. 4D). The recruitment site of DHX37 is close to UTP14, a protein that had been previously implicated in DHX37 recruitment and activation (30, 32, 33). As recruitment and activation of DHX37 need to be tightly controlled, UTP14 elements involved in either of these processes are themselves highly regulated and respond to the overall processing state of the human SSU processome.
In late states of SSU processome disassembly in yeast (Dis-C), an N-terminal helical segment of UTP14 (here referred to as UTP14R) recruits DHX37 close to its substrate U3 snoRNA near the 18S 3′ end (13). Positioning of UTP14R and access to its final binding site therefore require extensive control (Fig. 4 and fig. S19). In the pre-A1 state, UTP14R is sequestered on the UtpB subunit UTP6, and its final binding site is occupied by C1orf131, the human homolog of Faf1 that we have identified in the present study (Fig. 4D and fig. S20). In the post-A1 state, UTP14R is targeted to a different binding site near the rearranged UTP20 C-terminal segment. In this state, the final UTP14R binding site is now occupied by the C terminus of WDR46 (Utp7 in yeast) that has replaced C1orf131 to block premature binding of UTP14R (Fig. 4E). Upon dissociation of WDR46 during later stages of SSU processome disassembly, UTP14R can translocate to its final binding site to recruit DHX37. The ability to recruit DHX37 to its final location near its substrate therefore depends on a perfectly choreographed movement of the UTP14R segment and access to its binding site that is guided by large conformational changes within the SSU processome that signal distinct maturation stages (Fig. 4 and fig. S19).
A second layer of control is provided by the activation of DHX37 helicase activity. An activating segment of UTP14 (here referred to as UTP14Act) has been shown to stimulate the adenosine triphosphatase activity of DHX37 in vitro (30), yet how premature activation of DHX37 on the SSU processome is prevented has remained unclear. In the pre-A1 state, the 5′ ETS RNA threads through a narrow groove between PWP2 and WDR46 (Fig. 4D), and upon processing of the 5′ ETS from pre-A1 to post-A1, this same location is used to sequester UTP14Act. At the same time, a stabilizing segment of UTP14 (here referred to as UTP14S) stabilizes both PNO1 and the 18S 3′ end to help create half of the binding site that will later be used by UTP14R and the C-terminal domain of DHX37 during substrate engagement. The release of WDR46 during later stages of SSU processome disassembly therefore not only vacates the UTP14R binding site to enable DHX37 recruitment but also releases UTP14Act for subsequent activation of DHX37.
Together, our crystallographic and cryo-EM analyses reveal elegant layers of control that are used to keep DHX37 in an inactive state and regulate both its localization and activity as a function of larger conformational changes that are structurally read out by UTP14 (movie S6). Lastly, we observe that the presence of DHX37 in the post-A1 state is also necessary for the recruitment of the DIM1 methyltransferase, explaining the previously observed codependence of both proteins in depletion experiments of either protein in human cells (fig. S21) (33). The timing of DHX37 and DIM1 incorporation into the SSU processome is further coordinated with the dissociation of the NAT10-NOL10-AATF complex, as overlapping binding sites near eS6 ensure a mutually exclusive presence of these enzymes (fig. S21).
Protein-guided mechanism of cleavage at site A1
A key, irreversible event during the maturation of the small ribosomal subunit is the cleavage between the 5 ETS and 18S rRNA that generates the mature 5 end of the 18S rRNA. Within the core of the human SSU processome, cleavage at site A1 is orchestrated through protein-guided RNA movements that reposition the substrate RNA within the PIN domain nuclease UTP24 (34). Here, a set of helical elements originating from NOP14, IMP4, and the guanosine triphosphatase BMS1 undergo radical conformational changes to stabilize boxes A and A′ in the pre-A1 and post-A1 states (Fig. 5). In the pre-A1 state, the C terminus of BMS1 reaches across U3 snoRNA between boxes A and A′ to make contact with a protein cluster containing IMP4, IMP3, and C1orf131, while the C-terminal segment of NOP14 stabilizes box A′ in a lower position. The site of RNA cleavage, the 5′ end of the 18S rRNA, is positioned far away from the nuclease UTP24, which is additionally occluded by C1orf131 (Fig. 5A). In the post-A1 state, C1orf131 has disappeared and KRR1 has been replaced by PNO1. Box A′ is moved upward and stabilized by the NOP14 C-terminal segment that has changed its secondary structure from a previously continuous helix to a partially disrupted helix. Box A has been relocated closer to UTP24, and nucleotide 2 of the 18S rRNA is now seen in close proximity to the nuclease active site (Fig. 5B). The configuration of boxes A and A' is further stabilized by long helices of IMP4 and BMS1, both of which have undergone substantial conformational changes (movie S5).
Fig. 5. Protein-guided mechanism of cleavage at site A1.

(A) Catalytic core of state pre-A1 with U3 boxes A and A′ (pink) and base-pairing 18S (white). C1orf131 (light blue) occludes the nuclease UTP24 (purple), and nucleotide 2 of the 18S rRNA (red) is positioned far away. (B) In state post-A1, C1orf131 has disappeared and KRR1 has been replaced by PNO1 (yellow). Rearrangements in U3 box A and A′ allow nucleotide 2 (red) of the 18S rRNA to be in close proximity to the UTP24 nuclease active site (magnesium ion, green circle). (C) In state pre-A1, the UTP24 N terminus (purple) is sequestered via interactions with the 5′ hinge (pink), 5′ ETS (yellow), and the IMP3 N terminus (olive). Black arrows highlight structural changes within the N terminus of UTP24, the 5′ end of 18S rRNA, and other proteins that are required for A1 cleavage. (D) Destabilization of the 5′ ETS and accompanying changes in the post-A1 state allow the rearranged UTP24 N terminus to stabilize incoming substrate 18S rRNA.
During the transition from the pre-A1 state to the post-A1 state, the N-terminal segment of UTP24 reads out conformational changes that directly depend on the destabilization of the 5′ ETS (Fig. 5, C and D). In the pre-A1 state, the UTP24 N terminus is wedged between the 5′ hinge, the IMP3 N terminus, PWP2, and WDR46 (Fig. 5C). With the processing of the 5′ ETS in the post-A1 state, IMP3, PWP2, and WDR46 undergo conformational changes that prevent the UTP24 N terminus from binding in the same region. Instead, the N terminus of UTP24 now forms a lid on top of the nuclease active site, presumably to stabilize the incoming substrate 18S rRNA (Fig. 5D). The combined insights obtained from human SSU processomes in states pre-A1 and post-A1 highlight that upon exosome-mediated processing of the 5′ ETS, the movement of boxes A and A′ is guided by the coordinated structural adaptation of a number of proximal protein segments. In addition, the activation of UTP24 depends on the release of the inhibitory C1orf131 and the release of the UTP24 N terminus to act as part of the enzyme-substrate complex for cleavage at site A1.
Human diseases of the SSU processome
Mutations in both ribosomal proteins and assembly factors present in the human SSU processome have been associated with human diseases, so-called ribosomopathies (35), such as Diamond-Blackfan anemia, and several other childhood diseases, which can now be visualized in the context of the human SSU processome (Fig. 6). In >25% of patients suffering from the ribosomopathy Diamond-Blackfan anemia (36), mutations in eS19 have been identified, which are linked to the accumulation of a 21S preribosomal RNA precursor species (37, 38). In the human SSU processome, we have identified AROS (also known as RPS19BP1) as a chaperone that specifically mediates the integration of eS19 in state post-A1 (Fig. 6, A, B, and D, and fig. S22). The significance of AROS in the post-A1 state is further highlighted by previous biochemical data showing that small interfering RNA–mediated knockdown of AROS phenocopies the loss of eS19 (39) and that the AROS C-terminal domain is required for eS19 binding (40). The binding site for the AROS N terminus is created in state post-A1 upon the conformational changes in UTP20 during the transition from pre-A1 to post-A1, which are communicated through HEATR1 near U3 snoRNA. These changes result in the creation of a composite binding site between HEATR1 and U3, allowing AROS to thread along a similar trajectory as the unrelated protein Rrt14 in yeast (Fig. 6B, fig. S22, and movie S6). As eS19 is recognized by a conserved small C-terminal helix of AROS, any mutations within eS19 likely abrogate this interaction and thus the incorporation of eS19 into the SSU processome (Fig. 6, B and D). The structure of the human SSU processome further provides physiological context for the effects of proximal mutations found in uS12 and BMS1, which give rise to different pathologies (Fig. 6C) and illustrates how mutations found in Diamond-Blackfan anemia [ribosomal protein S7 (RPS7), RPS19, RPS24, RPS27, and RPS28], 5q- syndrome (RPS14), RPS23-related ribosomopathies, aplasia cutis congenita (BMS1), Bowen-Conradi syndrome (NEP1), and North American Indian childhood cirrhosis (UTP4) (36, 41-45) may affect SSU processome formation and maturation (Fig. 6).
Fig. 6. Human diseases of the SSU processome.

(A) SSU processome factors with disease-causing mutations are highlighted within state post-A1. MC, microcephaly; BWCNS, Bowen-Conradi syndrome; NAIC, North American Indian childhood cirrhosis; 5q-, 5q- syndrome; DBA, Diamond-Blackfan anemia; ACC, aplasia cutis congenita. (B) Detailed view of AROS (yellow, transparent surface) and its interacting proteins. DBA1-causing mutations in eS19 are colored red. (C) Detailed view of BMS1 (orange), uS12 (gray), and neighboring proteins. Disease-causing mutations in BMS1 (ACC) and uS12 (MC) are labeled and shown as red spheres. (D) A close-up view of the interface between eS19 (gray) and AROS (yellow, transparent surface). DBA1-causing mutations in eS19 are labeled and shown as red sticks.
Discussion
The recognition and controlled processing of large preribosomal RNA segments are the key functions of the human SSU processome. Here we provide high-resolution insights into the nucleolar maturation of human SSU processomes. These structural insights, together with complementary functional data, elucidate the mechanisms by which 50 ribosome assembly factors work in concert to precisely tune a strict chronology of assembly events in the nucleolus.
A key principle that emerges from this study is how changes in RNA folding are transmitted through conformational changes in assembly factors and how these conformational changes are subsequently recognized by downstream factors involved in ensuing pre-rRNA maturation steps. Two prominent examples of this principle are the conformation-specific recognition of human SSU processomes by the RNA exosome (Fig. 3) and the complex interplay of different placeholder proteins that precisely orchestrate localization and activation of the DEAH-box helicase DHX37 (Fig. 4). These concepts are broadly applicable to large assemblies such as maturing preribosomal particles or assembling spliceosomes.
Eukaryotic ribosome assembly is a very tightly controlled process, and our discovery that the exosome component EXOSC10 (Rrp6) lasso directly recognizes a particular state of the SSU processome highlights redundant pathways that intimately link the exosome with one of its key substrates, the SSU processome. At least two redundant links exist between the exosome and the SSU processome: First, the exosome-associated helicase Mtr4 can interact with SSU processome factors containing AIM motifs [Utp18 in yeast (29) and presumably TDIF2 in humans]. Second, the lasso of EXOSC10 (Rrp6) can directly bind to SSU processomes that have already lost NGDN (Lcp5) (Fig. 3). Whereas a loss of either of these links should be tolerated, the removal of both links should be deleterious, and this is indeed observed in yeast where Utp18 AIM mutants are synthetically lethal with an Rrp6 deletion (29).
On the basis of our data, a model emerges in which biomolecular condensate formation within the nucleolus enables human SSU processome maturation by concentrating ribosome assembly factors (Fig. 7A). The recognition of the SSU processome for exosome-mediated maturation is triggered by the removal of NGDN (Lcp5) (Fig. 7, B to D). With dual connections between the exosome and the SSU processome, the exosome uses its 3′-5′ exonuclease activity to remove the 5′ ETS from the 3′ end. With this trajectory, it is likely that the exosome triggers the major rearrangements that we observe in the SSU processome core. The exosome-mediated removal of the 5′ ETS RNA within the interior of the SSU processome will further drive drastic conformational changes of those assembly factors that are in direct contact with the 5′ ETS in state pre-A1 (WDR46, PWP2, UTP24, IMP4, BMS1, and NOP14, among others). These conformational changes include the assembly factors involved in the protein-guided cleavage at site A1 within the core of the SSU processome as well as conformational changes within the periphery of the SSU processome that contribute to the control of the helicase DHX37 before it engages U3 snoRNA during subsequent maturation steps.
Fig. 7. Model for nucleolar maturation of the human ribosomal small subunit.

(A) Illustration depicting the assembly of human SSU processomes within the nucleolus, with fibrillar center (FC, red), dense fibrillar component (DFC, yellow), and granular component (GC, blue). The expanded human 5′ ETS (yellow) is shown interacting with IDRs of nucleolar proteins. Processing of pre-18S rRNA progresses via different states of the SSU processome (black box). (B to D) Detailed views of three states indicating the chronology of events with components present in states pre-A1 (B), pre-A1* (C), and post-A1 (D). Components leaving and joining the particles are indicated, and inhibitory mechanisms are highlighted.
Our functional studies illuminate the evolution of eukaryotic ribosome assembly and the expansion of the 5′ ETS sequences in higher eukaryotes, which may explain the observed changes in morphology of the nucleolus toward a tripartite architecture in humans (fig. S13). As mature human small ribosomal subunits can be assembled from a minimal pre-rRNA with a greatly reduced 5′ ETS (Fig. 2 and fig. S14), these data suggest that there are two distinct functions of the human 5′ ETS: First, a minimal 5′ ETS is sufficient for ribosome assembly in cis, as a human SSU processome can form on this pre-rRNA and mature 40S ribosomes are generated (Fig. 2F). Second, the large extensions found in human 5′ ETS sequences likely drive ribosome assembly in trans by forming a biomolecular condensate with intrinsically disordered regions (IDRs) of highly concentrated ribosome assembly factors, the dense fibrillar component (DFC) (Fig. 7A). Therefore, the targeted degradation of the 5′ ETS by the RNA exosome eliminates large interaction partners within the DFC, which could liberate late human SSU processomes from the biomolecular condensate that they initiate.
Lastly, the structures of maturing human SSU processomes further provide physiological context for a range of disease-causing mutations in ribosomal proteins and assembly factors. The observation that AROS is directly involved in the recruitment of eS19 into the SSU processome suggests that, in the future, mutations within AROS may be found that give rise to a similar pathology as mutations within eS19 that are currently observed in patients suffering from Diamond-Blackfan anemia.
Materials and methods
Generation of a biallelically tagged NOC4L-GFP cell line
For the swift generation of a biallelic tagged NOC4L-GFP cell line, we developed an iterative genome editing platform called SNEAK PEEC (surface engineered fluorescence assisted kit with protein epitope enhanced capture), which combines CRISPR-Cas9 genome editing with cell-surface display. SNEAK PEEC links each precisely targeted knock-in with the expression of a distinct exogenous protein epitope on the cell surface, which allows clonal identification using fluorescent binders of each epitope. The system uses two DNA repair templates, each encoding a 3C protease cleavable green fluorescent protein (GFP) tag followed by a surface display sequence encoding a distinct protein epitope. For this study, the btuF vitamin B12 binding protein in Escherichia coli [Protein Data Bank (PDB) ID 5OVW] and capsid protein p24 from human immunodeficiency virus 1 (PDB ID 5O2U) were used as the surface display epitopes.
Both DNA repair template plasmids contained homology arms (left and right) that covered ~600 base pairs (bp) in either direction of the Cas9 cut site in the last exon of the NOC4L gene. Homology arms flanking a multiple cloning site (MCS) were first cloned into pUC57 vectors (GenScript, Piscataway, NJ). The entire knock-in sequence (3C-GFP-2A-display) was then introduced using NotI/PacI sites in the MCS. A plasmid expressing a single guide RNA (sgRNA) together with a high specificity Streptococcus pyogenes Cas9 variant [eSpCas9(1.1)] was a gift from F. Zhang (46) (Addgene plasmid # 71814). The 20-bp sgRNA target sequence (5′-AAACTCCAGTGGCACCG-GCT-3′, PAM: CGG) was cloned into this vector using a pair of BbsI sites. The sgRNA sequences were selected using an online resource, crispor.telfor.net (47).
Transfections were carried out on HEK 293-F cells (ThermoFisher Scientific, R79007) grown in 24-well plates (Falcon, 353047). Each well received 1 μg of total DNA, split in equimolar concentrations of each plasmid (Repair template 1, Repair template 2, Cas9 +sgRNA). All transfections were carried out in 500 μl of Opti-MEM medium/well (ThermoFisher Scientific, 31985070) using the commercial transfection reagent Lipofectamine 2000 (ThermoFisher Scientific, 11668019). Cells were transfected at 70 to 90% confluence, and 14 to 16 hours after transfection, cells were washed and resuspended in Freestyle 293 Expression Medium (ThermoFisher Scientific, 12338026) supplemented with 2% heat inactivated fetal bovine serum. Cells were allowed to recover, after which they were expanded to six-well plates (VWR, 10062-892) for at least 7 days.
For flow cytometry, cells were harvested from six-well plates alter a minimum of 7 days after transfection. Cells were first detached by gentle aspiration and washed 1× with 1X phosphate-buffered saline (PBS), 0.1% bovine serum albumin (BSA). They were then resuspended in 1X PBS plus 0.1% BSA at a concentration of 1 × 106 to 10 × 106 cells/ml. For cell surface staining, two fluorescently labeled nanobodies, each of which specifically binds to one of the surface display epitopes, were added to 200-μl cell suspensions at a final concentration of 10 nM. Cells were labeled on ice in the dark for 30 min, washed two times with 1X PBS plus 0.1% BSA and resuspended in 200 μl of the same buffer. Samples were filtered before cell sorting to remove clumps (BD Falcon, 352235). DAPI (4′,6-diamidino-2-phenyl-indole) was used to stain dead cells. For the identification of a biallelically tagged NOC4L-GFP cell line, sorting was used to first select cells expressing GFP, followed by selection of cells within this population that stained positive for both btuF and p24 surface display epitopes. Cell sorting was carried out using a BD FACSAria cell sorter (BD Biosciences) using FACSDiva Software (BD Biosciences). All necessary compensation was performed using single color controls before measurement and analysis of the data. FlowJo (FlowJo, LLC) was used for analysis of the data.
Sorted single cell clones were expanded for 2 weeks, after which they were screened via polymerase chain reaction (PCR) to confirm biallelic editing. Extraction of human genomic DNA was carried out using the QuickExtract DNA Extraction Solution (Lucigen, QE09050), per the manufacturer’s protocol. Thirty microliters of extracted solution was used per screening PCR reaction (50 μl final volume). The following PCR primers were used in this study (fig. S1H): PCR1, fwd (CCTTTCATCTGTAACC-TGCTGCGCC) and rev (GACTCCACGGGGCCA-ACTGTCTCAAGG); PCR2, fwd (CCTTTCATCT-GTAACCTGCTGCGCC) and rev (TGCTGTCAT-CATTTCCTCGAGCGTAGCACC).
Purification of human SSU processomes
Purification of the human SSU processome was carried out by means of sequential lysis. NOC4L-GFP cells were grown as suspension cultures, reaching a cell concentration between 4 × 106 and 5 × 106 cells/ml. Cell pellets were washed twice with cold 1X PBS and frozen in liquid nitrogen for storage at −80°C until use. Frozen cell pellets were resuspended in basic buffer [25 mM Hepes pH 7.6, 65 mM NaCl, 65 mM KCl, 10 mM MgCl2, 1 mM EDTA, 10% glycerol, 0.05% Triton, 1 mM dithiothreitol (DTT), 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM pepstatin, 1 mM E-64 protease inhibitor] and the resulting lysate centrifuged, and the supernatant (cytoplasmic fraction) discarded. Pellets were washed with basic buffer, the supernatants discarded, and the pellet was resuspended in active buffer [25 mM Hepes pH 7.6, 10 mM KCl, 10 mM MgCl2, 1 mM CaCl2, 1 mM EDTA, 1 mM spermidine, 5% glycerol, 0.1% Triton, 100 mM arginine, 25 mM adenosine triphosphate, 1 mM DTT, 0.5 mM PMSF, 1 mM pepstatin, 1 mM E-64 protease inhibitor, 800U RNase-free Dnasel (Worthington)]. The insoluble fraction was removed by centrifugation at 4°C, 40,000g for 20 min. The supernatant was incubated with anti-GFP nanobody beads (Chromotek) for 4 hours at 4°C, and the beads were washed 2× with wash buffer 1 (25 mM Hepes pH 7.6, 75 mM NaCl, 75 mM KCl, 5 mM MgCl2, 0.5 mM EDTA, 2.5% glycerol, 0.1% Triton, 100 mM arginine, 1 mM DTT, 0.5 mM PMSF, 1 mM pepstatin, 1 mM E-64). This was followed by 1x wash with wash buffer 2 [25 mM Hepes pH 7.6,75 mM NaCl, 75 mM KCl, 5 mM MgCl2, 0.5 mM EDTA, 2.5% glycerol, 0.05% Q2E8 (Anatrace, O330), 100 mM arginine]. The complex was eluted using 3C-protease cleavage, the integrity of particles confirmed using negative stain EM, and immediately followed by preparation of cryo-EM grids.
Cryo-EM grid preparation and data acquisition
Cryo-EM grids were prepared using a Vitrobot Mark IV robot (FEI Company) maintained at 12°C and 90% humidity. Three and a half microliters of the sample was applied to glow discharged Quantifoil R 2/2 grids coated with a layer of 2-nm ultrathin carbon (Au 400 2nm C, Electron Microscopy Sciences). After 70-s incubation, the sample was manually blotted while still in the Vitrobot chamber, and 3.5 μl of fresh sample was reapplied to the same grid. After 70-s incubation, the sample was blotted for 8 s using a blot force of 8, followed by plunging into liquid ethane.
Imaging was carried out on a Titan Krios electron microscope (FEI) equipped with an energy filter (slit width: 20 eV) and a K3 Summit detector (Gatan) operating at 300 kV with a nominal magnification of 64,000×. Using SerialEM (48), six datasets totaling 84,904 micrographs were collected with a defocus range of −0.7 to −2.7 mm and a super-resolution pixel size of 0.54 Å. Images with 45 subframes were collected using a total dose of 30.1 electrons per pixel per second (1.08 Å pixel size at the specimen) with an exposure time of 2.25 s and a total dose of 58 e-/Å2. A multishot imaging strategy was used to record seven micrographs per hole. The beam tilt was restricted to a single hole.
Cryo-EM data processing
Upon data collection, images were gain corrected, dose weighted, aligned, and binned to a pixel size of 1.08 Å using RELION’s implementation of a MotionCor2-like algorithm (49, 50). The defocus value of each micrograph was estimated using Gctf (51). A total of 9,297,626 particles were picked from six datasets comprising 84,904 micrographs using CrYOLO (52). The particles from each dataset were extracted at a pixel size of 4.32 Å and were separately subjected to a global search 3D classification in RELION 3.1 (53). An ab initio model generated in RELION 3.1 or a well-resolved 3D class from dataset 1 were used as initial references for 3D classification of dataset 1 and datasets 2 to 6, respectively (fig. S3). For each dataset, the particles from the best class were re-centered and reextracted at a pixel size of 1.08 Å and subjected to three rounds of CTF refinement and Bayesian polishing in RELION 3.1. The polished particles from each dataset were merged into a consensus particle stack of 824,089 particles. This particle stack was subjected to a 3D refinement resulting in a reconstruction of the human SSU processome at a global resolution of 2.68 Å. To separate different states present in the consensus reconstruction, three parallel 3D classifications were performed: one local search within eight classes with an angular search of 15°, and two classifications within 12 and 16 classes without alignment. A pre-A1 state containing the NAT10-NOL10 module and a post-A1 state containing DHX37 were discovered (fig. S3). Particles belonging to the pre-A1 or post-A1 state were merged separately and duplicate particles were removed. To further distinguish other possible states from the pre-A1 state, a focused classification on NGDN was performed and resulted in the separation of pre-A1 and pre-A1* states, characterized by the presence or absence of NGDN. States pre-A1, pre-A1*, and post-A1 were refined in RELION 3.1 to global resolutions of 3.51, 3.87, and 2.65 Å, respectively (fig. S3).
To facilitate accurate atomic model building of the entire human SSU processome, high-resolution and high-quality cryo-EM reconstructions of the different states were essential. Focused 3D classifications followed by focused 3D refinements on 15 different regions of the pre-A1 and post-A1 states were performed in RELION 3.1. The focused maps were further improved using phenix.resolve_cryo_em (54), which helped considerably with the model building (figs. S4 to S6). Ultimately, the 15 focused maps for each state were postprocessed in RELION 3.1 and combined into composite maps using phenix.combine_focused_maps (table S1) (55). The same procedure was used to generate composite half-maps that were used to calculate the final overall resolution of the composite pre-A1 (3.60 Å) and post-A1 (2.69 Å) maps. This strategy was not pursued for the pre-A1* state owing to its low number of particles.
All reported resolutions are based on the gold standard FSC-0.143 criterion (56), and FSC-curves were corrected using high-resolution noise substitution methods (57) in RELION 3.1. All reconstructions were corrected for the modulation transfer function of the detector and sharpened by applying a negative B-factor that was estimated using automated procedures in RELION 3.1 (53). Local resolution for the overall, focused, and composite maps was calculated using Blocres (58) within cryoSPARC v3 (59) (figs. S7 to S9).
Model building and refinement
A combination of homology modeling using Modeller (60), a few existing x-ray structures, artificial intelligence (AI)–based neural network methods (61), and de novo model building was used to build the entire human SSU processome. Model building was first done for the highest resolution map, the post-A1 state solved at 2.65 Å. The core of the map reaches local resolution up to 2.5 Å, and only a few parts have a resolution lower than 3.5 Å (figs. S8 and S9). Most of the human ribosomal protein atomic coordinates were extracted from the SARS-CoV-2-Nsp1-40S EM structure (PDB ID 6ZOJ), rigid-body docked in our EM density and manually adjusted in COOT (62). X-ray structures of some proteins contained within the human U3 snoRNP and of very few assembly factors were available and also docked into our EM map with manual adjustment in COOT (62). For the UtpA and UtpB subcomplexes and the rest of the assembly factors, homology models were generated with MODELLER (60) using templates from yeast SSU processome structures (PDB ID 5WLC or 6ZQD) or de novo using trRosetta (63). These homology models, together with AI-based models generated with Deep Tracer (61), served as a starting point for manual adjustments and manual building in COOT (62). The post-A1 final model was then used to build the pre-A1 and pre-A1* models. Proteins specific to the post-A1 were removed, and assembly factors specific to the pre-A1 and pre-A1* states were added and manually curated with the same procedure as for the post-A1 state. Conformational changes between the different states of many assembly factors were also observed and manually adjusted in COOT (62).
A complete list of templates and crystal structures used to build the final pre-A1, pre-A1*, and post-A1 models can be found in tables S2 and S3. The final models for the three states were real-space refined using phenix.real_space_refine (64) with secondary structure restraints for proteins and RNAs. The model refinement statistics can be found in table S4. The maps and models were visualized in either Chimera (65), ChimeraX (66), or PyMOL (67). The figures were generated using ChimeraX (66).
Expression and purification of yeast Dhr1 for crystallization
Crystallization of both human DHX37 and yeast Dhr1 was attempted, but only yeast Dhr1 yielded crystals. Yeast Dhr1 (residues 379 to 1185) was PCR amplified from Saccharomyces cerevisiae genomic DNA and cloned into an RSF-Duet1 vector containing an N-terminal His-14 SUMO tag (H14-SUMO) using BglII and XhoI restriction sites. This vector was transformed into RIL cells for expression. Large-scale growth was done in 2x Yeast-Tryptone (2xYT) medium at 37°C until an optical density (OD) of 1. At this point, expression was induced by the addition of 1 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG), and growth was continued at 20°C. Twenty hours after induction, cells were harvested by centrifugation at 8000g and resuspended in buffer A (20 mM Tris/HCl pH 7.6, 500 mM NaCl, 0.1% Triton X-100) supplemented with protease inhibitors (E64, pepstatin, and PMSF) and DNase/RNase. Cells were lysed at 4°C by passing the cell suspension once through a Constant Systems cell disruptor at 30 kPSI. Lysates were centrifuged at 40,000g for 45 min, and clarified lysates were applied to a His-Trap HP column (GE Lifesciences). The column was washed with wash buffer (20 mM Tris/HCl pH 7.6,30 mM imidazole, 500 mM NaCl) and then cleaved on-column with SUMO protease buffer (20 mM Tris/HCl pH 7.6, 500 mM NaCl, 15 μg/ml SUMO protease). Elutions were diluted to 125 mM NaCl with 20 mM Tris/HCl pH 7.6 and then loaded onto a HiTrap Heparin HP column (GE Lifesciences). The Heparin column was washed with two column volumes of 20 mM Tris/HCl pH 7.6 and 100 mM NaCl before being eluted with a salt gradient from 20 mM Tris/HCl pH 7.6, 100 mM NaCl to 20 mM Tris/HCl pH 7.6, 1 M NaCl. Fractions containing Dhr1 were pooled and concentrated before size exclusion chromatography on a HiLoad 16/600 Superdex 200 equilibrated with 20 mM Tris/HCl pH 7.6, 250 mM NaCl. Size exclusion fractions were concentrated to 8 mg/ml using a 30-kDa molecular weight cut-off Macrosep Advance centrifugal device (Pall Corporation).
Crystallization of yeast Dhr1 (residues 379 to 1185)
Purified yeast Dhr1 (residues 379 to 1185) at 8 mg/ml was supplemented with 1 mM adenosine 5′-diphosphate monopotassium salt and 1 mM magnesium chloride. Crystals were grown using the sitting drop vapor-diffusion method, where 1 μl of Dhr1 was mixed with 2 μl well solution containing 10.5% PEG 6000 and 0.1 M HEPES pH 7.0 at 4°C. Crystals were cryoprotected with a mix of 75% well solution and 25% PEG 400 before flash freezing in liquid nitrogen.
X-ray data collection and structure determination
X-ray data was collected from beam line 24-ID-E of the Advanced Photon Source and Argonne National Laboratories (Chicago, IL) at a wavelength of 0.97919 Å. The Dhr1 crystal was solved using molecular replacement with Prp43 subdomains (PDB ID 2XAU) as search models using PHENIX (68).
Model building and refinement
Model building was performed with COOT (69), and the structure was refined using PHENIX (68). Model statistics for the Dhr1 core are provided in table S5.
Recombinant human rDNA engineering
A full copy of the human rDNA sequence present on a BAC plasmid (clone JH4) (70) was extracted and cloned into an in-house modified pUC57 plasmid. The copy includes the Pol I promoter, 5′ ETS, 18S, ITS1, 5.8S, ITS2, 28S, 3′ ETS, and a shorter terminator comprising only five termination signals. By subcloning fusion-PCR amplicons into the rDNA, we were able to introduce distinct sequences into the expansion segments of the small (ES3S) and large subunits (ES15L). These sequences are not recognized within the human transcriptome and can be used to monitor the production of recombinant ribosomes in human cells transfected with the rDNA plasmid by Northern blot. To assess the role and requirement of different regions of the human 5′ ETS in the maturation of the small ribosomal subunit, we performed structure-based truncations into specific regions of the 5′ ETS that are not immediately stabilized within the SSU processome (Fig. 2). We first removed the 5′ region from the Pol I promoter to the A′ site, just before the ECM (Δ1: 39 to 422). Then, the region between the two base-paring sites (U3 3′ and 5′ hinges) was removed (Δ2: 893 to 1382). Finally, the remaining the large helical segment between sites A0 and A1 was removed (Δ3: 1722 to 3546). We also engineered combinations of truncations (Δ1-Δ2, Δ2-Δ3, and Δ1-Δ2-Δ3). All the plasmids were fully sequenced, and no mutations were detected.
Cell transfection for functional studies
Three micrograms of each plasmid harboring the different truncations, as well as the wild-type rDNA plasmid, were reverse transfected into human HEK 293-F cells using Lipofectamine 3000 reagent (1 M cells per transfection per well in six-well plates). The cells were incubated at 37°C, 8% CO2. After 48 hours, the cells were washed once with cold PBS, gently detached from the plate with cold PBS, and then pelleted at 100 g for 5 min. The pellets were flash frozen into liquid nitrogen and stored at −80°C until usage.
RNA extraction and Northern blot analysis
The RNA was extracted from the cells using Trizol/chloroform and isopropanol precipitation following standard procedures. Ten micrograms of total RNA from each sample was resolved on a 1.2% denaturing agarose gel. After complete migration, the RNA was transferred to a positively charged nylon membrane and fixed by ultraviolet cross-linking. The following 5′-32P-labeled oligonucleotide probes were used for Northern blot analysis: ITS1 probe (5′-GTGAGCACGACGTCACCACATCGATCGAAG-ATC-3′), 18S probe (5′- CGAGGATCGAGGC-TTT -3′), and 28S probe (5′-AGGTACACTCGA-GAGCTTCA-3′). The Millenium RNA marker (ThermoFisher Scientific) was used as a molecular weight ladder.
Sequence alignments and secondary structure prediction
Sequence alignments were performed using Clustal Omega (71) and analyzed using Consurf (72). Secondary structure predictions were performed using PSIPRED (73).
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Ebrahim, J. Sotiris, and H. Ng for help with grid screening and data collection at the Evelyn Gruss Lipper Cryo-Electron Microscopy Resource Center at The Rockefeller University. We thank V. L. Larionov at the Center for Cancer Research (NCI-NIH) for sharing the BAC containing the human rDNA sequence. Molecular graphics and analyses were performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases. We thank members of the Klinge laboratory for critical reading of this manuscript.
Funding:
A.V.B. is supported by an EMBO long-term fellowship (ALTF 711-2019). S.K. is supported by a NIH New Innovator Award (1DP2GM123459).
Footnotes
Competing interests: The Rockefeller University has filed a patent related to the described human genome editing platform, on which S.S. and S.K. are inventors.
Data and materials availability: The raw unaligned multiframe movies and aligned micrographs have been deposited in the Electron Microscopy Public Image Archive (EMPIAR-10781).The cryo-EM maps and atomic models have been deposited in the Electron Microscopy Data Bank (EMDB) and the Protein Data Bank (PDB): state pre-A1 (EMD-23936, EMD-23939, and PDB ID 7MQ8), state pre-A1* (EMD-23937 and PDB ID 7MQ9), and state post-A1 (EMD-23938, EMD-23940, and PDB ID 7MQA). All focused refinement maps have also been deposited in the EMDB (table S1). The atomic coordinates for the yeast Dhr1 core have been deposited to the Protein Data Bank under ID 7MQJ. Materials in the main text or the supplement are available from S.K. upon reasonable request.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Bohnsack KE, Bohnsack MT, Uncovering the assembly pathway of human ribosomes and its emerging links to disease. EMBO J. 38, e100278 (2019). doi: 10.15252/embj.2018100278; pmid: 31268599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lafontaine DLJ, Riback JA, Bascetin R, Brangwynne CP, The nucleolus as a multiphase liquid condensate. Nat. Rev. Mol. Cell Biol 22, 165–182 (2021). doi: 10.1038/s41580-020-0272-6; pmid: 32873929 [DOI] [PubMed] [Google Scholar]
- 3.Michot B, Bachellerie JP, Secondary structure of the 5′ external transcribed spacer of vertebrate pre-rRNA. Presence of phylogenetically conserved features. Eur. J. Biochem 195, 601–609 (1991). doi: 10.1111/j.1432-1033.1991.tb15743.x; pmid: 1999184 [DOI] [PubMed] [Google Scholar]
- 4.Hunziker M et al. , Conformational switches control early maturation of the eukaryotic small ribosomal subunit. eLife 8, e45185 (2019). doi: 10.7554/eLife.45185; pmid: 31206356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yao R-W et al. , Nascent pre-rRNA sorting via phase separation drives the assembly of dense fibrillar components in the human nucleolus. Mol. Cell 76, 767–783.e11 (2019). doi: 10.1016/j.molcel.2019.08.014; pmid: 31540874 [DOI] [PubMed] [Google Scholar]
- 6.Dragon F et al. , A large nucleolar U3 ribonucleoprotein required for 18S ribosomal RNA biogenesis. Nature 417, 967–970 (2002). doi: 10.1038/nature00769; pmid: 12068309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaker-Margot M, Hunziker M, Barandun J, Dill BD, Klinge S, Stage-specific assembly events of the 6-MDa small-subunit processome initiate eukaryotic ribosome biogenesis. Nat. Struct. Mol. Biol 22, 920–923 (2015). doi: 10.1038/nsmb.3111; pmid: 26479197 [DOI] [PubMed] [Google Scholar]
- 8.Zhang L, Wu C, Cai G, Chen S, Ye K, Stepwise and dynamic assembly of the earliest precursors of small ribosomal subunits in yeast. Genes Dev. 30, 718–732 (2016). doi: 10.1101/gad.274688.115; pmid: 26980190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kornprobst M et al. , Architecture of the 90S pre-ribosome: A structural view on the birth of the eukaryotic ribosome. Cell 166, 380–393 (2016). doi: 10.1016/j.cell.2016.06.014; pmid: 27419870 [DOI] [PubMed] [Google Scholar]
- 10.Chaker-Margot M, Barandun J, Hunziker M, Klinge S, Architecture of the yeast small subunit processome. Science 355, eaal1880 (2017). doi: 10.1126/science.aal1880; pmid: 27980088 [DOI] [PubMed] [Google Scholar]
- 11.Barandun J et al. , The complete structure of the small-subunit processome. Nat. Struct. Mol. Biol 24, 944–953 (2017). doi: 10.1038/nsmb.3472; pmid: 28945246 [DOI] [PubMed] [Google Scholar]
- 12.Cheng J, Kellner N, Berninghausen O, Hurt E, Beckmann R, 3.2-Å-resolution structure of the 90S preribosome before A1 pre-rRNA cleavage. Nat. Struct. Mol. Biol 24, 954–964 (2017). doi: 10.1038/nsmb.3476; pmid: 28967883 [DOI] [PubMed] [Google Scholar]
- 13.Cheng J et al. , 90S pre-ribosome transformation into the primordial 40S subunit. Science 369, 1470–1476 (2020). doi: 10.1126/science.abb4119; pmid: 32943521 [DOI] [PubMed] [Google Scholar]
- 14.Du Y et al. , Cryo-EM structure of 90S small ribosomal subunit precursors in transition states. Science 369, 1477–1481 (2020). doi: 10.1126/science.aba9690; pmid: 32943522 [DOI] [PubMed] [Google Scholar]
- 15.Lau B et al. , Structure of the maturing 90S pre-ribosome in association with the RNA exosome. Mol. Cell 81, 293–303. e4 (2021). doi: 10.1016/j.molcel.2020.11.009; pmid: 33326748 [DOI] [PubMed] [Google Scholar]
- 16.Ameismeier M, Cheng J, Berninghausen O, Beckmann R, Visualizing late states of human 40S ribosomal subunit maturation. Nature 558, 249–253 (2018). doi: 10.1038/s41586-018-0193-0; pmid: 29875412 [DOI] [PubMed] [Google Scholar]
- 17.Ameismeier M et al. , Structural basis for the final steps of human 40S ribosome maturation. Nature 587, 683–687 (2020). doi: 10.1038/s41586-020-2929-x; pmid: [DOI] [PubMed] [Google Scholar]
- 18.Tafforeau L et al. , The complexity of human ribosome biogenesis revealed by systematic nucleolar screening of Pre-rRNA processing factors. Mol. Cell 51, 539–551 (2013). doi: 10.1016/j.molcel.2013.08.011; pmid: 23973377 [DOI] [PubMed] [Google Scholar]
- 19.Badertscher L et al. , Genome-wide RNAi screening identifies protein modules required for 40S subunit synthesis in human cells. Cell Rep. 13, 2879–2891 (2015). doi: 10.1016/j.celrep.2015.11.061; pmid: 26711351 [DOI] [PubMed] [Google Scholar]
- 20.Farley-Barnes KI et al. , Diverse regulators of human ribosome biogenesis discovered by changes in nucleolar number. Cell Rep. 22, 1923–1934 (2018). doi: 10.1016/j.celrep.2018.01.056; pmid: 29444442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sondalle SB, Baserga SJ, Human diseases of the SSU processome. Biochim. Biophys. Acta 1842, 758–764 (2014). doi: 10.1016/j.bbadis.2013.11.004; pmid: 24240090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Natchiar SK, Myasnikov AG, Kratzat H, Hazemann I, Klaholz BP, Visualization of chemical modifications in the human 80S ribosome structure. Nature 551, 472–477 (2017). doi: 10.1038/nature24482; pmid: 29143818 [DOI] [PubMed] [Google Scholar]
- 23.Taoka M et al. , Landscape of the complete RNA chemical modifications in the human 80S ribosome. Nucleic Acids Res. 46, 9289–9298 (2018). doi: 10.1093/nar/gky811 pmid: 30202881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng J et al. , Thermophile 90S pre-ribosome structures reveal the reverse order of co-transcriptional 18S rRNA subdomain integration. Mol. Cell 75, 1256–1269.e7 (2019). doi: 10.1016/j.molcel.2019.06.032; pmid: 31378463 [DOI] [PubMed] [Google Scholar]
- 25.Kass S, Craig N, Sollner-Webb B, Primary processing of mammalian rRNA involves two adjacent cleavages and is not species specific. Mol. Cell. Biol 7, 2891–2898 (1987). doi: 10.1128/MCB.7.8.2891; pmid: 3670298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watkins NJ, Bohnsack MT, The box C/D and H/ACA snoRNPs: Key players in the modification, processing and the dynamic folding of ribosomal RNA. WIREs RNA 3, 397–414 (2012). doi: 10.1002/wrna.117; pmid: 22065625 [DOI] [PubMed] [Google Scholar]
- 27.Sun Q et al. , Molecular architecture of the 90S small subunit pre-ribosome. eLife 6, e22086 (2017). doi: 10.7554/eLife.22086; pmid: 28244370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wasmuth EV, Lima CD, The Rrp6 C-terminal domain binds RNA and activates the nuclear RNA exosome. Nucleic Acids Res. 45, 846–860 (2017). doi: 10.1093/nar/gkw1152; pmid: 27899565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thoms M et al. , The exosome is recruited to RNA substrates through specific adaptor proteins. Cell 162, 1029–1038 (2015). doi: 10.1016/j.cell.2015.07.060; pmid: 26317469 [DOI] [PubMed] [Google Scholar]
- 30.Boneberg FM et al. , Molecular mechanism of the RNA helicase DHX37 and its activation by UTP14A in ribosome biogenesis. RNA 25, 685–701 (2019). doi: 10.1261/rna.069609.118; pmid: 30910870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roychowdhury A et al. , The DEAH-box RNA helicase Dhr1 contains a remarkable carboxyl terminal domain essential for small ribosomal subunit biogenesis. Nucleic Acids Res. 47, 7548–7563 (2019). doi: 10.1093/nar/gkz529; pmid: 31188444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu J, Liu X, Anjos M, Correll CC, Johnson AW, Utp14 recruits and activates the RNA helicase Dhr1 to undock U3 snoRNA from the preribosome. Mol. Cell. Biol 36, 965–978 (2016). doi: 10.1128/MCB.00773-15; pmid: 26729466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choudhury P, Hackert P, Memet I, Sloan KE, Bohnsack MT, The human RNA helicase DHX37 is required for release of the U3 snoRNP from pre-ribosomal particles. RNA Biol. 16, 54–68 (2019). doi: 10.1080/15476286.2018.1556149; pmid: 30582406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wells GR et al. , The PIN domain endonuclease Utp24 cleaves pre-ribosomal RNA at two coupled sites in yeast and humans. Nucleic Acids Res. 44, 5399–5409 (2016). doi: 10.1093/nar/gkw213; pmid: 27034467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farley-Barnes KI, Ogawa LM, Baserga SJ, Ribosomopathies: Old concepts, new controversies. Trends Genet. 35, 754–767 (2019). doi: 10.1016/j.tig.2019.07.004; pmid: 31376929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ulirsch JC et al. , The genetic landscape of Diamond-Blackfan anemia. Am. J. Hum. Genet 103, 930–947 (2018). doi: 10.1016/j.ajhg.2018.10.027; pmid: 30503522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Draptchinskaia N et al. , The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat. Genet 21, 169–175 (1999). doi: 10.1038/5951; pmid: 9988267 [DOI] [PubMed] [Google Scholar]
- 38.Flygare J et al. , Human RPS19, the gene mutated in Diamond-Blackfan anemia, encodes a ribosomal protein required for the maturation of 40S ribosomal subunits. Blood 109, 980–986 (2007). doi: 10.1182/blood-2006-07-038232; pmid: 16990592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Knight JRP, Willis AE, Milner J, Active regulator of SIRT1 is required for ribosome biogenesis and function. Nucleic Acids Res. 41, 4185–4197 (2013). doi: 10.1093/nar/gkt129; pmid: 23462953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maeda N, Toku S, Kenmochi N, Tanaka T, A novel nucleolar protein interacts with ribosomal protein S19. Biochem. Biophys. Res. Commun 339, 41–46 (2006). doi: 10.1016/j.bbrc.2005.10.184; pmid: 16289379 [DOI] [PubMed] [Google Scholar]
- 41.Ebert BL et al. , Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature 451, 335–339 (2008). doi: 10.1038/nature06494; pmid: 18202658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Paolini NA et al. , A ribosomopathy reveals decoding defective ribosomes driving human dysmorphism. Am. J. Hum. Genet 100, 506–522 (2017). doi: 10.1016/j.ajhg.2017.01.034; pmid: 28257692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marneros AG, BMS1 is mutated in aplasia cutis congenita. PLOS Genet. 9, e1003573 (2013). doi: 10.1371/journal.pgen.1003573; pmid: 23785305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Armistead J et al. , Mutation of a gene essential for ribosome biogenesis, EMG1, causes Bowen-Conradi syndrome. Am. J. Hum. Genet 84, 728–739 (2009). doi: 10.1016/j.ajhg.2009.04.017; pmid: 19463982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chagnon P et al. , A missense mutation (R565W) in Cirhin (FLJ14728) in North American Indian childhood cirrhosis. Am. J. Hum. Genet 71, 1443–1449 (2002). doi: 10.1086/344580; pmid: 12417987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Slaymaker IM et al. , Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88 (2016). doi: 10.1126/science.aad5227; pmid: 26628643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Concordet J-P, Haeussler M, CRISPOR: Intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46, W242–W245 (2018). doi: 10.1093/nar/gky354; pmid: 29762716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mastronarde DN, Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol 152, 36–51 (2005). doi: 10.1016/j.jsb.2005.07.007; pmid: 16182563 [DOI] [PubMed] [Google Scholar]
- 49.Zivanov J et al. , New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018). doi: 10.7554/eLife.42166; pmid: 30412051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zheng etal SQ, MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). doi: 10.1038/nmeth.4193; pmid: 28250466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang K, Gctf: Real-time CTF determination and correction. J. Struct. Biol 193, 1–12 (2016). doi: 10.1016/j.jsb.2015.11.003; pmid: 26592709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52..Wagner T et al. , SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol 2, 218 (2019). doi: 10.1038/s42003-019-0437-z; pmid: 31240256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zivanov J, Nakane T, Scheres SHW, Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 7, 253–267 (2020). doi: 10.1107/S2052252520000081; pmid: 32148853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Terwilliger TC, Ludtke SJ, Read RJ, Adams PD, Afonine PV, Improvement of cryo-EM maps by density modification. Nat. Methods 17, 923–927 (2020). doi: 10.1038/s41592-020-0914-9; pmid: 32807957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liebschner D et al. , Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. D 75, 861–877 (2019). doi: 10.1107/S2059798319011471; pmid: 31588918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosenthal PB, Henderson R, Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol 333, 721–745 (2003). doi: 10.1016/j.jmb.2003.07.013; pmid: 14568533 [DOI] [PubMed] [Google Scholar]
- 57.Chen S et al. , High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135, 24–35 (2013). doi: 10.1016/j.ultramic.2013.06.004; pmid: 23872039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heymann JB, Guidelines for using Bsoft for high resolution reconstruction and validation of biomolecular structures from electron micrographs. Protein Sci. 27, 159–171 (2018). doi: 10.1002/pro.3293; pmid: 28891250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA, cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). doi: 10.1038/nmeth.4169; pmid: 28165473 [DOI] [PubMed] [Google Scholar]
- 60.Webb B, Sali A, Protein structure modeling with MODELLER. Methods Mol. Biol 2199, 239–255 (2021). doi: 10.1007/978-1-0716-0892-0_14; pmid: 33125654 [DOI] [PubMed] [Google Scholar]
- 61.Pfab J, Phan NM, Si D, DeepTracer for fast de novo cryo-EM protein structure modeling and special studies on CoV-related complexes. Proc. Natl. Acad. Sci. U.S.A 118, e2017525118 (2021). doi: 10.1073/pnas.2017525118; pmid: 33361332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Casañal A, Lohkamp B, Emsley P, Current developments in Coot for macromolecular model building of Electron Cryomicroscopy and Crystallographic Data. Protein Sci. 29, 1069–1078 (2020). doi: 10.1002/pro.3791; pmid: 31730249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang J et al. , Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. U.S.A 117, 1496–1503 (2020). doi: 10.1073/pnas.1914677117; pmid: 31896580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Afonine PV, Poon BK, Read RJ, Sobolev OV, Terwilliger TC, Urzhumtsev A, Adams PD, Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Biol. Crystallogr 74, 531–544 (2018). doi: 10.1107/S2059798318006551; pmid: 29872004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pettersen EF et al. , UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612 (2004). doi: 10.1002/jcc.20084; pmid: 15264254 [DOI] [PubMed] [Google Scholar]
- 66.Pettersen EF et al. , UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82 (2021). doi: 10.1002/pro.3943; pmid: 32881101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.The PyMOL Molecular Graphics System, Version 2.0 (Schrödinger, LLC, 2017). [Google Scholar]
- 68.Adams PD et al. , Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH, PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221 (2010). doi: 10.1107/S0907444909052925; pmid: 20124702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emsley P, Cowtan K, Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132 (2004). doi: 10.1107/S0907444904019158; pmid: 15572765 [DOI] [PubMed] [Google Scholar]
- 70.Kim J-H et al. , Variation in human chromosome 21 ribosomal RNA genes characterized by TAR cloning and long-read sequencing. Nucleic Acids Res. 46, 6712–6725 (2018). doi: 10.1093/nar/gky442; pmid: 29788454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sievers F et al. , Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol 7, 539 (2011). doi: 10.1038/msb.2011.75; pmid: 21988835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ashkenazy H et al. , ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350 (2016). doi: 10.1093/nar/gkw408; pmid: 27166375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Buchan DWA, Jones DT, The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 47, W402–W407 (2019). doi: 10.1093/nar/gkz297; pmid: 31251384 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.