

To the Editor:
Cryopyrin-associated periodic syndrome (CAPS) is a rare congenital autoinflammatory disease caused by mutations in the NLRP3 gene. Previously, CAPS was assumed to include 3 distinct diseases: familial cold-induced autoinflammatory syndrome, Muckle-Wells syndrome, and neonatal-onset multisystem inflammatory disease (also known as chronic infantile neurologic, cutaneous, and articular syndrome). The general clinical features of CAPS include recurrent fever, skin rashes, joint pain, and systemic inflammation.1
In clinical practice, there is a significant delay between disease onset and diagnosis of CAPS. The median diagnosis delay was 7.3 years (range, 0.3–76 years) according to a report by an international registry on autoinflammatory diseases in the context of the Eurofever project.2 Delayed diagnosis may result in growth retardation, economic and psychological pressure for the patient and family, and antibiotic abuse.
Here, we report a case of a 42-month-old girl who had periodic fever for 20 months and was treated as an infectious disease without any improvement; she was ultimately diagnosed as having CAPS.
CASE PRESENTATION
The patient presented with an urticaria-like rash over her face and trunk on the second day after her birth. The rash worsened in the summer and improved in the winter, but never disappeared (Figs. A and B). She had recurrent fever beginning at 23 months of age, with a maximum body temperature of 39.5°C. The patient was treated with various antibiotics for more than 1 month, but no improvement was noted. Subsequently, she experienced sudden loss of consciousness and a staring-eye gaze lasting for 1 minute, without headache or vomiting. Laboratory tests showed that she had increased peripheral white blood cell counts (17.75 × 109/L; 57% neutrophils and 35.4% lymphocytes), anemia with hemoglobin level of 90 g/L, and increased C-reactive protein levels (44.5 mg/L). She had normal liver and kidney functions. Her immunoglobulin levels and CD4- and CD8-positive cell counts were normal. To rule out the possibility of intracranial infection, lumbar puncture was performed. Examination of cerebrospinal fluid (CSF) revealed an increased open pressure of 160 mm H2O, 97 white blood cells/μL (20% lymphocytes and 80% neutrophils), greater than 3000 mg/L protein, 1.3 mmol/L glucose, 1.08 U/L adenosine deaminase (reference range, 0–19.6 U/L), and 175 U/L lactate dehydrogenase. Cerebrospinal fluid smears and cultures were negative for bacteria, mycobacteria, and fungi. Chest computed tomography (CT) scans showed bilateral pneumonia (Fig. C). Cranial CT scans showed widened sulci and gyri and enlarged lateral ventricles and third ventricle of the cerebrum (Fig. D). Bacterial meningitis was suspected, and empirical antibiotics, including meropenem, cefepime, vancomycin, and linezolid, were administered. The patient's condition remained unchanged; there was no obvious absorption of lung lesions, and she became irritable. Examination of CSF after more than 1 month of treatment revealed 210 white blood cells/μL (32% lymphocytes and 68% neutrophils), greater than 1857 mg/L protein, 0.94 mmol/L glucose, 3 U/L adenosine deaminase, and 157 U/L lactate dehydrogenase; all etiological evidence was obtained. She tested positive on the T-SPOT.TB tests; thus, tuberculosis and tuberculous meningitis were suspected. A diagnostic antituberculosis regimen with isoniazid, rifampin, pyrazinamide, and ethambutol was given. However, gastric fluid was negative for acid-fast smear and Mycobacterium culture. After 6 weeks of treatment, the patient's symptoms persisted and CSF examination results showed no improvement. Additional bone marrow acid-fast smear and Mycobacterium culture were performed and gave negative results. When glucocorticoids (prednisone, 7.5 mg/d, 0.75 mg/kg per day) were added to the regimen, the patient's condition improved dramatically. Moreover, once the glucocorticoids were withdrawn, the symptoms returned. Repeated gastric fluid acid-fast smear and Mycobacterium culture remained negative after 14 months of treatment. After 15 months of antituberculosis treatment, the fever and rash were not improved, and the patient's growth and development were delayed significantly.
FIGURE.
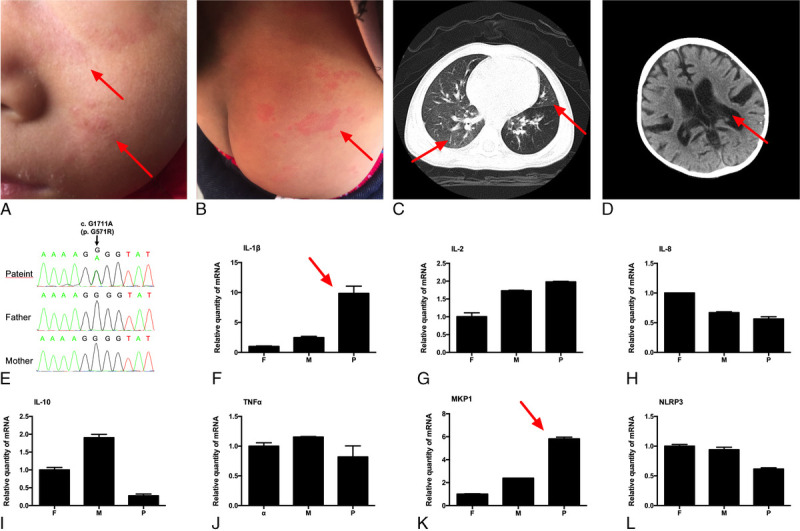
Urticaria-like rash over the patient's face and trunk (A and B). ChestCT showing bilateral pneumonia (C). CranialCT scans showingwidened sulci and gyri and enlarged lateral ventricles (D). Geneticmutation in NLRP3 of the patient (E) and relative mRNA expression of (F) IL-1β, (G) IL-2, (H) IL-8, (I) IL-10, (J) TNF-α, (K) MKP-1, and (L) NLRP3 of the patient and her parents. F indicates father; M, mother; P, patient.
After the patient was admitted to our department, we reviewed all her medical records and the response to glucocorticoid treatment caught our attention. We applied next-generation sequencing using a targeted panel of 535 genes to screen primary immunodeficiency genes in this patient. The heterozygous point mutation G571R (G1711A, chromosome 1:247588456) in exon 3 of the NLRP3 gene was identified; this was a de novo mutation that was not detected in her parents. We further confirmed the mutation by Sanger sequencing (Fig. E). In addition, the plasma level of interleukin (IL)-1β in the patient was increased to 22.3 ± 0.2 pg/mL, whereas IL-1β levels in both her parents were below the lower limit of detection (4 pg/mL) of the enzyme-linked immunosorbent assay kit. Real-time polymerase chain reaction showed that IL-1β mRNA levels were markedly higher in the blood cells from the patient than in those from her father and mother (Fig. F). The relative mRNA levels of other inflammatory cytokines, including IL-2, IL-8, IL-10, and tumor necrosis factor-α, remained normal in the patient compared with those in the healthy parents. Mitogen-activated protein kinase phosphatase-1 (MKP-1), a negative regulator of mitogen-activated protein kinase activity and an indirect marker of the host's inflammation level, was also upregulated in the patient (Figs. G–K). NLRP3 mRNA was not overexpressed in the patient (Fig. L).
Based on the combination of clinical symptoms, NLRP3 gene mutation, and increased IL-1β production in the blood, the patient was finally diagnosed as having CAPS. All antituberculosis drugs were discontinued. Although IL-1β inhibition is an effective therapeutic option,3 anakinra, rilonacept, and canakinumab are available in China. Therefore, we treated the patient with prednisolone (15 mg/d, 1.5 mg/kg per day). From the second day, the patient's body temperature returned to normal, her rash subsided, and her condition and appetite improved. Notably, the rash recurred when she had diarrhea or flu during prednisolone treatment, and it usually lasted for 4 to 5 days.
The NLRP3 gene encodes the protein cryopyrin; mutations in this gene lead to constitutive activation of caspase 1 and excessive IL-1β secretion and cause inflammatory manifestations. The G1711A mutation is a newly discovered nucleotide mutation in the NLRP3 gene; however, this mutation (previously described as G569R) was first reported in a patient with Muckle-Wells syndrome and later in a patient with neonatal-onset multisystem inflammatory disease.4 The severity of CAPS disease may be related to IL-1β production.4 It is important to precisely diagnose CAPS early and treat the disease immediately to prevent persistent inflammation-induced organ damage.
CONCLUSIONS
In our case, before the final diagnosis of CAPS, the patient had been misdiagnosed as suffering from infectious diseases and was incorrectly given different antibiotics for approximately 2 years. Physicians are expected to have sufficient knowledge on clinical symptoms of autoinflammatory diseases and list them as differential diagnoses when taken care of a patient who shows “infection-like” symptoms that cannot be explained by infection. Next, they should use available technologies, such as next-generation sequencing, to confirm the genetic diagnosis. Early diagnosis and timely treatment are of great importance to avoid irreversible and severe organ damage and disability due to autoinflammatory diseases.
Key Message
Cryopyrin-associated periodic syndrome is a rare congenital autoinflammatory disease that shows symptoms similar to those of most infections and can, therefore, be easily misdiagnosed. Genetic detection using next-generation sequencing can facilitate the early diagnosis and treatment of such rare genetic diseases.
Tao Li, MD, PhD
Shanghai Public Health Clinical CenterFudan UniversityShanghai, China
Jing Wu, PhD
Qiaoling Ruan, MD
Wenhong Zhang, MD, PhD
Department of Infectious DiseasesHuashan HospitalFudan UniversityShanghai, China
Wenhong Zhang, MD, PhD
Key Laboratory of MedicalMolecular VirologyShanghai Medical CollegeFudan UniversityShanghai, China
Ping Liu, MD
Ning Pei, MD
Xiuhong Xi, MD
Shuihua Lu, MD
Shanghai Public Health Clinical CenterFudan UniversityShanghai, Chinatubercle@shaphc.org
Ning Jiang, PhD
Department of Biostatistics andComputational Biology, SKLGSchool of Life SciencesFudan UniversityShanghai, Chinaningjiang@fudan.edu.cn
Footnotes
Tao Li, Jing Wu, and Qiaoling Ruan contributed equally to this work.
The authors declare no conflict of interest.
This study was approved by the ethics committee of Shanghai Public Health Clinical Center.
Written informed consent was obtained from the patient's mother for reporting this case and publishing any accompanying images.
This work was supported by the National Natural Science Foundation of China (81700014) and Shanghai Health and Family Planning System (grant no. 2016ZB01-01). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Authors' contributions: T.L., S.L., W.Z., P.L., N.P., and H.X. were involved in patient care. J.W. performed the gene expression assay. J.W. and Q.R. drafted most of the manuscript. W.Z., P.L., N.P., and H.X. were involved in reviewing the manuscript. N.J. organized the study and performed the bioinformatics analysis. All authors read and approved the final version of the manuscript.
Contributor Information
Tao Li, Email: litaokc@126.com.
Jing Wu, Email: jingee@fudan.edu.cn.
Qiaoling Ruan, Email: qlruan07@fudan.edu.cn.
Wenhong Zhang, Email: wenhongzhang_hs@126.com.
Wenhong Zhang, Email: wenhongzhang_hs@126.com.
Ping Liu, Email: 12429717@qq.com.
Ning Pei, Email: peining1125@163.com.
Xiuhong Xi, Email: xixiuhong@126.com.
Shuihua Lu, Email: Tubercl@163.com.
REFERENCES
- 1.Kuemmerle-Deschner JB. CAPS—pathogenesis, presentation and treatment of an autoinflammatory disease. Semin Immunopathol. 2015;37:377–385. [DOI] [PubMed] [Google Scholar]
- 2.Toplak N Frenkel J Ozen S, et al. An international registry on autoinflammatory diseases: the Eurofever experience. Ann Rheum Dis. 2012;71:1177–1182. [DOI] [PubMed] [Google Scholar]
- 3.Kone-Paut I, Galeotti C. Current treatment recommendations and considerations for cryopyrin-associated periodic syndrome. Expert Rev Clin Immunol. 2015;11:1083–1092. [DOI] [PubMed] [Google Scholar]
- 4.Turel O Goknar N Uzuner S, et al. CINCA syndrome in an infant presenting with hydrocephalus. Int J Rheum Dis. 2014;17:346–348. [DOI] [PubMed] [Google Scholar]