

Abstract
Purpose
Mutant hypothyroid mouse models have recently shown that thyroid hormone is critical for skeletal development during an important prepubertal growth period. Additionally, thyroid hormone negatively regulates total body fat, consistent with the well-established effects of thyroid hormone on energy and fat metabolism. Since bone marrow mesenchymal stromal cells differentiate into both adipocytes and osteoblasts and a relationship between bone marrow adipogenesis and osteogenesis has been predicted, we hypothesized thyroid hormone deficiency during the postnatal growth period increases marrow adiposity in mice.
Methods
Marrow adiposity in TH-deficient (Tshr−/−) mice treated with T3/T4, TH receptor β-specific agonist GC-1, or vehicle control was evaluated via dual-energy X-ray absorptiometry and osmium micro-computed tomography. To further examine the mechanism for thyroid hormone regulation of marrow adiposity, we used real-time RT-PCR to measure the effects of thyroid hormone on adipocyte differentiation markers in primary mouse bone marrow mesenchymal stromal cells and two mouse cell lines in vitro and in Tshr−/− mice in vivo.
Results
Marrow adiposity increased >20% (P < 0.01) in Tshr−/− mice at 3 weeks of age, and treatment with T3/T4 when serum thyroid hormone normally increases (day 5–14) rescued this phenotype. Furthermore, GC-1 rescued this phenotype equally well, suggesting this thyroid hormone effect is in part mediated via TRβ signaling. Treatment of bone marrow mesenchymal stromal or ST2 cells with T3 or GC-1 significantly increased expression of several brown/beige fat markers. Moreover, injection of T3/T4 increased browning-specific markers in white fat of Tshr−/− mice.
Conclusions
These data suggest that thyroid hormone regulation of marrow adiposity is mediated at least in part via activation of TRβ signaling.
Keywords: Thyroid hormone, Thyroid hormone receptor beta, Bone marrow stromal cells, Adiposity, Brown/beige fat, Bone density, Obesity
Introduction
Fat and bone are complex tissues which interact in many different ways. Despite extensive study, the relationship between fat and bone is far from exhaustively understood. Two of the most prevalent and potentially debilitating chronic diseases in the Western world are osteoporosis and obesity, and, although conventional wisdom would suggest that obesity protects against osteoporosis, these two diseases often afflict the same patients and may be causally related [1]. Since osteoblasts and adipocytes both are derived from common mesenchymal stem cell (MSC) precursors, a close relationship between these tissues has been predicted, particularly in the bone marrow, a site of direct interface between bone and fat. A simple shift in the differentiation of MSCs from osteoblasts to adipocytes would explain many of the observed changes in bone and marrow adipose tissue (MAT) during aging and metabolic disease states. For example, obesity has been associated with fracture risk in older men [2], and marrow adiposity seems to negatively correlate with cortical bone parameters in patients at varying ages [3, 4]. Furthermore, increased marrow adiposity correlates with increased fracture risk in the context of type I diabetes [5, 6]. Counterintuitively, type II diabetes is frequently characterized by an increased fracture risk even in the presence of above average bone mass [7]; the situation is made more complex by the fact that treatment with thiazolidinediones, a popular antidiabetic drug class, which acts by activating PPARγ, induces both marrow adipogenesis, and bone loss in rodents [8-10] and possibly in humans [11, 12]. However, a simple shift in differentiation from bone to fat does not explain all observations; for example, periods of peak MAT formation and skeletal growth coincide in humans [13], suggesting a more complex relationship between bone and MAT.
Thyroid hormone (TH) has received considerable attention as a regulator of bone and adipose tissues based on its established role in controlling energy metabolism and skeletal growth [14, 15]. For example, hypothyroidism during childhood is a well-known cause of cretinism, and we have recently shown that TH is critical for long bone growth in mice during a critical prepubertal window [16]. In addition, TH is a well-established regulator of basal metabolic rate, and hypothyroid patients often present clinically as overweight. Thus, TH signaling is of critical importance for possible therapeutic treatments of diseases of both bone and energy metabolism.
The effects of TH are mediated via activation of nuclear TH receptors (TRs), of which there are multiple isoforms (e.g., TRα and TRβ). TRs bind to TH response elements in the regulatory regions of target genes and modulate gene transcription in the presence of TH ligand via complex interactions with many other nuclear proteins, including corepressors, coactivators, and cointegrators, leading to chromatin remodeling [17]. While both TRα and TRβ are known to be expressed in adipocytes and osteoblast lineage cells, the issue of which TR mainly contributes to the TH effects on skeletal and adipose tissue metabolism remains to be settled.
In this study, we set out to investigate the effect of TH on the relationship of bone and fat in the bone marrow adipose tissue. In our previous studies, we showed that an increase in TH levels is predominantly responsible for bone accretion during the prepubertal growth period, and that TH-deficient mice exhibited greater adiposity at the end of this growth period [16]. Here, we have endeavored to characterize the relationships among TH, bone, and bone marrow adipose tissue by testing whether TH can reduce bone marrow adiposity and influence differentiation of bone marrow mesenchymal progenitors, and by identifying which TR is primarily involved in mediating the TH effects on bone marrow adipose tissue.
Materials and methods
Chemicals and reagents
T3, T4, insulin, dexamethasone, indomethacin, 3-isobutyl-1-methylxanthine (IBMX), osmium tetroxide, and potassium dichromate were purchased from Sigma (St. Louis, MO, USA). Recombinant human bone morphogenetic protein-4 (BMP-4) was purchased from ThermoFisher (catalog no. PHC9534). GC-1 was a gift from Professor Thomas S. Scanlan, Oregon Health & Science University, Portland, OR. Mouse marrow stromal cell line ST2 was obtained from American Type Culture Collection (Manassas, VA).
Mice
Tshr+/− heterozygous mice with a point mutation in the coding region of the thyroid stimulating hormone receptor (Tshr) gene were purchased from the Jackson Laboratory (Bar Harbor, ME, USA; CBy.RF-Tshrhyt/J, stock no. 000805). We received and maintain a colony of heterozygous Tshr+/− mice, which are phenotypically euthyroid and therefore used as controls. We bred heterozygotes to generate homozygous Tshr−/− mice in which serum TH levels are undetectable [18]. Mice from both genders were used, and both genders were pooled for analyses since mice at the ages studied (≤ postnatal day 21) are prepubertal and too young to display sexual dimorphism in the traits measured. Mice were housed at the VA Loma Linda Healthcare System Veterinary Medical Unit (Loma Linda, CA, USA) under standard approved laboratory conditions. All the procedures were performed with the approval of the Institutional Animal Care and Use Committee of the VA Loma Linda Healthcare System (permit number: 0029-01-13). For euthanasia, animals were exposed to CO2 prior to cervical dislocation.
TH/GC-1 rescue experiments
Tshr−/− mice were injected intraperitoneally with a combination of 1 μg T3 and 10 μg T4 or 0.2 μg TRβ-specific agonist GC-1 daily for 10 days from postnatal day 5 to day 14 or the same volume of vehicle (5 mM NaOH) as described [16, 19, 20]. Phenotypically euthyroid littermate Tshr+/− mice treated with vehicle were used as controls (henceforth referred to as euthyroid controls). At postnatal day 21, mice were euthanized. After euthanasia, bones were collected for dual-energy X-ray absorptiometry (DXA) and micro-computed tomography (μCT) analysis, and abdominal subcutaneous white fat and interscapular brown fat were collected for RNA extraction and real-time RT-PCR analysis (Fig. 1). Additionally, skeletal tissue (femoral and tibial epiphyses) from 7 day-old Tshr−/− injected daily with 0.2 μg GC-1 for 2 days was harvested for RNA extraction and real-time RT-PCR analysis.
Fig. 1.
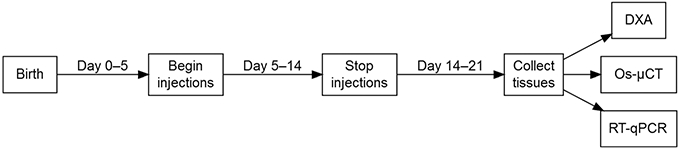
TH treatment regime and tissue collection in TH-deficient mice. Mice were injected on days 5–14 and tissues collected on day 21. Beginning at 5 days after birth, TH-deficient Tshr−/− mice were injected with T3/T4, TH receptor (TR) β-specific agonist GC-1, or vehicle control (NaOH) daily for 10 days. Euthyroid control mice (Tshr+/− littermates) were injected with vehicle control (NaOH) daily for 10 days. At postnatal day 14, injections were stopped, and all mice received standard laboratory care until postnatal day 21, at which time mice were euthanized and tissues were collected for analysis
Dual-energy X-ray absorptiometry
A PIXImus instrument (Lunar Corp, Madison, WI, USA) was used to evaluate bone mineral density (BMD) and percentage adiposity in femurs, tibiae, and lumbar vertebrae from mice treated with T3/T4, GC-1, or vehicle control. Data can be found in Online Resource 1.
Micro-computed tomography
MAT was visualized and quantified as described [21]. Briefly, femurs and tibiae were collected, cleaned, and fixed in 4% paraformaldehyde for 72 h at 4 °C. The bones were then washed and decalcified for 18 days in 14% ethylene-diaminetetraacetic acid at 4 °C. Decalcified bones were stained for lipid with a mixture of equal volumes 2% osmium tetroxide and 5% potassium dichromate for 60 h. After washing, the stained bones were imaged using μCT (viva CT40, Scanco Medical AG, Switzerland) with energy of 55 kVp, an integration time of 500 ms, and a maximum isometric voxel size of 10 μm.
Cell cultures
To study the molecular effects of T3 and GC-1 treatment on marrow mesenchymal stromal cells, which can differentiate into several tissues including white fat, brown fat, and bone, we used primary bone marrow mesenchymal stromal (BMMS) cells in addition to the established mouse marrow stromal ST2 cell line and the mouse embryonic stem cell line C3H10T1/2. BMMS cells were harvested from femurs and tibiae of 4-week-old wild type (C57BL/6J) mice and grown in six-well culture plates with α-minimum essential medium (α-MEM) supplemented with 10% fetal bovine serum (FBS), penicillin (100 units/mL), and streptomycin (100 μg/mL) to 80% confluence before beginning experiments. ST2 cells were grown in 6 cm culture dishes with α-MEM supplemented with 10% FBS, penicillin (100 units/mL), and streptomycin (100 μg/mL) to 70% confluence. C3H10T1/2 cells were grown to confluence in 6 cm culture dishes with DMEM supplemented with 10% FBS, penicillin (100 units/mL), and streptomycin (100 μg/mL).
Adipocyte differentiation
For gene expression data, primary BMMS cells or ST2 cells were synchronized with serum-free α-MEM containing 0.1% bovine serum albumin, penicillin, and streptomycin for 24 h before treatment with the adipocyte differentiation factors insulin (1 μM), dexamethasone (10 nM), and indomethacin (250 nM) for 1, 3 or 6 days. Experimental groups were treated with 0.1, 1, or 10 ng/mL (0.154, 1.54, or 15.4 nM) T3 or 0.1 nM GC-1 in addition to the adipocyte differentiation factors. For comparison of the effect of TH on marrow mesenchymal stromal cells under adipogenic conditions vs. osteogenic conditions, BMMS and ST2 cells were also treated with the same dose of T3 in the presence of 50 μg/mL (0.284 mM) ascorbic acid and 10 mM β-glycerophosphate instead of the adipocyte differentiation factors listed above.
C3H10T1/2 cells must be treated with bone morphogenetic protein-4 (BMP-4) in order to induce commitment to the adipocyte lineage [22]. Briefly, C3H10T1/2 cells were plated at a low density (20,000 cells per 6 cm dish) and grown for 6 days until confluence in the presence of 50 ng/mL BMP-4. Adipocyte differentiation was then induced using D-MEM containing 1% FBS, insulin (1 μM), dexamethasone (250 nM), and IBMX (0.5 mM) for 6 days followed by 1 or 6 days’ treatment with 10 ng/mL (15.4 nM) T3 or vehicle control.
Quantitative real-time RT-PCR
RNA samples were extracted from mouse abdominal white adipose tissue (WAT), mouse interscapular brown adipose tissue (BAT), bone, primary BMMS cells, ST2 cells, and C3H10T1/2 cells with TRI Reagent (MRC, Cincinnati, OH, USA) and the E.Z.N.A.® Total RNA Kit I (Omega Bio-tek, Norcross, GA, USA) according to the manufacturers’ instructions. An aliquot of RNA (300 ng) was reverse-transcribed using SuperScript II® reverse transcriptase (Invitrogen, Grand Island, NY). Quantitative real time RT-PCR was performed as previously described [23]. The delta-delta-Ct method was used to calculate relative gene expression with the housekeeping gene PPIA serving as an internal control as previously described [23]. The primer sequences used for real time RT-PCR are listed in Table 1, and Ct values can be found in Online Resource 2.
Table 1.
Mouse primer sequences for real-time RT-PCR
| Gene | Forward | Reverse |
|---|---|---|
| ADPN | 5′-GATGCAGGTCTTCTTGGTCCTA | 5′-AGCGAATGGGTACATTGGGA |
| ALP | 5′-ATGGTAACGGGCCTGGCTACA | 5′-AGTTCTGCTCATGGACGCCGT |
| ASC-1 | 5′-GGGTGGCACTCAAGAAAGAG | 5′-AGTGTTCCAGGACACCCTTG |
| CEBPA | 5′-AAACAACGCAACGTGGAGAC | 5′-TGTCCAGTTCACGGCTCAG |
| Cd137 | 5′-CGTGCAGAACTCCTGTGATAAC | 5′-GTCCACCTATGCTGGAGAAGG |
| Epsti1 | 5′-ACCCTGATAGCACCAAACGA | 5′-AGGTCTGCCAGTTCTTGCTC |
| HOXC8 | 5′-GTCTCCCAGCCTCATGTTTC | 5′-TCTGATACCGGCTGTAAGTTTG |
| HOXC9 | 5′-GCAGCAAGCACAAAGAGGAGAAG | 5′-GCGTCTGGTACTTGGTGTAGGG |
| Osx | 5′-TCCTCTCTGCTTGAGGAAGAAG | 5′-GAGTCCATTGGTGCTTGAGAAG |
| P2RX5 | 5′-CTGCAGCTCACCATCCTGT | 5′-CACTCTGCAGGGAAGTGTCA |
| PAT2 | 5′-GTGCCAAGAAGCTGCAGAG | 5′-TGTTGCCTTTGACCAGATGA |
| PGC-1α | 5′-GCAGCCAAGACTCTGTATGG | 5′-TTCCGATTGGTCGCTACACC |
| PPARγ | 5′-GAGGGCGATCTTGACAGGAA | 5′-GGATCGAAACTGGCACCCTT |
| PPIA | 5′-CCATGGCAAATGCTGGACCA | 5′-TCCTGGACCCAAAACGCTCC |
| PRDM16 | 5′-CAGCACGGTGAAGCCATTC | 5′-GCGTGCATCCGCTTGTG |
| TBX1 | 5′-GTCAAGGCTCCGGTGAAGAA | 5′-TGGAACGTGGGGAACATTCG |
| UCP1 | 5′-TGGAAAGGGACGACCCCTAA | 5′-CAGGAGTGTGGTGCAAAACC |
| Zfp516 | 5′-CTGAGGAAGTGGTGGATGACAG | 5′-CTGAGGAAGTGGTGGATGACAG |
| Zic1 | 5′-TTTCCCTGCCCGTTTCCTG | 5′-CCCTCGAACTCGCACTTGA |
ADPN adiponectin, ALP alkaline phosphatase, ASC-1 asc-type amino acid transporter 1, CEBPA CCAAT/enhancer-binding protein alpha, Epsti1 epithelial stromal interaction 1, HOXC8 homeobox C8, HOXC9 homeobox C9, Osx osterix, P2RX5 purinergic receptor P2X, ligand gated ion channel, 5, PAT2 proton assistant amino acid transporter-2, PGC-1α peroxisome proliferator-activated receptor gamma, coactivator 1 alpha, PPARγ peroxisome proliferator-activated receptor gamma, PPIA peptidylprolyl isomerase A, PRDM16 PR domain containing 16, TBX1 T-box 1, UCP1 uncoupling protein 1, Zfp516 zinc finger protein 516, Zic1 zinc finger protein of the cerebellum 1
Statistics
All values are presented as mean ± SEM. Student’s t-tests were performed using Microsoft Excel to compare treatment groups, and a P value less than 0.05 was considered statistically significant.
Results
TH rescues bone marrow adiposity
In order to determine if TH influences MAT, we compared bone marrow adiposity, measured by DXA, in three skeletal sites among TH-deficient Tshr−/− mice, TH-deficient mice injected with T3/T4, and euthyroid controls (Tshr+/− littermates, Fig. 2a; raw data in Online Resource 1). Compared to euthyroid controls, Tshr−/− mice exhibited significantly increased bone marrow adiposity at all three skeletal sites; 20 and 19% increases were observed in tibiae and femurs, respectively, while 60% increases were observed in the lumbar vertebrae. Furthermore, injection with T3/T4 for 10 days from postnatal day 5 to 14, when serum levels of TH are known to increase during the prepubertal growth period in mice, was sufficient to bring bone marrow adiposity down to levels seen in euthyroid controls or, in the case of femurs, levels even below those seen in euthyroid controls. This trend was confirmed using osmium-μCT to image MAT (Fig. 2b).
Fig. 2.

TH reduces bone marrow adiposity. TH-deficient Tshr−/− mice were injected with T3/T4, TR β-specific agonist GC-1, or vehicle control (5 mM NaOH) on postnatal days 5–14 and compared to euthyroid controls (Tshr+/− littermates) injected with NaOH. At postnatal day 21, bone marrow adiposity and BMD were measured using DXA and μCT. a Bone marrow adiposity, presented as mean fold-change vs. euthyroid controls ± SEM (n = 5–14). TH-deficient Tshr−/− mice have 20, 19, and 60% greater bone marrow adiposity than euthyroid controls at the tibiae, femurs, and vertebrae, respectively. Injection of T3/T4 or GC-1 on postnatal days 5–14 brought bone marrow adiposity down to or below the levels seen in euthyroid controls. Fold-change is vs. euthyroid controls (Tshr+/− NaOH). * = P < 0.05. b Osmium-μCT images showing greater bone marrow adiposity in Tshr−/− mice which is rescued by T3/T4 treatment. c Correlation analysis of bone marrow adiposity and BMD for all genotypes and treatments shows a significant negative correlation at the femurs and lumbar vertebrae
One possible explanation for increased bone MAT in response to TH deficiency and decreased bone MAT in response to TH replacement is that TH might cause a shift in differentiation of mesenchymal progenitor cells from adipocytes to osteoblasts. If this explanation is correct, we would expect a negative correlation between bone and MAT. In support of this explanation, we observed a significant negative correlation of bone adiposity and BMD in both lumbar vertebrae and femurs (Fig. 2c; Pearson’s correlation coefficients r = −0.5, P < 0.05 and r = −0.4, P < 0.05, respectively). However, the correlation between bone adiposity and BMD in tibiae was not significant (Pearson’s correlation coefficient r = −0.18, P = 0.33).
T3 increases brown/beige adipose markers in marrow cells
A possible explanation for the negative correlations we observed between MAT and BMD is that there is a balance between differentiation of marrow mesenchymal progenitor cells into adipocytes vs. osteoblasts. Under this explanation, TH might cause the balance to shift toward differentiation of osteoblasts rather than adipocytes. In support of this hypothesis, we observed that T3 can increase bone differentiation markers in primary BMMS cells cultured under osteogenic conditions (in the presence of ascorbic acid and β-glycerophosphate; Fig. 3a). This effect of T3 is consistent with our previously reported data [19]. To test whether this shift in differentiation is indeed the mechanism of our observed TH-mediated decrease in MAT, we used quantitative real-time RT-PCR to measure the expression of several marker genes in two in vitro models of marrow mesenchymal progenitors: primary mouse BMMS cells and the established mouse marrow stromal cell line ST2.
Fig. 3.
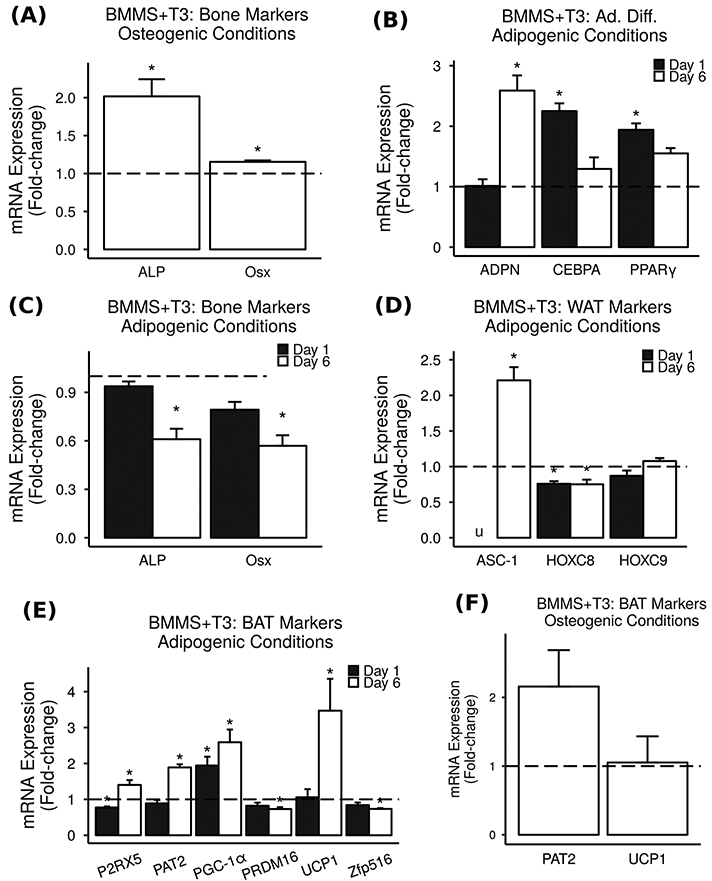
T3 increases brown/beige markers in BMMS cells. Primary BMMS cells were treated with 10 ng/mL (15.4 nM) T3, and mRNA expression was measured via real-time RT-PCR. Results are presented as mean fold-change vs. vehicle control ± SEM (n = 2–4). * = P < 0.05 vs. vehicle control, u = could not be measured due to the detection limits of the instrument. a Under osteogenic conditions, T3 significantly increased bone differentiation markers in BMMS cells. b Under adipogenic conditions, T3 significantly increased adipocyte differentiation markers in BMMS cells. c Under adipogenic conditions, T3 did not increase bone markers in BMMS cells. d Under adipogenic conditions, treatment with T3 for 1 or 6 days had relatively small effects on WAT markers in BMMS cells. e Under adipogenic conditions, T3 caused significant increases in BAT markers in BMMS cells. f Under osteogenic conditions, T3 did not increase BAT markers in BMMS cells
Primary BMMS cells cultured under adipogenic conditions and treated with T3 or vehicle for 1 or 6 days showed significant increases in adipocyte differentiation markers ADPN, CEBPA, and PPARγ (Fig. 3b). Furthermore, treatment with T3 did not increase osteoblast differentiation markers ALP and Osx in primary BMMS cells cultured under adipogenic conditions (Fig. 3c). Given our observation that TH decreases marrow adiposity, we expected treatment with T3 to decrease adipocyte markers and increase bone markers; however, we observed exactly the opposite of what we expected. Thus, a shift in differentiation of mesenchymal precursors from adipocytes to osteoblasts does not appear to be the primary mechanism of TH-mediated decrease in MAT.
An alternative explanation for the decreased MAT we observed in TH-treated mice is that TH causes an increase in fat and energy metabolism, thereby decreasing the amount of adipose tissue present. While TH can increase metabolism via a number of different mechanisms, one such mechanism of note is an increase in metabolically active brown/beige adipose tissue (BAT). An increase in the ratio of BAT to WAT in the bone marrow would result in a decrease in overall MAT volume, since BAT does not maintain large stores of lipids in the same way WAT does, and an increase in either WAT or BAT could explain the increased adipocyte differentiation markers we observed. In support of this hypothesis, WAT markers HOCX8 and HOXC9, but not ASC-1, were unaffected or decreased by T3 treatment (Fig. 3d). In addition, T3 treatment caused significant 1.4–3.5-fold increases in BAT markers PAT2, P2RX5, PGC-1α, and UCP1 in primary BMMS cells under adipogenic conditions (Fig. 3e). Interestingly, T3 did not significantly increase expression of BAT markers when BMMS cells were cultured under osteogenic conditions (Fig. 3f), suggesting that T3 may have opposite effects on BAT and bone differentiation depending on context.
In the established ST2 mouse mesenchymal marrow cell line, we confirmed that T3 did not increase bone markers under adipogenic conditions (Fig. 4a) but did increase adipocyte differentiation markers (Fig. 4b). In addition, we found that T3 treatment led to 1.1–1.8-fold increases in WAT markers HOXC8 and HOXC9 (Fig. 4c) and caused significant 1.1–14.4-fold increases in BAT markers P2RX5, PAT2, PGC-1α, PRDM16, and Zfp516 (Fig. 4d). An increase in both WAT and BAT markers in response to T3 treatment might be expected if T3 is inducing browning of the MAT rather than a direct increase in classical BAT differentiation. Furthermore, the effect of T3 on ST2 cell gene expression may be dose-dependent (Fig. 4e). While the overall trends observed in BMMS cells were confirmed in ST2 cells, the gene expression patterns in response to T3 treatment differed both in terms of which genes were most highly upregulated and in terms of the time point at which they were upregulated. These changes may be due to inherent differences between cultured primary cells and immortalized cell lines in addition to the developmental stage at which the primary cells were harvested from mice.
Fig. 4.
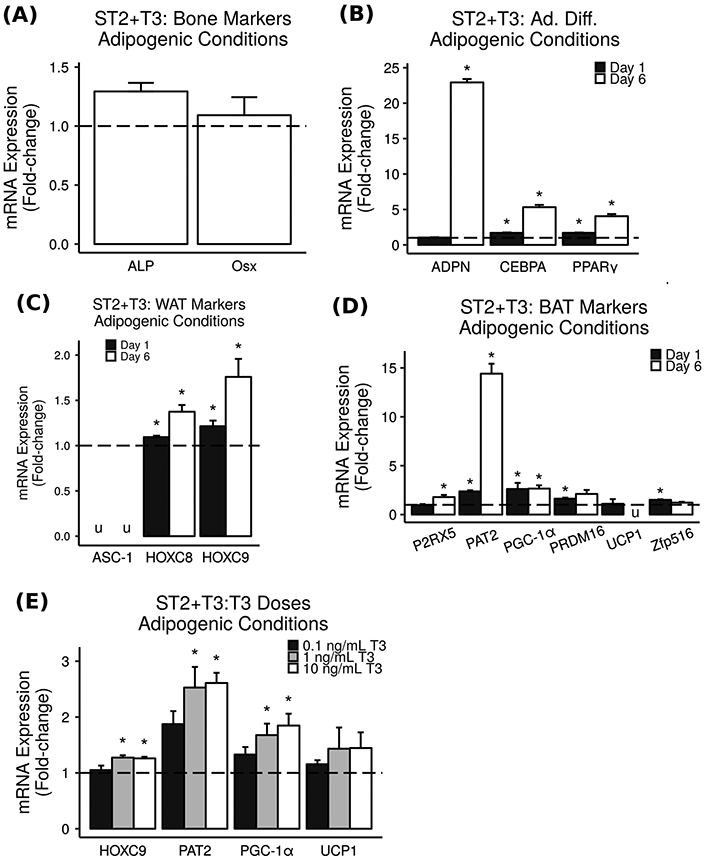
T3 increases BAT and WAT markers in ST2 cells. Mouse mesenchymal stromal cell line ST2 was treated with varying doses of T3 under adipogenic conditions, and mRNA expression was measured via real-time RT-PCR. Results are presented as mean fold-change vs. vehicle control ± SEM (n = 3–4). * = P < 0.05 vs. vehicle control, u = could not be measured due to the detection limits of the instrument. a Bone markers were not increased by 10 ng/mL (15.4 nM) T3 treatment in ST2 cells under adipogenic conditions. b Adipocyte differentiation markers were increased by 10 ng/mL (15.4 nM) T3 treatment in ST2 cells under adipogenic conditions. c WAT markers were increased by 10 ng/mL (15.4 nM) T3 treatment in ST2 cells. d BAT markers were significantly increased by 10 ng/mL (15.4 nM) T3 treatment in ST2 cells. e Significant increases in expression of WAT marker HOCX9 and BAT markers PAT2 and PGC-1α were observed when ST2 cells were treated for 1 day with 1 or 10 ng/mL (1.54 or 15.4 nM) T3 but not with the lower dose of 0.1 ng/mL (0.154 nM) T3
Furthermore, adipocyte-differentiated C3H10T1/2 cells treated with T3 showed significant increases in adipocyte differentiation markers ADPN, CEBPA, and PPARγ (Fig. 5a), WAT marker Asc-1 (Fig. 5b), and BAT markers PAT2, PGC-1α, UCP1, and Zfp516 (Fig. 5c). Notably, key BAT gene UCP1 was increased 24.9-fold.
Fig. 5.
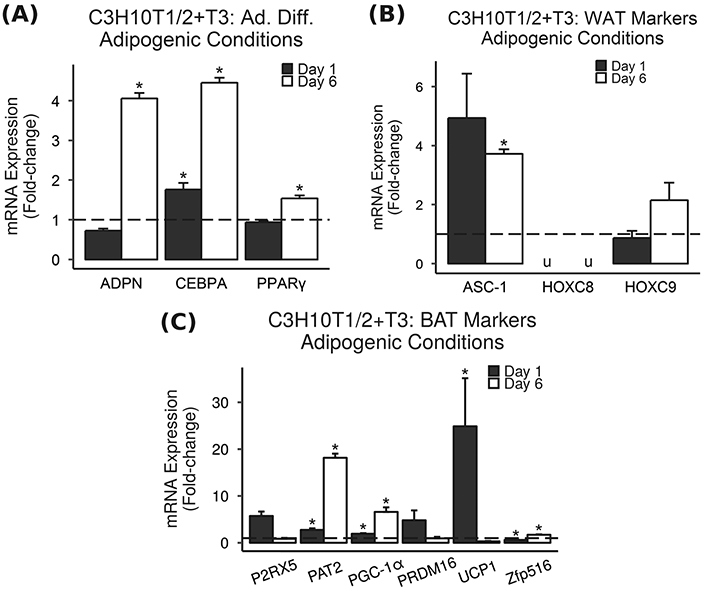
T3 increases BAT and WAT markers in C3H10T1/2 cells. Adipocyte-differentiated mouse C3H10T1/2 cells were treated with T3 under adipogenic conditions, and mRNA expression was measured via real-time RT-PCR. Results are presented as mean fold-change vs. vehicle control ± SEM (n = 3–4). * = P < 0.05 vs. vehicle control, u = could not be measured due to the detection limits of the instrument. a Adipocyte differentiation markers were increased by 10 ng/mL (15.4 nM) T3 treatment in C3H10T1/2 cells under adipogenic conditions. b WAT markers were increased by 10 ng/mL (15.4 nM) T3 treatment in C3H10T1/2 cells under adipogenic conditions. c BAT markers were significantly increased by 10 ng/mL (15.4 nM) T3 treatment in C3H10T1/2 cells under adipogenic conditions. Notably, key BAT gene UCP1 was increased 24.9-fold
TH increases brown and beige adipose markers in vivo
In order to investigate whether TH can cause browning of WAT into beige adipose tissue or direct differentiation of mesenchymal progenitors into classical BAT, we used quantitative real-time RT-PCR to measure gene expression in abdominal subcutaneous WAT (Fig. 6a, b) and interscapular BAT (Fig. 6c, d) from TH-deficient Tshr−/− mice treated with TH or vehicle and from euthyroid controls. WAT from vehicle-treated Tshr−/− mice significantly expressed less than 1% of important BAT marker UCP1 compared to WAT from euthyroid controls (Fig. 6a). In addition, expression of browning-specific markers Cd137, Epsti1, and TBX1 was also significantly decreased in WAT from Tshr−/− mice (Fig. 6b). Treatment with TH significantly increased expression of classical BAT markers PGC-1α, PRDM16, UCP1, and Zic1 (Fig. 6a) and browning-specific markers Cd137, Epsti1, and TBX1 (Fig. 6b) to levels well above those seen in WAT of euthyroid controls. The significant decreases of browning-specific markers in WAT from TH-deficient mice and the significant increases of browning-specific markers in WAT from TH-treated mice suggest that TH induces conversion of WAT into beige adipose tissue, although further study is needed to validate the functional relevance of the observed changes in expression levels of BAT and WAT markers to skeletal energy metabolism.
Fig. 6.
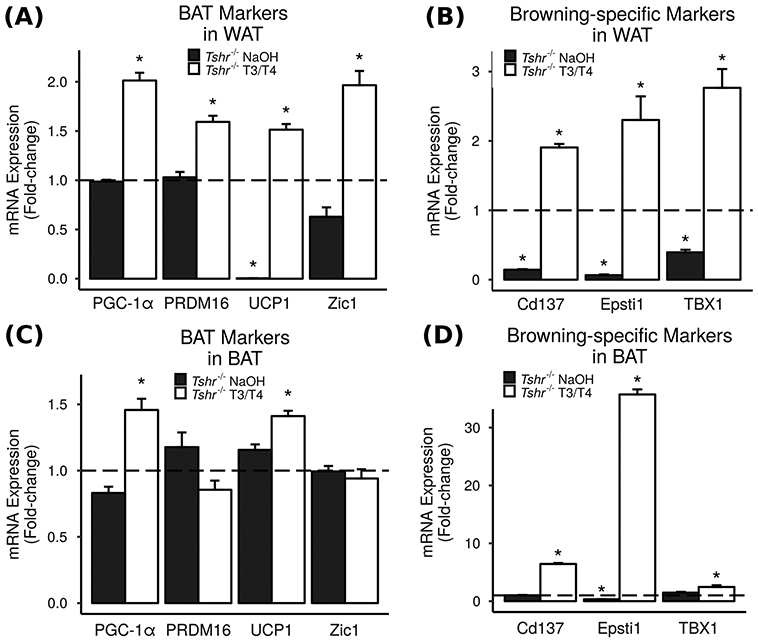
TH increases brown and beige adipose markers in vivo. Abdominal subcutaneous WAT and interscapular BAT were collected from TH-deficient Tshr−/− mice injected with T3/T4 or vehicle (NaOH), and compared to euthyroid controls (Tshr+/− littermates) by real-time RT-PCR. Results are presented as mean fold-change vs. euthyroid controls ± SEM (n = 4–6). * = P < 0.05 vs. euthyroid controls. a Classical BAT gene expression in WAT. b Browning-specific gene expression in WAT. c Classical BAT gene expression in BAT. d Browning-specific gene expression in BAT
The decrease observed in several brown and beige adipose markers in the WAT of TH-deficient mice was blunted in the BAT of these same mice (Fig. 6c, d). However, treatment with TH similarly increased expression of BAT markers PGC-1α, UCP1, Cd137, Epsti1, and TBX1 in the BAT of Tshr−/− mice compared to euthyroid controls (Fig. 6c, d).
GC-1 reduces bone marrow adiposity and increases BAT markers
Finally, to elucidate whether the TH effects on bone marrow adiposity are mediated via ligand binding TRα1 and/or TRβ1 receptors, we injected Tshr−/− mice with TRβ-specific agonist GC-1 and measured bone marrow adiposity. We found that GC-1 was at least as effective as T3/T4 at bringing bone marrow adiposity levels down to or below levels seen in euthyroid controls (Fig. 2a). In addition, we treated primary BMMS cells and ST2 cells with GC-1 and observed significant increases in BAT markers P2RX5, PAT2, PGC-1α, and PRDM16 (Fig. 7a, b) which are similar in magnitude to the increases caused by treatment with T3, suggesting that TRβ may be responsible for the browning effects of TH. Furthermore, TH-deficient Tshr−/− mice injected with GC-1 showed a highly significant 149-fold increase in skeletal tissue UCP1 expression compared to Tshr−/− injected with vehicle control (Fig. 7c). The magnitude of this increase further suggests that TRβ signaling may contribute to TH-induced browning.
Fig. 7.
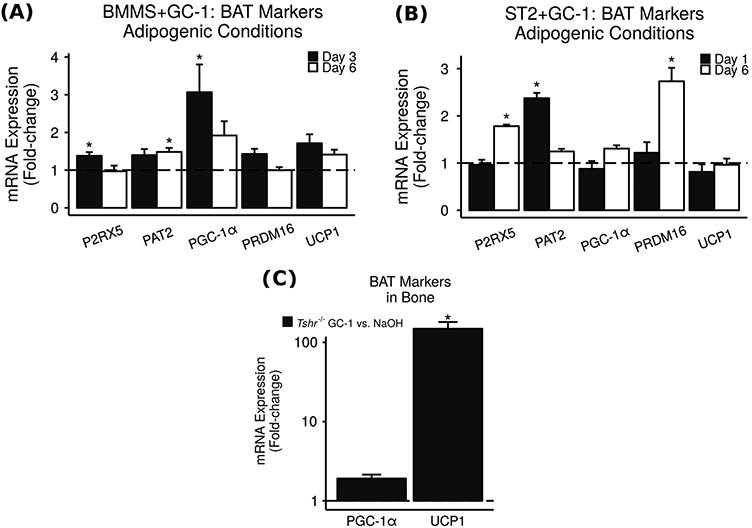
GC-1 increases BAT markers in vitro and in vivo. In culture, BMMS cells (a) and mouse mesenchymal stromal cell line ST2 (b) were treated with 0.1 nM TR β-specific agonist GC-1, and mRNA expression was measured via real-time RT-PCR. Results are presented as mean fold-change vs. vehicle control ± SEM (n = 3–4). * = P < 0.05 vs. vehicle control. Expression levels of bBAT marker genes P2RX5, PAT2, PGC-1α, and PRDM16 were significantly increased. c In vivo, 7 day-old TH-deficient Tshr−/− mice were injected daily with 0.2 μg TRβ-specific agonist GC-1 or vehicle control for 2 days. Skeletal tissues (femoral and tibial epiphyses) were removed and mRNA expression measured using real-time RT-PCR. Results are presented as mean fold-change vs. vehicle control ± SEM (n = 4–8). * = P < 0.05 vs. vehicle control. Key BAT gene UCP1 was increased 149-fold
Discussion
THs have been extensively studied as regulators of both bone [15, 16, 19] and adipose tissue [24]. In this study, we examined the effect of TH on an area of interface between those two tissue types: bone marrow adipose tissue. We have previously reported the critical role of TH in longitudinal bone growth and bone mass accretion as reflected by reduced bone length and both cortical and trabecular bone mass in TH-deficient Tshr−/− mice at the end of the prepubertal growth period [16, 19]. In working with these mice, we also noticed increased fat mass in the TH-deficient mice.
Indeed, we confirmed this observation by measuring bone marrow adiposity which was not only decreased in response to TH treatment but also showed a significant negative correlation with BMD in the pooled data from vehicle- and TH-treated Tshr−/− mice (Fig. 2). There appear to be differences among the responses of the MAT at the three skeletal sites we measured (femur, tibia, and lumbar vertebrae) to TH and GC-1 treatment. For example, marrow adiposity levels in femurs of Tshr−/− mice injected with TH were significantly less than marrow adiposity levels in femurs of euthyroid controls while there was no statistical difference in marrow adiposity levels at the tibiae or lumbar vertebrae between TH-injected mice and euthyroid controls. Injection with GC-1, however, significantly decreased marrow adiposity levels at all three skeletal sites to below the levels seen in euthyroid controls. Further studies with greater numbers of mice and different doses of T3/T4 and GC-1 are needed to determine the nature of these possible site-specific effects of TH signaling on bone marrow adiposity.
Since both osteoblasts and adipocytes are derived from the same MSC progenitors, we first hypothesized that the ability of TH to increase bone mass and decrease fat mass might be due to a shift in the differentiation of MSCs from adipocytes to osteoblasts. This is an especially attractive hypothesis since we have previously observed increased osteoblast differentiation in response to TH treatment [19]. Interestingly, our gene expression data suggest that, rather than causing a shift in differentiation of BMMS cells from adipocytes to osteoblasts, T3 treatment may cause a shift in BMMS differentiation toward brown/beige adipocytes. Since T3 treatment caused marked increases in BAT marker genes, with small changes in WAT markers (Fig. 3-5), an increase in the ratio of BAT to WAT may contribute to explaining our observations given that BAT occupies a much smaller volume than lipid-storing WAT.
BAT differs from WAT in a number of characteristics. Whereas WAT primarily stores lipids in large fat droplets, BAT is much more metabolically active, using UCP1 to dissipate energy from lipids in non-shivering thermogenesis [25]. T3 is known to upregulate UCP1, and the pituitary-thyroid-adrenal axis is involved in the mammalian response to cold exposure which leads to recruitment and activation of BAT [26]. An increase in metabolic rate along with expression of thermogenic BAT genes could explain our observation of less lipogenesis in BMMS cells treated with T3 since increased metabolism would decrease the amount of adipose tissue present, and BAT takes up less volume than WAT. TH induction of BAT could involve a shift in the differentiation of the MSCs directly into classical BAT, which develops through myogenic precursors, and/or so-called browning of WAT in which WAT becomes beige adipose tissue, taking on many characteristics of BAT. This browning may involve the activation and proliferation of a subpopulation of beige adipose precursors resident within the WAT; alternatively, browning may include transdifferentiation of white adipocytes directly into BAT-like thermogenic beige adipocytes. Indeed, although small deposits of BAT have been demonstrated in adult humans, the preponderance of adipose tissue is WAT; browning of WAT therefore may have greater therapeutic potential than simple activation of small stores of BAT. The question of browning is difficult to answer in vitro since many of the genetic markers which would distinguish beige from BAT are not expressed in tissue culture [27]. However, our measurement of gene expression in WAT of TH-deficient mice in vivo (Fig. 6) revealed that, in addition to causing an increase in classic BAT genes such as UCP1 and PGC-1α, treatment with TH led to an increase in TBX1, Cd137, and Epsti1, genes which have been validated as browning-specific markers [27]. These data suggest that TH induction of BAT genes in bone marrow adipose tissue may be the result of browning of WAT
Finally, we investigated which of the TH receptors (TRs) mediates these effects of TH on bone marrow adipose tissue. TH has a wide spectrum of effects on tissues throughout the body; thus, therapeutic manipulation of TH signaling must be targeted in order to avoid unwanted adverse effects. GC-1, a TRβ-specific agonist also known as sobetirome, can be used to help distinguish between the effects of TRα and TRβ signaling. Although GC-1 can bind to both TRα and TRβ, it has a greater affinity for TRβ; the in vivo and in vitro doses of GC-1 which we used in these studies are based upon previous studies in which we have determined that the effects of GC-1 at these doses are primarily due to TRβ signaling [28]. We observed that GC-1 is able to replicate many of the effects of TH on bone marrow adipose tissue, including decreased bone marrow adiposity in vivo and increased BAT gene expression in vitro and in vivo (Fig. 7). This observation is in accordance with previous studies implicating T3/TRβ signaling in the upregulation of UCP1 in adipose tissue [29] and with a recent study demonstrating browning of WAT in response to GC-1 [30]. Furthermore, GC-1 has been shown to increase energy expenditure and decrease weight gain in rats, and although the magnitude of this effect was smaller than the effect of T3, GC-1 did not cause adverse effects such as decreased skeletal muscle mass which were observed with T3 treatment [31]. Indeed, after extensive testing in rodent models, GC-1 has undergone early-stage human clinical trials as an LDL cholesterol-lowering agent, reducing LDL levels while demonstrating none of the adverse cardiovascular or other effects associated with thyrotoxicosis [32-37].
In conclusion, we have investigated the role of TH in the regulation of and induction of browning genes in bone marrow adipose tissue. Further studies are needed to confirm the functional significance of the upregulation of browning genes in MAT in response to TH treatment in the context of skeletal energy metabolism. Additionally, studies utilizing mouse models with target knockout of TRα1 and TRβ1 are needed to confirm the extent to which TRβ1 mediates these effects on MAT. However, browning of white fat has enormous therapeutic potential in the treatment of metabolic conditions such as obesity, and further understanding of molecular mechanisms for browning of bone marrow adipose tissue via activation of TRβ signaling may play a role in identifying future therapeutic strategies for these conditions.
Supplementary Material
Acknowledgements
This work was supported by funding from National Institutes of Health grant AR048139 to SM, Senior Research Career Scientist Award to SM from the US Department of Veterans Affairs, and NIH grant 2R25 GM060507 (LLU CHDMM/IMSD). All work was performed in facilities provided by the US Department of Veterans Affairs. The authors thank Ms. Heather Watt, Ms. Catrina Alarcon, Ms. Sheila Pourteymoor, Dr. Andy Cheng, and Mr. Charles Abreu for technical assistance.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s12020-017-1265-x) contains supplementary material, which is available to authorized users.
Conflict of interest The authors declare that they have no conflict of interest.
References
- 1.Devlin MJ, Rosen CJ, Lancet Diabetes Endocrinol. 3, 141 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nielson CM, Marshall LM, Adams AL, LeBlanc ES, Cawthon PM, Ensrud K, Stefanick ML, Barrett-Connor E, Orwoll ES, Osteoporotic Fractures in Men Study Research Group. J. Bone Miner. Res 26, 496 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wren TAL, Chung SA, Dorey FJ, Bluml S, Adams GB, Gilsanz V, J. Clin. Endocrinol. Metab 96, 782 (2011) [DOI] [PubMed] [Google Scholar]
- 4.Gao Y, Zong K, Gao Z, Rubin MR, Chen J, Heymsfield SB, Gallagher D, Shen W, J. Clin. Densitom 18, 203 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCabe LR, J. Cell Biochem 102, 1343 (2007) [DOI] [PubMed] [Google Scholar]
- 6.McCabe L, Zhang J, Raehtz S, Crit. Rev. Eukaryot. Gene Expr 21, 187 (2011) [DOI] [PubMed] [Google Scholar]
- 7.Leslie WD, Rubin MR, Schwartz AV, Kanis JA, J. Bone Miner. Res 27, 2231 (2012) [DOI] [PubMed] [Google Scholar]
- 8.Rzonca SO, Suva LJ, Gaddy D, Montague DC, Lecka-Czernik B, Endocrinology 145, 401 (2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Syversen U, Stunes AK, Gustafsson BI, Obrant KJ, Nordsletten L, Berge R, Thommesen L, Reseland JE, BMC Endocr. Disord 9, 10 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lecka-Czernik B, Ackert-Bicknell C, Adamo ML, Marmolejos V, Churchill GA, Shockley KR, Reid IR, Grey A, Rosen CJ, Endocrinology 148, 903 (2007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grey A, Beckley V, Doyle A, Fenwick S, Horne A, Gamble G, Bolland M, Eur. J. Endocrinol. Eur. Fed. Endocr. Soc 166, 1087 (2012) [DOI] [PubMed] [Google Scholar]
- 12.Harsløf T, Wamberg L, Møller L, Stødkilde-Jørgensen H, Ringgaard S, Pedersen SB, Langdahl BL, J. Clin. Endocrinol. Metab 96, 1541 (2011) [DOI] [PubMed] [Google Scholar]
- 13.Moore SG, Dawson KL, Radiology 175, 219 (1990) [DOI] [PubMed] [Google Scholar]
- 14.Mullur R, Liu Y-Y, Brent GA, Physiol. Rev 94, 355 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wojcicka A, Bassett JHD, Williams GR, Biochim. Biophys. Acta 1830, 3979 (2013) [DOI] [PubMed] [Google Scholar]
- 16.Xing W, Govoni KE, Donahue LR, Kesavan C, Wergedal J, Long C, Bassett JHD, Gogakos A, Wojcicka A, Williams GR, Mohan S, J. Bone Miner. Res 27, 1067 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ortiga-Carvalho TM, Sidhaye AR, Wondisford FE, Nat. Rev. Endocrinol 10, 582 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beamer WJ, Eicher EM, Maltais LJ, Southard JL, Science 212, 61 (1981) [DOI] [PubMed] [Google Scholar]
- 19.Xing W, Cheng S, Wergedal J, Mohan S, J. Bone Miner. Res 29, 2262 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bassett JHD, Williams AJ, Murphy E, Boyde A, Howell PGT, Swinhoe R, Archanco M, Flamant F, Samarut J, Costagliola S, Vassart G, Weiss RE, Refetoff S, Williams GR, Mol. Endocrinol 22, 501 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scheller EL, Troiano N, Vanhoutan JN, Bouxsein MA, Fretz JA, Xi Y, Nelson T, Katz G, Berry R, Church CD, Doucette CR, Rodeheffer MS, Macdougald OA, Rosen CJ, Horowitz MC, Methods Enzymol. 537, 123 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang Q-Q, Otto TC, Lane MD, Proc. Natl. Acad. Sci. USA 101, 9607 (2004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheng S, Zhao SL, Nelson B, Kesavan C, Qin X, Wergedal J, Mohan S, Xing W, PLoS One 7, e32887 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Obregon M-J, Front. Physiol 5, 479 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cannon B, Nedergaard J, Physiol. Rev 84, 277 (2004) [DOI] [PubMed] [Google Scholar]
- 26.Lee P and Greenfield JR, Mol. Cell. Endocrinol 418 Pt 2, 184 (2015) [DOI] [PubMed] [Google Scholar]
- 27.de Jong JMA, Larsson O, Cannon B, and Nedergaard J, Am. J. Physiol. Endocrinol. Metab 308, E1085 (2015) [DOI] [PubMed] [Google Scholar]
- 28.Xing W, Aghajanian P, Goodluck H, Kesavan C, Cheng S, Pourteymoor S, Watt H, Alarcon C, Mohan S, Am. J. Physiol. Endocrinol. Metab 310, E846 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee J-Y, Takahashi N, Yasubuchi M, Kim Y-I, Hashizaki H, Kim M-J, Sakamoto T, Goto T, Kawada T, Am. J. Physiol. Cell. Physiol 302, C463 (2012) [DOI] [PubMed] [Google Scholar]
- 30.Lin JZ, Martagón AJ, Cimini SL, Gonzalez DD, Tinkey DW, Biter A, Baxter JD, Webb P, Gustafsson J-Å, Hartig SM, Phillips KJ, Cell Rep. 13, 1528 (2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Villicev CM, Freitas FRS, Aoki MS, Taffarel C, Scanlan TS, Moriscot AS, Ribeiro MO, Bianco AC, Gouveia CHA, J. Endocrinol 193, 21 (2007) [DOI] [PubMed] [Google Scholar]
- 32.Johansson L, Rudling M, Scanlan TS, Lundåsen T, Webb P, Baxter J, Angelin B, Parini P, Proc. Natl. Acad. Sci. USA 102, 10297 (2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scanlan TS, Heart Fail. Rev 15, 177 (2010) [DOI] [PubMed] [Google Scholar]
- 34.Tancevski I, Demetz E, Eller P, Recent Pat. Cardiovasc. Drug Discov 6, 16 (2011) [DOI] [PubMed] [Google Scholar]
- 35.Lin JZ, Martagón AJ, Hsueh WA, Baxter JD, Gustafsson J-Å, Webb P, Phillips KJ, Endocrinology 153, 6136 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kannisto K, Rehnmark S, Slätis K, Webb P, Larsson L, Gåfvels M, Eggertsen G, Parini P, Atherosclerosis 237, 544 (2014) [DOI] [PubMed] [Google Scholar]
- 37.Lammel Lindemann J, Webb P, Expert Opin. Ther. Targets 20, 145 (2016) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.