

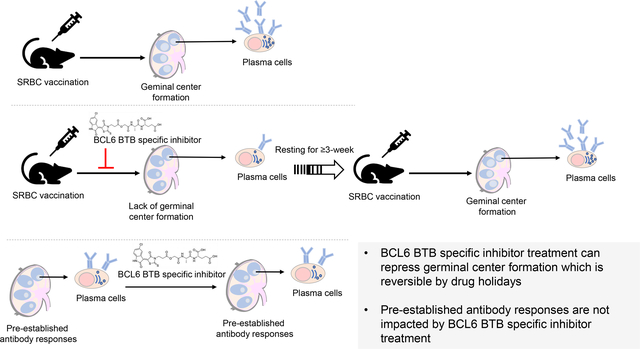
Abstract
Inhibition of the BCL6 BTB domain results in killing Diffuse Large B-cell Lymphoma (DLBL) cells, reducing the T-cell dependent germinal center (GC) reaction in mice, and reversing GC hyperplasia in nonhuman primates. The available BCL6 BTB-specific inhibitors are poorly water soluble thus limiting their absorption in vivo and our understanding of therapeutic strategy targeting GC. We synthesized a prodrug (AP-4-287) from a potent BCL6 BTB inhibitor (FX1) with improved aqueous solubility and pharmacokinetics (PK) in mice. We also evaluated its in vivo biological activity on humoral immune responses using the sheep red blood cells (SRBC)-vaccination mouse model. AP-4-287 had a significant higher aqueous solubility and was readily converted to FX1 in vivo after intraperitoneally (i.p.) administration, but a shorter half-life in vivo. Importantly, AP-4-287 treatment led to a reversible effect on (1) the reduction in the frequency of splenic Ki67+ CD4+ T cells, Tfh cells, and GC B cells; (2) lower GC formation following vaccination; and (3) a decrease in the titers of antigen-specific IgG and IgM antibodies. Our study advances the pre-clinical development of drug targeting BCL6 BTB domain for the treatment of diseases that are associated with abnormal BCL6 elevation.
Keywords: BCL6, Tfh, T cell activation, germinal center reaction, T-cell dependent immune response
Graphical Abstract
Introduction
The B-cell lymphoma 6 (BCL6) protein is an evolutionarily conserved zinc finger protein which recruits co-repressors (e.g., SMRT, N-CoR, and BCoR) to transcriptionally repress cell cycle checkpoint genes (e.g., TP53, CDKN1A, ART, and CHECK1) and attenuate the apoptotic response to DNA damage upon environmental stimuli [1–3]. The function of BCL6 protein is adapted by the immune system as a mechanism to tolerate B cell somatic hypermutations during germinal center (GC) reaction across vertebrates [1]. BCL6 is a well-known master transcription factor dictating GC reaction with high abundance in GC B cells and Tfh CD4+ T cells [4, 5]. Constitutive BCL6 expression in vivo leads to increase Tfh differentiation and its dysregulation is associated with development of diffuse large B-cell lymphoma (DLBCL) [6, 7]. Dysregulation of BCL6 is also found in other GC-associated malignancies (e.g., Burkitt lymphoma, follicular lymphoma and Hodgkin lymphoma) [8–11]. Most recently, upregulation of BCL6 protein has been identified as a potential feature of pan-cancer cells with its expression serving as a stress tolerance mechanism to survive from host apoptotic response to DNA damage [1, 2]. Therefore, BCL6 has been proposed as a therapeutic target for treating B-cell lymphoma or diseases associated with abnormal elevations in BCL6 expression [9, 12–14].
Recruitment and binding of co-repressors through the BTB domain of BCL6 is essential in mediating GC reaction. Recently, a novel BTB-specific BCL6 inhibitor (FX1) was identified using the Site Identification by Ligand Competitive Saturation (SILCS) approach [14, 15]. FX1 has a higher affinity than its natural ligand (SMRT), and impedes BCL6 from binding to its repressor proteins [14, 15]. FX1 can effectively act against large B-cell lymphoma cells ex vivo and in vivo, as well as inhibiting T-cell dependent immune responses in mice without any toxic effect [14, 15]. We also recently showed that FX1 treatment could lead to a reversal of lymphoid hyperplasia in rhesus macaque [16]. However, FX1 or 79-6 as the available BCL6 BTB-specific inhibitors both have limited development potential due to their high lipophilicity, poor aqueous solubility and poor bioavailability, which restricts their in vivo use [14] as BCL6 BTB-specific inhibitors. Although BCL6 degrading compound was found to be superior than BCL6 inhibition [17], poor solubility and poor bioavailability remains an issue.
In this study, we described and tested biological modulation of humoral responses by a synthesized prodrug (AP-4-287) of the potent BCL6 BTB inhibitor (FX1). We found improved aqueous solubility ex vivo and its in vivo activity in modulating humoral immune responses using the sheep red blood cells (SRBC)-vaccination mouse model.
Results
Synthesis of AP-4-287, a Prodrug of FX1.
We sought to improve aqueous solubility and biodistribution while preserving biological activity by converting FX1 into a pro-drug. A prodrug for FX1 was expected to improve the bioavailability of the active “drug” [18] after undergoing metabolic transformation in vivo to release the active drug species. In our studies, we synthesized an amino acid-based ester prodrug of FX1 (named AP-4-287, Fig. 1A) to target amino acid transporters. Amino acid transporters are membrane bound solute carrier (SLC) transporters that are abundantly expressed in many cells to enhance the uptake and utilization of nutrients in the cell [19, 20].
Figure 1. The Chemical Synthesis Process, Properties and Pharmacokinetic Analysis of Optimized Prodrug (AP-4-287).
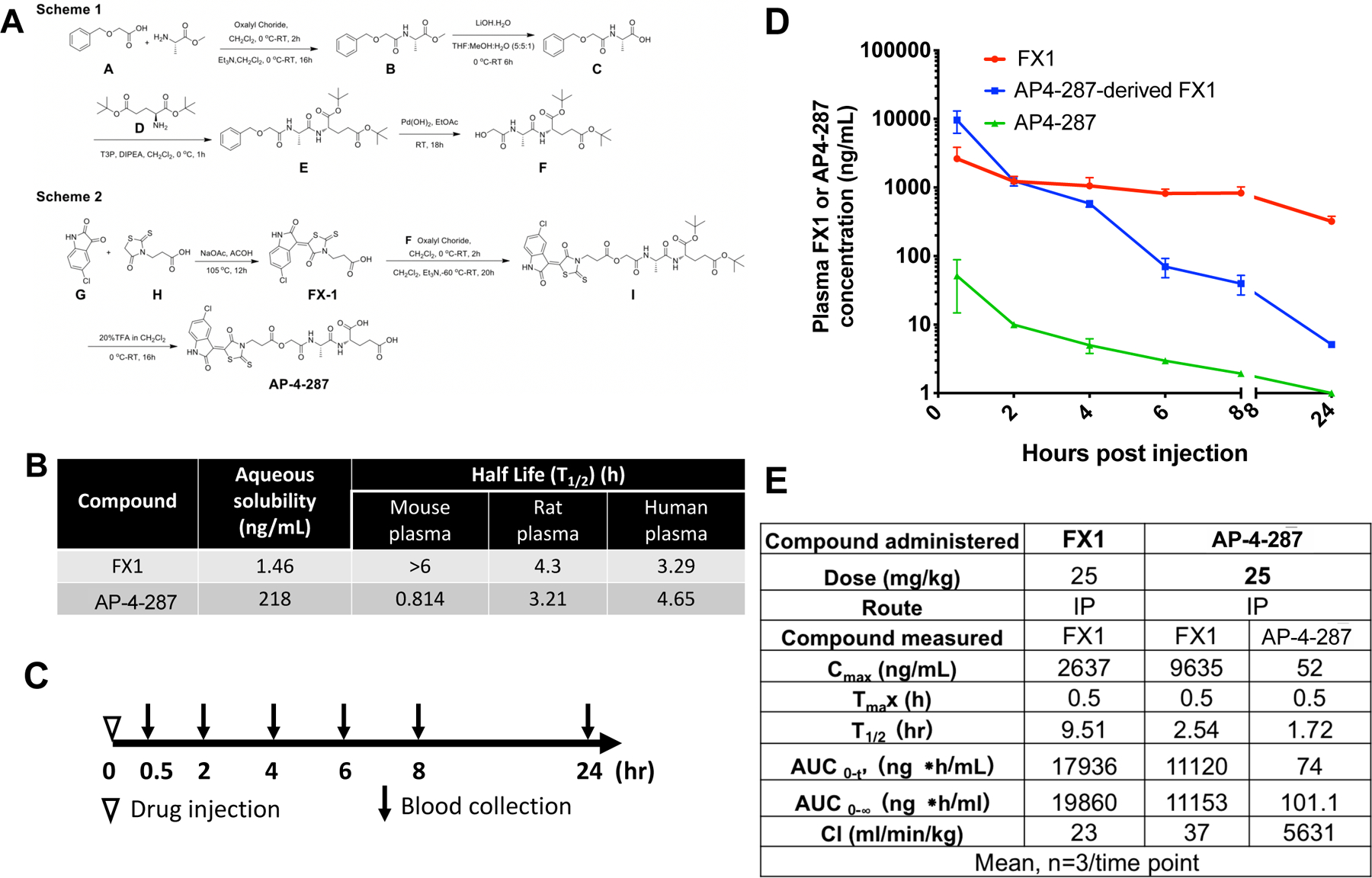
(A) Summary for the chemical synthesis of AP-4-287. The FX1 Pro-drug, AP-4-287, was synthesized using the synthetic routes shown in Scheme 1 and Scheme 2. In Scheme 1 the key intermediate F was synthesized from the precursors, 2-(benzyloxy) acetic acid, A, and L-alanine methyl ester which were coupled to form the amide, B. Lithium hydroxide ester hydrolysis provided the acid, C, which was coupled to di-tert-butyl-L-glutamate, D, to form compound E. Palladium catalyzed hydrogenation to remove the O-benzyl protecting group provided F. In Scheme 2, 5-chloroisatin, G, reacted with 3-(4-oxo-2-thioxothiazolidin-3-yl) propanoic acid, H, under basic conditions to provide FX1. FX1 was then treated with oxalyl chloride to generate the acid chloride of H which reacted with alcohol, F, to generate the O-tert-butyl protected pro-drug, I. Deprotection of the tertiary butyl esters under acidic conditions provided AP-4-287. (B) The aqueous solubility and ex vivo plasma stability of FX1 and AP-4-287 (average of triplicates in one experiment). (C) Study design for comparing pharmacokinetics of FX1 and AP-4-287 in mice. We administered 18 CD-1 mice with FX1 at 25mg/kg (in 100uL vehicle), and additional 18 CD-1 mice with AP-4-287 at 25mg/kg in the same volume of vehicle as FX1. Blood samples from three mice receiving FX1 or AP-4-287 were collected at 0.5, 2, 4, 6, 8 and 24hr after injection. We also administered three mice with 100uL vehicle and collected blood at 24hr to be used as negative control. (D) Plasma FX1 and AP-4-287 concentration were measured by HPLC-MS/MS assay for those animals receiving AP-4-287 (3 animals per timepoint in one experiment). For those animals receiving FX1, we only measured plasma FX1 concentration. Error bars represent mean ± SD (E) Data summary for the pharmacokinetics of AP-4-287 and FX1.
AP-4-287 had solubility in PBS buffer of 218 ng/mL presenting higher which is an ~150 folds increase in aqueous solubility compared to FX1 (Fig. 1B). AP-4-287 is an ester pro-drug with a shorter half-life (T1/2) in mouse plasma than in rat or human plasma based on the in vitro plasma stability assay, when it is hydrolyzed to FX1. While increased in solubility, AP-4-287 has similar stability in human plasma to FX1 with T1/2 in human plasma of 4.65 and 3.29-hour, respectively (Fig. 1B). In contrast to human plasma, in vitro plasma T1/2 for FX1 had a significantly longer stability over AP-4-287 in mouse plasma (Fig. 1B).
Pharmacokinetic Analysis of FX1 and AP-4-287 in Mice.
To compare the pharmacokinetic plasma exposure of active drug (FX1) as compared to prodrug (AP-4-287), we performed one i.p. administration of FX1 or AP-4-287 (25 mg/kg in 100μL) into CD-1 mice and collected blood at 0.5-, 2-, 4-, 6-, 8- and 24-hour post injection (Fig 1C). The in vivo derived plasma samples were analyzed by HPLC-MS/MS to identify the fraction of FX1 or AP-4-287 at each timepoint. Intraperitoneal administration of AP-4-287 retained very low concentrations of AP-4-287 in plasma suggesting that AP-4-287 was rapidly converted to FX1 soon after drug administration. Rapid conversion of prodrug to FX1 was consistent with peak FX1 levels observed at 0.5-hour after administration. Overall, AP-4-287 administration presented higher peak FX1 (Cmax=9635 ng/mL for those receiving AP-4-287; Cmax=2637 ng/mL for those receiving FX1), but shorter half-life for FX1 (T1/2=2.54-hour for those receiving AP-4-287; T1/2=9.51-hour for those receiving FX1) (Fig 1D&E) consistent with in vitro murine plasma stability tests. Therefore, the Cmax of the prodrug was ~3.65 folds than that of FX1 (Figure 1E). The FX1 exposure levels (AUC0-t) within 24-hour were 11120 ng.h/mL when receiving AP-4-287 administration and 17936 ng.h/mL when receiving FX1 administration (Fig 1D&E). Both drugs were cleared within 24hr after drug administration (Fig 1D). The in vivo T1/2 for AP-4-287 was 1.72hr and AUC0-t was 74 ng.h/mL, supporting the rapid conversion from AP-4-287 converted to FX1.
AP-4-287 Repressed SRBC-mediated Germinal Center Reaction.
To evaluate the in vivo activity of AP-4-287, we performed an 8-day course daily treatment with 25mg/kg, 15mg/kg or 5 mg/kg AP-4-287 or vehicle 3 days after SRBC vaccination. We tested a maximal dose of 25mg/kg dosed three times a day based on bioactive doses of FX1 described at 80mg/kg [14] and the in vivo half-life (T1/2) of AP4-287 determined at ~1/3 FX1 T1/2. The mice were then necropsied 11 days after SRBC vaccination to examine the frequency of Tfh and GC B by flow cytometry method and GC formation by H&E and immunohistochemistry staining (see Fig 2A for the experimental schema). The frequency of CXCR5+PD1hiCD4+ Tfh (Fig 2B) and GL7+CD95+B220+ activated B cells (Fig 2E) were significantly lower in the animals receiving AP-4-287 comparing to their counterparts from the animals receiving vehicle. The reduction of Tfh and activated B cells due to the treatment with AP-4-287 was in a dose-response manner (Fig 2 C & F) supporting the specificity of AP-4-287 in repressing GC reaction from SRBC vaccination. Furthermore, we confirmed that AP-4-287 treatment reduced frequency of the GC induced by SRBC vaccination with H&E staining method (Fig 2H, upper panels) and immunohistochemistry staining using anti-B220 antibody to identify B cells (Fig 2H, lower panels). As a control for effects by FX1 if administered directly at 80 mg/kg FX1 (dose used in Gardenas M G [14]), we observe that FX1 dosing can lower GC B but it did not reduce the frequency of CD4+ Tfh within GCs (Fig 2 D&G).
Figure 2. AP-4-287 repressed germinal center formation & reduced Tfh CD4+ T cells in SRBC-vaccinated mice.
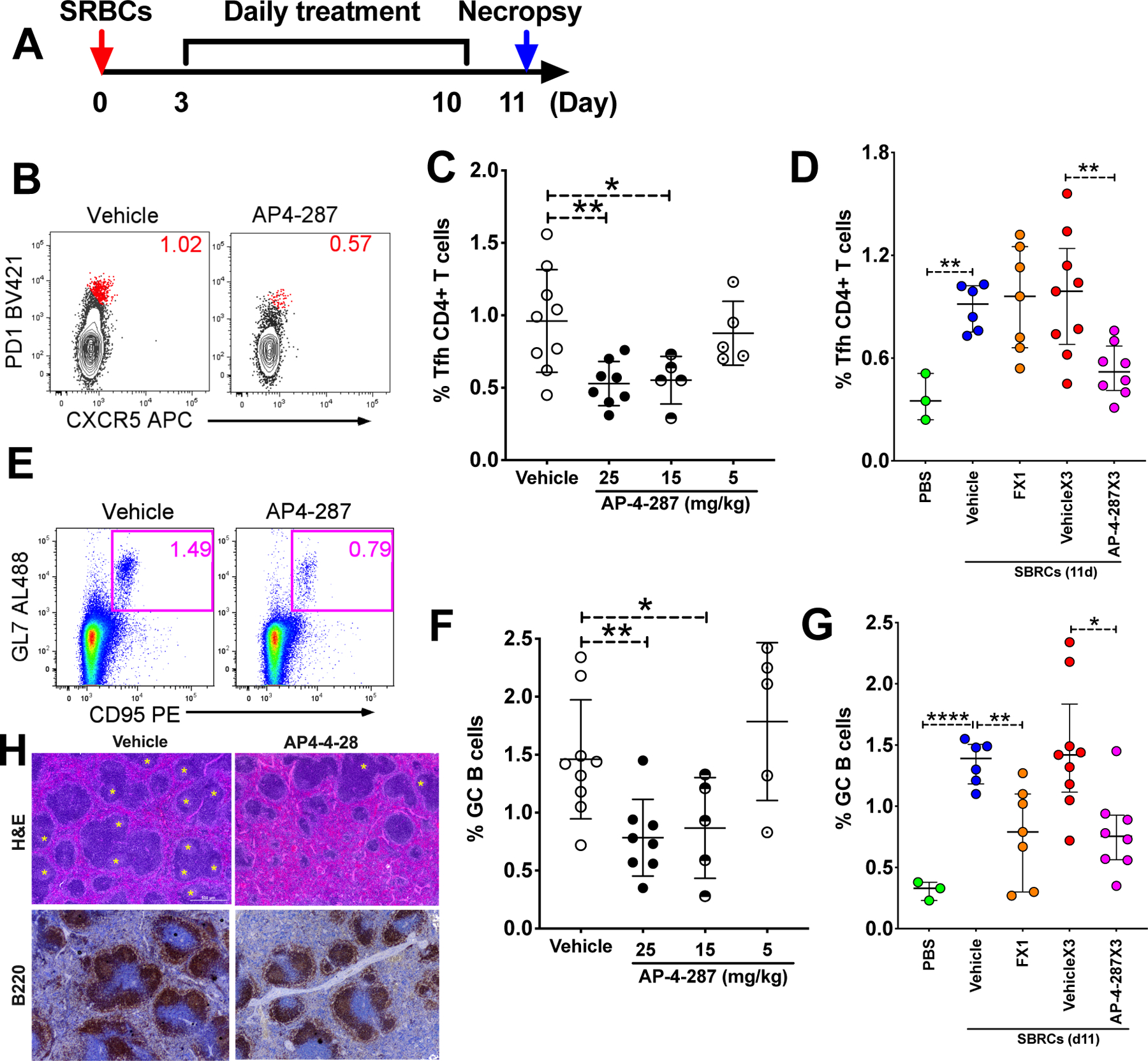
(A) Study design for evaluating AP-4-287 in repressing GC reaction in a sheep red blood cell (SRBC) vaccinated T cell-dependent mice model. All mice in the study received 100ul 10% SRBC i.p. on day 0 and underwent resting for 3 days. An 8-days course of AP-4-287 treatment (25mg/kg in 100uL vehicle three times daily with 5hr apart) or vehicle treatment started by day 4. By day 11, all mice were euthanized for collecting necropsy specimens including blood and spleen. (B) Representative flow plot showing AP-4-287 reduced the frequency of splenic PD1hiCXCR5+Tfh CD4+ T cells. (C) AP-4-287 reduced the frequency of Tfh CD4+ T cells in a dose-dependent manner. (D) Data obtained from cytometry analysis showed that the frequency of Tfh CD4 T cells in the mice receiving 80mg/kg FX1, or 25mg/kg AP4-287 or vehicle. (E) Representative flow plot showing AP-4-287 reduced GC B cells (B220+GL7+CD95+) in the spleen. (F) AP-4-287 reduced GC B cells in a dose-dependent manner. (G) Data obtained from cytometry analysis showed that the frequency of Tfh CD4 T cells in the mice receiving 80mg/kg FX1, 25mg/kg AP4-287 or vehicle. (H) Representative images (4x magnification) stained with H&E staining method and IHC using antibody against B220 showing AP-4-287 reduced GC formation in the spleens of SRBC-vaccinated mice. Welch-test was performed to compare the frequencies of Tfh and GC B cells between two groups. * p<0.05; ** p<0.01; ****p<0.0001. All data are from 2–3 independent experiments with 3 mice per experiment, except for PBS treatment in Figure C right and E right.
AP-4-287 Selectively Repressed De novo, but not pre-established, Humoral Immune Responses.
To corroborate that in vivo AP-4-287 could reduce antigen-specific antibody responses, we analyzed vaccination-induced SRBC-specific IgM and IgG1 levels, as well as ELISA assay to quantify pan IgG and IgM. At 11 days after vaccination, we observed that the MFI of anti-mouse IgG1 or IgM labeled SRBC incubated with plasma from 25mg/kg AP-4-287-treated animals were lower than their counter parts incubated with plasma from vehicle-treated animals (Fig 3A & D). Lower SRBC-specific IgM and IgG1 was also observed when using plasma from mice receiving AP-4-287 at 5mg/kg or 15mg/kg (Fig 3B & E), as well as in mice receiving FX1 (Supplemental Fig S3 A&B). Importantly, there was no difference in the total titer of plasma pan IgG and IgM between vehicle- and AP-4-287-treated groups (Fig 3C & F) or FX1 (Supplemental Fig S3 C&D), ruling out a pan inhibition of antibody production with drug exposure.
Figure 3. AP-4-287 repressed SRBC-specific antibodies in the vaccinated mice.
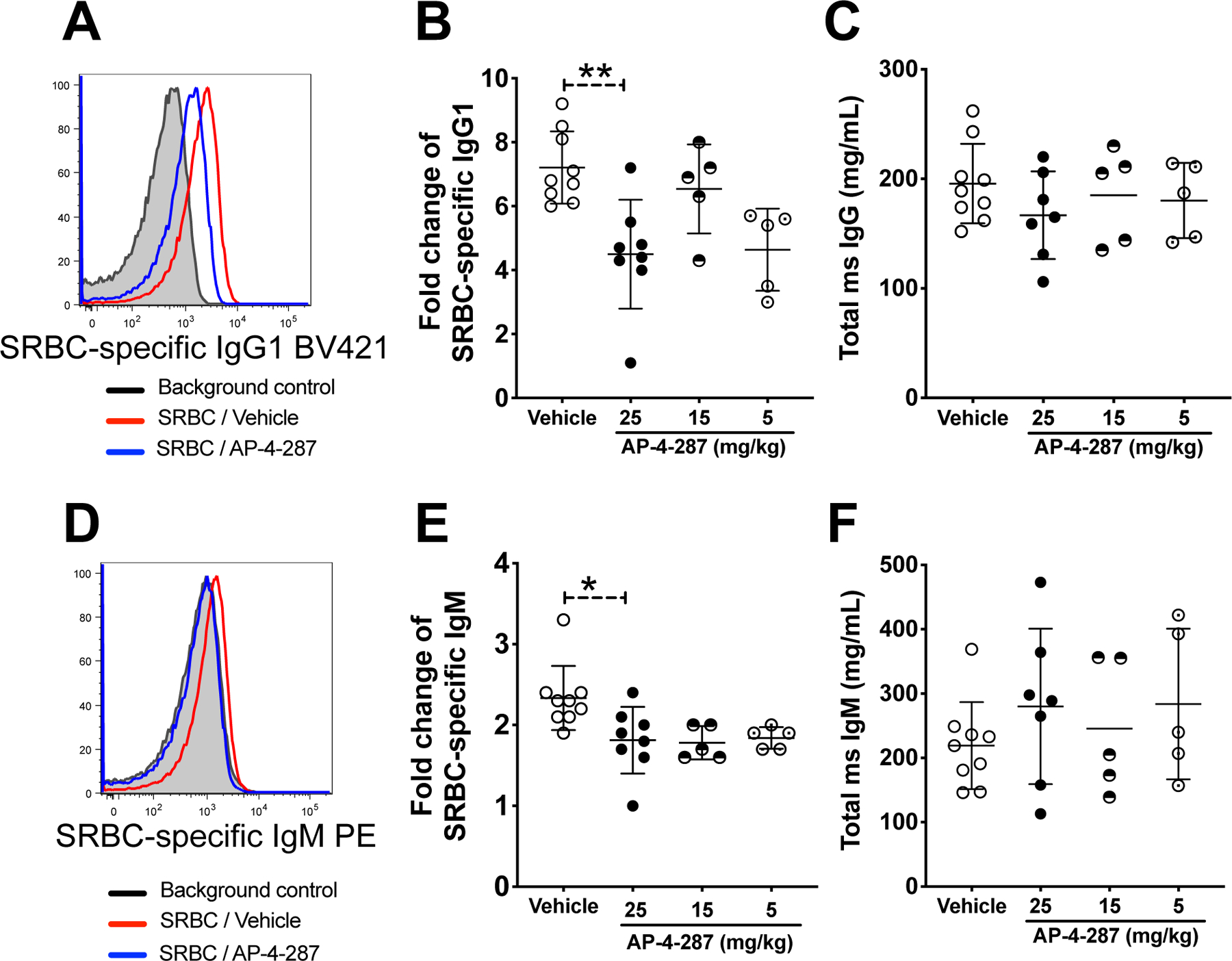
(A) Representative flow plot showing the mean fluorescent intensity (MFI) of SRBC incubated with plasma from SRBC-vaccinated mice received either vehicle or AP-4-287 treatment and BV421-conjugated anti-mouse IgG1. (B) AP-4-287 reduced the production of SRBC-specific IgG1 in a dose-dependent manner. (C) AP-4-287 did not change the production of pan IgG in vaccinated mice measured with ELISA method. (D) Representative flow plot showing the mean fluorescent intensity (MFI) of SRBC incubated with plasma from SRBC-vaccinated mice received either vehicle or AP-4-287 treatment and PE-conjugated anti-mouse IgM. (E) AP-4-287 reduced the production of SRBC-specific IgM in a dose-dependent manner. (F) AP-4-287 did not change the production of pan IgM in vaccinated mice measured with ELISA method. We used Welch-test to compare the results between two groups. * p<0.05; ** p<0.01. All data are from 2–3 independent experiments with 3–5 mice per experiment.
AP-4-287 Reduced T cell activation and Tfh differentiation in the GC.
Based on the observation that FX1 reduced T cell activation ex vivo [21] and in vivo [16], we analyzed the frequency of Ki67+CD4+ T cells by flow cytometry and the expression of Ki67+ cells by immunohistochemistry staining in treated mice. Our data confirmed that the frequency of Ki67+CD4+ T cells from the spleen of mice receiving AP-4-287 was lower when compared to those from vehicle-treated mice (Fig 4A). The AP-4-287-mediated reduction in frequency of splenic Ki67+ CD4+ T was notable at 25mg/kg but absence when dosing at 15mg/kg or 5mg/kg (Fig 4B). As expected, immunocytochemistry showed the location of Ki67+ cells was primarily in the GC with a reduction clearly noted in AP-4-287 treated mice (Fig 4C). At 25mg/kg AP-4-287, the frequency of CD4+ Tfh precursor (CXCR5− PD1+; Fig 4D) also trended to be lower (Fig 4E). Together, our data supports that AP-4-287 can lower the activation / differentiation of GC Tfh CD4+T cells.
Figure 4. AP-4-287 repressed Ki67+ CD4+ T cells in the GC and trended to lower Tfh precursor cells in SRBC-vaccinated mice.
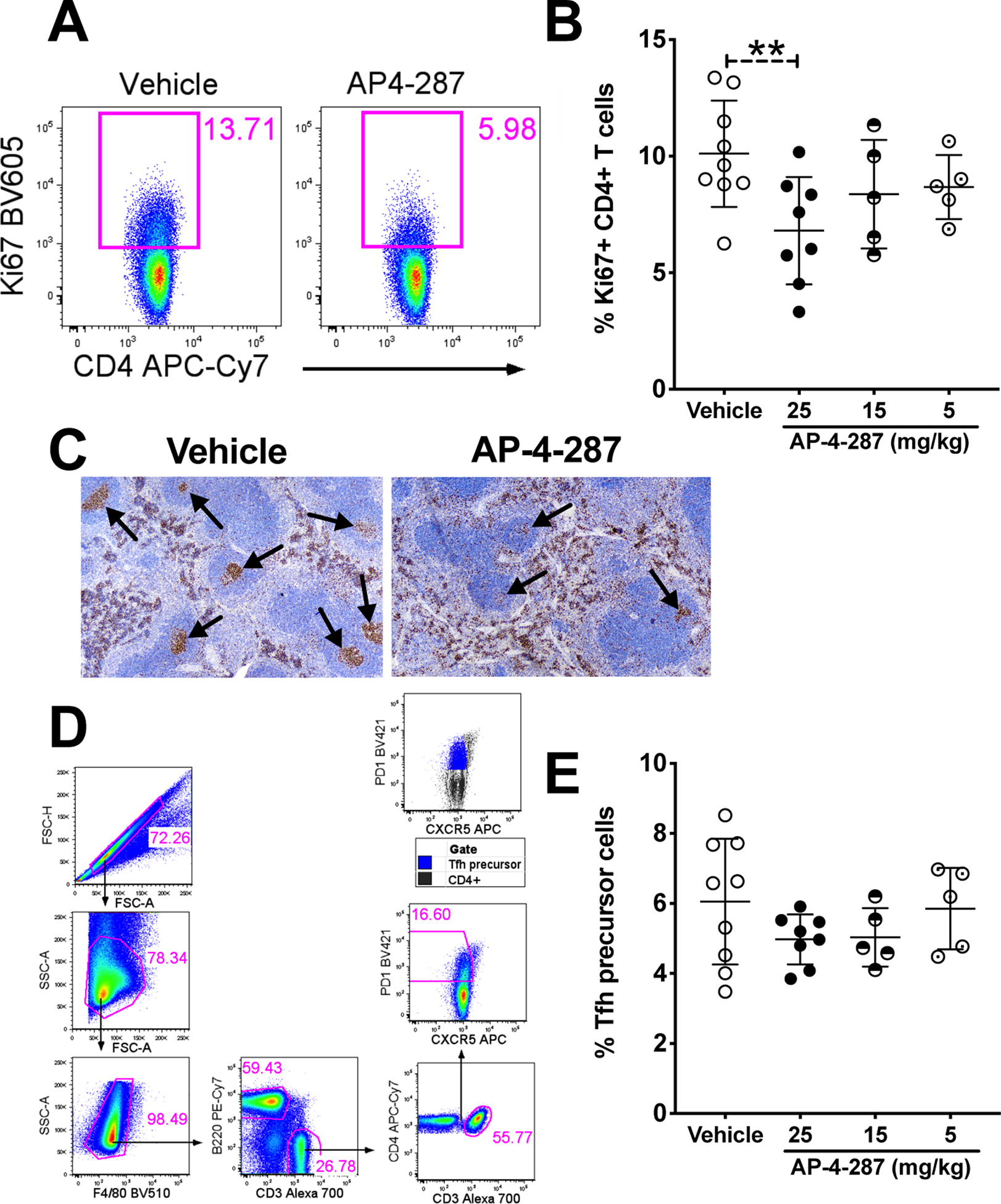
(A) Representative flow plot showing AP-4-287 reduced Ki67+CD4+ T cells in SRBC-vaccinated mice. (B) AP-4-287 reduced the frequency of Ki67+CD4+ T cells in a dose-dependent manner. (C) Representative IHC images (4x magnification) showing AP-4-287 reduced Ki67+CD4+ cells in the GC of spleen from SRBC-vaccinated mice. (D) Gating strategy for analyzing CXCR5−PD1dimTfh precursor CD4+ T cells. (E) AP-4-287 trended to reduce Tfh precursor CD4+ T in a dose-dependent manner. Welch-test was performed to compare the results between two groups. * p<0.05; ** p<0.01. All data are from 2–3 independent experiments with 3–5 mice per experiment.
AP-4-287-mediated inhibitory effect on GC formation and antibody responses are reversible.
We performed an additional study to test the effects of adding a 2nd SRBC vaccination 3-week after the last dose of vehicle or AP-4-287 treatment (Fig 5A) in order to test the reversibility of targeting the BCL6 BTB domain. The spleen and blood samples were harvested at 11 days after 2nd SRBC vaccination for analysis of splenic cell phenotype (Tfh & activated B cells) and the antibody responses in the plasma (SRBC-specific IgG1 and IgM) as above. We observed the 2nd SRBC-vaccination induced comparable level of Tfh CD4+ T cell regardless of prior treatment with vehicle or AP-4-287 (Fig 5B). The levels of GC B cells and SRBC-specific IgG1 from AP-4-287-treated groups after 2nd SRBC-vaccination were similar to their counterparts from vehicle-treated groups after SRBC vaccination once (Fig 5C & D), but highest in the group that received vehicle treatment and the 2nd SRBC-vaccination (Fig 5C & D). By contrast, SRBC-IgM were comparable in both groups after 2nd SRBC vaccination (Fig 5E). The Ki67+CD4+ T cells and CD4+Tfh precursor cells were also similar in both groups and reached a level that is higher than their counter parts in the group that SRBC vaccination once and vehicle treatment (Fig 5F & G). In summary, these data supported that AP-4-287-mediated GC reaction and antibody responses could be recovered following drug discontinuation without any sustained disruption of future humoral responses.
Figure 5. AP-4-287-mediated repression on germinal center reaction and antibody responses in SRBC-vaccinated mice were reversible.
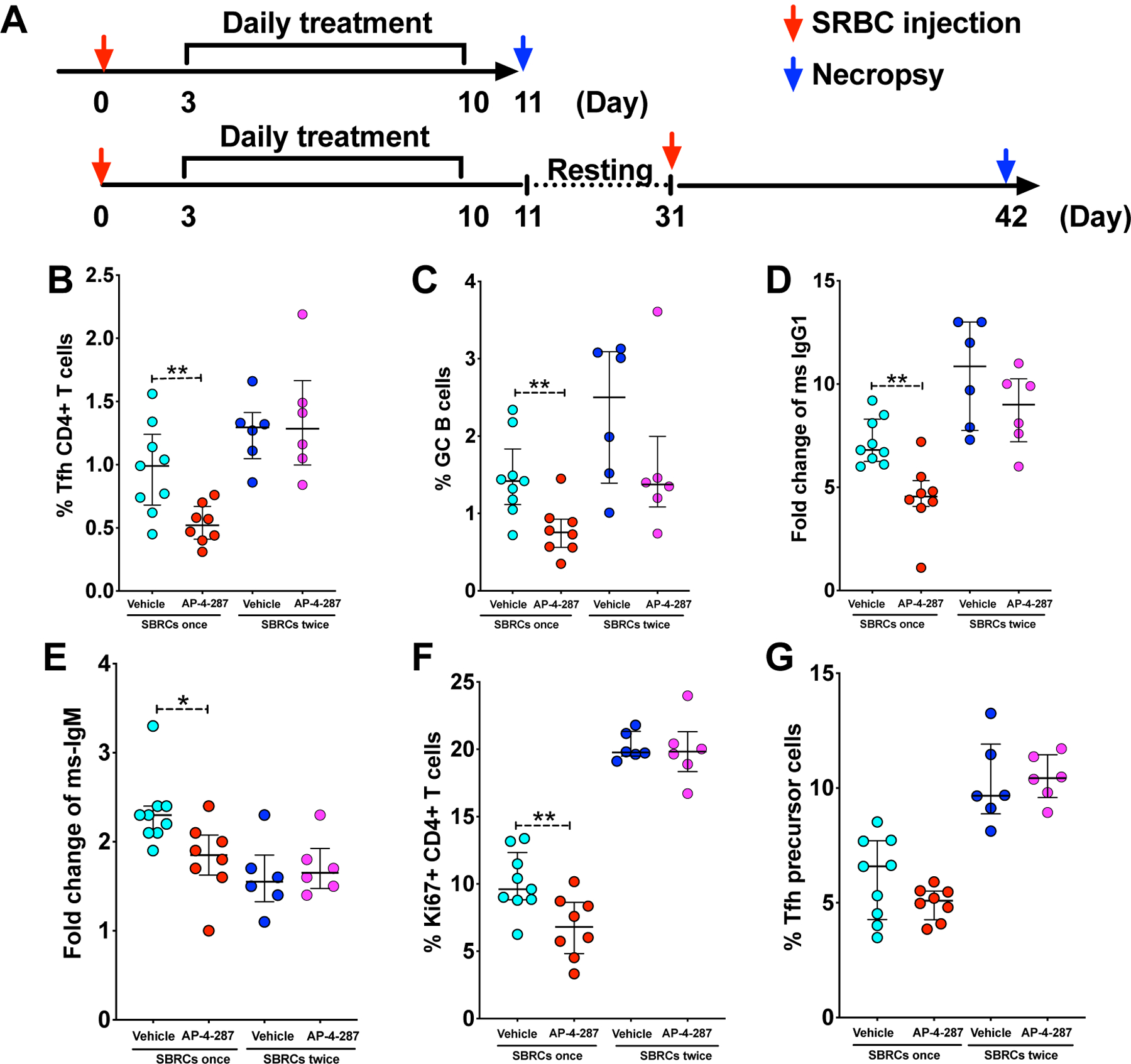
(A) Study design to test if 3-week resting period will allow the mice immune system to recover and responses to 2nd SRBC-vaccination. All mice in the study received the 1st SRBC vaccination, 3-day resting and an 8-days course of AP-4-287/vehicle treatment as previously. Then half of the mice receiving AP-4-287 or vehicle was euthanized and for necropsy sample collection; the rest half underwent a resting period of 3-week and received 2nd SRBC vaccination following with necropsy at 11 days later. (B-G) The level of splenic Tfh (B), splenic activated GC B (C), SRBC-specific IgG1 (D), SRBC-specific IgM (E), Ki67+CD4+ T cells (F) and Tfh precursor cells (G) were measured by flow cytometry. Welch-test was performed to compare the results between two groups. * p<0.05; ** p<0.01. All data are from 3–5 independent experiments with 3–5 mice per experiment.
In addition, in vivo safety of AP-4-287 was supported by our observation that administration of AP-4-287 did not cause histological changes in the brain, heart, lung, liver and kidney (Supplemental Fig S4), or decrease the level of pan-IgG /IgM, or permanent damage of host humoral immune response.
Discussion
We demonstrate that FX1 prodrug, AP-4-287, as a novel BTB-specific inhibitor, increases aqueous solubility and can be readily converted to FX1 in vivo with retained activity against GC formation. Specifically, AP-4-287 exposure can lead to: (1) reduction in the frequency of splenic Ki67+ CD4+ T cells, Tfh cells, and GC B cells; (2) decrease in GC formation following SRBC-vaccination; and a (3) decline in the titers of SRBC-specific IgG and IgM antibodies without any concurrent adverse effect on pre-established humoral immune responses. The frequency of splenic GC CD4+ Tfh in SRBC-vaccinated mice positively correlated with the frequency of GC B cells in the GC, the activation of splenic CD4+ T cells (Ki67+), and the level of SRBC-specific IgM and IgG (Supplemental Fig S5). We also show that targeting the BCL6 BTB domain with AP-4-287 is reversible after a washout period.
The in vivo safety of AP-4-287 was supported by our observation that administration of AP-4-287 did not cause histological changes in the brain, heart, lung, liver and kidney (Supplemental Fig S5), or decrease the level of pan IgG /IgM (surrogates for pre-established humoral immune responses), or permanent damage of host humoral immune response. Due to a shorter half-life (T1/2) in mice sera and subsequent PK analysis, AP-4-287 administration required 3 times daily for a course of 8-day to achieve similar effect as FX1 but with lower dose. It is important to note that stability of AP-4-287 are different between mouse and human sera with greater stability by AP-4-287 observed in presence of human sera. Therefore, the in vivo dosing strategy used in this study on testing biological effects in mice likely need to be adjusted for the PK analysis for AP-4-287 in humans. While prodrug design targets cell surface amino acid transporters remains to be determined if AP-4-287 is a direct substrate for amino transporters such as PEPT1 [22].
BCL6 can bind to the promoter of >3000 genes and transcriptionally inhibit >1000 genes inclusive of genes encoding cell cycle proteins [23]. Aside from the anti-GC effect and similarity to FX1, we also observed that AP-4-287 lowered the expression of the cell cycle protein Ki67 in CD4+ T cells [21]. Ki67 is generally deemed an activation or proliferation marker. Future experiments are needed to identify the specific gene(s) and proteins that are affected by AP-4-287 treatment in order to address its mechanism of action in vivo and potential utility in diseases associated with abnormal T cell activation. Relevant to normal responses, activation and induction of antigen-specific Tfh is crucial for the vaccine-mediated antibody response. A recent study showing that blocking BCL6, interleukin-21 (IL-21), ICOS, and CD40 signaling led to diminished anti-HA antibody production [24] stresses that any anti-BCL6 strategy may impose a risk of vaccine failure. While our data directly support the role of BCL-6 in the generation of vaccine responses as AP-4-287 can effectively inhibit responses we also show that once drug exposure is removed, vaccine responses can return to normal supporting full reversibility of drug effects.
As BCL6 transcriptionally regulates the differentiation of GC Tfh, B cell activation, and GC formation [14], it is expected that AP-4-287 could potentially target diseases beyond lymphomas that are associated with elevated GC activity (e.g. autoimmune diseases, graft-versus-host-disease) [25] and/or have pathogenic roles for Tfh such as retention of persistent HIV following antiretroviral therapy (ART). BCL6+CD4+ Tfh are highly susceptible to HIV infection and thought to harbor replication-competent HIV reservoirs within GC after ART [26–28]. It remains to be determined if AP-4-287 could be beneficial if added to ART as BCL6 inhibition by FX1 was able to lower the frequency of Tfh CD4+ T and Tfh precursor cells in vivo as tested in non-human primates [29] and can reduce HIV replication when added in vitro [21].
Our study has several limitations and future studies will be needed to address the questions below. First, the PK data from mouse model will likely not predict the PK in humans as noted above. Second, long-term immunotherapy effects of chronic AP-4-287 exposure remain to be determined. Third, we only evaluated effect of AP-4-287 on humoral immune responses with SRBC model which probably will have limited coverage of antigen specific responses.
Materials and Methods
Chemical synthesis of FX1 prodrug, AP-4-287.
The FX1 Pro-drug, AP-4-287, was synthesized using the synthetic routes summarized in Fig 1A. In Scheme 1 the key intermediate F was synthesized from the precursors, 2-(benzyloxy) acetic acid (A), and L-alanine methyl ester which were coupled to form the amide (B). Lithium hydroxide ester hydrolysis provided the acid (C), which was coupled to di-tert-butyl-L-glutamate (D), to form compound E. Palladium catalyzed hydrogenation to remove the O-benzyl protecting group provided F. In Scheme 2, 5-chloroisatin (G), reacted with 3-(4-oxo-2-thioxothiazolidin-3-yl) propanoic acid (H), under basic conditions to provide FX1. FX1 was then treated with oxalyl chloride to generate the acid chloride of H which reacted with alcohol, F, to generate the O-tert-butyl protected pro-drug (I, AP-4-287). Deprotection of the tertiary butyl esters under acidic conditions provided AP-4-287. The purity of AP-4-287 and intermediate compound were evaluated by NMR and LCMS Spectra (supplemental materials).
Aqueous solubility test.
The aqueous solubility of candidate BCL6 BTB-specific inhibitors, FX1 and its prodrug AP-4-287, was performed at the Alliance Pharm (Malvern, PA). Briefly, 1 mL of PBS buffer (pH 7.4) was added to approximately 10 mg of test compound. The solutions were sonicated for 30 minutes and then vortexed at a low speed for 30 minutes. The solutions then sat at room temperature for at least 16 hours. Next, the solutions were filtered through a 0.22-micron filter followed by diluting 1:2, 1:10, 1:100, and 1:1,000, 1:10,000, and 1:100,000 in triplicate in acetonitrile (ACN): water [1:1, volume/volume (v/v)]. The final sample solutions were mixed with internal standard followed by LC MS/MS analysis. Solubility was determined according to the calibration curve generated from at least 5 concentration standards. The concentrations for FX1 standards were 0.5, 1, 2, 10, 25 and 50 ng/mL; the concentrations for AP-4-287 standards were 2, 10, 25, 50, 100 and 200 ng/mL.
In vitro plasma stability assay.
The in vitro plasma stability of candidate BCL6 BTB-specific inhibitors was performed at the Alliance Pharm (Malvern, PA). Briefly, the inhibitors were dissolved in DMSO and dilute into 0.1mM with DMSO: water (1:1, v/v). Mouse, rat and human plasma purchased from qualified vendors were thaw and pre-warm in the water bath (37 °C) prior incubation with compounds at 100:1 ratio (495 μL plasma with 5μL compound working solution) in the incubator (37°C, 5% CO2) with shaking at 200rpm. At 0, 15, 30, 60, and 120 minutes later, 50 μL mixture were collected and mixed with stop solution (ACN containing 100ng/mL tolbutaminde) at 5:1 (v/v) by vortex at 17 rpm for 3 min. The samples were centrifuged at 3500 rpm for 15 minutes. Finally, 100 μL supernatant were collected into a fresh plate and analyzed by HPLC-MS/MS. The data analysis was performed using the natural logarithm of the peak area ratio (analyte peak area/IS peak area) is plotted against time and the following equation is fit to the data: Ct=C0*e−kt (k is the elimination rate constant). The half-life (T1/2) is calculated using the equation: T1/2=0.693/k.
Mice pharmacokinetic (PK) study and blood collections.
A total of 39 male adult CD-1 mice (8 weeks old) were used in the pharmacokinetic (PK) study at the animal facility of Alliance Pharm (Malvern, PA) under the protocol #112754 approved by the IACUC committee at the Wistar Institute. Eighteen mice received one administration of FX1 at 25mg/kg in 100 μL vehicle that comprised 30% propylene glycol, 65% PEG-400, 5% Dextrose (5%) intraperitoneally (i.p.) and underwent six blood collection with three mice at each time point (0.5, 2, 4, 6, 8 and 24 hours later. Fig 1C); another eighteen mice received one administration of AP-4-287 at 25mg/kg in 100 μL vehicle i.p. and underwent six blood collections as above; the rest three mice received 100 μL vehicle and underwent blood collection at 24hr later as the negative control. Plasma samples were fractionated for PK analysis of AP-4-287 and/or FX1 level by HPLC-MS/MS (Alliance Pharm, Malvern, PA).
Sheep red blood cell (SRBC) vaccination study, BCL6 BTB-specific inhibitor treatment, and sample collections.
Male adult C57/BL6 mice (8–12 weeks old) from Jackson Laboratory (Bar Harbor, ME) were used following the IACUC approved at the Wistar Institute (protocol #112754). To establish the SRBC vaccination protocol (Supplemental Fig S1), we first performed i.p. administration with 100uL PBS or 100uL 10% SRBC (LAMPIRE Biological Laboratories, Inc. Pipersville, PA) and collect blood and spleen at 7 days or 11 days later. The optimal time point for monitoring the host immune response turned out to be 11 days post vaccination, which was applied in all the studies later on. FX1 was administered three days after SRBC vaccination at a dose of 80mg/kg in 100uL vehicle once daily for an 8-day treatment course; AP-4-287 treatment was performed using a similar schedule as FX1 except that doses 25, 10 and 5mg/kg in 100 uL vehicle at three times daily (TID) with 5 hours apart were used. Vehicle was administered at volume and schedule as control for FX1 or AP-4-287 treatment. At 11 days after SRBC vaccination, the mice that received vehicle, FX1 or AP-4-287 treatment were euthanized with CO2, plasma was collected from the blood, the spleen was collected for cell isolation and histology studies. The mice torsos (with open chest, brain and stomach) were fixed with 10% formalin-phosphate buffer (Thermo Fisher, MA) for 48hr and kept in 70% ethanol (Thermo Fisher, MA) prior collecting tissues (e.g. intestine, heart, lung, kidney, brain et al) for histology analysis. We also performed secondary SRBC vaccination after resting the animals for 3 weeks, followed by necropsy of the animals at 11 days later to exam if the impact from treatment with BCL6 BTB-specific inhibitors could be recovered. The blood, plasma, and tissue sample collections were similar described above.
Mouse spleen tissue harvest and mononuclear cell isolation.
Spleen tissues were obtained and split into 2 parts after removing adipose tissues. Half was used for mononuclear cell isolation and preserved in 3 mL complete medium comprised of RPMI1640 (Cellgro; Manassas, VA) supplemented with 10% fetal bovine serum (Gibco, cat no. 26140-079; Grand Island, NY, USA), 100 IU/mL of penicillin/streptomycin (EMD Millipore, Billerica, MA) and 2 mM of L-glutamine (Cellgro; Manassas, VA); the other half was used for making paraffin blocks and preserved in 10% formalin-phosphate buffer (Thermo Fisher). Mononuclear cell isolation from spleen tissue was prepared by meshing the cells through a 70μM cell strainer (BD; San Jose, USA) with a sterile 3 mL syringe plunger (BD; San Jose, USA). The cell suspensions underwent centrifugation at 2000 rpm for 10 minutes (Beckman, Allegra X-12R, Brea, CA, USA) and washed with 10mL complete medium (PBS containing 2% FBS). Two million splenic mononuclear cells (SMC) were used for flow staining, and the rest cells were suspended in BAMBANKER™ serum-free cell culture freezing medium (Wako Laboratory Chemicals, cat no. 302-14681; Richmond, VA) and stored in liquid nitrogen until further analyses.
Flow cytometry and data analysis.
Two million of SMC were stained for flow cytometry as previously described to analyze the expression of surface and intracellular markers as well as cellular composition using the 4-laser FACS LSRII-14 (Becton Dickinson; San Jose, USA)[30], as well as following the guideline for the use of flow cytometry reported by Cossarizza et al [31]. Antibodies used in these analyses were purchased from either BD or Biolegend and included anti-mouse CXCR5 (clone 2G8), anti-BCL6 (clone K112-91), anti-mouse PD1 (clone RMP-130), anti-mouse CD95 (clone SA367H8), anti-mouse B220 (clone RA3-6B2), anti-mouse CD4 (clone RM4-5), anti-mouse CD3e (clone 500A2), anti-mouse F4/80 (clone BM8), anti-mouse Ki67 (clone 16A8). 7AAD were used to stain nuclei. A minimum of 50,000 CD4+ T cells were collected, and post-acquisition analysis performed by Y.C. using FlowJo (Version 9.6, TreeStar) software. The data gating strategy is shown in Supplemental Fig S2.
Histochemical staining and imaging.
The spleen and other tissues (e.g. intestine, heart, lung, kidney, brain et al) were processed into paraffin blocks for H&E staining. Briefly, tissue was fixed in 10% neutral-buffered formalin, sectioned to 5 μm thickness, and stained using the hematoxylin and eosin (H&E) staining method at the Wistar Histology Core. The images of stained tissue sections were captured under regular light microscopy at 100x magnification at the Wistar Imaging Core. Spleen tissue sections were used to examine the germinal center formation after SRBC vaccination and treatment with vehicle, FX1 or AP-4-287. We also performed histology analysis of other tissues (e.g. live, heart, lung, kidney, brain et al) to determine the integrity of multiple organs associated with drug administration.
Immuno-histochemistry staining and imaging.
Immuno-histochemistry staining was performed by the staff at the Wistar Histology Core. Briefly, the spleen tissue sections were stained with anti-mouse Ki67 (polyclonal, #Ab15580; Abcam, Cambridge, UK) or anti-CD220 (clone RA3-6B2, Thermo Fisher) prior incubation with HRP-conjugated secondary antibody and substrate to produce a brown color for detection of tissue antigen. The images of stained tissue sections were captured under regular light microscopy as previously described at the Wistar Imaging Core. Adobe Photoshop software (Version 7.0; Adobe Systems; Mountain View, CA) was used to process and assemble the images.
Mouse pan antibody and SRBC-specific antibody detection.
Detection of mouse pan IgM and IgG was performed using an ELISA method with the kit from ThermoFisher Scientific following the manufacturer instructions. Evaluation of the titer of SRBC-specific IgM and IgG1 was performed according to the method developed by Ellen J. McAllister and colleagues[32]. Briefly, 20 μL 2% SRBC were incubated with 5 μL pre-diluted mouse plasma (1:5 dilution using PBS) for 5min following with an additional 20-minute incubation with fluorescence conjugated anti-mouse IgM (clone RMM-1) or IgG1 (clone RMG1-1) (Biolegend, San Diego, CA). The stained SRBC was washed with 1mL PBS, re-suspended with 1 mL 1% paraformaldehyde (PFA) (Electron Microscope Science, Hatfield, PA) and proceeded with analysis using the 4-laser FACS LSRII-14 (Becton Dickinson; San Jose, USA). Isotype antibodies were used to stain SRBC after incubation with mouse plasma as negative control. The mean fluorescent intensity (MFI) of SRBC that were incubated with plasma from vaccinated and vehicle- or AP-4-287-treated animals was obtained and normalized to the MFI of SRBC incubated with unvaccinated mice to compare the changes in titer of SRBC-specific antibodies (Data were presenting as fold changes in the figure).
Statistical analysis.
Data is summarized as mean and standard deviation. Unpaired t-test with Welch correction was used to compare difference between two groups. Spearman test was used for correlation analysis. Data were analyzed and graphed using GraphPad Prism v8.0c software (San Diego, CA). Two-sided P <0.05 was considered significant.
Supplementary Material
Acknowledgements:
We thank Aubrey Leso and James Hayden from the Wistar Imaging Shared Resource for capturing the images from the H&E stained slides. We are grateful for the technical assistance from Fangping Chen from the Wistar Histology Core for H&E and immunohistochemistry staining, and Denise DiFrancesco, Emily Conicello, Marcia Houston-Leslie and Malachi Weight from the Wistar Animal Facility for the mice drug injections and necropsy sample collections. We are grateful for the Shared Resources Facilities at the Wistar Institute supported by a Cancer Center Support Grant (P30CA010815). This study was supported by The BEAT-HIV Delaney Collaboratory (UM1AI126620), and Wistar Cancer Center Grant (P30 CA10815). This work was supported by the following grants: U01AI110434; P30 AI 045008; P30 CA10815 and the Robert I. Jacobs Fund of The Philadelphia Foundation. LJM is also supported by the Herbert Kean, M.D., Family Professorship. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Conflict-of-interest disclosure: The authors declare no commercial or financial conflict of interests.
Data availability statement:
The data that supports the findings of this study are available in the supplementary material of this article.
Reference
- 1.Fernando TM, Marullo R, Pera Gresely B, Phillip JM, Yang SN, Lundell-Smith G, Torregroza I, Ahn H, Evans T, Gyorffy B, Prive GG, Hirano M, Melnick AM and Cerchietti L, BCL6 Evolved to Enable Stress Tolerance in Vertebrates and Is Broadly Required by Cancer Cells to Adapt to Stress. Cancer Discov 2019. 9: 662–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ranuncolo SM, Wang L, Polo JM, Dell’Oso T, Dierov J, Gaymes TJ, Rassool F, Carroll M and Melnick A, BCL6-mediated attenuation of DNA damage sensing triggers growth arrest and senescence through a p53-dependent pathway in a cell context-dependent manner. J Biol Chem 2008. 283: 22565–22572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Basso K, Saito M, Sumazin P, Margolin AA, Wang K, Lim WK, Kitagawa Y, Schneider C, Alvarez MJ, Califano A and Dalla-Favera R, Integrated biochemical and computational approach identifies BCL6 direct target genes controlling multiple pathways in normal germinal center B cells. Blood 2010. 115: 975–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kitano M, Moriyama S, Ando Y, Hikida M, Mori Y, Kurosaki T and Okada T, Bcl6 protein expression shapes pre-germinal center B cell dynamics and follicular helper T cell heterogeneity. Immunity 2011. 34: 961–972. [DOI] [PubMed] [Google Scholar]
- 5.Nurieva RI, Chung Y, Martinez GJ, Yang XO, Tanaka S, Matskevitch TD, Wang YH and Dong C, Bcl6 mediates the development of T follicular helper cells. Science 2009. 325: 1001–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnston RJ, Poholek AC, DiToro D, Yusuf I, Eto D, Barnett B, Dent AL, Craft J and Crotty S, Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 2009. 325: 1006–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cattoretti G, Pasqualucci L, Ballon G, Tam W, Nandula SV, Shen Q, Mo T, Murty VV and Dalla-Favera R, Deregulated BCL6 expression recapitulates the pathogenesis of human diffuse large B cell lymphomas in mice. Cancer Cell 2005. 7: 445–455. [DOI] [PubMed] [Google Scholar]
- 8.Ueda C, Akasaka T and Ohno H, Non-immunoglobulin/BCL6 gene fusion in diffuse large B-cell lymphoma: prognostic implications. Leuk Lymphoma 2002. 43: 1375–1381. [DOI] [PubMed] [Google Scholar]
- 9.Cardenas MG, Oswald E, Yu W, Xue F, MacKerell AD Jr. and Melnick AM, The Expanding Role of the BCL6 Oncoprotein as a Cancer Therapeutic Target. Clin Cancer Res 2017. 23: 885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lo Coco F, Ye BH, Lista F, Corradini P, Offit K, Knowles DM, Chaganti RS and Dalla-Favera R, Rearrangements of the BCL6 gene in diffuse large cell non-Hodgkin’s lymphoma. Blood 1994. 83: 1757–1759. [PubMed] [Google Scholar]
- 11.Pasqualucci L, Migliazza A, Basso K, Houldsworth J, Chaganti RS and Dalla-Favera R, Mutations of the BCL6 proto-oncogene disrupt its negative autoregulation in diffuse large B-cell lymphoma. Blood 2003. 101: 2914–2923. [DOI] [PubMed] [Google Scholar]
- 12.Cerchietti L and Melnick A, Targeting BCL6 in diffuse large B-cell lymphoma: what does this mean for the future treatment? Expert Rev Hematol 2013. 6: 343–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang F, Jin Y and Wei Y, MicroRNA-187 induces diffuse large B-cell lymphoma cell apoptosis via targeting BCL6. Oncol Lett 2016. 11: 2845–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cardenas MG, Yu W, Beguelin W, Teater MR, Geng H, Goldstein RL, Oswald E, Hatzi K, Yang SN, Cohen J, Shaknovich R, Vanommeslaeghe K, Cheng H, Liang D, Cho HJ, Abbott J, Tam W, Du W, Leonard JP, Elemento O, Cerchietti L, Cierpicki T, Xue F, MacKerell AD Jr. and Melnick AM, Rationally designed BCL6 inhibitors target activated B cell diffuse large B cell lymphoma. J Clin Invest 2016. 126: 3351–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beguelin W, Teater M, Gearhart MD, Calvo Fernandez MT, Goldstein RL, Cardenas MG, Hatzi K, Rosen M, Shen H, Corcoran CM, Hamline MY, Gascoyne RD, Levine RL, Abdel-Wahab O, Licht JD, Shaknovich R, Elemento O, Bardwell VJ and Melnick AM, EZH2 and BCL6 Cooperate to Assemble CBX8-BCOR Complex to Repress Bivalent Promoters, Mediate Germinal Center Formation and Lymphomagenesis. Cancer Cell 2016. 30: 197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cai Y, Watkins MA, Xue F, Ai Y, Cheng H, Midkiff CC, Wang X, Alvarez X, Poli ANR, Salvino JM, Veazey RS and Montaner LJ, BCL6 BTB-specific inhibition via FX1 treatment reduces Tfh cells and reverses lymphoid follicle hyperplasia in Indian rhesus macaque (Macaca mulatta). J Med Primatol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kerres N, Steurer S, Schlager S, Bader G, Berger H, Caligiuri M, Dank C, Engen JR, Ettmayer P, Fischerauer B, Flotzinger G, Gerlach D, Gerstberger T, Gmaschitz T, Greb P, Han B, Heyes E, Iacob RE, Kessler D, Kolle H, Lamarre L, Lancia DR, Lucas S, Mayer M, Mayr K, Mischerikow N, Muck K, Peinsipp C, Petermann O, Reiser U, Rudolph D, Rumpel K, Salomon C, Scharn D, Schnitzer R, Schrenk A, Schweifer N, Thompson D, Traxler E, Varecka R, Voss T, Weiss-Puxbaum A, Winkler S, Zheng X, Zoephel A, Kraut N, McConnell D, Pearson M and Koegl M, Chemically Induced Degradation of the Oncogenic Transcription Factor BCL6. Cell Rep 2017. 20: 2860–2875. [DOI] [PubMed] [Google Scholar]
- 18.Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Jarvinen T and Savolainen J, Prodrugs: design and clinical applications. Nat Rev Drug Discov 2008. 7: 255–270. [DOI] [PubMed] [Google Scholar]
- 19.Perland E and Fredriksson R, Classification Systems of Secondary Active Transporters. Trends Pharmacol Sci 2017. 38: 305–315. [DOI] [PubMed] [Google Scholar]
- 20.Cha YJ, Kim ES and Koo JS, Amino Acid Transporters and Glutamine Metabolism in Breast Cancer. Int J Mol Sci 2018. 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cai Y, Abdel-Mohsen M, Tomescu C, Xue F, Wu G, Howell BJ, Ai Y, Sun J, Azzoni L, Le Coz C, Romberg N and Montaner LJ, BCL6 Inhibitor-Mediated Downregulation of Phosphorylated SAMHD1 and T Cell Activation Are Associated with Decreased HIV Infection and Reactivation. J Virol 2019. 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wuelfing WP, Kwong E and Higgins J, Identification of suitable formulations for high dose oral studies in rats using in vitro solubility measurements, the maximum absorbable dose model, and historical data sets. Mol Pharm 2012. 9: 1163–1174. [DOI] [PubMed] [Google Scholar]
- 23.Hatzi K and Melnick A, Breaking bad in the germinal center: how deregulation of BCL6 contributes to lymphomagenesis. Trends Mol Med 2014. 20: 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aljurayyan A, Puksuriwong S, Ahmed M, Sharma R, Krishnan M, Sood S, Davies K, Rajashekar D, Leong S, McNamara PS, Gordon S and Zhang Q, Activation and Induction of Antigen-Specific T Follicular Helper Cells Play a Critical Role in Live-Attenuated Influenza Vaccine-Induced Human Mucosal Anti-influenza Antibody Response. J Virol 2018. 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vinci P and Hanash AM, BCL6 inhibition: a chronic GVHD twofer. Blood 2019. 133: 4–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paiardini M and Lichterfeld M, Follicular T helper cells: hotspots for HIV-1 persistence. Nat Med 2016. 22: 711–712. [DOI] [PubMed] [Google Scholar]
- 27.Miles B and Connick E, TFH in HIV Latency and as Sources of Replication-Competent Virus. Trends Microbiol 2016. 24: 338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fukazawa Y, Lum R, Okoye AA, Park H, Matsuda K, Bae JY, Hagen SI, Shoemaker R, Deleage C, Lucero C, Morcock D, Swanson T, Legasse AW, Axthelm MK, Hesselgesser J, Geleziunas R, Hirsch VM, Edlefsen PT, Piatak M Jr., Estes JD, Lifson JD and Picker LJ, B cell follicle sanctuary permits persistent productive simian immunodeficiency virus infection in elite controllers. Nat Med 2015. 21: 132–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cai Y, Watkins MA, Xue F, Ai Y, Cheng H, Midkiff CC, Wang X, Alvarez X, Poli ANR, Salvino JM, Veazey RS and Montaner LJ, BCL6 BTB-specific inhibition via FX1 treatment reduces Tfh cells and reverses lymphoid follicle hyperplasia in Indian rhesus macaque (Macaca mulatta). J Med Primatol 2020. 49: 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hasegawa A, Liu H, Ling B, Borda JT, Alvarez X, Sugimoto C, Vinet-Oliphant H, Kim WK, Williams KC, Ribeiro RM, Lackner AA, Veazey RS and Kuroda MJ, The level of monocyte turnover predicts disease progression in the macaque model of AIDS. Blood 2009. 114: 2917–2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cossarizza A, Chang HD, Radbruch A, Acs A, Adam D, Adam-Klages S, Agace WW, Aghaeepour N, Akdis M, Allez M, Almeida LN, Alvisi G, Anderson G, Andra I, Annunziato F, Anselmo A, Bacher P, Baldari CT, Bari S, Barnaba V, Barros-Martins J, Battistini L, Bauer W, Baumgart S, Baumgarth N, Baumjohann D, Baying B, Bebawy M, Becher B, Beisker W, Benes V, Beyaert R, Blanco A, Boardman DA, Bogdan C, Borger JG, Borsellino G, Boulais PE, Bradford JA, Brenner D, Brinkman RR, Brooks AES, Busch DH, Buscher M, Bushnell TP, Calzetti F, Cameron G, Cammarata I, Cao X, Cardell SL, Casola S, Cassatella MA, Cavani A, Celada A, Chatenoud L, Chattopadhyay PK, Chow S, Christakou E, Cicin-Sain L, Clerici M, Colombo FS, Cook L, Cooke A, Cooper AM, Corbett AJ, Cosma A, Cosmi L, Coulie PG, Cumano A, Cvetkovic L, Dang VD, Dang-Heine C, Davey MS, Davies D, De Biasi S, Del Zotto G, Dela Cruz GV, Delacher M, Della Bella S, Dellabona P, Deniz G, Dessing M, Di Santo JP, Diefenbach A, Dieli F, Dolf A, Dorner T, Dress RJ, Dudziak D, Dustin M, Dutertre CA, Ebner F, Eckle SBG, Edinger M, Eede P, Ehrhardt GRA, Eich M, Engel P, Engelhardt B, Erdei A, Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). Eur J Immunol 2019. 49: 1457–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McAllister EJ, Apgar JR, Leung CR, Rickert RC and Jellusova J, New Methods To Analyze B Cell Immune Responses to Thymus-Dependent Antigen Sheep Red Blood Cells. J Immunol 2017. 199: 2998–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article.