

Abstract
Objective:
Children with autism spectrum disorder (ASD) commonly exhibit the symptoms of a wide range of non-ASD psychiatric disorders including depression. The aim of the present study was to characterize the association of two functional single nucleotide polymorphisms (SNPs) (rs6311, rs6314) in the serotonin 2A receptor gene (HTR2A) with severity of depression symptoms in children with ASD. These SNPs were previously shown to be associated with depression symptom severity and response to SSRIs in adults with diagnosed depressive disorder.
Methods:
Parents completed a validated DSM-IV-referenced rating scale for depressive symptoms in 104 children with ASD. Depression symptom severity was compared across rs6311 and rs6314 genotypes, measured from genomic DNA of affected children.
Results:
Youths homozygous for the G allele of rs6311 had significantly more severe depression symptoms than GA and AA genotypes (p=0.025), and the effects size (eta-squared) was small in this ASD sample (ηp2=0.047) but somewhat larger when controlling for severity of generalized anxiety disorder symptoms (p=.006, ηp2=0.072). When analyses were restricted to Caucasians, results were essentially the same as for the entire sample (p=.004, ηp2=0.086). There were no significant associations with rs6314 (CC vs. T carriers).
Conclusions:
The functional rs6311 polymorphism in HTR2A, associated with differential HTR2A mRNA expression in other studies, may modulate depression symptom severity in children with ASD. These tentative, hypothesis-generating findings require replication with larger independent samples.
Keywords: autism, autism spectrum disorder, depression, HTR2A, rs6311, rs6314
INTRODUCTION
Children with autism spectrum disorder (ASD) commonly exhibit the symptoms of one or more additional psychiatric disorder, and depression is among the most common co-occurring conditions (Gadow et al, 2012; Magnusson and Constantino, 2011). Gadow et al (2005) previously found that severity of depression symptoms in children with ASD was comparable to children referred for child psychiatry outpatient evaluation and much greater than typically developing peers. In another study with a different group of children with ASD, 20% were rated socially or academically impaired by depression symptoms according to parents (Kaat et al, 2013). These findings are consistent with the results of others who also assessed impairing depression (Leyfer et al, 2006; Simonoff et al, 2008; Witwer and Lecavalier, 2010). Depression appears to be a life-long vulnerability in ASD, as studies of adolescents and adults with the disorder also report high rates of current (37%) (Bakken et al, 2010) and lifetime (70%) (Lugnegård et al, 2011) depression, which contributes to caregiver burden (Cadman et al, 2012).
The propensity of individuals with ASD to suffer from depression compels further research delineating its underlying biological mechanisms including putative genetic factors that regulate drug targets for treating depression such as selective serotonin re-uptake inhibitors (SSRIs). Serotonin dysregulation has long been implicated in depression (Lapin and Oxenkrug, 1969; López-Muňoz and Alamo, 2009) as well as ASD (Gadow et al., 2013; Schain and Freedman, 1961; Whitaker-Azmitia, 2001). For example, both disorders have evidence for altered levels of platelet serotonin, in which approximately 30% of individuals with ASD have platelet hyperserotonemia (Cook et al., 1993), whereas individuals with major depression exhibit reduced platelet serotonin levels (Alvarez et al, 1999; Mück-Seler et al, 1996, 2004; Pivac et al, 2003; Ruljancic et al, 2013).
The serotonin 2A receptor is a major component of serotonergic signaling in the brain and periphery, including platelets, where it promotes aggregation and mediates release of intracellular calcium stores when activated. This aggregation and/or calcium response can be used as an indirect ex vivo measure of 5-HT2A function. Platelet function profiles in individuals with ASD or major depression differ with respect to 5-HT2A function, with depressed patients exhibiting greater aggregation response or intracellular calcium release (Eckert et al, 1993; Kusumi et al, 1994; Shimbo et al, 2002) and individuals with ASD a reduced response relative to controls (Hranilović et al, 2009; McBride et al, 1989). Furthermore, studies using both positron emission tomography (PET) and single-photon emission computed tomography (SPECT) have found in vivo evidence of reduced 5-HT2A receptor density in the cortex of individuals diagnosed with Asperger’s syndrome (Murphy et al, 2006), first-degree relatives of individuals with ASD (Goldberg et al, 2009), and in individuals with major depression (Attar-Lévy et al, 1999; Biver et al, 1997; Sheline et al, 2004; Yatham et al, 2000). Consistent with these in vivo findings Thanseem et al (2012) found reduced HTR2A gene expression in post mortem brain tissue from individuals with ASD. However, findings for the aforementioned measures in ASD and depressed populations are mixed, and occasionally suggest opposing 5-HTR2A function (Franke et al, 2000; Girgis et al, 2011; Meltzer et al, 1999; Meyer et al, 2001; Uebelhack et al, 2006) but nevertheless support the value of further study into genetic factors underlying co-morbid ASD and depression.
The serotonin 2A receptor, encoded by the HTR2A gene, harbors common functional genetic variants that contribute to a wide range of disorders (Smith et al, 2013), including depression (Narasimhan and Lohoff, 2012) and ASD (Cho et al, 2007; Hranilović et al, 2010). These common functional variants include rs6311 (−1438G>A), which we previously reported reduces the expression of an untranslated region of HTR2A mRNA, and rs6314 (His452Tyr), which changes the encoded amino acid and alters the potency and second-messenger signaling of the receptor in response to agonists in vitro (Davies et al, 2006; Hazelwood et al, 2004). We and others previously reported that rs6311(A) is associated with depression (Attar-Lévy et al, 1999; Christiansen et al, 2007; Kamata et al, 2011; Kim et al, 2012; Smith et al, 2013), although this association is not consistently observed (Choi et al, 2004; Frisch et al, 1999; Minov et al, 2001; Oswald et al, 2003). There is also evidence of an association with seasonal affective disorder (Arias et al, 2001; Lee et al, 2006) and response to antidepressant medication (both therapeutic and untoward) in patients with major depressive disorder (Kato and Serretti, 2010; Minov et al, 2001; Narasimhan and Lohoff, 2012; Wilkie et al, 2009). rs6311 was previously shown to be associated with autism, either alone (Hranilović et al, 2010) or a haplotype containing both rs6311 and rs6313 (T102C) (Cho et al, 2007) although some studies do not find associations (Guhathakurta et al, 2009; Veenstra-VanderWeele et al, 2002. rs6311 and rs6313 are in near complete linkage disequilibrium (Spurlock et al, 1998), perhaps explaining clinical associations with rs6313, despite lack of functional evidence for this SNP. Previous studies found no association between rs6314 and ASD risk (Guhathakurta et al, 2009; Hranilović et al, 2010; Veenstra-VanderWeele et al, 2002) or depression (Minov et al, 2001). However, this SNP has been associated with antidepressant treatment outcome (Wilkie et al, 2009).
There is strong evidence that the HTR2A region harbors risk variants for ASD and depression, with varying amounts of influence depending on the specific attributes of the populations studied. The general aim of the present study was to examine behavioral variation (i.e., depression symptoms) within the ASD clinical phenotype. More specifically, given our own á priori evidence of (a) the pervasiveness of depression symptoms in children with ASD (Gadow et al, 2005, 2012, Kaat et al, 2013), (b) regulatory activity for rs6311 and the nonsynonymous coding SNP rs6314 (Smith et al, 2013), and (c) association with depression in non-ASD samples (Smith et al, 2013) and the findings of others supporting an association of rs6311 with ASD (Hranilović et al, 2010) and depression (Narasimhan and Lohoff, 2012), we examined relations of the aforementioned SNPs with depression severity in children with ASD using a well-validated DSM-IV-referenced rating scale. In view of evidence supporting shared genetic risk factors in psychiatric disorders (Cross Disorder Group, 2013) and in particular serotonin dysregulation in both disorders (Gadow et al, 2013; Lapin and Oxenkrug, 1969; López-Muňoz and Alamo, 2009; Schain and Freedman, 1961; Whitaker-Azmitia, 2001), we predicted that depressogenic risk factors may constitute a second-hit in this serotonin-biased clinical phenotype in a subgroup of vulnerable individuals.
MATERIALS AND METHODS
Participants
Participants were recruited from referrals to a university hospital developmental disabilities specialty clinic located on Long Island, New York. All youth (N=104) between 4 and 14 years old with the prerequisite measures and a diagnosis of ASD included were included in the present study. Demographic characteristics were as follows: age (M=7.6; SD=2.7), gender (87% male), ethnicity (90% Caucasian), IQ (74%≥70), socioeconomic status assessed with Hollingshead’s (1975) index of occupational and educational social status (M=41.9; SD=11.1), single-parent household (9%), and current psychotropic medication use (25%). A subsample of youth (n=67) participated in prior studies of other gene variants (Gadow et al, 2008; Roohi et al, 2009). This study was approved by a university Institutional Review Board, informed consent was obtained, and appropriate measures were taken to protect patient (and rater) confidentiality.
As part the initial clinic evaluation, parents (usually the mother) completed behavior rating scales and a background information questionnaire. Diagnoses of ASD were confirmed by an expert diagnostician and based on five sources of information: (a) comprehensive developmental history, (b) clinician interview with child and caregiver(s), (c) direct observations of the child, (d) prior evaluations, and (e) review of validated ASD rating scales including the Child Symptom Inventory-4 (CSI-4) (Gadow and Sprafkin, 2002), which evidenced high sensitivity and specificity in identifying children with ASD in two independent studies (DeVincent et al, 2009; Gadow,Schwartz et al, 2008). Most youth (81%) were also evaluated with the Autism Diagnostic Observation Schedule (Lord et al, 2000) and/or Autism Diagnostic Interview-Revised (Rutter et al, 2003). Those who were not evaluated with these measures had well-documented prior history of ASD diagnoses and functional impairment associated with ASD symptoms. Over one third (36%) of these youth had T scores >65 for parents’ ratings of major depressive disorder.
Depression Ratings
The CSI-4 is a behavior rating scale that assesses the symptoms of DSM-IV disorders. Symptom severity is scored (never=0, sometimes=1, often=2, and very often=3). Confirmatory factor analysis supports the internal construct validity of DSM-IV syndromes in children with ASD including depression (Lecavalier et al, 2009). Numerous studies indicate that the CSI-4 major depressive episode subscale demonstrates satisfactory psychometric properties in community-based normative, clinic-referred non-ASD, and ASD samples (Gadow and Sprafkin, 2011). CSI-4 scores show little relation to age, IQ, or SES.
Data Analysis and Statistics
Depression scores were normally distributed (M=5.5, SD=3.6; range=2–17; Skewness=0.24; Kurtosis=0.14). Chi-square tests (categorical variables), correlations (continuous variables), and ANOVAs (combined categorical and continuous variables) were used to test associations of demographic characteristics (age, gender, ethnicity, IQ level [<70 vs. ≥70], SES, single-parent household, and currently receiving psychotropic medication or special education) with genotype groups as well as the dependent variable to identify potential covariates. Genotype groups were not significantly different for any of these variables; therefore, they were not used as co-variates in subsequent analyses. One way ANOVAs were conducted to examine main effects of genotype groups (rs6311, rs6314). Severity of generalized anxiety disorder symptoms was not related to genotype group and was used as a covariate in secondary analyses (Miller and Chapman, 2001). In addition to p-values, we report partial eta-squared (ηp2) to gauge the magnitude of group differences (i.e., percentage of variance in dependent variables accounted for by independent variables). A rule of thumb for determining the magnitude of ηp2 suggests the following: 0.01–0.06=small, 0.06–0.14=moderate, and >0.14 = large (Cohen, 1988).
Genotyping
Genomic DNA (25ng) from the children, isolated from peripheral blood cells or buccal swabs, was PCR-amplified 30 cycles in a standard 3-step reaction (95° denature, 58° annealing, 72° extension) using 2x Taq polymerase master mix (New England Biolabs, Inc.; NEB) and fluorescently-labeled primers surrounding the SNPs of interest. The resultant amplicons were cut with restriction enzymes that only recognize the ancestral reference alleles for rs6311 (G) and rs6314 (C). Following digestion, the lengths of the fluorescently-labeled amplicons were resolved on an ABI3730 DNA Analyzer (Life Technologies). For rs6311, a positive control enzyme cut site (underlined) was designed into the forward primer by a single T→C replacement (bold) in the primer sequence (5-TTCCACTCCGGACACAAACACTGT-3) and paired with a fluorescently-labeled reverse primer (5-[6FAM]CCCATTAAGGTAGGTAAGTGGCACTGT), resulting in a 151bp amplicon. Following digestion with HpaII (NEB), the positive control fragment with the SNP allele was 143bp, while the ancestral G allele fragment was 108bp. For rs6314, amplification with the forward (5-[HEX] AGCCAACTTCAAATGGGACAA-3) and reverse primer (5-CCTATCACACACAGCTCACCTTTT-3) resulted in a 157bp amplicon for SNP allele and an 85bp fragment for ancestral (C) allele cut by BsmI (NEB). Examples of the genotypes represented by the fragment lengths following enzyme digestion are displayed in Figure 1. Linkage disequilibrium between the two SNPs was calculated using SNP & Variation Suite 7 (Golden Helix, Bozeman, MT).
Figure 1.
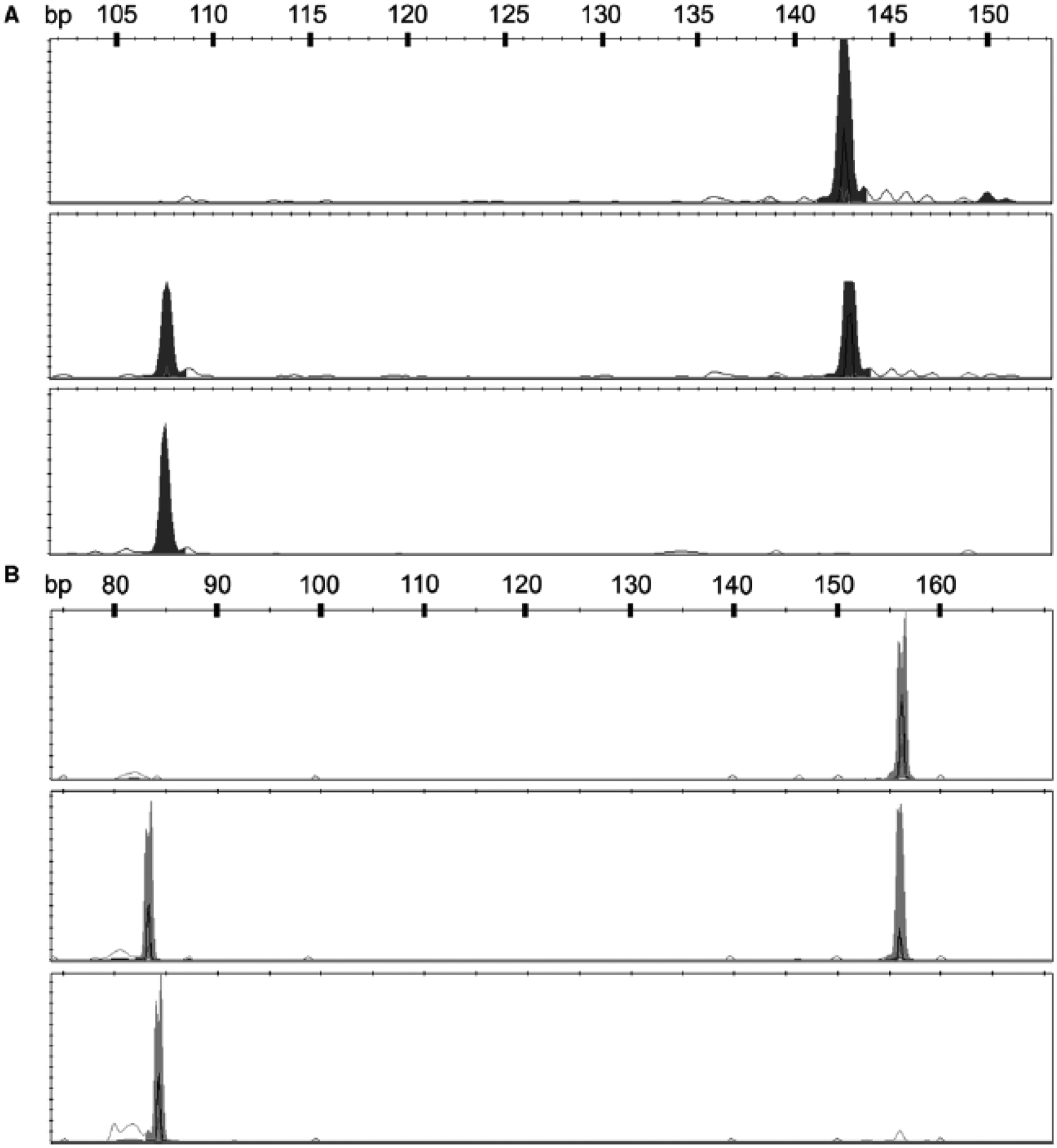
Representative fragments following restriction enzyme digestions for genotyping rs6311 and rs6314. A. Following digestion with HpaII, rs6311 heterozygous samples were represented by two peaks (top panel), while homozygous SNP allele “A” subjects (middle panel) or ancestral allele “G” subjects (bottom panel) were represented by single peaks at 143bp and 108bp, respectively. Still apparent in the middle panel, but not interfering with genotype calls is residual undigested full-length 151bp amplicons. B. Digestion of amplicons containing rs6314 with BsmI resulted in the same pattern where homozygous ancestral “C” allele subjects (bottom panel) or SNP “T” allele subjects (top panel) resulted in single peaks of 85bp and 157bp, respectively, and heterozygous samples having both peaks (middle panel).
RESULTS
Linkage disequilibrium (LD) between rs6311 and rs6314 is consistent with LD data reported by the International HapMap project (http://hapmap.ncbi.nlm.nih.gov/) given our mixed population (D′=0.47, R2=0.03). The biallelic distribution of rs6311 genotypes (frequencies/percents) were G/G (25/24%), G/A (59/57%), and A/A (20/19%), which does not deviate from HWE (X2 = 1.96, p>0.05). Preliminary analyses indicated that the mean depression scores for G/A (M=5.07) and AA (M=5.10) groups were nearly identical, and they were therefore combined into one group; thus 25 (24%) of the children fell into the “high mRNA expressing” G/G group, and 79 (76%) were in the “low mRNA expressing” A+ group. There was a significant main effect of genotype (F=5.20, p=0.025), with the G/G group receiving more severe depression ratings than the A+ group (Figure 2), and the effect size was small (ηp2=0.049). Mean scores of 7 and 5 correspond to T scores of 84 and 71, respectively, in typically developing boys (Gadow and Sprafkin, 2002). Controlling for severity of generalized anxiety disorder symptoms (F=7.87, p=0.006), the effect size was in the moderate range (ηp2=0.072). When analyses were restricted to Caucasians, results were essentially the same as for the entire sample (F=8.61, p=.004, ηp2=0.086).
Figure 2.
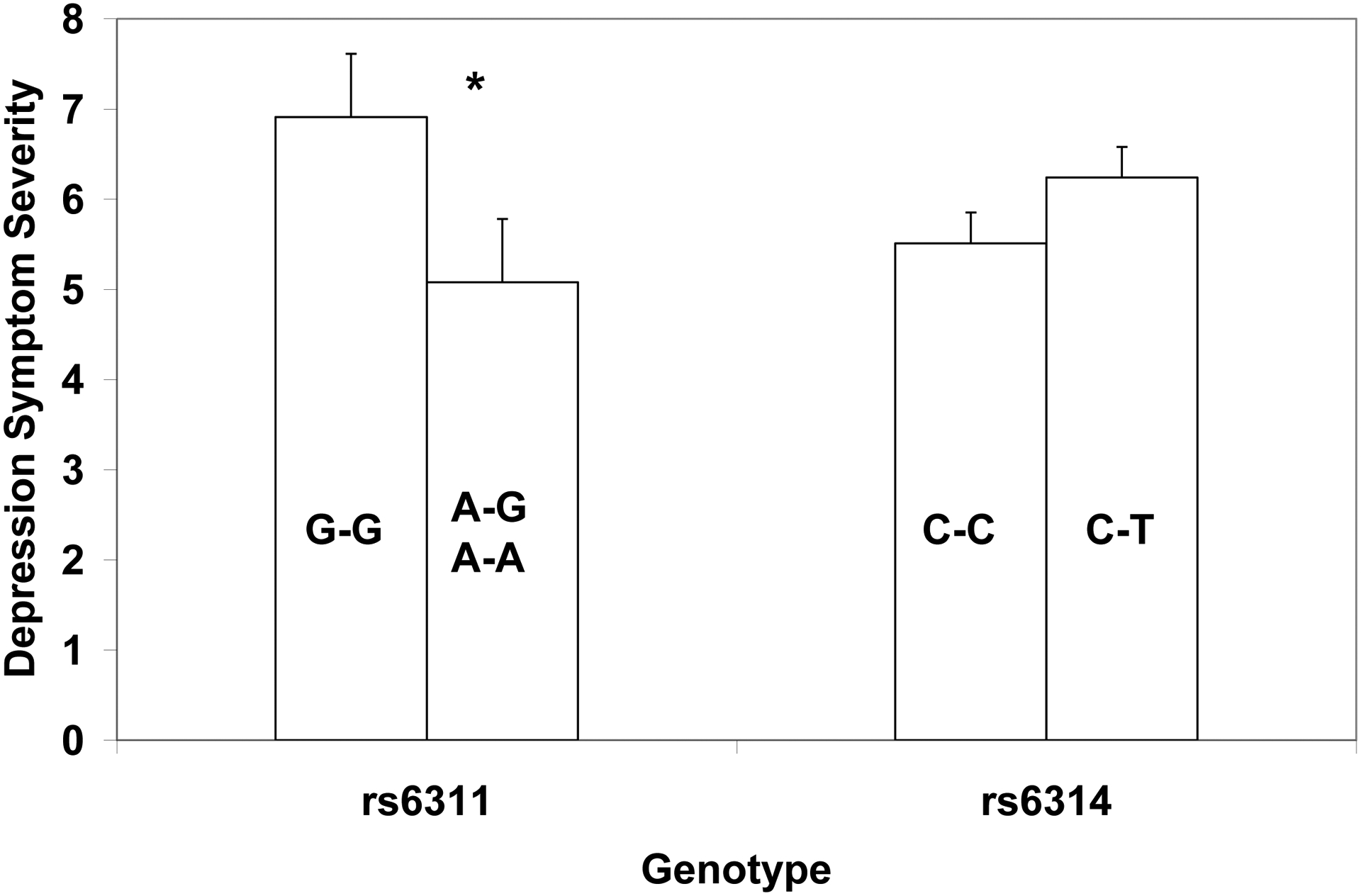
Association of depression symptom severity (mean score and standard error) with HTR2A single nucleotide polymorphisms rs6311 and rs6314. Genotype group differences were significant (* p=0.025) for rs6311 with the “high expressing” G/G group exhibiting more severe symptoms that the “low expressing” A+ group. No significant genotypic differences were observed for rs6314.
The biallelic distribution of rs6314 genotypes (frequencies/percents) were C/C (83/80%), C/T (17/16%), and T/T (4/4%), which deviates from HWE (X2 = 5.36, p<0.05). This could be the result of a small mixed ethnicity population considering differences in the distribution of rs6314 minor allele frequency among individuals of European (6.2%) versus African (16.4%) geographic ancestry in HapMap CEU and YRI populations, respectively. In support of this interpretation, HWE was not significant when analysis was confined to Caucasians (X2 = 2.18, p>0.05). Owing to the small number of children homozygous for the T allele, we compared with youths homozygous for the C allele (C/C) with heterozygotes. Although heterozygotes had more severe depression symptoms than the CC group (Figure 2), group differences were not significant, with or without controlling for severity of generalized anxiety disorder symptoms. Nevertheless, the T/T group had very low depression scores (M=2.6; SD=0.75), but larger samples will be necessary to examine this further. When analyses (controlling for generalized anxiety) were limited to Caucasians, results were essentially the same (F=1.15, p=.286, ηp2=0.013).
DISCUSSION
Depression is a significant clinical concern in individuals with ASD, but relatively little is known about its phenomenology, clinical features, or risk factors in this population, and there are no controlled clinical trials. Results of the present study indicate an association between HTR2A SNP rs6311 and severity of major depressive disorder symptoms in children with ASD where children homozygous for the G allele had more severe symptoms than carriers of the A allele, and the effect size was in the moderate range when co-varying severity of generalized anxiety (ηp2=0.072). The notion that a genetic risk factor for depression in the gene encoding the serotonin 2A receptor may modulate the severity of depression symptoms in children with ASD is consistent with research supporting serotonin dysregulation in both depression (Lapin and Oxenkrug, 1969; López-Muňoz and Alamo, 2009) and ASD (Gadow et al, 2013; Schain and Freedman, 1961; Whitaker-Azmitia, 2001). More specifically, prior research suggests that rs6311 may be associated with depression severity (Smith et al, 2013), seasonal affective disorder (Arias et al, 2001; Lee et al, 2006), and response to antidepressants (Kato and Serretti, 2010; Narasimhan and Lohoff, 2012) but findings are mixed (Jin et al, 2013; Johansson et al, 2001; Molnar et al, 2010). With regard to ASD, a study of Croatian children and adults with ASD and controls found the G allele and the G-G genotype to be overrepresented in the former (Hranilović et al, 2010). We examined associations between HTR2A genotypes and severity of ASD symptoms in the present sample, and results were negative for ASD global severity scores as well as scores for each of the three subdomains of ASD symptoms. In a recently completed study, we replicated our previously published finding with regard to the functionality of rs6311 in brain tissue from typically developing adults in ASD brains from the Autism Tissue Program. In addition, we found under-transmission of rs6311/A in a cohort of 158 trios (unpublished data).
Inconsistencies in genotype-phenotype associations across studies are not uncommon, even within the same sample. For example, the results of a recent study comparing association, maternal effects, and parent-of-origin effects data analysis strategies for another serotonergic gene variant, the 5-HTTLPR, in a large sample of children and adults with autism revealed different outcomes for each approach as well as for geographic ancestry (Kistner-Griffin et al, 2011). The authors note that different mechanisms may be associated with each effect. In addition, we have found that different alleles of the 5-HTTLPR to be associated with the symptoms of different disorders as well as informant discrepancy (Gadow et al, 2013). Other potential sources of heterogeneity in association studies of genes involved in serotonin signaling are (a) gene X environment interactions evidenced by the differential sensitivity of one allele to both positive and negative environmental experiences compared with the another allele of the same gene (Belsky et al, 2009), including rs6313 (Belsky and Pluess, 2009), and (b) heterogeneity in co-occurring psychiatric symptoms such as depression. Whether one or more of these variables explains differences in outcomes across HTR2A rs6311 studies is not known, but they warrant consideration in future research.
Given the high minor allele frequency for rs6311 in all HapMap populations (>30%), we find it most likely that this SNP modulates phenotypic presentation of depression (or ASD), rather than exerting a causative effect. However, an abundance of potential confounding factors and uncertainties about the biologic substrates of genotype-phenotype relations precludes a compelling explanation of its mechanisms. Nevertheless, there are a number of interesting possibilities. One of the most compelling explanations for co-morbidity is shared risk factors, and this is well illustrated by the findings of a recent genome-wide association showing that a range of SNPs were associated with two or more of five different major psychiatric disorders including autism and depression (Cross Disorder Group, 2013). As we have noted previously (Gadow et al, 2013), there is evidence for a possible involvement of circadian system regulatory genes in autism (Hu et al, 2009) and depression (Kronfeld-Schor and Einat, 2012), the effect of seasonal light cycles on serotonin signaling in the brain mediated by the circadian system (Ciarlegio et al, 2011), and higher rates of autism (Lee et al, 2008) and depression (Pjrek et al, 2004) births in spring and early summer, although findings are mixed (Lee et al, 2008). There is also replicated evidence of seasonal fluctuations in brain 5-HTT binding (Willeit and Praschak-Rieder, 2010) as well as findings linking seasonal pattern in the first episode of major depression (higher for rs6313 C allele carriers, which is comparable to rs6311 G allele carriers) (Arias et al, 2001). Some investigators have found differentially lower 5-HT2A binding in several brain regions in people with ASD (Ciarlegio et al, 2011; Murphy et al, 2006;) but findings for depression are mixed (Savitz and Drevets, 2013). Mothers of children with ASD have higher rates of depression (and other psychopathology) than the general population (Daniels et al, 2008).
Another biologic system that warrants consideration with regard to shared risk factors and gene variants is the innate immune response. There is compelling evidence of elevated levels of pro-inflammatory cytokines in both a significant minority of individuals with major depression (Dowlati et al, 2010) and ASD (Vargas et al, 2005). Cytokines influence serotonin metabolism and signaling in the brain (Baganz and Blakely, 2013; Miller et al, 2013); immune system dysregulation is implicated in the pathogenesis or symptom modulation of both depression (Raison and Miller, 2013) and ASD (Depino, 2013; Goines and Ashwood, 2013); and serotonergic gene variants interact with these mechanisms as well as with immune system gene variants (Bull et al, 2009). Cytokine levels increase during stress (Maier, 2003; Slavich et al, 2010), and their synergistic actions may play an important role in depression (Anisman, 2009). Stress alters 5-HT2A expression in the brain of rats (Dwivedi et al, 2005), and there is some evidence that adults with the rs6311 G-G genotype are more reactive to stress (Fiocco et al, 2007). Individuals with pro-inflammatory bias may be particularly vulnerable to the adverse effects of environmental stressors known to trigger depression symptoms such as social rejection. For children with ASD who are aware of their social impediments, social rejection appears to contribute to depression symptoms (Magnusson and Constantino, 2011; Pouw et al, 2013), and this may be particularly difficult for youth with serotonin or pro-inflammatory bias, genetic risk factors, or some combination of these variables.
Although the strengths of this study include a relatively restricted age range of children with ASD, the examination of symptom phenotypes with a psychometrically sound measure, and focus on functional gene variants, there are limitations as well. The sample was small according to contemporary standards, which increases the probability of Type 1 error (false positives) as well Type 2 error (false negatives). For example, we did not observe a significant genotype-phenotype association for rs6314, but our results do not rule out this possibility although our obtained effect size was small. In addition, one must use caution in interpreting these associations as 5-HT2A receptor expression is susceptible to environmental factors (Oreland et al, 2009) and drugs that influence serotonin signaling (Hernandez and Sokolov, 2000; Willins et al, 1998), which could interact with genetic variants such as rs6311 to influence symptom severity. Some children were receiving psychotropic medication at the time their behavioral evaluations were being conducted. Although genotype groups did not differ in the percent receiving treatment, we nevertheless considered this variable as a potential covariate, but medication status was not linearly related to depression. However, as we have noted elsewhere (Kaat et al, 2013), it is unclear how this might have altered the general conclusions of the study because this clinical population appears to respond less satisfactorily to a range of psychotropic medications compared with non-ASD patients (Siegel and Beaulieu, 2012), and there is no compelling evidence for their efficacy in the treatment of depression. As we have noted previously (Gadow et al 2013), it would have been ideal to obtain all assessments off medication. However, withdrawal from treatment solely for the purpose of obtaining ratings raises a number of concerns. Equally problematic is their exclusion, which could potentially bias the sample with regard to the very behaviors that were the object of study. It is widely acknowledged that a many variables impact psychiatric symptom expression, and genetic variants are but one possibility. Although our results are to some extent consistent with the extant literature, they are presented here primarily for their heuristic value and as a suggestion for continued study and should not be considered as hypothesis confirming.
Summary
Children with HTR2A rs6311 G-G genotype had more severe symptoms of depression than A allele carriers, but this finding should be considered tentative pending replication in larger, independent samples. This finding suggests that genes involved in serotonin neurotransmission may be modulators of depression in this clinical population and may warrant consideration as potential indicators of response to intervention. For example, rs6311 is associated with response to SSRI’s in adults with major depressive disorder (Kato and Serretti, 2010), and depression is a major clinical concern in higher-functioning adults with ASD (Cadman et al, 2012), but to date there are no controlled trials indicating efficacy for SSRI’s or any other drug for depression in ASD. Owing to diagnostic, symptom, and etiologic heterogeneity as well as heterogeneity in co-morbidity, fully-realized models of pathogenesis and personalized medicine will likely incorporate a variety of biomarkers, some of which may be gene variants involved in serotonin signaling.
Acknowledgements
This study was supported in part by the Matt and Debra Cody Center for Autism and Developmental Disorders, charitable contributions, and a grant from the National Institute of Health General Medical Sciences U01 GM092655 (Wolfgang Sadee). The authors wish to thank Dr. John Pomeroy, Stony Brook University (Department of Pediatrics) for supervising the clinical diagnoses and Dr. Carla DeVincent, Stony Brook University (Department of Radiology) for managing data collection.
Conflicts of Interest and Source of Funding:
K.D. Gadow (Shareholder, Checkmate Plus, publisher of the Child Symptom Inventory-4); R.M. Smith (NONE); J. Pinsonneault (NONE). This study was supported in part by a grant from the National Institute of Health General Medical Sciences U01 GM092655 (Wolfgang Sadee, PI).
Contributor Information
Kenneth D. Gadow, Department of Psychiatry and Behavioral Sciences, Stony Brook University, Stony Brook, NY 11794-8790
Ryan M. Smith, Department of Pharmacology, Program in Pharmacogenomics, The Ohio State University Wexner Medical Center, Columbus, OH 43210
Julia K. Pinsonneault, Department of Pharmacology, Program in Pharmacogenomics, The Ohio State University Wexner Medical Center, Columbus, OH 43210
REFERENCES
- Alvarez JC, Gluck N, Arnulf I, et al. 1999. Decreased platelet serotonin transporter sites and increased platelet inositol triphosphate levels in patients with unipolar depression: effects of clomipramine and fluoxetine. Clin Pharmacol Ther. 66:617–24. [DOI] [PubMed] [Google Scholar]
- Anisman H 2009. Cascading effects of stressors and inflammatory immune system activation: implications for major depressive disorder. Rev Psychiatr Neurosci. 34:4–20. [PMC free article] [PubMed] [Google Scholar]
- Arias B, Gutiérrez B, Pintor L, et al. 2001. Variability in the 5-HT2A receptor gene is associated with seasonal pattern in major depression. Mol Psychiatry. 6:239–242. [DOI] [PubMed] [Google Scholar]
- Attar-Lévy D, Martinot JL, Blin J, et al. 1999. The cortical serotonin2 receptors studied with positron-emission tomography and [18F]-setoperone during depressive illness and antidepressant treatment with clomipramine. Biol Psychiatry. 45:180–6. [DOI] [PubMed] [Google Scholar]
- Baganz NL, Blakely RD. 2013. A dialogue between immune system and brain, spoken in the language of serotonin. ACS Chemical Neurosci. 4;48–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakken TL, Helverschou SB, Eilertsen DE, et al. 2010. Psychiatric disorders in adolescents and adults with autism and intellectual disability: A representative study in one county in Norway. Res Dev Disabil. 31:1669–1677. [DOI] [PubMed] [Google Scholar]
- Belsky J, Jonassaint C, Pluess M, et al. 2009. Vulnerability genes or plasticity genes? Mol Psychiatry. 14:746–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsky J, Pluess M. 2009. Beyond diathesis stress: Differential susceptibility to environmental influences. Psych Bull. 135;885–908. [DOI] [PubMed] [Google Scholar]
- Biver F, Wikler D, Lotstra F, et al. 1997. Serotonin 5-HT2 receptor imaging in major depression: focal changes in orbito-insular cortex. Br J Psychiatry. 171:444–448. [DOI] [PubMed] [Google Scholar]
- Bull SJ, Huezo-Diaz P, Binder EB, et al. 2009. Functional polymorphisms in the interleukin-6 and serotonin transporter genes, and depression and fatigue induced by interferon-α and ribavirin treatment. Mol Psychiatry. 14:1095–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadman T, Eklund H, Howley D, et al. 2012. Caregiver burden as people with autism spectrum disorder and attention-deficit/hyperactivity disorder transition into adolescence and adulthood in the United Kingdom. J Am Acad Child Adolesc Psychiatry. 51:879–888. [DOI] [PubMed] [Google Scholar]
- Cho IH, Yoo HJ, Park M, et al. 2007. Family-based association study of 5-HTTLPR and the 5-HT2A receptor gene polymorphisms with autism spectrum disorder in Korean trios. Brain Res. 1139:34–41. [DOI] [PubMed] [Google Scholar]
- Choi MJ, Lee HJ, Lee HJ, et al. 2004. Association between major depressive disorder and the −1438A/G polymorphism of the serotonin 2A receptor gene. Neuropsychobiology. 49:38–41. [DOI] [PubMed] [Google Scholar]
- Christiansen L, Tan Q, Iachina M, et al. 2007. Candidate gene polymorphisms in the serotonergic pathway: influence on depression symptomatology in an elderly population. Biol Psychiatry. 61:223–30. [DOI] [PubMed] [Google Scholar]
- Ciarlegio CM, Resuehr HES, McMahon DG. 2011. Interactions of the serotonin and circadian systems: Nature and nurture in rhythms and blues. Neuroscience. 197:8–16. [DOI] [PubMed] [Google Scholar]
- Cohen J 1988. Statistical Power Analysis for the Behavioral Sciences (2nd Ed). Mahwah, NJ: Lawrence Erlbaum. [Google Scholar]
- Cook EH Jr, Arora RC, Anderson GM, et al. 1993. Platelet serotonin studies in hyperserotonemic relatives of children with autistic disorder. Life Sci. 52:2005–2015. [DOI] [PubMed] [Google Scholar]
- Cross Disorder Group of the Psychiatric Genetics Consortium. 2013. Identification of risk loci with shared effects on five major psychiatric disorders: a genomic-wide analysis. Lancet, published online February 28, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels JL, Forssen U, Hultman CM, et al. 2008. Parental psychiatric disorders associated with autism spectrum disorders in the offspring. Pediatrics. 121:e1537–e1362. [DOI] [PubMed] [Google Scholar]
- Davies MA, Setola V, Strachan RT, et al. 2006. Pharmacologic analysis of non-synonymous coding h5-HT2A SNPs reveals alterations in atypical antipsychotic and agonist efficacies. Pharmacogenomics J. 6:42–51. [DOI] [PubMed] [Google Scholar]
- Depino AM. 2013. Peripheral and central inflammation in autism spectrum disorders. Mol Cell Neurosci. 53;69–76. [DOI] [PubMed] [Google Scholar]
- DeVincent CJ, Gadow KD. 2009. Relative clinical utility of three Child Symptom Inventory-4 scoring algorithms for differentiating children with autism spectrum disorder versus attention-deficit hyperactivity disorder. Autism Res. 2:312–321. [DOI] [PubMed] [Google Scholar]
- Dowlati Y, Herrmann N, Swardfager W, et al. 2010. A meta-analysis of cytokines in major depression. Biol Psychiatry. 67;446–457. [DOI] [PubMed] [Google Scholar]
- Dwivedi Y, Mondal AC, Payappagoudar GV, et al. 2005. Differential regulation of serotonin (5HT)2A receptor mRNA and protein levels after single and repeated stress in a rat brain: role in learned helplessness behavior. Neuropharmacology. 48:204–214. [DOI] [PubMed] [Google Scholar]
- Eckert A, Gann H, Riemann D, et al. 1993. Elevated intracellular calcium levels after 5-HT2 receptor stimulation in platelets of depressed patients. Biol Psychiatry. 34:565–568. [DOI] [PubMed] [Google Scholar]
- Fiocco AJ, Joober R, Poirer J, et al. 2007. Polymorphism of the 5-HT2A receptor gene: association with stress-related indices in healthy middle-aged adults. Frontiers Behav Neurosci. 1:article #3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke L, Schewe HJ, Müller B, et al. 2000. Serotonergic platelet variables in unmedicated patients suffering from major depression and healthy subjects: relationship between 5HT content and 5HT uptake. Life Sci. 67:301–315. [DOI] [PubMed] [Google Scholar]
- Frisch A, Postilnick D, Rockah R, et al. 1999. Association of unipolar major depressive disorder with genes of the serotonergic and dopaminergic pathways. Mol Psychiatry. 4:389–392. [DOI] [PubMed] [Google Scholar]
- Gadow KD, DeVincent CJ, Pomeroy J, et al. 2005. Comparison of DSM-IV symptoms in elementary school-aged children with PDD versus clinic and community samples. Autism. 9:392–415. [DOI] [PubMed] [Google Scholar]
- Gadow KD, DeVincent CJ, Siegal VI, et al. 2013. Allele-specific associations of 5-HTTLPR/rs25531 with ADHD and autism spectrum disorder. Prog Neuro-Psychopharmacol Biol Psychiatry. 40:292–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadow KD, Guttmann-Steinmetz S, Rieffe C, et al. 2012. Depression symptoms in boys with autism spectrum disorder and comparison samples. J Autism Dev Disord. 42:1353–1363. [DOI] [PubMed] [Google Scholar]
- Gadow KD, Roohi J, DeVincent CJ, et al. 2008. Association of ADHD, tics, and anxiety with dopamine transporter (DAT1) genotype in autism spectrum disorder. J Child Psychol Psychiatry. 49:1331–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadow KD, Schwartz J, DeVincent C, et al. 2008. Clinical utility of autism spectrum disorder scoring algorithms for the Child Symptom Inventory. J Autism Dev Disord. 38:419–427. [DOI] [PubMed] [Google Scholar]
- Gadow KD, Sprafkin J. 2002. Child Symptom Inventory-4 Screening and Norms Manual. Stony Brook, NY: Checkmate Plus. [Google Scholar]
- Gadow KD, Sprafkin J. 2011. The Symptom Inventories: An Annotated Bibliography [On-line]. Stony Brook, NY: Checkmate Plus. Available: www.checkmateplus.com. [Google Scholar]
- Girgis RR, Slifstein M, Xu X, et al. 2011. The 5-HT(2A) receptor and serotonin transporter in Asperger’s disorder: A PET study with [(1)(1)C]MDL 100907 and [(1)(1)C]DASB. Psychiatry Res. 194:230–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goines PE, Ashwood P. 2013. Cytokine dysregulation in autism spectrum disorders (ASD): Possible role of the environment. Neurotoxicol Teratol. 36:67–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg J, Anderson GM, Zwaigenbaum L, et al. 2009. Cortical serotonin type-2 receptor density in parents of children with autism spectrum disorders. J Autism Dev Disord. 39:97–104. [DOI] [PubMed] [Google Scholar]
- Guhathakurta S, Singh AS, Sinha S, et al. 2009. Analysis of serotonin receptor 2A gene (HTR2A): Association study with autism spectrum disorder in the Indian population and investigation of the gene expression in peripheral blood leukocytes. Neurochem Int. 55:754–759. [DOI] [PubMed] [Google Scholar]
- Hazelwood LA, Sanders-Bush E. 2004. His452Tyr polymorphism in the human 5-HT2A receptor destabilizes the signaling conformation. Mol Pharmacol. 66:1293–1300. [PubMed] [Google Scholar]
- Hernandez I, Sokolov BP. 2000. Abnormalities in 5-HT2A receptor mRNA expression in frontal cortex of chronic elderly schizophrenics with varying histories of neuroleptic treatment. J Neurosci Res. 59:218–225. [PubMed] [Google Scholar]
- Hollingshead AB. 1975. Four Factor Index of Social Status. Department of Sociology, Yale University: New Haven, CT. [Google Scholar]
- Hranilović D, Blazevic S, Babic M, et al. 2010. 5-HT2A receptor gene polymorphisms in Croatian subjects with autistic disorder. Psychiatry Res.178:556–558. [DOI] [PubMed] [Google Scholar]
- Hranilović D, Bujas-Petković Z, Tomicić M, et al. 2009. Hyperserotonemia in autism: activity of 5HT-associated platelet proteins. J Neural Transm. 116:493–501. [DOI] [PubMed] [Google Scholar]
- Hu VW, Sarachana T, Kim KS, et al. 2009. Gene expression profiling differentiates autism case-controls and phenotypic variants of autism spectrum disorders: Evidence for circadian rhythm dysfunction in severe autism. Autism Res. 2:78–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C, Wenwei X, Yuan J, et al. 2013. Meta-analysis of association between the −1438A/G (rs6311) polymorphism of the serotonin 2A receptor gene and major depressive disorder. Neurol Res. 35:7–14. [DOI] [PubMed] [Google Scholar]
- Johansson C, Smedh C, Partonen P, et al. 2001. Seasonal affective disorder and serotonin-related polymorphisms. Neurobiol Dis. 8:351–357 [DOI] [PubMed] [Google Scholar]
- Kaat AJ, Gadow KD, Lecavalier L. 2013. Psychiatric symptom impairment in children with autism spectrum disorders. J Abnorm Child Psychol. 41:959–969. [DOI] [PubMed] [Google Scholar]
- Kamata M, Suzuki A, Yoshida K, et al. 2011. Genetic polymorphisms in the serotonergic system and symptom clusters of major depressive disorder. Affect Disord. 135:374–6. [DOI] [PubMed] [Google Scholar]
- Kato M, Serretti A. 2010. Review and meta-analysis of antidepressant pharmacogenetic findings in major depressive disorder. Mol Psychiatry. 15:473–500. [DOI] [PubMed] [Google Scholar]
- Kim JM, Stewart R, Bae KY, et al. 2012. Serotonergic and BDNF genes and risk of depression after stroke. J Affect Disord. 36:833–840. [DOI] [PubMed] [Google Scholar]
- Kistner-Griffin E, Brune C, Davis LK, et al. 2011. Parent-of-origin effects of the serotonin transporter gene associated with autism. Am J Med Genet B Neuropsychiatr Genet. 156:139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronfeld-Schor N, Einat H. 2012. Circadian rhythms and depression: Human psychopathology and animal models. Neuropharmacology. 62:101–114. [DOI] [PubMed] [Google Scholar]
- Kusumi I, Koyama T, Yamashita I. 1994. Serotonin-induced platelet intracellular calcium mobilization in depressed patients. Psychopharmacology (Berl). 113(3–4):322–327. [DOI] [PubMed] [Google Scholar]
- Lapin JP, Oxenkrug GF. 1969. Intensification of the central serotonergic processes as a possible determinal of the thymoleptic effect. Lancet. 1:132–136. [DOI] [PubMed] [Google Scholar]
- Lecavalier L, Gadow KD, DeVincent CJ, et al. 2009. Validation of DSM-IV model of psychiatric syndromes in children with autism spectrum disorder. J Autism Dev Disord. 39:278–289. [DOI] [PubMed] [Google Scholar]
- Lee H-J, Sung S-M, Lim S-W, et al. 2006. Seasonality associated with the serotonin 2A receptor −1438 A/G polymorphism. J Affect Disord. 95:145–148. [DOI] [PubMed] [Google Scholar]
- Lee LC, Newschaffer CJ, Lessler JT, et al. 2008. Variation in season of birth in singleton and multiple births concordant for autism spectrum disorder. Pediatr Perinat Epidemiol. 22:172–179. [DOI] [PubMed] [Google Scholar]
- Leyfer OT, Folstein SE, Bacalman S, et al. 2006. Comorbid psychiatric disorders in children with autism: Interview development and rates of disorders. J Autism Dev Disord. 36:849–861 [DOI] [PubMed] [Google Scholar]
- López-Muňoz F, Alamo C. 2009. Monoaminergic neurotransmission: The history of the discovery of antidepressants from 1950s until today. Curr Pharm Des. 15:1563–1586. [DOI] [PubMed] [Google Scholar]
- Lord C, Risi S, Lambrecht L, et al. 2000. The autism diagnostic observation schedule-generic: A standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord. 30:205–223. [PubMed] [Google Scholar]
- Lugnegård T, Hallerbäck MU, Gillberg C. 2011. Psychiatric comorbidity in young adults with a clinical diagnosis of Asperger syndrome. Res Dev Disabil. 32:1910–1917. [DOI] [PubMed] [Google Scholar]
- Magnusson KM, Constantino JN. 2011. Characterization of depression in children with autism spectrum disorders. J Dev Behav Pediatr. 32:332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier SF. 2003. Bi-directional immune-brain communication: Implications for understanding stress, pain, and cognition. Brain Behav Immun. 17:69–85. [DOI] [PubMed] [Google Scholar]
- McBride PA, Anderson GM, Hertzig ME, et al. 1989. Serotonergic responsivity in male young adults with autistic disorder. Results of a pilot study. Arch Gen Psychiatry. 46:213–21. [DOI] [PubMed] [Google Scholar]
- Meltzer CC, Price JC, Mathis CA, et al. 1999. PET imaging of serotonin type 2A receptors in late-life neuropsychiatric disorders. Am J Psychiatry. 156:1871–1878. [DOI] [PubMed] [Google Scholar]
- Meyer JH, Kapur S, Eisfeld B, et al. 2001. The effect of paroxetine on 5-HT(2A) receptors in depression: an [(18)F]setoperone PET imaging study. Am J Psychiatry. 158:78–85. [DOI] [PubMed] [Google Scholar]
- Miller AH, Haroon E, Raison CL, et al. 2013. Cytokine targets in the brain: Impact on neurotransmitters and neurocircuits. Depres Anxiety. 30:297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GA, Chapman JP. 2001. Misunderstanding analysis of covariance. J Abnorm Psychol. 110:40–48. [DOI] [PubMed] [Google Scholar]
- Minov C, Baghai TC, Schüle C, et al. 2001. Serotonin-2A-receptor and -transporter polymorphisms: lack of association in patients with major depression. Neurosci Lett. 303:119–22. [DOI] [PubMed] [Google Scholar]
- Molnar E, Lazary J, Benko A., et al. 2010. Seasonality and winter-type seasonal depression are associated with the rs731779 polymorphism of the serotin-2A receptor gene. Eur Neuropsychphmacol. 20:655–662. [DOI] [PubMed] [Google Scholar]
- Mück-Seler D, Jakovljević M, Pivac N. 1996. Platelet 5-HT concentrations and suicidal behaviour in recurrent major depression. J Affect Disord. 39:73–80. [DOI] [PubMed] [Google Scholar]
- Mück-Seler D, Pivac N, Mustapic M, et al. 2004. Platelet serotonin and plasma prolactin and cortisol in healthy, depressed and schizophrenic women. Psychiatry Res. 127:217–26. [DOI] [PubMed] [Google Scholar]
- Murphy DGM, Daly E, Schmitz N, et al. 2006. Cortical serotonin 5-HT2A receptor binding and social communication in adults with Asperger’s syndrome: An in vivo SPECT study. Am J Psychiatry. 163:934–936. [DOI] [PubMed] [Google Scholar]
- Narasimhan S, Lohoff FW. 2012. Pharmacogenetics of antidepressant drugs: current clinical practice and future directions. Pharmacogenomics. 13:441–464. [DOI] [PubMed] [Google Scholar]
- Oreland S, Pickering C, Gokturk L, et al. 2009. Two repeated maternal separation procedures differentially affect brain 5-hydroxytryptamine transporter and receptors in young and adult male and female rats. Brian Res. 1305:S37–S49. [DOI] [PubMed] [Google Scholar]
- Oswald P, Souery D, Massat I, et al. 2003. Lack of association between the 5HT2A receptor polymorphism (T102C) and unipolar affective disorder in a multicentric European study. Eur Neuropsychopharmacol. 13:365–368. [DOI] [PubMed] [Google Scholar]
- Pivac N, Mück-Seler D, Sagud M, et al. 2003. Long-term sertraline treatment and peripheral biochemical markers in female depressed patients. Prog Neuropsychopharmacol Biol Psychiatry. 27:759–65. [DOI] [PubMed] [Google Scholar]
- Pjrek E, Winkler D, Heiden A, et al. 2004. Seasonality of birth in seasonal affective disorder. J Clin Psychiatry. 65:1389–1393. [DOI] [PubMed] [Google Scholar]
- Pouw LBC, Rieffe C, Stockmann L, et al. 2013. The link between emotion regulation, social functioning, and depression in boys with ASD. Res Autism Spect Disord. 7:549–556. [Google Scholar]
- Raison CL, Miller AH. 2013. The evolutionary significance of depression in pathogen host defense (PATHOS-D). Mol Psychiatry. 18:15–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roohi J, DeVincent CJ, Hatchwell E, et al. 2009. Association of a monoamine oxidase-A gene promoter polymorphism with ADHD and anxiety in boys with autism spectrum disorder. J Autism Dev Disord. 39:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruljancic N, Mihanovic M, Cepelak I, et al. 2013. Platelet serotonin and magnesium concentrations in suicidal and non-suicidal depressed patients. Magnes Res. 26:9–17. [DOI] [PubMed] [Google Scholar]
- Rutter M, LeCouteur A, Lord C. 2003. Autism Diagnostic Interview-Revised. Los Angeles, CA: Western Psychological Services. [Google Scholar]
- Savitz JB, Drevets WC. 2013. Neurorecepto imaging in depression. Neurobiol Dis. 52:49–65. [DOI] [PubMed] [Google Scholar]
- Schain RJ, Freedman DX. 1961. Studies on 5-hydroxyindole metabolism in autistic and other mentally retarded children. J Pediatr. 58:315–320. [DOI] [PubMed] [Google Scholar]
- Sheline YI, Mintun MA, Barch DM, et al. 2004. Decreased hippocampal 5-HT(2A) receptor binding in older depressed patients using [18F]altanserin positron emission tomography. Neuropsychopharmacology. 29:2235–2241. [DOI] [PubMed] [Google Scholar]
- Shimbo D, Child J, Davidson K, et al. 2002. Exaggerated serotonin-mediated platelet reactivity as a possible link in depression and acute coronary syndromes. Am J Cardiol. 89:331–3. [DOI] [PubMed] [Google Scholar]
- Siegel M, Beaulieu AA. 2012. Psychotropic medications in children with autism spectrum disorders: a systematic review and synthesis for evidence-based practice. J Autism Dev Disord. 42:1592–1605.\ [DOI] [PubMed] [Google Scholar]
- Simonoff E, Pickles A, Charman T, et al. 2008. Psychiatric disorders in children with autism spectrum disorders: Prevalence, comorbidity, and associated factors in a population-derived sample. J Am Acad Child Adolesc Psychiatry. 47, 921–929. [DOI] [PubMed] [Google Scholar]
- Slavich GM, Way BM, Eisenberger NI, et al. 2010. Neural sensitivity to social rejection is associated with inflammatory responses to social stress. PNAS. 107:14817–14822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RM, Papp AC, Webb A, et al. 2013. Multiple regulatory variants modulate expression of the serotonin 2A receptor gene (HTR2A) in human cortex. Biol Psychiatry. 73:546–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurlock G, Heils A, Holmans P, et al. 1998. A family based association study of T102C polymorphism in 5HT2A and schizophrenia plus identification of new polymorphisms in the promoter. Mol Psychiatry. 3:42–49. [DOI] [PubMed] [Google Scholar]
- Thanseem I, Anitha A, Nakamura K, et al. 2012. Elevated transcription factor specificity protein 1 in autistic brains alters the expression of autism candidate genes. Biol Psychiatry. 71:710–418. [DOI] [PubMed] [Google Scholar]
- Uebelhack R, Franke L, Herold N, et al. 2006. Brain and platelet serotonin transporter in humans-correlation between [123I]-ADAM SPECT and serotonergic measurements in platelets. Neurosci Lett. 406:153–8. [DOI] [PubMed] [Google Scholar]
- Vargas DL, Nascimbene C, Krishnan C, et al. 2005. Neurological activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 57:67–81. [DOI] [PubMed] [Google Scholar]
- Veenstra-VanderWeele J, Kim SJ, Lord C, et al. 2002. Transmission disequilibrium studies of the serotonin 5-HT2A receptor gene (HTR2A) in autism. Am J Med Genet. 114:277–283. [DOI] [PubMed] [Google Scholar]
- Whitaker-Azmitia PM. 2001. Serotonin and brain development: Role in human developmental diseases. Brain Res Bull. 56:479–485. [DOI] [PubMed] [Google Scholar]
- Wilkie MJ, Smith G, Day RK, et al. 2009. Polymorphisms in the SLC6A4 and HTR2A genes influence treatment outcome following antidepressant therapy. Pharmacogenomics J. 9:61–70. [DOI] [PubMed] [Google Scholar]
- Willeit M, Praschak-Rieder N. 2010. Imaging the effects of genetic polymorphisms on radioligand binding in the living human brain: A review on genetic neuroreceptor imaging of monoaminergic systems in psychiatry. NeuroImage. 53:878–892. [DOI] [PubMed] [Google Scholar]
- Willins DL, Alsayegh L, Berry SA, et al. 1998. Serotonergic antagonist effects on trafficking of serotonin 5-HT2A receptors in vitro and in vivo. Ann NY Acad Sci. 861:121–127. [DOI] [PubMed] [Google Scholar]
- Witwer AN, Lecavalier L. 2010. Validity of comorbid psychiatric disorders in youngsters with autism spectrum disorders. J Dev Phys Disabil. 22:367–380. [Google Scholar]
- Yatham LN, Liddle PF, Shiah IS, et al. 2000. Brain serotonin2 receptors in major depression: a positron emission tomography study. Arch Gen Psychiatry. 57:850–858. [DOI] [PubMed] [Google Scholar]