

Abstract
Heat shock protein 90 (Hsp90) is a molecular chaperone that facilitates the maturation of its client proteins including protein kinases, transcription factors, and steroid hormone receptors which are structurally and functionally diverse. These client proteins are involved in various cellular signaling pathways, and Hsp90 is implicated in various human diseases including cancer, inflammation, and diseases associated with protein misfolding; thus making Hsp90 a promising target for drug discovery. Some of its client proteins are well-known cancer targets. Instead of targeting these client proteins individually, however, targeting Hsp90 is more practical for cancer drug development. Efforts have been invested in recognizing potential drugs for clinical use that inhibit Hsp90 activity and result in the prevention of Hsp90 client maturation and dampening of subsequent signaling cascades. Here, we discuss current assays and technologies used to find and characterize Hsp90 inhibitors that include biophysical, biochemical, cell-based assays and computational modeling. This review highlights recent discoveries that N-terminal isoform-selective compounds and inhibitors that target the Hsp90 C-terminus that may offer the potential to overcome some of the detriments observed with pan Hsp90 inhibitors. The tools and assays summarized in this review should be used to develop Hsp90-targeting drugs with high specificity, potency, and druglike properties that may prove immensely useful in the clinic.
Keywords: Hsp90, Drug discovery, Drug targets, Human diseases, Molecular chaperone
1. Introduction
Molecular chaperones are proteins that are important for the folding of client protein substrates, as well as the degradation of misfolded proteins (Biebl & Buchner, 2019). Hsp90 (heat shock protein 90) is one of the most well-recognized members of this family and is an ATP-dependent molecular chaperone (Trepel, Mollapour, Giaccone, & Neckers, 2010). Hsp90 is also one of the most abundant proteins within the cell and binds ATP at the N-terminal region wherein it hydrolyzes ATP to provide the requisite energy to fold and release its client protein substrates (Biebl & Buchner, 2019). More than 400 Hsp90-dependent clients have been identified, many of which are involved in important biological functions such as signaling cascades, DNA damage repair, protein trafficking, hormone receptor activation, innate immunity, and many more (Taipale, Jarosz, & Lindquist, 2010; Trepel et al., 2010; Whitesell & Lindquist, 2005). The Hsp90 machinery controls client protein function by accelerating the client's conformational maturation, which allows for ligand binding and/or the formation of biologically active complexes (Schopf, Biebl, & Buchner, 2017).
Four isoforms of Hsp90 have been identified in humans: Hsp90α and Hsp90β are both cytosolic, whereas GRP94 (94 kDa glucose-related protein) is present in the endoplasmic reticulum and TRAP1 (tumor necrosis factor-associated protein 1) is localized to the mitochondria (Csermely, Schnaider, Soti, Prohászka, & Nardai, 1998; Sreedhar, Kalmár, Csermely, & Shen, 2004). Structurally, an Hsp90 monomer consists of three domains; the N-terminal ATPase domain (NTD), which is critical for ATP binding and hydrolysis; the middle domain, which is important for binding clients and the ɣ-phosphate of ATP; and the carboxy-terminal domain (CTD), which is important for Hsp90 dimerization (Harris, Shiau, & Agard, 2004; Prodromou et al., 1997) (Fig. 1A and B).
Fig. 1.

Approaches to identifying ligand binding to the Hsp90 based on structural information. (A) Cartoon representation of human Hsp90 homodimer structure with ATP bound NTD (Red), middle domain (Blue), and C-terminal domain (Green) (PDB:5FWM). (B) Cartoon representation of human Hsp90 monomer structure with ATP bound NTD (Red), middle domain (Blue), and C-terminal domain (Green) (PDB:5FWM).
The extreme C-terminus of Hsp90 consists of a Met-Glu-Glu-Val-Asp (MEEVD) motif that is important for binding co-chaperones that contain a tetratricopeptide repeat (TPR) (Prodromou, 2017). It has been shown that both the N- and C-termini contain ATP binding sites, however, only the NTD manifests ATPase activity (Söti, Rácz, & Csermely, 2002).
In the absence of ATP, Hsp90 remains in an open conformation, whereas ATP binding causes Hsp90 to undergo a conformational change that results in a closed conformation at the N-terminus (Ratzke, Nguyen, Mayer, & Hugel, 2012). This conformational change affects all three domains and brings the NTDs into close proximity. The CTD is critical for the dimerization of two monomers and it also contributes to NTD dimerization and ATPase activity (Prodromou et al., 2000). ATP hydrolysis at the N-terminus provides the necessary energy for the Hsp90-dependent protein folding process (Burlison, Neckers, Smith, Maxwell, & Blagg, 2006). The N-terminal ATP binding site must interact with the middle domain for ATP hydrolysis to occur (Schopf et al., 2017). In fact, Hsp90 activates and facilitates the proper folding of its client proteins through an ATPase cycle that has been well studied (Garg, Khandelwal, & Blagg, 2016) (Fig. 2A). In brief, client proteins are brought to the dimerized Hsp90 chaperone by Hsp70, Hsp40, and other factors to form an early, client-bound Hsp90 complex. Transfer of the client is mediated by the adapter protein, Hop/Sti1, which stabilizes the open, client-bound conformation of the complex. ATP binding to the Hsp90 NTD induces a transition to a more closed conformation, wherein the two Hsp90 N-termini are brought into close proximity. p23/Sba1 stabilizes this closed conformation of Hsp90 and promotes the dissociation of Hop/Sti1 from the complex. The energy derived from ATP hydrolysis provides the energy used for the proper folding, activation, and release of the client protein, which is followed by the disassociation of p23/Sba1 and allows for the return of Hsp90 to its open homodimeric conformation (Garg et al., 2016) (Fig. 2A). During this process, co-chaperones serve to regulate the Hsp90 protein folding machinery (Prodromou, 2017). Some co-chaperones bind the NTD and/or MD, such as CDC37 which is crucial for the stabilization of Hsp90- kinase complexes (Bickel & Gohlke, 2019). Other co-chaperones that contain a TPR domain, including HOP and p23, bind the C-terminus of Hsp90 (Nelson, Huffman, & Smith, 2003; Patwardhan et al., 2013; Wegele, Wandinger, Schmid, Reinstein, & Buchner, 2006). In general, co-chaperones without TPR domains bind the NTD and/or MD of Hsp90 (Mayer & Le Breton, 2015; Röhl, Rohrberg, & Buchner, 2013). A number of co-chaperone proteins, including protein phosphatase (Pp5) and Aha1, have been shown to accelerate the Hsp90 ATPase cycle, although these are not necessary for basal Hsp90 ATPase activity (Shelton, Koren, & Blair, 2017b). Hsp90 ATPase activity is vital for its function in vivo and any disruption halts the normal Hsp90-mediated protein folding process (Mishra & Bolon, 2014; Obermann, Sondermann, Russo, Pavletich, & Hartl, 1998; Panaretou et al., 1998).
Fig. 2.

Schematic representation showing the use of Hsp90 inhibitors and their role in human diseases. (A) Stepwise (I-VIII) protein folding mechanism by Hsp90 as described (Garg et al., 2016). (B) Hsp90 drug discovery process. Created with BioRender.com
Several Hsp90 inhibitors that bind the Hsp90 N-terminal ATP binding site have been identified, as they prevent Hsp90 from completing the protein folding cycle. These inhibitors destabilize the Hsp90 heteroprotein complex and prevent ATP binding and hydrolysis, which results in the degradation of client proteins via the ubiquitin-proteosome pathway (Garcia-Carbonero, Carnero, & Paz-Ares, 2013; Ou, Tan, Xie, Yu, & Tan, 2014) (Fig. 2A). As a result, Hsp90 inhibitors transform the protein folding machinery into a process in which Hsp90-dependent clients are degraded (Burlison et al., 2006). Hsp90 client proteins regulate critical cellular functions, such as cell division, cell migration, and cell death. Many of these Hsp90 client proteins become mutated and/or over-expressed during malignancy, further enhancing their dependency upon Hsp90. Some examples of Hsp90-dependent oncogenes include c-MET, the serine/threonine kinase Raf-1, the oncogenic tyrosine kinase v-Src, the mutated oncogene Bcr/Abl, receptor tyrosine kinase of the erbB family, as well as the steroid hormone receptors and numerous transcription factors such as hypoxia-inducible factor-1a and tumor suppressor p53 (Koga, Kihara, & Neckers, 2009; Tatokoro, Koga, Yoshida, & Kihara, 2015). As a result of Hsp90 inhibition, these client proteins are ubiquitinylated and targeted for degradation via the proteasome (Garg et al., 2016; Koga et al., 2009; Whitesell & Lindquist, 2005; Zuehlke & Johnson, 2010). In tumors, Hsp90 exists as a multi-chaperone complex and exhibits ~200 fold higher affinity for ATP than homodimeric Hsp90. In normal tissue, Hsp90 exists in an uncomplexed form that has a lower affinity for both ATP and competitive inhibitors, which results in a large therapeutic window for the development of new anti-cancer agents (Kamal et al., 2003). In addition, Hsp90 is overexpressed in various cancers, which further increases the opportunity to selectively inhibit tumor-derived Hsp90 (Chatterjee & Burns, 2017). Unfortunately, most of the Hsp90 inhibitors that underwent clinical evaluation for cancer have been terminated due to a lack of efficacy and/or detrimental side effects. Due to its suitability as a cancer target, researchers have sought to identify inhibitors of Hsp90 that do not exhibit adverse effects or induce resistance mechanisms. In addition, clinical trials with Hsp90 N-terminal pan inhibitors produced both off- and on-target Hsp90-related toxicities, and consequently, there is a need to develop inhibitors with a more acceptable therapeutic profile (Garcia-Carbonero et al., 2013) (Aherne et al., 2003) (Fig. 2B).
Hsp90 also plays a critical role in the modulation of the heat shock response (HSR), which is a pro-survival mechanism that enables a cell to respond to a wide range of cellular insults/stress. Under normal conditions, Hsp90 suppresses the transcriptional activity of heat shock factor 1 (HSF-1) by existing as an Hsp90-Hsf1 complex. However, in response to stress, Hsf-1 is disassociated from Hsp90, undergoes trimerization, and then translocates to the nucleus to initiate transcription of Hsp70, Hsp90, and other heat shock proteins that can alleviate cellular stress and refold denatured proteins (Prodromou, 2016). Disruption of the Hsp90 protein folding machinery by Hsp90 inhibitors leads to the dissociation of HSF-1 from Hsp90, resulting in induction of the pro-survival HSR. It has been hypothesized that HSR induction can accelerate the disaggregation of misfolded proteins and increase the protein level of various molecular chaperones, including Hsp70 and Hsp90. Consequently, HSR induction may be beneficial for the treatment of neurodegenerative disorders such as Alzheimer's and Parkinson's diseases (Luo, Sun, Taldone, Rodina, & Chiosis, 2010; Paul & Mahanta, 2014; Urban, Dobrowsky, & Blagg, 2012; Whitesell, Bagatell, & Falsey, 2003), as previous studies have demonstrated that Hsp90 inhibitors inhibited amyloid-beta (Aβ) aggregation and inhibited amyloid-beta (Aβ) formation (Luo & Le, 2010; Ou et al., 2014). Another report suggested that inhibition of Hsp90 significantly decreases disease-associated phosphorylation of tau species via proteasomal degradation (Koren et al., 2009). While Hsp90's role in HSR activation may be exploited to treat conditions such as neurodegeneration, the HSR represents a considerable obstacle to overcome while pharmacologically targeting Hsp90 to treat cancer. Specifically, pan Hsp90 inhibition also induces the HSR to upregulate Hsp90 along with other cellular proteins. As a result of the increased expression of Hsp90, higher doses of the inhibitor are required to achieve a therapeutic effect. However, this dose-escalation pushes the patient towards the MTD and elicits detrimental effects. In recent years, Hsp90 inhibitors that do not induce the HSR have been sought and discovered,
Hsp90 is not only a contributor to cancer and neurodegeneration, but it also plays a key role in other human diseases too. Recent reports suggested that Hsp90 can be inhibited to reduce ischemia/reperfusion injury, (Der Sarkissian et al., 2020) while others have shown that Hsp90 is utilized by parasites as a response to cellular stress (Rochani, Singh, & Tatu, 2013). Hsp90 plays a critical role in the growth of various pathogens including Candida, Giardia, Plasmodium, and Trypanosoma (Rochani et al., 2013); and as a result, Hsp90 has emerged as a target for the treatment of infectious diseases as well (Rochani et al., 2013). Due to its involvement in various diseases, there is a strong interest to identify small molecules that can inhibit specific Hsp90 functions. Some N-terminal Hsp90 inhibitors with different scaffolds are shown in Fig. 3A. Some of these reported N-terminal Hsp90 inhibitors have demonstrated clinical efficacy, but off-target effects and/or Hsp90- related toxicities exist for many (Garcia-Carbonero et al., 2013; Neckers & Workman, 2012; Scaltriti, Dawood, & Cortes, 2012).
Fig. 3.

Representative structures with different scaffolds of (A) N-terminal and (B) C-terminal inhibitors of Hsp90.
Due to toxicities associated with the drugs undergoing evaluation, medicinal chemistry campaigns have recently focused on the development of Hsp90β isoform-selective inhibitors as a method to increase efficacy, while reducing toxicity and on-target issues related to pan inhibition of all four isoforms (Khandelwal et al., 2018) (Fig. 4B). Parallel efforts have also been invested to find C-terminal inhibitors that do not induce the HSR. (Fig. 3B). Unfortunately, no co-crystal structures are available for the CTD of Hsp90 in complex with an inhibitor. However, based on multiple computational models, several C-terminal inhibitors have been designed and synthesized. One such C-terminal inhibitor, KU-596, manifests strong neuroprotective activities in preclinical studies and is currently undergoing clinical trials for the treatment of neuropathy (Neckers et al., 2018) (Fig. 3B). Compounds have also been discovered that disrupt interactions between Hsp90 and it's co-chaperones (Fig. 4A). In summary, Hsp90 inhibitors can regulate a wide variety of chaperone function (e.g. blocking ATP hydrolysis at the N-terminal domain, inhibiting the Hsp90 C-terminal dimerization process, disrupting the interaction between Hsp90 and co-chaperones) and consequently, can produce various downstream effects. However, there is no FDA approved Hsp90 inhibitor to treat cancer or any other human disease at present. Therefore, new methods that can be used to elucidate alternative mechanistic inhibitors are needed. In this review, a summary of assays and technologies that can be used to identify and characterize Hsp90 inhibitors for future development is outlined.
Fig. 4.
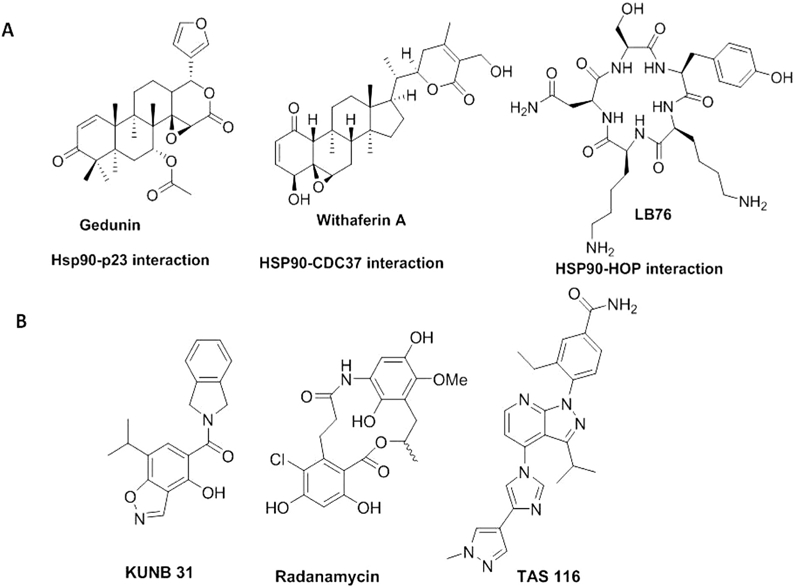
Representative structures of Hsp90-co-chaperone interaction disruptors (A) and Hsp90 isoform-selective inhibitors (B).
2. Assays and technologies to identify and characterize Hsp90 inhibitors
There are many approaches to identify and characterize Hsp90 inhibitors that focus on any of the three domains that can be targeted to disrupt chaperone function. However, most inhibitors discovered thus far inhibit the N-terminal ATPase domain, however, small molecules that target the C-terminal domain of Hsp90 have also been identified. Some of the C-terminal inhibitors disrupt Hsp90 function by preventing interactions between Hsp90 and its co-chaperones, which are critical for the maturation of most client proteins. However, inhibitors of the middle domain have been discovered that also inhibit Hsp90 ATPase activity, but do not disrupt Hsp90/co-chaperone interactions, and/or promote client protein degradation (Zhang, Ho, & Wong, 2018a). Most of the N-terminal inhibitors were discovered through biochemical assays or computational studies and later evaluated in cell based assays. Consequently, most of the assays that were developed for the identification of N-terminal inhibitors have been miniaturized into a high-throughput screening (HTS) format. A representative list of compounds that have been identified and/or characterized as Hsp90 inhibitors using these different assays is provided (Fig. 5). Since the majority of these assays exhibit both advantages and disadvantages, a combination is generally most useful to elucidate the mechanism of inhibition (Table 1). The following biochemical and biophysical assays have been developed to identify and characterize some of the well-known Hsp90-ligand interactions.
Fig. 5.
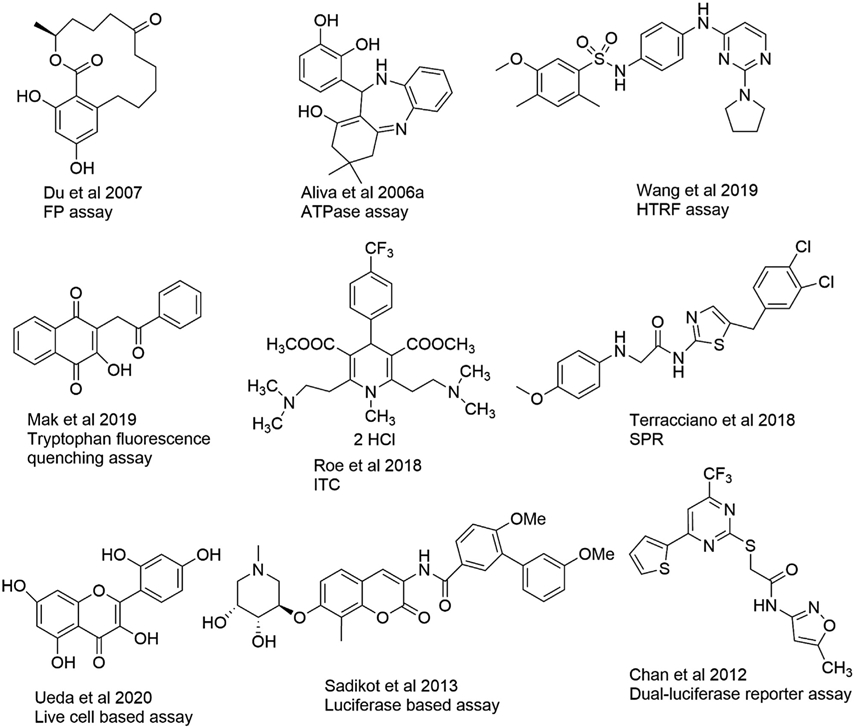
Representative structures of different Hsp90 inhibitors identified or characterized by the assays described.
Table 1.
Summary of different assays used to discover and/or characterize Hsp90 inhibitors.
| Assay | Description | Reference |
|---|---|---|
| FP | -HTS amenable -Useful for PAN inhibitor and/or isoform-selective inhibitor screening -Used for N-terminal Hsp90 inhibitor screening for compounds that bind at the ATP binding site |
(Du et al., 2007; Khandelwal et al., 2018; Kim et al., 2004; Lea & Simeonov, 2011; Wang et al., 2019) |
| ATPase assay | -HTS amenable -Used to screen and characterize inhibitors of Hsp90 ATPase activity |
(Avila, Hadden, et al., 2006a; Avila, Kornilayev, & Blagg, 2006b; Bartolini et al., 2013; Rowlands et al., 2004; Wang et al., 2019) |
| AlphaScreen | -HTS amenable -Homogeneous robust assays with a high signal-to-background ratio -Useful in screening small molecule C-terminal Hsp90 inhibitors |
(Rahimi et al., 2018; Rahimi & McAlpine, 2019; Yi et al., 2009) |
| TR-FRET assay | -HTS amenable Used to find disruptors of Hsp90 and Hsp90-Cdc37 interactions -Used for several SAR studies |
(Wang et al., 2019; Wang et al., 2020; Zhou et al., 2004) |
| Tryptophan fluorescence quenching | -Simple method to characterize protein-ligand interactions and calculate dissociation constants -Used to characterize Hsp90 ligands by calculation of dissociation constants |
(Huang et al., 2017; Mak et al., 2019) |
| NMR | -A well-known method to characterize protein-ligand interactions and screen fragment libraries. -Used to characterize Hsp90 and ligand interactions or screen fragment-based small molecules. -−2D HSQC NMR method is most popular to study protein-ligand interactions |
(Casale et al., 2014; Hagn et al., 2011; Raingeval et al., 2019; Roughley & Hubbard, 2011) |
| Structure-based drug design | -HTS in silico, inexpensive -Used to find novel Hsp90 ligands. -Crystal structures help researchers to rationally design Hsp90 inhibitors -Useful for isoform-selective inhibitor design (Roughley & Hubbard, 2011) |
(Dutta Gupta et al., 2015; Khandelwal et al., 2018; Lee et al., 2015; Stebbins et al., 1997) |
| ITC | -Label-free method useful for protein-ligand interactions and quantifying thermodynamic parameters -Used to study Hsp90 and ligand interactions -Used to characterize the nature of these interactions (e.g. binding affinity, entropic contributions, and enthalpic contributions) |
(Garnier et al., 2002; Murray et al., 2010; Roe et al., 2018) |
| SPR | -Useful for studying direct protein-ligand interaction studies -Used to find dissociation constants and kinetic parameters -Used to screen Hsp90 ligands for C-terminal Hsp90 affinity |
(Terracciano et al., 2018; Zhou et al., 2004) |
| Thioflavin-T assay | -Used to screen for small molecule inhibitors of Hsp90-mediated tau aggregation -Used to screen small molecule Hsp90 inhibitors that may be useful for treating neurodegenerative diseases |
(Shelton, Baker, et al., 2017a) |
| Luciferase based assays | -Cancer cell lines that transgenically express luciferase are used to develop in-cell Hsp90-dependent luciferase refolding assays -Used to screen novobiocin analogs with varying degrees of activity -Used to discover the mechanism of a C-terminal domain inhibitor of Hsp90 -A dual-luciferase reporter system has been used in a high-throughput screening format to identify Hsp90 inhibitors in cell cultures and live mice -Used to screen compounds with multimodality molecular imaging to identify Hsp90 inhibitors |
(Chan et al., 2012; Rahimi & McAlpine, 2019; Sadikot et al., 2013; Wang, Koay, & McAlpine, 2017) |
| Other cell-based assays and live cell-based assays | -Used to discover new small molecules that either bind to the N-terminal ATP-binding domain or C-terminus of Hsp90. -Include traditional techniques such as western blotting, real-time PCR, and protein co-immunoprecipitations -Seahorse assays can be useful for neurodegeneration studies |
(Khandelwal et al., 2018; Pugh et al., 2020; Shelton, Baker, et al., 2017a; Ueda et al., 2020; Zhang et al., 2019; Zhang, You, et al., 2018b) |
| Mass spectroscopy | -Used to characterize interactions between ligands and Hsp90 -Used to determine the binding site novobiocin analogs at the Hsp90 C-terminus |
(Matts et al., 2011; Riccardi Sirtori et al., 2015; Xu et al., 2016) |
2.1. Fluorescence polarization assay
Fluorescence polarization (FP) is a homogeneous technique that enables one to quantify molecular interactions in solution (Lea & Simeonov, 2011). It is a powerful tool for studying molecular affinity that can be quantified via the ratio of ligand-bound to a target versus unbound. This approach has been widely used by various researchers to characterize Hsp90 inhibitory ligands. Choisis and coworkers developed a fluorescence polarization assay for Hsp90 that used a fluorescent geldanamycin ligand, which was very robust and shown to exhibit good reproducibility, and applicable to a high-throughput setting (Du et al., 2007; Kim et al., 2004). FP assays have been widely used by members of the proteostasis community in an effort to identify new inhibitors of Hsp90 (Wang et al., 2019), as well as to characterize isoform-selective inhibitors against all four Hsp90 paralogs (Khandelwal et al., 2018). A graphical representation of the assay is shown in Fig. 6A.
Fig. 6.

Principles of the FP assay and the TR-FRET assay. (A) A graphical representation of the principal Hsp90 fluorescence polarization assay. (B) A graphical representation of the TR-FRET assay as described herein (Zhou et al., 2004). Created with BioRender.
2.2. ATPase assay
Hsp90 is an ATP hydrolyzing enzyme and the energy derived from ATP hydrolysis is used to fold client proteins and then subsequent release them from the complex (Fig. 7A). Consequently, Hsp90 chaperone function is dependent upon ATP hydrolysis, and the inhibition of ATPase activity represents a successful method to discover inhibitors (Bartolini, Wainer, Bertucci, & Andrisano, 2013; Rowlands et al., 2004). High- throughput screening (HTS) assays have been developed to identify inhibitors of Hsp90 ATPase activity (Rowlands et al., 2004). One assay is based upon the detection of inorganic phosphate, which is generated upon enzymatic cleavage of ATP to form a phosphomolybdate complex with malachite green (MG)–a reagent used for colorimetric determination (Rowlands et al., 2004). Other methods have also been developed that utilize the phosphate ions to trigger an enzymatic cascade that results in the formation of resorufin, which is fluorescent and can be readily measured and quantified (Avila et al., 2006a; Avila, Kornilayev, & Blagg, 2006b). Assays for ATPase activity have been widely used by various researchers to screen for novel Hsp90 inhibitors and/or to characterize the effect on ATPase activity (Wang et al., 2019).
Fig. 7.

Principles of the ATPase assay and the ThT assay. (A) A graphical presentation of the Hsp90 based ATPase assay. (B) A graphical presentation of the ThT assay. Created with BioRender.
2.3. AlphaScreen assay
AlphaScreen technology has been utilized in high-throughput screening assays to identify small molecule inhibitors for a wide variety of biomolecular interactions (Yasgar, Jadhav, Simeonov, & Coussens, 2016). The alpha screen represents a homogeneous and robust method that provides a high signal-to-noise ratio, which makes this technology appropriate for high-throughput screening and confirmation of inhibitory activity (Yasgar et al., 2016). Amplified Luminescence Proximity Homogeneous Assay (AlphaScreenTM) has been used to identify small molecules that disrupt interactions between Hsp90-TRP2A and HOP (Yi et al., 2009). This particular assay is based on the interaction between the 20-mer C-terminal peptide of Hsp90 and the TPR2A domain of HOP. Using this technique, researchers screened 76,134 compounds, while producing a Z' of 0.77, and ultimately led to the discovery of small molecules that disrupt this interaction (Yi et al., 2009). Another C-terminal assay has also been developed wherein recombinant and truncated Hsp90 C-terminus and PPID proteins are used to elucidate disruptors of interactions between PPID and the Hsp90 C-terminus (BPS Biosciences, San Diego, CA) (Rahimi et al., 2018; Rahimi & McAlpine, 2019).
2.4. Time-resolved fluorescence energy transfer assay
Time-resolved fluorescence energy transfer (TR-FRET), also known as HTRF (Homogeneous Time-resolved fluorescence) is a homogeneous and robust assay used for drug targeting studies (Degorce et al., 2009) and is based on the fluorescent energy transfer between a donor fluorophore and an acceptor fluorophore via time-resolution (Degorce et al., 2009; Sittampalam, Kahl, & Janzen, 1997). High-throughput screening (HTS) TR-FRET assays have been developed to screen for inhibitors of Hsp90. T Zhou and coworkers developed a robust TR-FRET assay for use in a 1536-well plate format that utilized biotinylated geldanamycin and the His-tagged Hsp90 N-terminal domain (Zhou et al., 2004) (Fig. 6B). Another HTRF assay has been used to identify disruptors of Hsp90-Cdc37 interactions using glutathione S-transferase (GST)–tagged Cdc37, His-tagged Hsp90, and the corresponding anti-GST and anti-His antibodies (Wang et al., 2019; Wang et al., 2020).
2.5. Intrinsic tryptophan fluorescence quenching assay
Intrinsic tryptophan fluorescence is attributed to the aromatic amino acids–primarily tryptophan, which can be measured via excitation at 295 nm. The resulting emitted fluorescence can then be measured and quantified. These assays are based on the observation that the emission profile of tryptophan residues are altered when the local hydrophobic environment changes due to denaturation or conformational effects that are induced by protein-ligand or protein-protein interactions (Akbar, Sreeramulu, & Sharma, 2016). This assay has been used to characterize Hsp90 modulators, and to calculate dissociation constants based on the observed fluorescence induced in a dose-dependent manner (Mak, Chand, Reynisson, & Leung, 2019). The drawback associated with this simple technique is that the compounds under investigation may also exhibit fluorescence properties, which can interfere with this assay.
2.6. NMR spectroscopic techniques
Nuclear Magnetic Resonance (NMR) is a well-known technique that is used to characterize protein-ligand interactions or to screen fragment libraries against protein targets (Harner, Mueller, Robbins, & Reily, 2017). Fragment-based drug discovery approaches are becoming increasingly popular in drug discovery programs that aim to identify small molecules that interact with a protein (Kuo, 2011). The most common ligand-observed 1H NMR experiments are the saturation-transfer difference (STD) experiments and the waterLOGSY NMR protocols–both of which measure protein-ligand binding constants; however, waterLOGSY is reported to be more sensitive when compared to STD experiments (Antanasijevic, Ramirez, & Caffrey, 2014). Not surprisingly, these methods have been used to characterize Hsp90 and its ligand interactions as well as to screen fragment-based libraries against Hsp90 (Raingeval et al., 2019; Roughley & Hubbard, 2011). The 19F NMR method is also commonly used to study protein-ligand interactions when the small molecule contains a fluorine atom. This method has been used by Casale and coworkers to identify a novel class of Hsp90 inhibitors using fluorine chemical shift anisotropy and exchange for screening NMR (FAXS-NMR) (Casale et al., 2014). Another popular NMR method is the 2D HSQC NMR method, which is primarily used to study protein-ligand interactions, can be used to identify specific residues on a protein that are important for the interaction and also to determine dissociation constant values (Huang et al., 2017). This multidimensional NMR approach has been used to identify the previously unknown binding site of Hsp90, wherein small molecules bind and disrupt Hsp90-Cdc37 interactions (Wang et al., 2019). A related approach was utilized to characterize the interaction between Hsp90 and p53 using NMR spectroscopy (Hagn et al., 2011).
2.7. Crystallography and structure-based drug design
Crystal structures provide researchers the opportunity to rationally design inhibitors or to perform virtual screens for Hsp90 modulators. Structure-guided drug design represents an efficient method to predict active inhibitors in a cost-effective manner, and can also be used to derive structure-activity relationships studies (Dutta Gupta et al., 2015). High-resolution crystal structures of Hsp90 in complex with nucleotides, ligands, and/or co-chaperones have been solved and provide a foundation to explain ligand binding interactions (Khandelwal et al., 2018; Stebbins et al., 1997). The structure-based design of new Hsp90 inhibitors has been sought utilizing crystal structures of Hsp90 bound to first-generation ligands, which ultimately led to the discovery of isoform-selective inhibitors of Hsp90 (Khandelwal et al., 2018). For example, overlays of the co-crystal structures between Hsp90α and Hsp90β bound to radicicol revealed three water molecules that play an important role in distinguishing these two isoforms and their potential ligands binding modes (Khandelwal et al., 2018; Lee et al., 2015). Computational modeling studies such as QSAR and molecular dynamics (MD) simulations were also used to design Hsp90 inhibitors (Abbasi, Sadeghi-Aliabadi, & Amanlou, 2018; Kung et al., 2011; Yan, Grant, & Richards, 2008). Structure based design strategies used by Kung and coworkers led to the discovery of pyrrolopyrimines as Hsp90 inhibitors and determined that water molecules that are present in the binding site play an important role in determining inhibitor potency (Kung et al., 2011). By MD simulation studies, Yan and co-workers found that several conserved water molecules present in the Hsp90α binding site participate in interactions between Hsp90α and PU3. These water molecules are difficult to replace, and are critical for ligand binding (Yan et al., 2008). Docking studies have also been used to identify small molecule disruptors Hsp90-Cdc37 interactions (Wang et al., 2019). While no crystal structure exists for Hsp90 bound with C-terminal inhibitors, the interaction is well characterized and supported by computational studies. Several Hsp90 crystal structures are available either alone or in complex with N-terminal inhibitors that bind the ATP binding site of Hsp90.
2.8. Isothermal titration calorimetry (ITC) and surface plasmon resonance (SPR)
Isothermal titration calorimetry (ITC) measures the energy provided or required to facilitate interactions between a protein and ligand, and detects changes in heat upon ligand binding and consequently, yields thermodynamic properties that can be used to calculate affinity (Renaud et al., 2016). ITC has been used to study interactions between Hsp90 and several ligands as well as their binding affinities and entropic/ enthalpic contributions to binding (Roe et al., 2018) (Garnier et al., 2002; Murray et al., 2010).
Like ITC, surface plasmon resonance (SPR), is also a label-free method that typically requires less protein, and can be used to measure protein-ligand dissociation constants or other kinetic parameters (Renaud et al., 2016). SPR has been used to identify Hsp90 inhibitors, and some authors have discovered structurally unrelated ligands that bind the Hsp90 C-terminus (Terracciano et al., 2018). Interactions between Hsp90 and N-terminal inhibitors have also been investigated (Zhou et al., 2004).
2.9. Thioflavin T (ThT) assays
Thioflavin T (ThT) is a benzothiazole dye that has been used historically as a stain for amyloid beta-sheet fibril formation (Fig. 7B). The fibrillar-bound dye can be selectively excited at 450 nm, which leads to fluorescence emission at 482 nm (Khurana et al., 2005). Specifically, ThT has been shown to bind to beta-sheet-dense tau aggregates during fibrillization (Sui, Liu, & Kuo, 2015; Xue, Lin, Chang, & Guo, 2017). A similar assay has been developed to monitor changes in sarkosyl-insoluble tau aggregates that occur upon incubation of P301L tau with combinations of Hsp90 and various co-chaperones (Shelton et al., 2017a). The assay has been further modified to identify small molecules that inhibit Hsp90-stimulated tau aggregation. Furthermore, the seeding of Hsp90-mediated tau fibrils can be followed via TEM imaging (Shelton, Baker, et al., 2017a).
2.10. Cell-based assays
Cell-based assays are physiologically more relevant than biochemical or biophysical assays, but are usually more complex in the context of drug discovery. This is further complicated in the case of the Hsp90 chaperome family, whom participate in these more complex cellular settings. Despite these challenges, several cell-based assays have been used to identify or characterize Hsp90 inhibitors, which are summarized below:
2.10.1. Luciferase-based assays
Following heat denaturation, the refolding of firefly luciferase is an Hsp90-dependent process. Therefore, a panel of cancer cell lines that transgenically express luciferase have been utilized to develop an in-cell and Hsp90-dependent rematuration luciferase assay (Sadikot et al., 2013). A similar assay was used to screen novobiocin analogs against Hsp90, which revealed varying degrees of activity amongst the compounds (Sadikot et al., 2013). A luciferase refolding assay was also used to characterize the C-terminal ligand-binding domain of Hsp90, which ultimately led to the discovery of a molecule that disrupted complex formation and inhibited luciferase rematuration (as detected by a reduction in luminescence signal) (Rahimi & McAlpine, 2019). A dual-luciferase reporter assay system has also been used in a high-throughput screen to identify Hsp90 inhibitors from cell culture and via live mice (Chan et al., 2012) (Fig. 8A). The authors in this study screened compounds that exhibited multimodality molecular imaging and were able to reveal several Hsp90 inhibitors that inhibit specific chaperone-protein interactions (Chan et al., 2012).
Fig. 8.

Principles of cell based assays. (A) The split Renila luciferase (RL) complementation system to discover Hsp90 inhibitors. A schematic illustration showing Hsp90/p23 interactions using the split RL complementation system. The two interacting proteins, p23 and Hsp90, are fused to the inactive N-RL and C-RL portions of RL through a peptide linker. In the presence of ATP and the substrate coelenterazine, the system produces light. Hsp90 inhibitors prevent ATP binding to Hsp90, reduce RL activity and light output (Chan et al., 2012). (B) Cell-based ligand-screening system by the ligand directed N-acyl-N-alkyl sulfonamide (LDNASA) chemistry (Ueda et al., 2020). The labeling reagent consists of PU-H71, fluorescein diacetate, and the NASA reactive group. Created with Biorender.
2.10.2. Live-cell based assays and other cell-based assays
Recently, Udea et al. reported a novel live-cell based assay to identify Hsp90 inhibitors and were able to discover a new small molecule and ligand-binding event at the N-terminal ATP binding domain of Hsp90 within live cells (Ueda, Tamura, & Hamachi, 2020) (Fig. 8B).
Traditional cell-based techniques have also been used to characterize Hsp90 and ligand or co-chaperone interactions and include western blots, anti-proliferation assays, real-time PCR, and co-immunoprecipitation assays to understand both the mechanism of ligand interaction as well as the client protein levels in normal versus transformed cells (Khandelwal et al., 2018; Shelton, Baker, et al., 2017a; Wang et al., 2019; Zhang, Banerjee, Davis, & Blagg, 2019). Oxidative stress in neuropathic cells can lead to mitochondrial dysfunction and contribute to the pathogenesis of several degenerative neuropathies. As a result, a Seahorse assay was established to observe changes in mitochondrial oxygen consumption rate and maximal respiratory capacity with an XF96 Extracellular Flux Analyzer in the presence of Hsp90 inhibitors (Zhang, You, Dobrowsky, & Blagg, 2018b). A number of assays have also been developed to assess the neuroprotective activity manifested by small-molecule Hsp90 modulators (Zhang, You, et al., 2018b). Early studies evaluated the ability of Hsp90 inhibitors to protect cultured neurons from various forms of neurotoxins in cell viability assays. Subsequent assays were then employed to assess the effect of Hsp90 inhibitors on JNK inhibition/ activation and prevention of neuronal cell death.
2.11. Mass spectroscopic-based analysis of ligand binding to Hsp90
Mass spectroscopy-based methods have also been used to identify small molecule binding sites within the C-terminal domain of Hsp90. Matts and coworkers prepared photolabile derivatives of novobiocin and identified a putative C-terminal Hsp90 binding site using LSMS/MS spectroscopy (Matts et al., 2011). The stability of proteins from rates of oxidation (SPROX) mass spectroscopy-based approach was used to measure the affinity that geldanamycin-manifested towards Hsp90 interactions, which were found to be consistent with previously published data (Strickland et al., 2013; West, Tang, & Fitzgerald, 2008; Xu, Wallace, & Fitzgerald, 2016). Likewise, Sirtori and coworkers used an automated flow injection ESI-MS method to screen a fragment-based library against Hsp90 (Riccardi Sirtori et al., 2015).
2.12. Conclusions and future directions
Hsp90 is a molecular chaperone that has tremendous potential as a drug target for cancer and other human diseases. Interestingly, Hsp90 inhibitors are ~200-fold more selective for the high-affinity Hsp90 species that is present in transformed cells, as compared to normal cells, which results in a large therapeutic window. However, there is currently no FDA approved Hsp90 inhibitor available to treat cancer, because most of the N-terminal inhibitors that have undergone clinical trials produced both off- and on-target toxicities. Therefore, there remains an opportunity to develop a small molecule that can inhibit Hsp90 without the side effects associated with classical N-terminal inhibition. Since Hsp90 is inhibited by compounds with diverse chemical structures and sizes, much opportunity remains. The most recent advancements in the Hsp90 drug discovery area led to isoform-selective inhibitors that may overcome the detriments associated with pan inhibition of Hsp90. In fact, some of the recently discovered isoform-specific Hsp90 inhibitors have exhibited properties that appear to overcome these concerns and are likely to progress towards clinical investigation. Similarly, Hsp90 C-terminal inhibitors represent another opportunity to develop Hsp90 inhibitors that do not invoke the heat shock response. The assays mentioned herein are well-established and can be adapted for the high-throughput screening of individual isoforms. In summary, initial screening for Hsp90 ligands is largely conducted using in vitro recombinant assays or in silico virtual screenings. Subsequent studies are then carried out in cell-based assays and/or animal models. A summary of different assays that have been used to expedite the discovery and/or characterization of Hsp90 inhibitors is outlined in Table 1 for brevity.
Each of the assays described in this review have advantages and disadvantages. The FP assay is homogenous, sensitive, and suitable for high-throughput screening, but requires a large quantity of protein. It is also fast, easy-to-operate, and cost-effective. However, because this is a fluorescent based assay, fluorescent compounds may interfere with the assay.TR-FRET or AlphaScreen assays are also HTS amenable, homogeneous, sensitive, rapid, reliable, robust, and require small amounts of protein. However, these assays are costly, as the donor/acceptor beads are expensive. The tryptophan fluorescence quenching assay is a simple technique for measuring the binding constant between a protein and its ligand or partner. However, drawbacks include possible interference by fluorescent compounds and the intrinsic requirement for several tryptophan residues to be present within a protein. ITC and SPR are both label-free methods. SPR provides kinetic parameters, whereas ITC produces thermodynamic parameters. Both techniques measure direct protein-ligand interactions. However, large quantities of protein are required for ITC and this assay is generally low throughput. On the other hand, SPR requires small quantities of protein, but requires the protein to be immobilized. Structure based drug design is not suitable for high-throughput screening and is very inexpensive, however, this approach can be useful for isoform-selective inhibitor design. The thioflavin T assay is relatively inexpensive and can be adapted for high throughput screening, but does require large quantities of protein. Crystallography also requires large amounts of protein as well as specialized scientists to obtain crystals and to perform data analysis. This technique also provides high resolution structural information for both the apo and ligand-bound conformations of the protein. Cell-based assays can be more physiologically relevant than biochemical assays, and can be made suitable for high-throughput screening. However, compounds can manifest off-target effects and the compounds may show variability based on the conditions used.
The discovery of small molecule Hsp90 modulators with good druglike properties remain a challenging task and through minimization of the side effects, these new drugs could be clinical useful. N-terminal isoform-selective compounds, inhibitors that target the Hsp90 C-terminus, as well as isoform-selective C-terminal compounds may offer the potential to overcome some of these detriments observed with the pan Hsp90 inhibitors. The assays described herein may be suitable for the discovery of new Hsp90 inhibitors and will likely be useful for the preclinical development of compounds that target individual isoforms. Ultimately, it is important to understand the specific roles played by each isoform and/or their co-chaperones in various disease states. The tools and assays summarized in this review can be used to develop Hsp90-targeting drugs with high specificity, potency, and drug-like properties.
Acknowledgements
BSJB is supported by The National Institutes of Health [CA213586] [N5075311]. BMK is a fellow of the Chemistry-Biochemistry-Biology Interface (CBBI) Program at the University of Notre Dame, supported by the NIH Training Grant T32GM075762 from the National Institutes of General Medical Sciences.
Abbreviations:
- Hsp90
heat shock protein 90
- GRP94
94 kDa glucose-related protein
- TRAP-1
tumor necrosis factor-associated protein 1
- ATP
Adinosine triphosphate
- CDC37
Cell division cycle 37
- HSF-1
Heat shock factor 1
- TPR
tetratricopeptide repeat
- c-MET
tyrosine protein kinase Met
- ThT
Thioflavin T
- SPR
surface plasmon resonance
- ITC
isothermal titration calorimetry
- HSF-1
heat shock factor 1
- HSR
heat shock response
- FP
fluorescence polarization
- HTS
high-throughput screening
- MG
malachite green
- TR-FRET
Time-resolved fluorescence energy transfer
- GST
glutathione S-transferase
- NMR
nuclear magnetic resonance
- MD
molecular dynamics
- TEM
transmission electron microscopy
Footnotes
Declaration of Competing Interest
Authors report no conflict of interest to declare.
References
- Abbasi M, Sadeghi-Aliabadi H, & Amanlou M (2018). 3D-QSAR, molecular docking, and molecular dynamic simulations for prediction of new Hsp90 inhibitors based on isoxazole scaffold. Journal of Biomolecular Structure & Dynamics 36, 1463–1478. [DOI] [PubMed] [Google Scholar]
- Aherne W, Maloney A, Prodromou C, Rowlands MG, Hardcastle A, Boxall K, … Workman P (2003). Assays for HSP90 and inhibitors. Methods in Molecular Medicine 85, 149–161. [DOI] [PubMed] [Google Scholar]
- Akbar SM, Sreeramulu K, & Sharma HC (2016). Tryptophan fluorescence quenching as a binding assay to monitor protein conformation changes in the membrane of intact mitochondria. Journal of Bioenergetics and Biomembranes 48, 241–247. [DOI] [PubMed] [Google Scholar]
- Antanasijevic A, Ramirez B, & Caffrey M (2014). Comparison of the sensitivities of WaterLOGSY and saturation transfer difference NMR experiments. Journal of Biomolecular NMR 60, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila C, Hadden MK, Ma Z, Kornilayev BA, Ye QZ, & Blagg BS (2006a). High-throughput screening for Hsp90 ATPase inhibitors. Bioorganic & Medicinal Chemistry Letters 16, 3005–3008. [DOI] [PubMed] [Google Scholar]
- Avila C, Kornilayev BA, & Blagg BS (2006b). Development and optimization of a useful assay for determining Hsp90’s inherent ATPase activity. Bioorganic & Medicinal Chemistry 14, 1134–1142. [DOI] [PubMed] [Google Scholar]
- Bartolini M, Wainer IW, Bertucci C, & Andrisano V (2013). The rapid and direct determination of ATPase activity by ion exchange chromatography and the application to the activity of heat shock protein-90. Journal of Pharmaceutical and Biomedical Analysis 73, 77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickel D, & Gohlke H (2019). C-terminal modulators of heat shock protein of 90 kDa (HSP90): State of development and modes of action. Bioorganic & Medicinal Chemistry 27, 115080. [DOI] [PubMed] [Google Scholar]
- Biebl MM, & Buchner J (2019). Structure, function, and regulation of the Hsp90 machinery. Cold Spring Harbor Perspectives in Biology 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burlison JA, Neckers L, Smith AB, Maxwell A, & Blagg BS (2006). Novobiocin: Redesigning a DNA gyrase inhibitor for selective inhibition of hsp90. Journal of the American Chemical Society 128, 15529–15536. [DOI] [PubMed] [Google Scholar]
- Casale E, Amboldi N, Brasca MG, Caronni D, Colombo N, Dalvit C, … Isacchi A, et al. (2014). Fragment-based hit discovery and structure-based optimization of aminotriazoloquinazolines as novel Hsp90 inhibitors. Bioorganic & Medicinal Chemistry 22, 4135–4150. [DOI] [PubMed] [Google Scholar]
- Chan CT, Reeves RE, Geller R, Yaghoubi SS, Hoehne A, Solow-Cordero DE, … Gambhir SS (2012). Discovery and validation of small-molecule heat-shock protein 90 inhibitors through multimodality molecular imaging in living subjects. Proceedings of the National Academy of Sciences of the United States of America 109, E2476–E2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S, & Burns TF (2017). Targeting heat shock proteins in cancer: A promising therapeutic approach. International Journal of Molecular Sciences 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csermely P, Schnaider T, Soti C, Prohászka Z, & Nardai G (1998). The 90-kDa molecular chaperone family: Structure, function, and clinical applications. A comprehensive review. Pharmacology & Therapeutics 79, 129–168. [DOI] [PubMed] [Google Scholar]
- Degorce F, Card A, Soh S, Trinquet E, Knapik GP, & Xie B (2009). HTRF: A technology tailored for drug discovery - a review of theoretical aspects and recent applications. Current Chem Genomics 3, 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der Sarkissian S, Aceros H, Williams PM, Scalabrini C, Borie M, & Noiseux N (2020). HSP90 inhibition and multi-target approach to maximize cardioprotection in ischemic injury. British Journal of Pharmacology 110, 1861–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Moulick K, Rodina A, Aguirre J, Felts S, Dingledine R, … Chiosis G (2007). High-throughput screening fluorescence polarization assay for tumor-specific Hsp90. Journal of Biomolecular Screening 12, 915–924. [DOI] [PubMed] [Google Scholar]
- Dutta Gupta S, Bommaka MK, Mazaira GI, Galigniana MD, Subrahmanyam CV, Gowrishankar NL, & Raghavendra NM (2015). Molecular docking study, synthesis and biological evaluation of Mannich bases as Hsp90 inhibitors. International Journal of Biological Macromolecules 80, 253–259. [DOI] [PubMed] [Google Scholar]
- Garcia-Carbonero R, Carnero A, & Paz-Ares L (2013). Inhibition of HSP90 molecular chaperones: Moving into the clinic. The Lancet Oncology 14, e358–e369. [DOI] [PubMed] [Google Scholar]
- Garg G, Khandelwal A, & Blagg BS (2016). Anticancer inhibitors of Hsp90 function: Beyond the usual suspects. Advances in Cancer Research 129, 51–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnier C, Lafitte D, Tsvetkov PO, Barbier P, Leclerc-Devin J, Millot JM, … Peyrot V (2002). Binding of ATP to heat shock protein 90: Evidence for an ATP-binding site in the C-terminal domain. The Journal of Biological Chemistry 277, 12208–12214. [DOI] [PubMed] [Google Scholar]
- Hagn F, Lagleder S, Retzlaff M, Rohrberg J, Demmer O, Richter K, … Kessler H (2011). Structural analysis of the interaction between Hsp90 and the tumor suppressor protein p53. Nature Structural & Molecular Biology 18, 1086–1093. [DOI] [PubMed] [Google Scholar]
- Harner MJ, Mueller L, Robbins KJ, & Reily MD (2017). NMR in drug design. Archives of Biochemistry and Biophysics 628, 132–147. [DOI] [PubMed] [Google Scholar]
- Harris SF, Shiau AK, & Agard DA (2004). The crystal structure of the carboxy-terminal dimerization domain of htpG, the Escherichia coli Hsp90, reveals a potential substrate binding site. Structure 12, 1087–1097. [DOI] [PubMed] [Google Scholar]
- Huang R, Ayine-Tora DM, Muhammad Rosdi MN, Li Y, Reynisson J, & Leung IKH (2017). Virtual screening and biophysical studies lead to HSP90 inhibitors. Bioorganic & Medicinal Chemistry Letters 27, 277–281. [DOI] [PubMed] [Google Scholar]
- Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, & Burrows FJ (2003). A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 425, 407–410. [DOI] [PubMed] [Google Scholar]
- Khandelwal A, Kent CN, Balch M, Peng S, Mishra SJ, Deng J, … Cohen M, et al. (2018). Structure-guided design of an Hsp90β N-terminal isoform-selective inhibitor. Nature Communications 9, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana R, Coleman C, Ionescu-Zanetti C, Carter SA, Krishna V, Grover RK, … Singh S (2005). Mechanism of thioflavin T binding to amyloid fibrils. Journal of Structural Biology 151, 229–238. [DOI] [PubMed] [Google Scholar]
- Kim J, Felts S, Llauger L, He H, Huezo H, Rosen N, & Chiosis G (2004). Development of a fluorescence polarization assay for the molecular chaperone Hsp90. Journal of Biomolecular Screening 9, 375–381. [DOI] [PubMed] [Google Scholar]
- Koga F, Kihara K, & Neckers L (2009). Inhibition of cancer invasion and metastasis by targeting the molecular chaperone heat-shock protein 90. Anticancer Research 29, 797–807. [PubMed] [Google Scholar]
- Koren J, Jinwal UK, Lee DC, Jones JR, Shults CL, Johnson AG, … Dickey CA (2009). Chaperone signalling complexes in Alzheimer’s disease. Journal of Cellular and Molecular Medicine 13, 619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung PP, Sinnema PJ, Richardson P, Hickey MJ, Gajiwala KS, Wang F, … Maegley K, et al. (2011). Design strategies to target crystallographic waters applied to the Hsp90 molecular chaperone. Bioorganic & Medicinal Chemistry Letters 21, 3557–3562. [DOI] [PubMed] [Google Scholar]
- Kuo LC (2011). Fragment-based drug design - tools, practical approaches, and examples. Preface Methods Enzymol 493 (xxi–xxii). [DOI] [PubMed] [Google Scholar]
- Lea WA, & Simeonov A (2011). Fluorescence polarization assays in small molecule screening. Expert Opinion on Drug Discovery 6, 17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Park HK, Jeong H, Lim J, Lee AJ, Cheon KY, … Kim ND, et al. (2015). Development of a mitochondria-targeted Hsp90 inhibitor based on the crystal structures of human TRAP1. Journal of the American Chemical Society 137, 4358–4367. [DOI] [PubMed] [Google Scholar]
- Luo GR, & Le WD (2010). Collective roles of molecular chaperones in protein degradation pathways associated with neurodegenerative diseases. Current Pharmaceutical Biotechnology 11, 180–187. [DOI] [PubMed] [Google Scholar]
- Luo W, Sun W, Taldone T, Rodina A, & Chiosis G (2010). Heat shock protein 90 in neurodegenerative diseases. Molecular Neurodegeneration 5, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak OW, Chand R, Reynisson J, & Leung IKH (2019). Identification of isoform-selective ligands for the middle domain of heat shock protein 90 (Hsp90). International Journal of Molecular Sciences 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matts RL, Dixit A, Peterson LB, Sun L, Voruganti S, Kalyanaraman P, … Blagg BS (2011). Elucidation of the Hsp90 C-terminal inhibitor binding site. ACS Chemical Biology 6, 800–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MP, & Le Breton L (2015). Hsp90: Breaking the symmetry. Molecular Cell 58, 8–20. [DOI] [PubMed] [Google Scholar]
- Mishra P, & Bolon DN (2014). Designed Hsp90 heterodimers reveal an asymmetric ATPase-driven mechanism in vivo. Molecular Cell 53, 344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray CW, Carr MG, Callaghan O, Chessari G, Congreve M, Cowan S, Coyle JE, Downham R, Figueroa E, Frederickson M, et al. (2010). Fragment-based drug discovery applied to Hsp90. Discovery of two lead series with high ligand efficiency. Journal of Medicinal Chemistry 53, 5942–5955. [DOI] [PubMed] [Google Scholar]
- Neckers L, & Workman P (2012). Hsp90 molecular chaperone inhibitors: Are we there yet? Clinical Cancer Research 18, 64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckers L, Blagg B, Haystead T, Trepel JB, Whitesell L, & Picard D (2018). Methods to validate Hsp90 inhibitor specificity, to identify off-target effects, and to rethink approaches for further clinical development. Cell Stress & Chaperones 23, 467–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson GM, Huffman H, & Smith DF (2003). Comparison of the carboxy-terminal DP- repeat region in the co-chaperones Hop and Hip. Cell Stress & Chaperones 8, 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermann WM, Sondermann H, Russo AA, Pavletich NP, & Hartl FU (1998). In vivo function of Hsp90 is dependent on ATP binding and ATP hydrolysis. The Journal of Cell Biology 143, 901–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou JR, Tan MS, Xie AM, Yu JT, & Tan L (2014). Heat shock protein 90 in Alzheimer’s disease. BioMed Research International 2014, 796869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaretou B, Prodromou C, Roe SM, O’Brien R, Ladbury JE, Piper PW, & Pearl LH (1998). ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. The EMBO Journal 17, 4829–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patwardhan CA, Fauq A, Peterson LB, Miller C, Blagg BS, & Chadli A (2013). Gedunin inactivates the co-chaperone p23 protein causing cancer cell death by apoptosis. The Journal of Biological Chemistry 288, 7313–7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul S, & Mahanta S (2014). Association of heat-shock proteins in various neurodegenerative disorders: Is it a master key to open the therapeutic door? Molecular and Cellular Biochemistry 386, 45–61. [DOI] [PubMed] [Google Scholar]
- Prodromou C (2016). Mechanisms of Hsp90 regulation. The Biochemical Journal 473, 2439–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodromou C (2017). Regulatory mechanisms of Hsp90. Biochemistry & Molecular Biology Journal 3, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, & Pearl LH (1997). Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 90, 65–75. [DOI] [PubMed] [Google Scholar]
- Prodromou C, Panaretou B, Chohan S, Siligardi G, O’Brien R, Ladbury JE, … Pearl LH (2000). The ATPase cycle of Hsp90 drives a molecular “clamp” via transient dimerization of the N-terminal domains. The EMBO Journal 19, 4383–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh KW, Zhang Z, Wang J, Xu X, Munthali V, Zuo A, & Blagg BSJ (2020). From bacteria to cancer: A benzothiazole-based DNA gyrase B inhibitor redesigned for Hsp90 C-terminal inhibition. ACS Medicinal Chemistry Letters 11, 1535–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimi MN, & McAlpine SR (2019). Protein-protein inhibitor designed de novo to target the MEEVD region on the C-terminus of Hsp90 and block co-chaperone activity. Chem Commun (Camb) 55, 846–849. [DOI] [PubMed] [Google Scholar]
- Rahimi MN, Buckton LK, Zaiter SS, Kho J, Chan V, Guo A, … Lawler MF, et al. (2018). Synthesis and structure-activity relationships of inhibitors that target the C- terminal MEEVD on heat shock protein 90. ACS Medicinal Chemistry Letters 9, 73–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raingeval C, Cala O, Brion B, Le Borgne M, Hubbard RE, & Krimm I (2019). 1D NMR WaterLOGSY as an efficient method for fragment-based lead discovery. Journal of Enzyme Inhibition and Medicinal Chemistry 34, 1218–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratzke C, Nguyen MN, Mayer MP, & Hugel T (2012). From a ratchet mechanism to random fluctuations evolution of Hsp90’s mechanochemical cycle. Journal of Molecular Biology 423, 462–471. [DOI] [PubMed] [Google Scholar]
- Renaud JP, Chung CW, Danielson UH, Egner U, Hennig M, Hubbard RE, & Nar H (2016). Biophysics in drug discovery: Impact, challenges and opportunities. Nature Reviews. Drug Discovery 15, 679–698. [DOI] [PubMed] [Google Scholar]
- Riccardi Sirtori F, Caronni D, Colombo M, Dalvit C, Paolucci M, Regazzoni L, … Fogliatto G (2015). Establish an automated flow injection ESI-MS method for the screening of fragment based libraries: Application to Hsp90. European Journal of Pharmaceutical Sciences 76, 83–94. [DOI] [PubMed] [Google Scholar]
- Rochani AK, Singh M, & Tatu U (2013). Heat shock protein 90 inhibitors as broad spectrum anti-infectives. Current Pharmaceutical Design 19, 377–386. [DOI] [PubMed] [Google Scholar]
- Roe MS, Wahab B, Török Z, Horváth I, Vigh L, & Prodromou C (2018). Dihydropyridines allosterically modulate Hsp90 providing a novel mechanism for heat shock protein co-induction and neuroprotection. Frontiers in Molecular Biosciences 5, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röhl A, Rohrberg J, & Buchner J (2013). The chaperone Hsp90: Changing partners for demanding clients. Trends in Biochemical Sciences 38, 253–262. [DOI] [PubMed] [Google Scholar]
- Roughley SD, & Hubbard RE (2011). How well can fragments explore accessed chemical space? A case study from heat shock protein 90. Journal of Medicinal Chemistry 54, 3989–4005. [DOI] [PubMed] [Google Scholar]
- Rowlands MG, Newbatt YM, Prodromou C, Pearl LH, Workman P, & Aherne W (2004). High-throughput screening assay for inhibitors of heat-shock protein 90 ATPase activity. Analytical Biochemistry 327, 176–183. [DOI] [PubMed] [Google Scholar]
- Sadikot T, Swink M, Eskew JD, Brown D, Zhao H, Kusuma BR, … Holzbeierlein JM, et al. (2013). Development of a high-throughput screening cancer cell-based luciferase refolding assay for identifying Hsp90 inhibitors. Assay and Drug Development Technologies 11, 478–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaltriti M, Dawood S, & Cortes J (2012). Molecular pathways: Targeting hsp90αwho benefits and who does not. Clinical Cancer Research 18, 4508–4513. [DOI] [PubMed] [Google Scholar]
- Schopf FH, Biebl MM, & Buchner J (2017). The HSP90 chaperone machinery. Nature Reviews. Molecular Cell Biology 18, 345–360. [DOI] [PubMed] [Google Scholar]
- Shelton LB, Baker JD, Zheng D, Sullivan LE, Solanki PK, Webster JM, … Koren J, et al. (2017a). Hsp90 activator Aha1 drives production of pathological tau aggregates. Proceedings of the National Academy of Sciences of the United States of America 114, 9707–9712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton LB, Koren J, & Blair LJ (2017b). Imbalances in the Hsp90 chaperone machinery: Implications for tauopathies. Frontiers in Neuroscience 11, 724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sittampalam GS, Kahl SD, & Janzen WP (1997). High-throughput screening: Advances in assay technologies. Current Opinion in Chemical Biology 1, 384–391. [DOI] [PubMed] [Google Scholar]
- Söti C, Rácz A, & Csermely P (2002). A nucleotide-dependent molecular switch controls ATP binding at the C-terminal domain of Hsp90. N-terminal nucleotide binding unmasks a C-terminal binding pocket. The Journal of Biological Chemistry 277, 7066–7075. [DOI] [PubMed] [Google Scholar]
- Sreedhar AS, Kalmár E, Csermely P, & Shen YF (2004). Hsp90 isoforms: Functions, expression and clinical importance. FEBS Letters 562, 11–15. [DOI] [PubMed] [Google Scholar]
- Stebbins CE, Russo AA, Schneider C, Rosen N, Hartl FU, & Pavletich NP (1997). Crystal structure of an Hsp90-geldanamycin complex: Targeting of a protein chaperone by an antitumor agent. Cell 89, 239–250. [DOI] [PubMed] [Google Scholar]
- Strickland EC, Geer MA, Tran DT, Adhikari J, West GM, DeArmond PD, … Fitzgerald MC (2013). Thermodynamic analysis of protein-ligand binding interactions in complex biological mixtures using the stability of proteins from rates of oxidation. Nature Protocols 8, 148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui D, Liu M, & Kuo MH (2015). In vitro aggregation assays using hyperphosphorylated tau protein. Journal of Visualized Experiments 95, 1–9 e51537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M, Jarosz DF, & Lindquist S (2010). HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nature Reviews. Molecular Cell Biology 11, 515–528. [DOI] [PubMed] [Google Scholar]
- Tatokoro M, Koga F, Yoshida S, & Kihara K (2015). Heat shock protein 90 targeting therapy: State of the art and future perspective. EXCLI Journal 14, 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terracciano S, Russo A, Chini MG, Vaccaro MC, Potenza M, Vassallo A, … Bruno I (2018). Discovery of new molecular entities able to strongly interfere with Hsp90 C-terminal domain. Scientific Reports 8, 1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trepel J, Mollapour M, Giaccone G, & Neckers L (2010). Targeting the dynamic HSP90 complex in cancer. Nature Reviews. Cancer 10, 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda T, Tamura T, & Hamachi I (2020). Development of a cell-based ligand-screening system for identifying Hsp90 inhibitors. Biochemistry 59, 179–182. [DOI] [PubMed] [Google Scholar]
- Urban MJ, Dobrowsky RT, & Blagg BS (2012). Heat shock response and insulin- associated neurodegeneration. Trends in Pharmacological Sciences 33, 129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Koay YC, & McAlpine SR (2017). Redefining the phenotype of heat shock protein 90 (Hsp90) inhibitors. Chemistry 23, 2010–2013. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhang L, Li L, Jiang J, Zheng Z, Shang J, Wang C, Chen W, Bao Q, Xu X, et al. (2019). Small-molecule inhibitor targeting the Hsp90-Cdc37 protein-protein interaction in colorectal cancer. Sci Adv 5 (eaax2277). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Jiang J, Zhang L, Zhang Q, Zhou J, Li L, … You Q (2020). Discovery and optimization of small molecules targeting the protein-protein interaction of heat shock protein 90 (Hsp90) and cell division cycle 37 as orally active inhibitors for the treatment of colorectal cancer. Journal of Medicinal Chemistry 63, 1281–1297. [DOI] [PubMed] [Google Scholar]
- Wegele H, Wandinger SK, Schmid AB, Reinstein J, & Buchner J (2006). Substrate transfer from the chaperone Hsp70 to Hsp90. Journal of Molecular Biology 356, 802–811. [DOI] [PubMed] [Google Scholar]
- West GM, Tang L, & Fitzgerald MC (2008). Thermodynamic analysis of protein stability and ligand binding using a chemical modification- and mass spectrometry-based strategy. Analytical Chemistry 80, 4175–4185. [DOI] [PubMed] [Google Scholar]
- Whitesell L, & Lindquist SL (2005). HSP90 and the chaperoning of cancer. Nature Reviews. Cancer 5, 761–772. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Bagatell R, & Falsey R (2003). The stress response: Implications for the clinical development of hsp90 inhibitors. Current Cancer Drug Targets 3, 349–358. [DOI] [PubMed] [Google Scholar]
- Xu Y, Wallace MA, & Fitzgerald MC (2016). Thermodynamic analysis of the Geldanamycin-Hsp90 interaction in a whole cell lysate using a mass spectrometry-based proteomics approach. Journal of the American Society for Mass Spectrometry 27, 1670–1676. [DOI] [PubMed] [Google Scholar]
- Xue C, Lin TY, Chang D, & Guo Z (2017). Thioflavin T as an amyloid dye: Fibril quantification, optimal concentration and effect on aggregation. Royal Society Open Science 4, 160696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan A, Grant GH, & Richards WG (2008). Dynamics of conserved waters in human Hsp90: Implications for drug design. Journal of Royal Society Interface 5(Suppl. 3), S199–S205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasgar A, Jadhav A, Simeonov A, & Coussens NP (2016). AlphaScreen-based assays: Ultra-high-throughput screening for small-molecule inhibitors of challenging enzymes and protein-protein interactions. Methods in Molecular Biology 1439, 77–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F, Zhu P, Southall N, Inglese J, Austin CP, Zheng W, & Regan L (2009). An AlphaScreen-based high-throughput screen to identify inhibitors of Hsp90-cochaperone interaction. Journal of Biomolecular Screening 14, 273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang FZ, Ho DH, & Wong RH (2018a). Triptolide, a HSP90 middle domain inhibitor, induces apoptosis in triple manner. Oncotarget 9, 22301–22315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, You Z, Dobrowsky RT, & Blagg BSJ (2018b). Synthesis and evaluation of a ring-constrained Hsp90 C-terminal inhibitor that exhibits neuroprotective activity. Bioorganic & Medicinal Chemistry Letters 28, 2701–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Banerjee M, Davis RE, & Blagg BSJ (2019). Mitochondrial-targeted Hsp90 C-terminal inhibitors manifest anti-proliferative activity. Bioorganic & Medicinal Chemistry Letters 29, 126676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou V, Han S, Brinker A, Klock H, Caldwell J, & Gu XJ (2004). A time-resolved fluorescence resonance energy transfer-based HTS assay and a surface plasmon resonance-based binding assay for heat shock protein 90 inhibitors. Analytical Biochemistry 331, 349–357. [DOI] [PubMed] [Google Scholar]
- Zuehlke A, & Johnson JL (2010). Hsp90 and co-chaperones twist the functions of diverse client proteins. Biopolymers 93, 211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]