

Abstract
Objective:
Enhancer aberrations are beginning to emerge as a key epigenetic feature of colorectal cancers, however, a comprehensive knowledge of chromatin state patterns in tumor progression, heterogeneity of these patterns and imparted therapeutic opportunities remain poorly described.
Design:
We performed comprehensive epigenomic characterization by profiling 222 chromatin profiles from 69 (33 colorectal adenocarcinomas, 4 adenomas, 21 matched normal tissues and 11 colon cancer cell lines) by profiling 6 histone modification marks: H3K4me3 for Pol II-bound and CpG-rich promoters, H3K4me1 for poised enhancers, H3K27ac for enhancers and transcriptionally active promoters, H3K79me2 for transcribed regions, H3K27me3 for polycomb repressed regions and H3K9me3 for heterochromatin.
Results:
We demonstrate that H3K27ac-marked active enhancer state could distinguish between different stages of CRC progression. By epigenomic editing, we present evidence that gains of tumor-specific enhancers for crucial oncogenes, such as ASCL2 and FZD10, was crucial for excessive proliferation. Consistently, combination of MEK plus bromodomain (BET) inhibition was found to have synergistic effects in CRC patient-derived xenograft (PDX) models. Probing inter-tumor heterogeneity, we identified four distinct enhancer subtypes (EpiC), three of which correlate well with previously defined transcriptomic subtypes (CMSs). Importantly, CMS2 can be divided into two EpiC subgroups with significant survival differences. Leveraging such correlation, we devised a combinatorial therapeutic strategy of enhancer-blocking bromodomain inhibitors with pathway-specific inhibitors (PARPi, EGFRi, TGFβi, mTORi and SRCi) for EPIC groups.
Conclusion:
Our data suggest that the dynamics of active enhancer underlies colorectal cancer progression and the patient-specific enhancer patterns can be leveraged for precision combination therapy.
INTRODUCTION
Colorectal cancer (CRC) is the third most common cancer in the world. Most cases of CRC originate from premalignant lesions, mainly adenomas, which can progress into malignant adenocarcinomas1. Genomic alterations in colorectal adenocarcinomas frequently occur in APC (83%), TP53 (60%), KRAS (43%), and BRAF (10%) genes2–6. However, targeted therapy for these mutational subgroups is not well established. This calls for a comprehensive approach to identify unique signatures derived from various molecular characteristics of tumors including genomic and epigenomic components which, along with clinical features, can be used to introduce clinically feasible targeted therapeutic strategies.
One such effort toward precision therapy was led by the CRC subtyping consortium, which identified four consensus molecular subtypes (CMSs) with distinct activation patterns of specific oncogenic signaling pathways using gene expression datasets3. Briefly, these transcriptomic subgroups include the immune subtype (CMS1), canonical subtype (CMS2), metabolic subtype (CMS3), and mesenchymal subtype (CMS4)7. Although distal cis-regulatory elements are known to play a central role in controlling transcription patterns, the bona fide contributions of these elements in defining CMSs are not understood. Importantly, epigenetic processes are reversible and can provide actionable targets for therapeutic development, such as bromodomain inhibitors (BRDi) that target super-enhancer elements8. The benefits of targeting the CRC epigenome have already been demonstrated by associations of the CpG island methylator phenotype (CIMP) with BRAF mutations, for which efforts are underway to devise combinatorial strategies targeting methylation and the BRAF oncogene3. Consistent with this finding, we and others have provided evidence that epigenomic alterations at the level of DNA methylation play crucial roles in CRC initiation9 and oncogenic function10.
While several studies have profiled enhancer elements in CRC cell lines and few patient samples11–14, the enhancer aberrations derived from a relatively larger number of primary untreated patient samples will be highly useful in defining the heterogeneity of the enhancer patterns between patients for precision therapy. Furthermore, a comprehensive understanding of other chromatin state alterations and their functional contribution to tumor progression is mostly lacking. Therefore, it is of immense importance to define the relationship between chromatin aberrations and genomic and transcriptomic features in tumors.
METHODS
Details of all methods are provided in the Supplementary Material.
RESULTS
To characterize the epigenomic makeup of CRC, we first generated ChIP-seq (chromatin immunoprecipitation followed by massive parallel sequencing)-based profiles for six histone marks including H3K4me1 and H3K27ac, associated with enhancers and transcriptionally active promoters; H3K4me3, associated with PolII-bound and CpG-rich promoters; H3K79me2, associated with transcribed regions; H3K9me3, associated with heterochromatin regions; and H3K27me3, associated with Polycomb repression in a total of 16 CRCs and 17 matching adjacent normal colonic mucosa samples (Supplementary Table S1). With the use of the ChromHMM algorithm15, we trained a 10-state, 12-state, and 15-state model (for samples having all the six marks; Supplementary Table S1) consisting of combinatorial patterns of specific histone modifications known as chromatin states, which are linked with the transcriptional output of the associated genomic loci and regulate the recruitment of various components of the transcriptional machinery. We chose a 10-state model for in-depth investigation because it represented biologically interpretable combinatorial patterns that were not repetitive or ambiguous (Fig. 1A and Supplementary Fig. S1A). We annotated these chromatin states based on the content of each state and its average relative distribution around known genomic features (Supplementary Fig. S1B). To define the chromatin states that best differentiate between normal and tumors, we developed a statistical approach, termed ChromXplorer (see Methods; Supplementary Fig. S1C), that utilizes the ChromDiff16 framework for all genomic loci instead of a gene-centric approach. The ChromXplorer-based dimensionality reduction approach showed the “active enhancer” state, but not others, to be the most discriminatory state in all three investigated models (Fig. 1B and Supplementary Fig. S1D–S1F). Analysis of singular histone marks indicated that the active enhancer mark, defined by H3K27ac, also led to clustering of the samples according to their phenotype (Supplementary Fig. S2A), whereas some of the other histone marks such as H3K79me2, H3K9me3, and H3K27me3 showed weaker clustering patterns due to variations among tumor samples (Supplementary Fig. S2A), suggesting de novo gain or loss of H3K27ac patterns as an important distinguishing feature of CRC.
Fig. 1. H3K27ac-identified global enhancer pattern could differentiate colorectal cancer from normal colon.
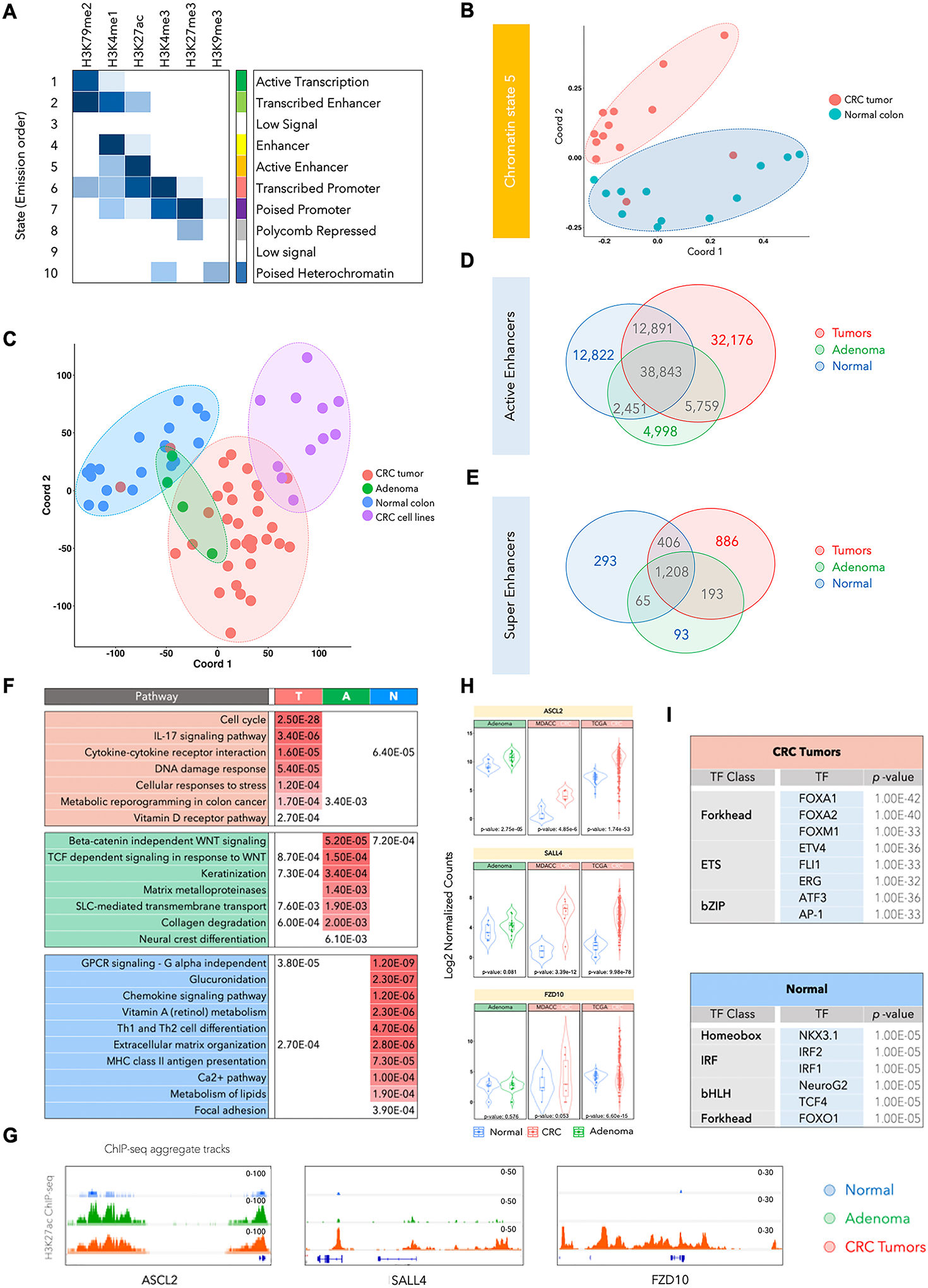
A. Emission probabilities of ChromHMM-based 10-chromatin state calls, which is based on the global distribution of combinatorial patterns of 6 histone marks (H3K4me1, H3K27ac, H3K4me3, H3K27me3, H3K79me2, and H3K9me3) from 13 adenocarcinomas and 12 adjacent normal tissues. Each row is representative of a combinatorial chromatin state pattern, which is annotated on the right based on the constituent histone mark and neighborhood plot (Supplementary Fig. S1B). Columns represent each histone mark. Colors represent the frequency of occurrence of that mark in the corresponding chromatin state on a scale of 0 (white) to 1 (blue).
B. Multidimensional scaling (MDS) plot for chromatin state 5 between normal tissues (blue) and adenocarcinoma samples (red).
C. MDS plot for global H3K27ac patterns in the adenomas (A), adenocarcinomas (T), adjacent normal colon (N) and cancer cell lines. Adenoma samples are spread over normal and tumor samples and cannot be distinguished with use of H3K27ac. Red, Green, Blue and Purple colors were used for Tumor, Adenoma, Normal Colon and CRC cell lines respectively.
D. Venn diagram showing overlaps between total number of H3K27ac-defined active enhancer peaks in normal samples (blue), adenomas (green) and CRC tumors (red).
E. Venn diagram showing overlaps between total number of super-enhancer peaks (called using ROSE) in normal samples (blue), adenomas (green) and CRC tumors (red).
F. List of significantly enriched pathways in genes targeted by enhancers specific to CRC tumors (T), adenomas (A), and normal colon tissue (N). q-values are shown and are color-coded based on the level of significance.
G. Integrative Genomics Viewer (IGV) images showing enrichment of H3K27ac peaks around ASCL2, SALL4 and FZD10 genes using aggregate ChIP-seq profiles of normal colon, adenoma and CRC tumors.
H. Violin plots showing mRNA expression levels for ASCL2, SALL4 and FZD10 in adenomas, the adenocarcinoma cohort used in this study (labeled as MDACC) and TCGA adenocarcinoma cohort. The reported p-values were calculated using the DESeq252.
I. List of enriched transcription factor (TF) motifs in CRC- or normal tissue-specific active enhancers. Motifs are identified using HOMER27 on the basis of the shared H3K27ac peak in each group.
In light of this, we expanded the cohort by profiling the H3K27ac mark in additional colorectal tumors (n = 17), adenomas (n = 4), normal crypts (n = 4), and cancer cell lines (n = 11) to characterize the enhancer patterns through the various stages of progression. Interestingly, distinct enhancer patterns appeared to define each progression stage, suggesting that these enhancer patterns may evolve as the disease progresses (Fig. 1C). Furthermore, the established CRC cell lines formed a separate cluster with three cell lines that partially overlapped with colorectal adenocarcinoma tumors (Fig. 1C). Similarly, computation of correlation matrix identified a higher similarity within normal samples as opposed to tumor samples which segregated into two subgroups, indicative of greater heterogeneity between adenocarcinoma specimens (Supplementary Fig. S2B).
To identify recurrent cancer-specific peaks, shared H3K27ac peaks in all tumor samples were summed up and overlapped with shared normal or shared adenoma enhancers. Our analysis identified 32,176 tumor-specific (T) enhancers, 12,822 normal-specific (N) enhancers, and 4,998 adenoma-specific (A) enhancers (Fig. 1D). Notably, 5,759 enhancers were present in both adenomas and adenocarcinomas, but not in normal tissue, whereas a much higher number (32,176) were specific to adenocarcinomas only, thereby, suggesting that several enhancers are gained in de novo manner in the adenocarcinoma stage. Furthermore, the average intensity of enhancer peaks was found to progressively increase in adenomas and colorectal tumors (Supplementary Fig. S2C). Similar to typical enhancers, we found super-enhancer elements (SE), that are thought to promote oncogene expression, to be upregulated in adenocarcinomas (Fig. 1E). We identified 886 adenocarcinoma-specific, 93 adenoma-specific, and 193 shared super-enhancers between adenomas and adenocarcinomas using ROSE algorithm17 (Fig. 1E).
Active enhancers specific to each disease progression stage marked important driver genes that were identified based on the presence of enhancers in their proximal regulatory regions as previously defined18 19 (Fig. 1F and Supplementary Table S2). For example, regulators and mediators of the cell cycle, DNA damage, stress and cytokine and metabolism harbored gains in a tumor-specific manner. Conversely, enhancers for regulators of T-cell differentiation, MHC class II antigen presentation, vitamin A metabolism, chemokine signaling, and focal adhesion, among others, were specifically present only in normal tissues and were lost during progression. Enhancers common to premalignant adenomas and tumors were enriched around regulators of cell fate, collagen degradation, metabolic transport, and WNT signaling (Fig. 1F). We noted that the enrichment of enhancers/super-enhancers in the proximity of key WNT signaling regulators such as ASCL2, SLL4, FZD10, CTNNB1 (β-catenin), AXIN2, LEF-1, and a subset of these correlated with their progressive increase in gene expression (Fig. 1G–H, Supplementary Fig. S3A–C and Supplementary Table S2). This suggests that the genes identified to harbor active enhancers or super-enhancers near their vicinity or in regions interacting with the gene body/promoters are likely to contribute to the carcinogenic properties. Enhancers are epigenetic platforms for binding of various transcription factors (TFs), and motif enrichment analysis has suggested that specific classes of TFs such as forkhead (FOXA1, FOXA2, and FOXM1), ETS (ETV4, ERG, and FLI1), and bZIP (ATF3 and AP-1) are overrepresented in the tumor-specific active enhancers (Fig. 1I and Supplementary Fig. S3D). Importantly, homeobox (NKX3.1), IRF (IRF1, IRF2), bHLH (NeuroG2, TCF4), and FOXO1 motifs were enriched in active enhancers that were lost during tumor progression (Fig. 1I and Supplementary Fig. S3D).
Inactivation of tumor-specific enhancers leads to the transcriptional and phenotypical switch
To test the functional role of some of the specifically activated tumor-specific enhancers, we first ranked the top differentially activated enhancers (Fig. 2A and Supplementary Table S3; see Methods). We chose to functionally test some of the top enhancers from this list that were located in the vicinity of two WNT signaling genes: ASCL2 and FZD10. ASCL2 is a known driver of intestinal stem cell phenotype20 and a predicted oncogene in CRCs21 whereas, FZD10 is a transmembrane protein that acts as a WNT receptor driving downstream signaling, which is a key signaling event in CRCs7. Moreover, established CRC cell lines harbored a significant percent of tumor-specific H3K27ac peaks (Supplementary Fig. S4A–B). We used dCas9-KRAB and specific gRNAs to suppress the activity of target enhancer loci in SW480 cells (Supplementary Fig. S4C), a well-characterized model with mutations in APC, KRAS, and TP5320. For ASCL2, we chose to focus on a distal super-enhancer that was present in the adenomas and adenocarcinomas, but not normal tissues (Fig. 2B, 1H, and Supplementary Fig. S3A). HiChIP and 3C experiments confirmed the direct interaction of this super-enhancer with the ASCL2 gene promoter (Fig. 2B and Supplementary Fig.S4D). We noted that all three gRNAs targeting this distal super-enhancer region were able to suppress the H3K27ac signal in their respective target regions (Fig. 2C). These gRNAs also downregulated gene expression (Fig. 2D) and downstream proliferation of SW480 cells, suggesting that the distal super-enhancer acts as a functional driver event in this cell line (Fig. 2E). For the FZD10 locus, we noted that gRNAs targeting E1, E5, and E6 regions reduced the H3K27ac signal which was consistent with the observations from HiChIP experiments showing the interaction of these enhancers with the promoter of this gene (Supplementary Figs. S4E–4F). Epigenetic suppression of these enhancers also downregulated gene expression (Supplementary Fig. S4F) and decreased cell proliferation (Supplementary Fig. S4H), suggesting multi-enhancer regulation of FZD10 transcription and the contribution of this locus to the proliferative properties of CRC cells.
Fig. 2. Inactivation of top tumor-specific enhancers leads to transcriptional downregulation and loss of cell proliferation.
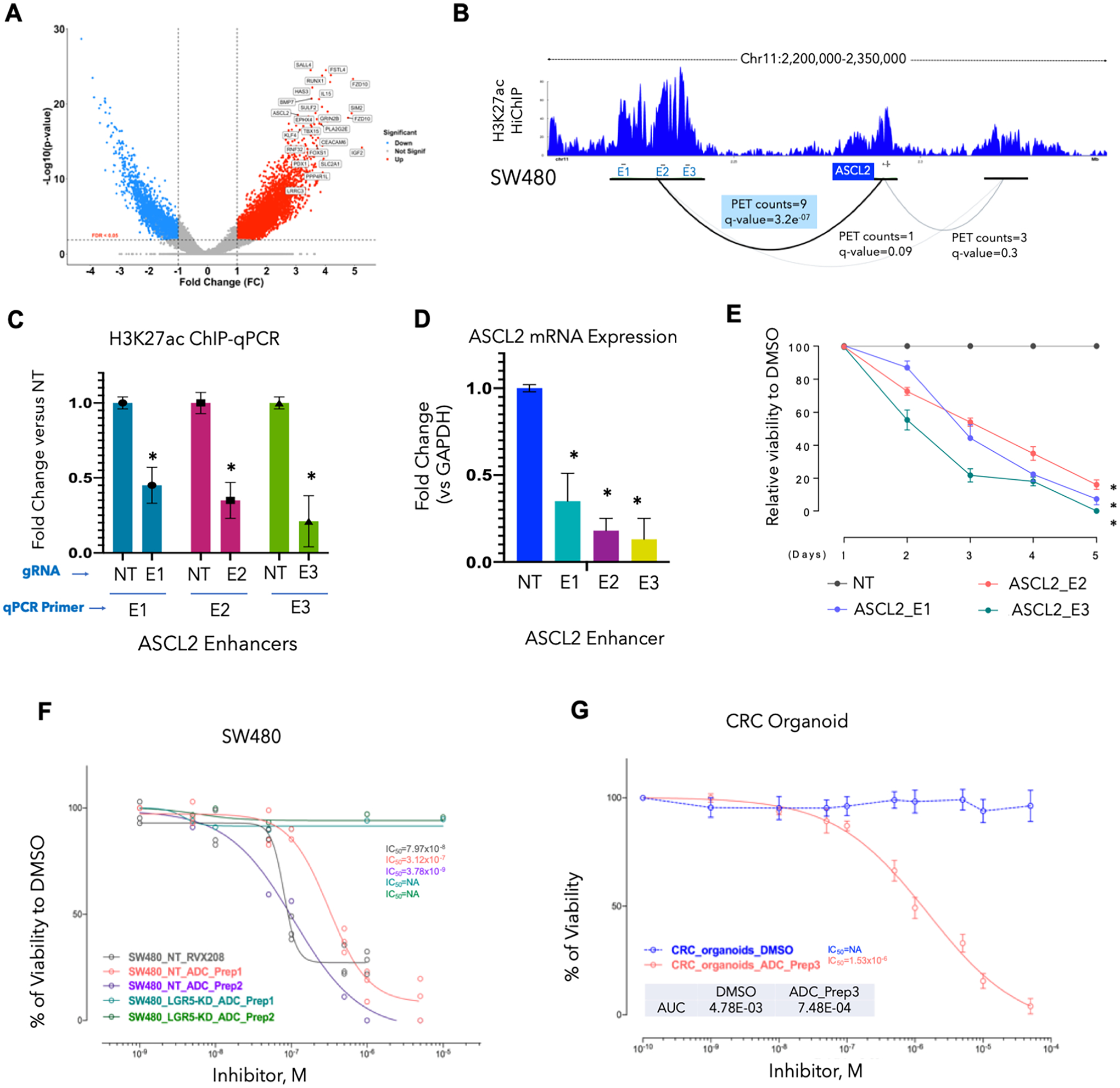
A. Volcano plot showing top CRC-enriched typical enhancers ranked based on fold-change and p-value. Top de novo gained enhancers among tumor (n = 33) as compared to adjacent normal (n = 15) samples are annotated according to their associated genes. Thresholds of log2FC > 1 and FDR < 0.05 are used to annotate de novo enhancer peaks. ASCL2 and FZD10 enhancers are among this subset of highly enriched regions. There are two top ranked enhancer peaks around FZD10 gene.
B. Interaction map of active enhancers (E) with the gene promoter (P) around the ASCL2 locus as determined in SW480 cell line by HiChIP. Interactions were observed in a super-enhancer (E1–E3) region located ~70kb upstream of ASCL2 promoter (PET counts=9, q-value=3.2e−07). These regions are among the highly enriched tumor-specific enhancers and were present in multiple CRC cell lines (Supplementary Fig. S4A).
C. Bar graph showing relative enrichment of H3K27ac as derived from a ChIP-qPCR experiment on E1–E3 for ASCL2 super-enhancer (from panel B) in SW480 cells harboring specific or non-targeting (NT) gRNAs to these enhancers. Y-axis represents fold change in intensity of H3K27ac signal for each enhancer in gRNA harboring cell in comparison to the cells harboring NT gRNA. Asterisk * denotes p-value < 0.05.
D. Bar graph showing relative ASCL2 mRNA expression in E1–E3 gRNAs for ASCL2 super-enhancer harboring SW480 cells in comparison to parental cells (ASCL2_NT).
E. Growth curve showing relative viability of SW480 cells harboring E1–E3 for ASCL2 super-enhancer in comparison to parental cell (ASCL2_NT) showing lower rates of proliferation in cells with ASCL2-inactivated enhancers.
F. Growth curves for LGR5-silenced SW480 cells (SW480LGR5-KD) in comparison to the parental cells upon treatment with increasing concentrations of two preparations of antibody-drug conjugate (ADC) with bromodomain inhibitor (RVX208) conjugated to LGR5 antibody (ADC-Prep1 and ADC-Prep2) and RVX-208 alone. Y-axis shows percent viability of treated cells in comparison to control (DMSO) treated cells.
G. Growth curves for a patient-derived CRC organoid upon treatment with LGR5-RVX208 conjugate (ADC). Y-axis shows percent viability of treated cells in comparison to control (DMSO) treated cells.
Although these studies provide evidence for the functionality of individual enhancers, a holistic approach would be to block many such enhancers using BRDi, which are being actively tested in clinical trials in solid tumors. Since ASCL2 and WNT pathways have established roles in regulating stem cell function in both normal and cancer tissues7 20 21, we asked whether targeting enhancer-blocking agents to stem cells, using the LGR5 antibody, would result in suppressing the growth of the cells. Hence, we synthesized an antibody-drug conjugate (ADC) by using LGR5 antibody and the RVX208 which is a BRDi in phase 3 trials for cardiovascular indications22. Indeed, LGR5-RVX208 (Supplementary Fig. S5A) ADCs specifically blocked the growth of SW480 cells or patient-derived human colon organoids in an LGR5 expression–dependent fashion (Fig. 2F–2G and Supplementary Figs. S5B–E). These studies established that some of the activated enhancers in tumors may play functional roles in imparting pro-tumorigenic properties to CRC.
Combination of MEK and bromodomain inhibitors reduces tumor growth in a subset of PDXs
Treatment of four different patient-derived xenograft (PDX) models (C1138, F3053, F3008, and B8131) with BRDi (iBET-151) caused strong tumor reduction in one model but showed no significant difference in 3 of 4 models, suggesting a context-dependent impact of enhancer blockade (Fig. 3A–B). Since chromatin aberrations are believed to be a permissive event for activation of pro-tumorigenic transcriptional signature upon oncogenic signaling, we reasoned that blockade of enhancers may be more effective in combination with inhibition of key oncogenic driver signaling pathways. We noted that a number of upstream regulators of the MEK pathway, including EGFR, NRAS, KRAS, FGFR3, MAPK3K1, MAPK11, MAPK12, MAP3K20, MAP3K10, AKT2, IRS2, FGF1, FGF3, FGF19, and FGF21, gained enhancers in colorectal tumors in comparison to adjacent normal tissues or adenomas (Supplementary Fig. S6A). Therefore, we analyzed whether enhancer-blocking BRDi may synergize with MEKi, which are actively being used in CRC clinical trials, to enhance therapeutic efficacy. Indeed, the treatment of four different PDX models (C1138, F3053, F3008, and B8131) showed significant synergy between MEKi (trametinib or binimetinib) and BRDi in 3 of these 4 models (Fig. 3A–B and Supplementary Fig.S6B). Although monotherapy with MEKi showed moderate activity on tumor growth in all of the models, significant tumor regression was noted in B8131, and two models (F3052 and 3008) showed slower growth in combinatorial treatment with BRDi.
Fig. 3. BRDi plus MEKi shows differential responses in different PDX models.
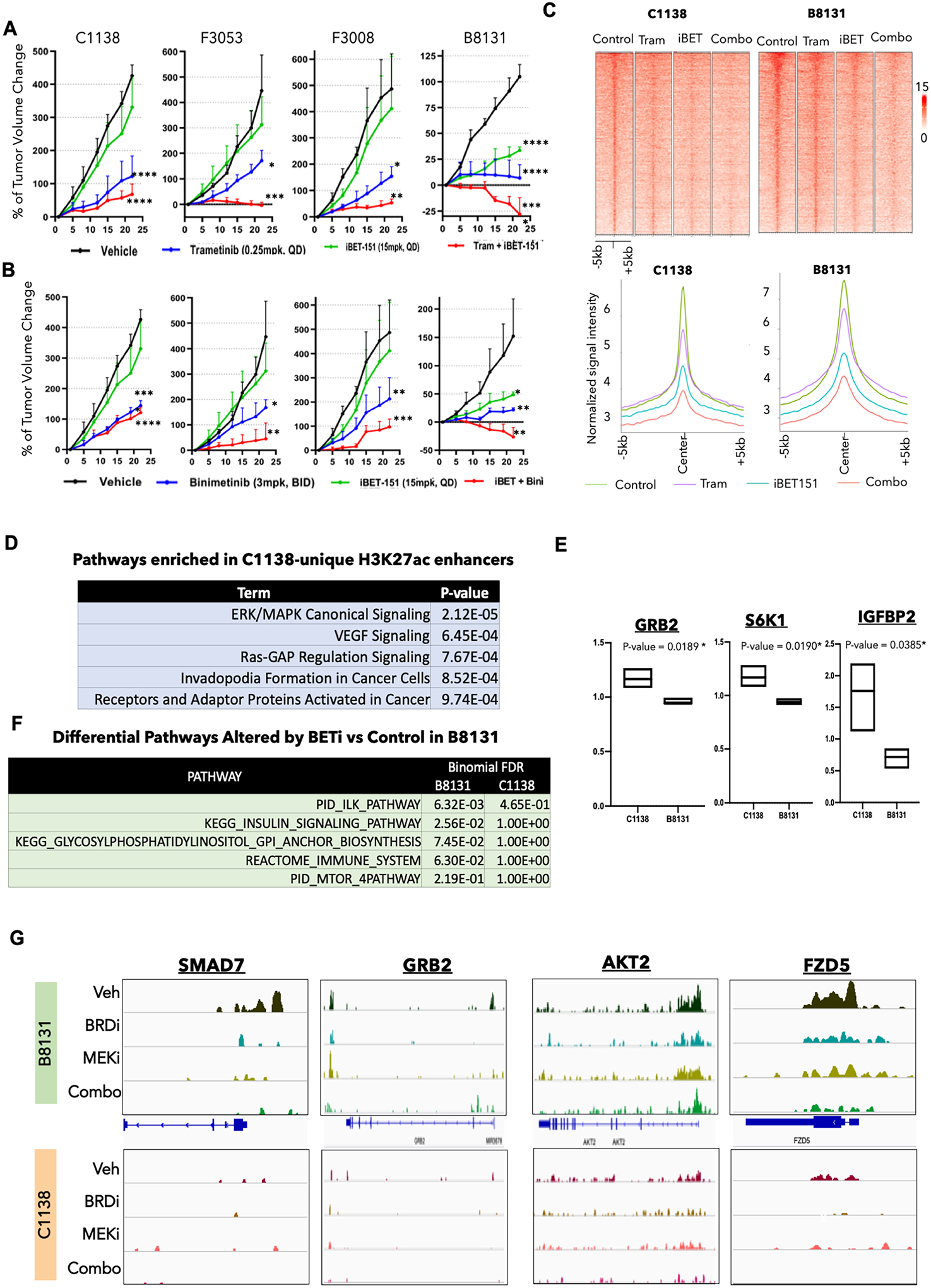
A. Tumor volume curves for C1138, F3053, F3008, and B8131 on treatment with MEK inhibitors (trametinib), BRD inhibitor (i-BET151), and a combination of MEK and BRD inhibitors (trametinib + i-BET151) along with the control vehicle group (n = 3–5 for each arm). P-values represent pairwise t test comparison between the experimental arm to vehicle treatment. *= p < 0.05; ** = p < 0.01; *** = p < 0.001; **** = p < 0.0001. Please refer to Supplementary Fig. S6B for all pair-wise comparisons of the p-values.
B. Tumor volume curves for C1138, F3053, F3008, and B8131 on treatment with MEK inhibitor (binimetinib), BRD inhibitor (i-BET151), and a combination of MEK and BRD inhibitors (binimetinib + iBET151) along with the control vehicle group (n = 3–5 for each arm). P-values represent pairwise t test comparison between the experimental arm to vehicle treatment. * = p < 0.05; ** = p < 0.01; *** = p < 0.001; **** = p < 0.0001. Please refer to Supplementary Fig. S6B for all pair-wise comparisons of the p-values.
C. Heatmap (top panel) and average intensity plot (bottom panel) for H3K27ac peaks in vehicle, trametinib, iBET-151 and combination (iBET-151 plus trametinib) treated PDXs, B8131 (responder) and C1138 (non-responder).
D. Elsevier pathways (identified by Enricher) enriched in gene targets of H3K27ac peaks unique to C1138 versus B8131 PDXs.
E. Box plots showing protein expression (RPPA) of GRB2, S6K1 and IGFBP2 enriched in C1138 versus B8131 PDXs.
F. Differential pathways between those enriched in gene targets of lost H3K27ac-peaks in iBET-151 versus control treated B8131 and C1138.
G. IGV snapshots of H3K27ac peaks around SMAD7, GRB2, AKT2 and FZD5 in vehicle, trametinib, iBET-151 and combination (iBET-151 plus trametinib) treated B8131 and C1138.
To understand the epigenetic mechanism behind differential responses to BRDi and the combination therapy with MEKi, we performed ChIP-Seq for H3K27ac in Vehicle, BRDi (iBET-151), MEKi (Trametinib) and BRDi plus MEKi (iBET-151 plus Trametinib) treated tumors for C1138 and B8131 which exhibited poor or strong responses to the combination therapy, respectively. We noted that while MEKi had a modest impact on intensity of H3K27ac enhancer peaks, BRDi as well combination highly reduced total number of H3K27ac peaks as well as average intensity in comparison to control sample (Fig. 3C and Supplementary Fig. S6C). Comparison of H3K27ac peaks between C1138 and B8131 tumors showed the enrichment of various growth signaling pathways, including canonical MAPK pathway, in uniquely enriched peaks in C1138 in comparison to B8131 (Fig. 3D, Supplementary Fig. S6D and Supplementary Table S4). Consistently, RPPA data from the PDX models showed increase in GRB2, S6 kinase and IGFBP2 expression in C1138 tumors in comparison to B8131 (Fig. 3E). Pathway analysis of gene targets of H3K27ac peaks that were depleted upon BRDi treatment in B8131 (responder), but not in C1138 (non-responder), shows multiple pathways implicated in resistance to MEKi such as mTOR, WNT and insulin signaling (Fig. 3F and Supplementary Table S4)23. These included important regulators of these pathways such as GRB2, AKT2, SMAD7, FZD5, SMAD3, RAC1, 4EBP1, ORS2, and MAP3K10 (Fig. 3G and Supplementary Fig. S6E). Overall, these data highlight the heterogenous responses of various PDX models to enhancer blockade and suggest that pre-existing enhancer states may underlie responses to monotherapy of bromodomain inhibitors and MEKi/BRDi combination therapy.
Distinct enhancer-driven EPIgenome-based Classification (EpiC) clusters of CRC
As exemplified by the MEKi + BRDi combination treatment results, all CRC tumors do not respond to the blanket therapies equally. This motivated us to perform molecular characterization of CRC tumors based on their epigenomic content, which can be potentially useful for patient stratification and subsequent assignment of each subtype to particular combination treatment with epigenetic compounds.
We classified adenomas and adenocarcinomas by using a consensus non-negative matrix factorization (NMF)-based clustering method that uses the enriched H3K27ac peaks. Based on the cophenetic and dispersion scores, we identified four distinct clusters of tumors (Fig. 4A, Supplementary Fig. S7A S7B, and Supplementary Table S5), hereafter referred to as EPIgenome-based Classification (EpiC) clusters: EpiC1, EpiC2, EpiC3, and EpiC4. The average intensity of enhancer peaks within these clusters varied such that EpiC2 and EpiC4 enhancers had significantly higher peaks than the other two clusters had (Supplementary Fig. S7C–S7D). EpiC3 clusters had the least overall peak intensity (Supplementary Fig. S7C–D). The distribution of H3K27ac peaks in regard to the gene body was similar across EpiC clusters (Supplementary Fig. S7E). EpiC clusters also did not associate with any known batches (Supplementary Fig. S8A). To interrogate the contribution of different cell types to EpiC classification, we overlapped EpiC-specific enhancers with those from 91 different cell types/datasets from ENCODE project (Supplementary Fig. S8B–G)24. This analysis showed significant overlap of EpiC1-specific enhancers with those from immune cells (Supplementary Figs. S8B–S8G), whereas enhancers unique to other three EpiCs only showed modest overlap to those from other non-colon cell types (Supplementary Figs. S8B, S8D–G) suggesting while EpIC1 classification may be convoluted from infiltration of immune cells, other EpiCs are likely derived from tumor-cell intrinsic enhancers.
Fig. 4. Reclassification of colorectal cancer tumors based on their global enhancer distribution leads to identification of enhancer-based subtypes.
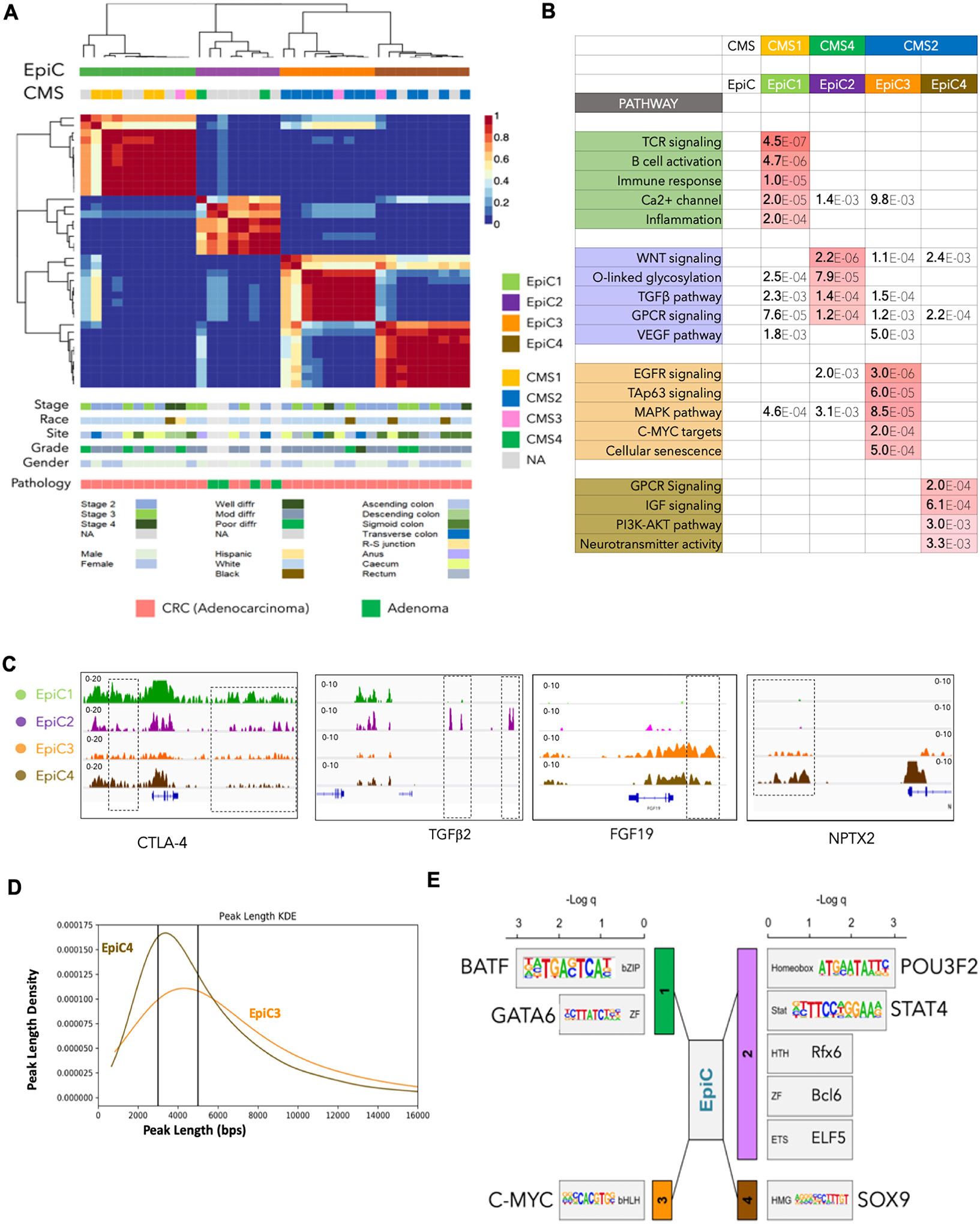
A. Non-negative matrix factorization (NMF) clustering of CRC adenocarcinoma (n = 33) and adenoma (n = 4) samples identified four EPIgenome-based Classification (EpiC) clusters of CRC, shown in the color-coded matrix on the top panel. Consensus molecular subtype (CMS) classification of each tumor sample is identified by using a 498 gene–based random forest approach and is overlaid with the EpiC clusters. Clinical data corresponding to the tumor samples in each cluster, including tumor stage, site, pathological grade, race, and gender of the patients, are provided in the lower panels.
B. Table Showing enriched major signaling pathways for genes targeted by EpiC-specific enhancers using GREAT18. False Discovery Rate (FDR) values are color-coded.
C. IGV images showing enrichment of H3K27ac peaks around CTLA4 for EpiC1, TGFβR3 in EpiC2, FGF19 in EpiC3, and NPTX2 in EpiC4 using aggregate ChIP-seq profiles of EpiC1, EpiC2, EpiC3 and EpiC4 group tumors.
D. KDE (Kernel Density Estimate) plot with peak lengths (x-axis) and densities (y-axis) of EpiC3 and EpiC3-specific enhancers. Peaks with length more than 5kbs were annotated as Broad whereas those with < 3kb in length were called as Sharp.
E. Motif enrichment analysis for enhancers unique to different EpiC groups using HOMER27.
Previously, four CMSs had been defined on the basis of transcriptomic signatures of more than four thousand CRC samples3 as the CMS1 (MSI immune subtype), CMS2 (canonical subtype), CMS3 (metabolic subtype), and CMS4 (mesenchymal subtype). We classified the tumors used in our study into these CMSs, for which we had gene expression data, by using the CMS classifier25, and based on the corresponding trained model3, we computed their EpiC subtype. Interestingly, we determined that epigenomic clusters (EpiCs) significantly overlap their assigned CMSs (Fig. 4A). All of the samples classified as CMS1 (immune) subtype overlapped with the EpiC1 subtype. Although only two patients had been assigned to the CMS4 (mesenchymal) subtype, strikingly, all four adenoma samples along with these two patients clustered as the EpiC2 subtype. It has been shown that adenoma lesions have mesenchymal properties and may progress to adenocarcinoma due to aberrations in the Ras signaling pathway26. Moreover, consensus NMF clustering divided the canonical subtype or CMS2 into two novel subsets of epigenomic CRC clusters, EpiC3 and EpiC4 (Fig. 4A). Of note, the metabolic subtype or CMS3 is not detectable with the use of enhancer-based epigenomic clustering and is scattered in multiple subtypes.
Interestingly, pathway analysis based on the gene targets of enhancers (see Methods) that are specifically enriched in each EpiC subtype showed similarity to those known to be CMS-specific3 (Fig. 4B). Enhancers specific to EpiC1, which overlaps significantly with CMS1, were in the proximity of genes regulating immune response, B- and T-cell activation, and inflammation. EpiC2, which overlaps with CMS4 and interestingly contains all adenoma samples, regulates the TGFβ, WNT, and VEGF pathways (Fig. 4B). CMS2 CRC tumors were distributed into two epigenetic subtypes EpiC3 and EpiC4. EpiC3 enhancers enriched on targets of c-MYC, EGFR, MAPK, and TAp63 signaling, whereas EpiC4 enhancers activated genes involved in GPCR, IGF, PI3K-AKT and neurotransmitter signaling (Fig. 4B and Supplementary Table S6). Examples of important genes near the EpiC-specific enhancers are shown in Fig. 4C and Supplementary Fig. S9. We also noted that EpiC3 enhancers were more spread (or “broad”) in nature whereas EpiC4 enhancers were narrower in their width (or “Sharp”) (Fig. 4D). Systematic analysis of broad (>5kb) and sharp (<3kb) peaks (see Methods) showed enrichment of broad peaks in EpiC3 versus sharp peaks in EpiC4 enhancers (Fig. 4D and Supplementary Fig. S9B) suggesting that likely two different epigenetic regulatory processes are responsible for establishment of the observed enhancer patterns in EpiC3 versus EpiC4 colorectal tumors.
To identify potential regulators of the EpiC-specific features, we performed motif analyses of enhancers specific to each EpiC subtype using HOMER27. Interestingly, distinct TF classes were enriched in each of the EpiC subtypes. bZIP (BATF) and ZF (GATA6) type TFs were enriched in EpiC1, and homeobox (BRN2 or POU3F2) and Stats (STAT4) were enriched in EpiC2; c-MYC (bHLH) was enriched in EpiC3, and HMGs (SOX9) were enriched in EpiC4 (Fig. 4E). Consistently, we noted that expression of TFs (except for POU3F2) was higher, specifically in the respective CMSs in TCGA samples that corresponded with EpiC subtypes (Supplementary Fig. S9C). Importantly, previous studies have implicated c-MYC28–31, GATA632–34, SOX935–37, and STAT438 39 in CRC progression. BRN2/POU3F2 has oncogenic roles in melanoma40 41, and BATF2, a BATF family member, has been implicated in CRC42. These data suggest that the potential origin of different EpiC subtypes may be due to the pioneering activity of TFs belonging to different classes of TF families.
Combinatorial treatments for EpiCs
The CMS classification is being tested as a prognostic indicator for use in targeted therapy in CRC. For example, the CMS1 and CMS4 subtype cell lines are more responsive to HSP90 inhibitor, whereas the CMS2 subtype shows more sensitivity to WNTi and EGFRi43 44. The CMS3 subtype is enriched in metabolic pathways and RAS mutations, but therapeutic vulnerabilities are unknown3.
Exploring the clinical implications of our findings, we used a panel of 11 signaling pathway inhibitors that are known to be involved in the CRC tumorigenesis and whose key regulators showed differential enhancer enrichments in their vicinity in different epigenomic subtypes of CRC. Therefore, we treated six CRC cell lines with the signaling pathway compounds along with a panel of 8 known epigenetic inhibitors to treat cell lines from different CMSs, previously annotated with use of a CMS classifier (Supplementary Figs. S10A, S10B, S11, S12, S13A, and S13B)43 44. We further analyzed both IC50 and AUC indices to choose compounds with the best potency as well as the highest efficiency (Fig. 5A–C, S10B, S13A). We used a combination of one signaling pathway inhibitor along with the most efficient epigenetic inhibitor according to the generated drug matrix in distinct CMSs. Based on the analysis of monotherapies, we tested combinations of BRDi + PARPi for CMS1/EpiC1, BRDi + EGFRi for CMS2/EpiC3–4, and BRDi + TGFbi for CMS4/EpiC2 and found the combinations to perform better in reducing cell growth in vitro (Fig. 5D–F and Supplementary Figs. S13B and S14A). Importantly, we observed drastically enhanced activities of these combinations in the in vivo experiments carried out in nude mice, compared with the results of experiments using monotherapies (Fig. 5G–I).
Fig. 5. Correlation between CRC molecular subtypes and EpiCs can predict combinatorial treatments with high clinical significance.

A–C. Summary of IC50 values for 19 small molecule inhibitors (Y-axis) in cell lines with similar expression features to CMS1/EPIC1 (HCT116 or SW480) (A), CMS2/EPIC3 or EPIC4 (T84) (B), and CMS4/EPIC2 (SW620) (C). −Log(IC50) values are demonstrated on X-axis for each of the compounds. Drugs with higher −Log(IC50) are highlighted and were further used in combinatorial experiments.
D–F. Growth curves showing responses of CMS-specific CRC cell lines for the drug combinations identified based on the IC50 and AUC indices (panels A–C and Figures S8–S11): CMS1 lines with PARPi (olaparib) + BRDi (iBET-151) (D), CMS2 with EGFRi (gefitinib) + BRDi (RVX-208) (E), and CMS4 with TGFβi (SB431542) + BRDi (ISOX-DUAL) (F).
G–L. Tumor volume curves for xenografts in NUDE mice generated from transplantation of EpiC1/CMS1 (G-H), EpiC3–4/CMS2 (I-J), and EpiC2/CMS4 (K-L) cell lines upon treatment with inhibitors of EGFR (gefitinib, 100mg/kg), PARP (olaparib, 50mg/kg), TGF-β (SB431542, 10mg/kg), BRDi (i-BET151, 15mg/kg), or the combination of EGFRi + BRDi, the combination of PARPi + BRDi, and the combination of TGF-βi + BRDi along with the control vehicle group. Mice were treated every other day. Best treatment response was observed in the PARPi + BRDi combination in EpiC1/CMS1, EGFRi + BRDi combination in EpiC3–4/CMS2, and TGF-βi + BRDi combination in EpiC2/CMS4 lines. * shows p-values <0.05, and ** shows p-values <0.01.
Importantly, these combinatorial therapies demonstrated specific activity to the corresponding CMS/EpiC and had a minor impact on other CMS/EpiC subtypes. Specifically, BRDi (iBET-151, 15mg/kg) + PARPi (Olaparib, 50mg/kg) showed more significant impact on CMS1/EpiC1 cells than CMS2/EpiC3–4 or CMS4/EpiC2 did; similarly, BRDi (iBET-151, 15mg/kg) + EGFRi (Gefitinib, 100mg/kg) impacted CMS2/EpiC3–4 cells specifically, and BRDi (iBET-151, 15mg/kg) + TGFβi (SB431542, 10mg/kg) reduced tumor growth of CMS4/EpiC2 cells specifically (Fig. 5G–I). Notably, we did not observe much loss in tumor weight upon treatment with these drug combinations suggesting little or no side effects of these therapies (Supplementary Fig. S14B).
CMS2 can be divided into two epigenetic subgroups with clinical significance
To determine if the observed dichotomy of CMS2 tumors into EpiC3 and EpiC4 in our cohort expanded to a larger set of CRC samples, we examined the clustering of CMS2-designated TCGA tumors45 based on the gene targets of EpiC3 and EpiC4 enhancers. NMF clustering of 115 CMS2 tumors (see Methods) identified 3 clusters (Supplementary Fig. S15A and Supplementary Table S7): Clusters 1, 2 and 3. Overlap of EpiC4 enhancer target genes with Cluster2-specific genes showed enrichment of EpiC4-specific pathways such as GPCR, PI3K pathway and NCAM mediated neurotransmitter signaling (including NPTX2 – a differentially expressed gene between EpiC3 and EpiC4) (Supplementary Figs. S15B–D). Overlap of EpiC3 enhancer target genes with Cluster3-specific genes showed enrichment of EpiC3-specific pathways such as EGFR signaling, MAPK, and c-MYC targets (Fig. 6A and S15B–C). On the contrary, EpiC3-target genes overlap with Cluster3-specific genes or EpiC4-target genes overlap with Cluster2-specific genes did not enrich in any pathways specific for EpiC3 or EpiC4 (Supplementary Fig. S15C). Cluster 1 specific genes were mostly enriched in immune pathways suggesting likely separation due to low tumor content (Supplementary Fig. S15C). Therefore, we designated Cluster2 (n = 37) as ‘EpiC4-like’ and Cluster3 (n = 63) as ‘EpiC3-like’. Importantly, EpiC3-like tumors showed poorer survival in comparison to EpiC4-like tumors (Fig. 6B and Supplementary Fig. S15D), thus providing clinical significance for enhancer-based classification of CRC tumors.
Fig. 6. Functional and clinical significance of EpiC3 and EpiC4 classification.

A. Kaplan-Meier plot of survival of CMS2 samples in the TCGA, comparing EpiC4-like patients (n = 37) and EpiC3-like patients (n = 63). The log-rank (Mantel-Cox) p-value is shown for the difference in survival.
B. Pathway analysis for genes in Clusters 2 and 3 from NMF clustering (see Supplementary Fig. S9) of 115 CMS2 TCGA CRC tumors that overlap with EpiC3 and EpiC4-unique genes (LogFC >0.5 and p-value < 0.05). Based on the pathway enrichment, Cluster 2 was annotated as “EpiC4-like” whereas Cluster 3 was annotated as “EpiC3-like”.
C. Violin plots showing protein expression of 4EBP1_pT70, PTEN, p70S6K1, SRC_pY527 and CKIT in EpiC3-like or EpIC4-like TCGA samples.
F. Tumor volume curves for xenografts in NUDE mice generated from transplantation of EpiC3 (left panels, SW948 and SW480) or EpiC4 (right panels, T84 and SW-403) cell lines upon treatment with inhibitors of mTOR (rapamycin, 4mg/kg), SRC/KIT (Dasatinib, 30mg/kg), BRDi (i-BET151, 15mg/kg), or the combination of Rapamycin + iBET-151 or the combination of Dasatinib + iBET-151 or the control vehicle group. Mice were treated every other day. Asterisks show p-values. * <0.05, ** <0.01 and *** <0.001.
To further enhance the clinical significance of EpIC subgroups, we investigated if EpiC3 versus EpiC4 epigenomic patterns may impart specific therapeutic strategies in these subgroups because they both are categorized within the same transcriptomic group of CMS2. To exploit the combination strategy of bromodomain inhibitors with specific pathway inhibitors in these EpiC subgroups, we examined the RPPA data between EpiC3-like and EpiC4-like TCGA patients. We noted loss of PTEN and concomitant increase of mTOR targets, 4EBP1 (pT70) and pS6 kinase, in EpiC3-like samples (Fig. 6C). On the other hand, EpiC4-like samples displayed increased levels of C-KIT and SRC phosphorylation (Fig. 6C). Based on this result and in line with the theme from previous results, we hypothesized that EpiC3-like tumor cells may be sensitive to mTOR inhibitors plus bromodomain inhibitors whereas EpiC4 group may be vulnerable to a combination of KIT/SRC inhibitors plus bromodomain inhibitors. For the functional analyses, we categorized previously CMS2-designated cell lines43 44 46 47 into EpiC3 (SW948 and SW480) and EpiC4 (SW403 and T84) groups based on NPTX2 enhancer pattern and gene expression (a feature of EpiC4 subgroup, Supplementary Fig. S15E) as well as RPPA data (Supplementary Fig. S15F–G). We then tested the sensitivity of the xenograft tumors from these cell lines to aforementioned combination therapy. Indeed, we found that combination of iBET-151 with Rapamycin (mTOR inhibitor) was able to suppress growth of xenograft tumors from EpiC3 cell lines whereas iBET-151 and Dasatinib (SRC/KIT inhibitor) combination was specifically highly effective in EpiC4 xenografts (Fig. 6D). Overall, these data provide specific therapeutic strategies for patients harboring specific epigenomic signature.
DISCUSSION
Our study provides the most comprehensive data on basic epigenetic elements, in line with definitions from Roadmap/ENCODE, for human CRC, pre-malignant adenomas, widely used cell lines, and normal crypts/tissues. Importantly, these tissues/cell lines have associated genomic, transcriptomic, drug-response, and clinical datasets available. Thus, these collective epigenomic maps elucidate the epigenomic makeup of CRC and are an essential resource, along with the previous datasets11–14, for the community to generate hypotheses regarding the roles of the epigenome in CRC progression. Based on this comprehensive resource, our unbiased chromatin state analyses identified the H3K27ac marked active enhancers are the most discriminatory epigenetic elements between the various histopathological stages of CRCs. Consistent with this, the number of enhancer peaks that differentiate adenomas, normal tissues, and tumors is very high, suggesting that enhancer activation is likely a major epigenetic aberration in CRC. We suggest that enhancer aberrations are a critical component of a permissive chromatin environment in cancer cells that allow oncogenic events/pathways to drive excessive tumor growth. In addition to the active enhancer state, we also noted the heterochromatin state (H3K9me3) to differentiate between tumors and normal tissues. This will need to be further examined for causative effects.
We identified superenhancers surrounding ASCL2 and FZD10 to be necessary for colorectal cancer cell growth in vitro (Figures 2 and S4). This is interesting in that it provides evidence for enhancer/superenhancer elements as being targets for therapeutics in the future, potentially with the evolution of CRISPR-based therapies. It is also imaginable that eRNAs made from such superenhancers may be targets for RNA-targeted therapeutics such as antisense oligonucleotides (ASOs)48. The advances in 3D epigenome studies, such as our HiChIP data, also suggest that one enhancer may target many genes and hence could prove to be a better therapeutic target than the presumed gene target itself. We found over >30,000 such enhancers to be uniquely activated in colorectal adenocarcinomas. Future studies will be needed to identify functional enhancers such as those surrounding ASCL2 and FZD10.
Most importantly, our work identifies heterogeneity in enhancer patterns between CRC patients and for the first time identifies epigenomic subtypes of CRC. We demonstrate that the EpiC subtypes correlate highly with the expression phenotype (CMS), in line with previous observations made in the pan-cancer TCGA ATAC-seq study49, except that the CMS2 subtype was bifurcated into two distinct EpiC subtypes, EpiC3 and EpiC4. How these EpiC-specific enhancer patterns are set-up and how they cooperate with genetic events for the transformation of normal cells into cancers with unique transcriptional signatures need to be studied in the future. This is an interesting observation since we hypothesize that EpiC3 is likely driven by a c-MYC–regulated transcriptional program, whereas EpiC4 is driven by SOX9. Importantly, EpiC4 showed activation of enhancers around important neuronal genes (Figure 3C and S8), such as NPTX250, which could be either due to neural invasion in this category of tumors51 or epigenetic derepression of neuronal features. We suggest that inhibitors that target the c-MYC program could be more useful in EpiC3 subtypes, whereas SOX9 may be a target in EpiC4 subtypes. Importantly, the two TCGA subsets with EpiC3 and EpiC4 like features showed significant difference in survival, hence providing a clinical significance of this bifurcation of the CMS2 subtype.
Finally, our data suggest that combination of bromodomain inhibitors as an umbrella approach for blockade permissive enhancer states along with specific driver signaling pathway inhibitors could be used in defined patient CRC populations, hence providing a paradigm for epigenome-focused personalized therapeutic strategy. Bromodomain inhibitor combined well with PARP inhibitors for EpiC1, TGFβ inhibitors for EpiC2, mTOR inhibitors for EpiC3, and SRC/kit inhibitors for EpiC4. This is an important observation as it suggests patient-stratification based on enhancer signatures as well as the use of epigenetic therapy in a patient-centric manner.
Supplementary Material
SUMMARY BOX.
1). What is already known about this subject?
Chromatin deregulation is an emerging hallmark of cancer. Recent studies have suggested aberrations in enhancer elements in colorectal cancer. However, if and how patient-specific enhancer features could be utilized for precision medicine is not well understood.
2). What are the new findings?
By comprehensive epigenome profiling, we show chromatin state evolution during tumorigenesis, define patient subgroups with different enhancer landscape and identify therapeutic strategies in these epigenetic subgroups.
3). How might it impact on clinical practice in the foreseeable future?
Along the theme of personalized medicine, we identify therapeutic strategies for each of these enhancer subgroups which are based on combination of bromodomain inhibitors and a targeted therapy. We demonstrate MEKi plus BRDi as a general therapeutic strategy in colorectal cancer which is now being pursued in the form of a clinical trial at our institution.
ACKNOWLEDGEMENTS
This work was supported by ACS research scholar award, CPRIT IIRA award (RP200390), a Career Development Award from MDACC GI SPORE to K.R. We thank Integromics group, the Advanced Technology Genomics Core Facility (NCI Grant CA016672(ATGC), the Research Animal Support Facility, the Advanced Microscopy Core Facility (funded by NIH S10 RR029552), Functional Genomics Core (NCI Cancer Center Support Grant (P30 CA016672) and MD Anderson Cancer Center Clinical Core. We thank Veena Kochat, Sharon Landers and Angela Bhalla for helpful discussions and suggestions.
Footnotes
COMPETING INTERESTS
All the authors declare no conflict of interests.
Code availability
The ChIP-seq analysis source code are available at the following webpage: https://doi.org/10.5281/zenodo.819971.
Data availability
ChIP-seq, RNA-seq and Hi-ChIP datasets that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) database with the accession codes as follows: GSE136889, GSE88945, GSE106500 and GSE136044.
REFERENCES
- 1.Strum WB. Colorectal Adenomas. The New England journal of medicine 2016;374(11):1065–75. doi: 10.1056/NEJMra1513581 [published Online First: 2016/03/18] [DOI] [PubMed] [Google Scholar]
- 2.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990;61(5):759–67. [DOI] [PubMed] [Google Scholar]
- 3.Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nature medicine 2015;21(11):1350–6. doi: 10.1038/nm.3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haan JC, Labots M, Rausch C, et al. Genomic landscape of metastatic colorectal cancer. Nature communications 2014;5:5457. doi: 10.1038/ncomms6457 [published Online First: 2014/11/15] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.TCGA. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487(7407):330–7. doi: 10.1038/nature11252 [published Online First: 2012/07/20] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yaeger R, Chatila WK, Lipsyc MD, et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer cell 2018;33(1):125–36 e3. doi: 10.1016/j.ccell.2017.12.004 [published Online First: 2018/01/10] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Terasaki H, Saitoh T, Shiokawa K, et al. Frizzled-10, up-regulated in primary colorectal cancer, is a positive regulator of the WNT - beta-catenin - TCF signaling pathway. Int J Mol Med 2002;9(2):107–12. [PubMed] [Google Scholar]
- 8.Loven J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013;153(2):320–34. doi: 10.1016/j.cell.2013.03.036 [published Online First: 2013/04/16] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rai K, Sarkar S, Broadbent TJ, et al. DNA demethylase activity maintains intestinal cells in an undifferentiated state following loss of APC. Cell 2010;142(6):930–42. doi: 10.1016/j.cell.2010.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tao Y, Kang B, Petkovich DA, et al. Aging-like Spontaneous Epigenetic Silencing Facilitates Wnt Activation, Stemness, and Braf(V600E)-Induced Tumorigenesis. Cancer cell 2019;35(2):315–28 e6. doi: 10.1016/j.ccell.2019.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akhtar-Zaidi B, Cowper-Sal-lari R, Corradin O, et al. Epigenomic enhancer profiling defines a signature of colon cancer. Science 2012;336(6082):736–9. doi: 10.1126/science.1217277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen AJ, Saiakhova A, Corradin O, et al. Hotspots of aberrant enhancer activity punctuate the colorectal cancer epigenome. Nature communications 2017;8:14400. doi: 10.1038/ncomms14400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hung S, Saiakhova A, Faber ZJ, et al. Mismatch repair-signature mutations activate gene enhancers across human colorectal cancer epigenomes. eLife 2019;8 doi: 10.7554/eLife.40760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnstone SE, Reyes A, Qi Y, et al. Large-Scale Topological Changes Restrain Malignant Progression in Colorectal Cancer. Cell 2020;182(6):1474–89 e23. doi: 10.1016/j.cell.2020.07.030 [published Online First: 2020/08/26] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nature methods 2012;9(3):215–6. doi: 10.1038/nmeth.1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yen A, Kellis M. Systematic chromatin state comparison of epigenomes associated with diverse properties including sex and tissue type. Nature communications 2015;6:7973. doi: 10.1038/ncomms8973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whyte WA, Orlando DA, Hnisz D, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013;153(2):307–19. doi: 10.1016/j.cell.2013.03.035 [published Online First: 2013/04/16] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLean CY, Bristor D, Hiller M, et al. GREAT improves functional interpretation of cis-regulatory regions. Nature biotechnology 2010;28(5):495–501. doi: 10.1038/nbt.1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao Q, Anyansi C, Hu X, et al. Reconstruction of enhancer-target networks in 935 samples of human primary cells, tissues and cell lines. Nature genetics 2017;49(10):1428–36. doi: 10.1038/ng.3950 [published Online First: 2017/09/05] [DOI] [PubMed] [Google Scholar]
- 20.Schuijers J, Junker JP, Mokry M, et al. Ascl2 acts as an R-spondin/Wnt-responsive switch to control stemness in intestinal crypts. Cell Stem Cell 2015;16(2):158–70. doi: 10.1016/j.stem.2014.12.006 [DOI] [PubMed] [Google Scholar]
- 21.Zhu R, Yang Y, Tian Y, et al. Ascl2 knockdown results in tumor growth arrest by miRNA-302b-related inhibition of colon cancer progenitor cells. PloS one 2012;7(2):e32170. doi: 10.1371/journal.pone.0032170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rout A, Sukhi A, Chaudhary R, et al. Investigational drugs in phase II clinical trials for acute coronary syndromes. Expert Opin Investig Drugs 2020;29(1):33–47. doi: 10.1080/13543784.2020.1708324 [DOI] [PubMed] [Google Scholar]
- 23.Kun E, Tsang YTM, Ng CW, et al. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat Rev 2021;92:102137. doi: 10.1016/j.ctrv.2020.102137 [published Online First: 2020/12/20] [DOI] [PubMed] [Google Scholar]
- 24.Consortium EP, Moore JE, Purcaro MJ, et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020;583(7818):699–710. doi: 10.1038/s41586-020-2493-4 [published Online First: 2020/07/31] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eide PW, Bruun J, Lothe RA, et al. CMScaller: an R package for consensus molecular subtyping of colorectal cancer pre-clinical models. Sci Rep 2017;7(1):16618. doi: 10.1038/s41598-017-16747-x [published Online First: 2017/12/02] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X, Halberg RB, Burch RP, et al. Intestinal adenomagenesis involves core molecular signatures of the epithelial-mesenchymal transition. J Mol Histol 2008;39(3):283–94. doi: 10.1007/s10735-008-9164-3 [published Online First: 2008/03/11] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heinz S, Benner C, Spann N, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular cell 2010;38(4):576–89. doi: 10.1016/j.molcel.2010.05.004 [published Online First: 2010/06/02] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee KS, Kwak Y, Nam KH, et al. Favorable prognosis in colorectal cancer patients with co-expression of c-MYC and ss-catenin. BMC Cancer 2016;16(1):730. doi: 10.1186/s12885-016-2770-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith DR, Goh HS. Overexpression of the c-myc proto-oncogene in colorectal carcinoma is associated with a reduced mortality that is abrogated by point mutation of the p53 tumor suppressor gene. Clinical cancer research : an official journal of the American Association for Cancer Research 1996;2(6):1049–53. [PubMed] [Google Scholar]
- 30.Erisman MD, Rothberg PG, Diehl RE, et al. Deregulation of c-myc gene expression in human colon carcinoma is not accompanied by amplification or rearrangement of the gene. Molecular and cellular biology 1985;5(8):1969–76. doi: 10.1128/mcb.5.8.1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rochlitz CF, Herrmann R, de Kant E. Overexpression and amplification of c-myc during progression of human colorectal cancer. Oncology 1996;53(6):448–54. doi: 10.1159/000227619 [DOI] [PubMed] [Google Scholar]
- 32.Belaguli NS, Aftab M, Rigi M, et al. GATA6 promotes colon cancer cell invasion by regulating urokinase plasminogen activator gene expression. Neoplasia 2010;12(11):856–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whissell G, Montagni E, Martinelli P, et al. The transcription factor GATA6 enables self-renewal of colon adenoma stem cells by repressing BMP gene expression. Nat Cell Biol 2014;16(7):695–707. doi: 10.1038/ncb2992 [DOI] [PubMed] [Google Scholar]
- 34.Martinelli P, Carrillo-de Santa Pau E, Cox T, et al. GATA6 regulates EMT and tumour dissemination, and is a marker of response to adjuvant chemotherapy in pancreatic cancer. Gut 2017;66(9):1665–76. doi: 10.1136/gutjnl-2015-311256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prevostel C, Blache P. The dose-dependent effect of SOX9 and its incidence in colorectal cancer. Eur J Cancer 2017;86:150–57. doi: 10.1016/j.ejca.2017.08.037 [DOI] [PubMed] [Google Scholar]
- 36.Prevostel C, Rammah-Bouazza C, Trauchessec H, et al. SOX9 is an atypical intestinal tumor suppressor controlling the oncogenic Wnt/ss-catenin signaling. Oncotarget 2016;7(50):82228–43. doi: 10.18632/oncotarget.10573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lu B, Fang Y, Xu J, et al. Analysis of SOX9 expression in colorectal cancer. Am J Clin Pathol 2008;130(6):897–904. doi: 10.1309/AJCPW1W8GJBQGCNI [DOI] [PubMed] [Google Scholar]
- 38.Cheng JM, Yao MR, Zhu Q, et al. Silencing of stat4 gene inhibits cell proliferation and invasion of colorectal cancer cells. J Biol Regul Homeost Agents 2015;29(1):85–92. [PubMed] [Google Scholar]
- 39.Slattery ML, Lundgreen A, Kadlubar SA, et al. JAK/STAT/SOCS-signaling pathway and colon and rectal cancer. Mol Carcinog 2013;52(2):155–66. doi: 10.1002/mc.21841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herbert K, Binet R, Lambert JP, et al. BRN2 suppresses apoptosis, reprograms DNA damage repair, and is associated with a high somatic mutation burden in melanoma. Genes & development 2019;33(5–6):310–32. doi: 10.1101/gad.314633.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simmons JL, Pierce CJ, Al-Ejeh F, et al. MITF and BRN2 contribute to metastatic growth after dissemination of melanoma. Sci Rep 2017;7(1):10909. doi: 10.1038/s41598-017-11366-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dai L, Cui X, Zhang X, et al. SARI inhibits angiogenesis and tumour growth of human colon cancer through directly targeting ceruloplasmin. Nature communications 2016;7:11996. doi: 10.1038/ncomms11996 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Sveen A, Bruun J, Eide PW, et al. Colorectal Cancer Consensus Molecular Subtypes Translated to Preclinical Models Uncover Potentially Targetable Cancer Cell Dependencies. Clinical cancer research : an official journal of the American Association for Cancer Research 2018;24(4):794–806. doi: 10.1158/1078-0432.CCR-17-1234 [published Online First: 2017/12/16] [DOI] [PubMed] [Google Scholar]
- 44.Linnekamp JF, Hooff SRV, Prasetyanti PR, et al. Consensus molecular subtypes of colorectal cancer are recapitulated in in vitro and in vivo models. Cell Death Differ 2018;25(3):616–33. doi: 10.1038/s41418-017-0011-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cancer Genome Atlas N Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487(7407):330–7. doi: 10.1038/nature11252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berg KCG, Eide PW, Eilertsen IA, et al. Multi-omics of 34 colorectal cancer cell lines - a resource for biomedical studies. Mol Cancer 2017;16(1):116. doi: 10.1186/s12943-017-0691-y [published Online First: 2017/07/08] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ronen J, Hayat S, Akalin A. Evaluation of colorectal cancer subtypes and cell lines using deep learning. Life Sci Alliance 2019;2(6) doi: 10.26508/lsa.201900517 [published Online First: 2019/12/04] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khvorova A, Watts JK. The chemical evolution of oligonucleotide therapies of clinical utility. Nature biotechnology 2017;35(3):238–48. doi: 10.1038/nbt.3765 [published Online First: 2017/03/01] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Corces MR, Granja JM, Shams S, et al. The chromatin accessibility landscape of primary human cancers. Science 2018;362(6413) doi: 10.1126/science.aav1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu C, Tian G, Jiang C, et al. NPTX2 promotes colorectal cancer growth and liver metastasis by the activation of the canonical Wnt/beta-catenin pathway via FZD6. Cell Death Dis 2019;10(3):217. doi: 10.1038/s41419-019-1467-7 [published Online First: 2019/03/06] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Amit M, Takahashi H, Dragomir MP, et al. Loss of p53 drives neuron reprogramming in head and neck cancer. Nature 2020;578(7795):449–54. doi: 10.1038/s41586-020-1996-3 [published Online First: 2020/02/14] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 2014;15(12):550. doi: 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ChIP-seq, RNA-seq and Hi-ChIP datasets that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) database with the accession codes as follows: GSE136889, GSE88945, GSE106500 and GSE136044.