

Abstract
Triple-negative breast cancer (TNBC) is the most aggressive subgroup of breast cancer, and patients with TNBC have few therapeutic options. Apoptosis resistance is a hallmark of human cancer, and apoptosis regulators have been targeted for drug development for cancer treatment. One class of apoptosis regulators is the inhibitors of apoptosis proteins (IAPs). Dysregulated IAP expression has been reported in many cancers, including breast cancer, and has been shown to be responsible for resistance to chemotherapy. Therefore, IAPs have become attractive molecular targets for cancer treatment. Here, we first investigated the anti-tumor efficacy of birinapant (TL32711), a biindole-based bivalent mimetic of second mitochondria-derived activator of caspases (SMAC), in TNBC. We found that birinapant as a single agent has differential anti-proliferation effects in TNBC cells. We next assessed whether birinapant has a synergistic effect with commonly used anti-cancer drugs, including entinostat (class I histone deacetylase inhibitor), cisplatin, paclitaxel, voxtalisib (PI3K inhibitor), dasatinib (Src inhibitor), erlotinib (epidermal growth factor receptor inhibitor), and gemcitabine, in TNBC. Among these tested drugs, gemcitabine showed a strong synergistic effect with birinapant. Birinapant significantly enhanced the anti-tumor activity of gemcitabine in TNBC both in vitro and in xenograft mouse models through activation of the intrinsic apoptosis pathway via degradation of cIAP2 and XIAP, leading to apoptotic cell death. Our findings demonstrate the therapeutic potential of birinapant to enhance the anti-tumor efficacy of gemcitabine in TNBC by targeting the IAP family of proteins.
Keywords: Birinapant, gemcitabine, synergistic effect, apoptosis, TNBC
Introduction
Triple-negative breast cancer (TNBC), which lacks druggable expression levels of estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2, is the most aggressive subgroup of breast cancer. TNBC comprises 15-20% of breast cancers but accounts for 30-40% of U.S. breast cancer deaths. These poor outcomes are associated with TNBC’s tendency to relapse and metastasize (1,2). Currently, not many therapeutic options are available for patients with TNBC, and effective therapeutic approaches are needed to prolong survival and reduce the mortality rate of these patients.
Apoptosis resistance is a hallmark of cancer. Apoptosis can be initiated by either the intrinsic signaling pathway (mitochondrial apoptotic pathway) through activation of caspase 9 or the extrinsic signaling pathway (death-receptor apoptotic pathway) through activation of caspase 8 (3,4). Defects in apoptosis can lead to malignant transformation, tumor metastasis, and drug resistance (4–6). Therefore, drug development targeting key regulators of apoptosis has become an attractive strategy for cancer treatment.
Many proteins have been reported to have pro- or anti-apoptotic activity in cells, and abnormal expression of these proteins has been associated with carcinogenesis by suppression of apoptosis (7,8). Several key regulators of apoptosis have been identified and targeted for drug development for cancer therapy. One class of such regulators is the inhibitors of apoptosis proteins (IAPs). IAPs are characterized by the presence of a baculovirus IAP repeat (BIR) protein domain (5). There are eight known IAPs: NAIP (BIRC1), cIAP1 (BIRC2), cIAP2 (BIRC3), X-linked IAP (XIAP/BIRC4), survivin (BIRC5), Apollon (BRUCE/BIRC6), Livin/MLIAP (BIRC7), and IAP-like protein 2 (BIRC8) (5). IAPs inhibit apoptosis by suppressing caspase activity by binding to the active sites of caspases via the caspases’ conserved BIR domains, which degrades active caspases or blocks interaction of caspases with their substrates (5). Dysregulated expression of IAPs has been reported in many cancers (9–11) and has been linked to resistance to chemotherapy (6). Thus, IAPs hold notable potential as targets in the development of cancer therapy. IAP-targeted approaches include antisense oligonucleotides (12,13) and small-molecule inhibitors targeting BIR domains of XIAP, survivin, MLIAP, and cIAP1/cIAP2 (14,15). Among IAPs, XIAP is reportedly the most potent inhibitor of apoptosis and is required for survival of cells that are resistant to therapeutic agents (16–18). XIAP effectively inhibits both the intrinsic and extrinsic pathways of apoptosis by binding and inhibiting upstream caspases 9 and 8 and downstream caspases 3 and 7 (19,20). Inhibition of XIAP using antisense oligonucleotides improved the anti-tumor efficacy of radiotherapy and chemotherapy in vitro and in vivo in various cancers (12,13,21–24). These studies demonstrate the targeting potential of IAPs in cancer therapy both at a molecular level and preclinically.
The second mitochondria-derived activator of caspases (SMAC) is an endogenous antagonist of IAPs. Upon apoptotic stimulation, SMAC is released from mitochondria and then binds via its N-terminal AVPI tetrapeptide to the BIR domain on IAPs (6,25). The interaction between SMAC and IAPs results in caspase activation and subsequent apoptotic cell death. The SMAC-mediated functional inhibition of IAPs has been emulated in the development of small-molecule inhibitors that mimic the IAP binding motif of SMAC and inhibit IAP protein functions (6,14,17,26). Birinapant (TL32711), a biindole-based bivalent SMAC mimetic, has high affinity to the BIR3 domains of cIAP1, cIAP2, and XIAP and to the single BIR domain of MLIAP. Upon binding to these sites, birinapant has the ability to cause rapid degradation of TRAF2-bound cIAP1 and cIAP2, which leads to inhibition of tumor necrosis factor (TNF)-mediated NF-κB activation (26). Upon TNF stimulation, birinapant also promotes caspase 8/RIPK1 complex formation, resulting in activation of downstream caspases (26). Although the anti-tumor efficacy of birinapant as a single agent in many cancer cells is mainly dependent on the levels of TNFα in the cells, an IAP-dependent but TNFα-independent mechanism has also been observed (17,27). In patient-derived xenograft mouse models of human ovarian cancer, melanoma, and colorectal cancer, intraperitoneal administration of birinapant inhibited tumor growth with no evidence of toxicity (26). In addition, birinapant showed synergistic anti-tumor effects with several widely used chemotherapeutic agents in various cancers (28–30). These studies demonstrate that birinapant can effectively activate apoptotic signaling by targeting IAP proteins and has potential applications for the treatment of multiple tumor malignancies.
Gemcitabine is a difluorinated pyrimidine analog of deoxycytidine that replaces cytidine during DNA replication, which inhibits elongation of the replicating DNA strand and leads to apoptosis (31,32). Gemcitabine is commonly used for the treatment of bladder, pancreatic, ovarian, breast, and non-small cell lung cancers (32). Here, we show the therapeutic potential of birinapant in combination with gemcitabine in TNBC. While birinapant was not highly effective at inhibiting cancer cell proliferation in the tested TNBC cell lines as a single agent, it exhibited strong synergistic cytotoxicity when combined with gemcitabine in TNBC cells, both in vitro and in vivo. We also found that birinapant enhanced the anti-tumor effectiveness of gemcitabine through activation of the intrinsic apoptotic pathway by targeting IAP proteins. Our findings convincingly demonstrate that the combination of birinapant and gemcitabine can be an effective therapeutic strategy for TNBC.
Materials and Methods
Cell culture and reagents
HCC38, HCC70, HCC1937, MDA-MB-231, MDA-MB-157, MDA-MB-436, MDA-MB-468, HS578T, and BT-20 human TNBC cells and MCF10A human breast epithelial cells were purchased from American Type Culture Collection (Manassas, VA, USA). SUM149 and SUM159 TNBC cells were purchased from Asterand Bioscience (Detroit, MI, USA). KTB6 human breast epithelial cells were a generous gift of Dr. Harikrishna Nakshatri (Indiana University, Bloomington, IN, USA). HCC38, HCC70, and HCC1937 cells were maintained in Roswell Park Memorial Institute 1640 medium (Life Technologies Inc., Carlsbad, CA, USA). MDA-MB-231, MDA-MB-157, MDA-MB-436, MDA-MB-468, HS578T, and BT-20 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM)/F-12 medium (Life Technologies Inc.). Both culture media were supplemented with 10% fetal bovine serum (FBS; GenDEPOT, Katy, TX, USA) and 1% antibiotic/antimycotic (Sigma-Aldrich Co. LLC, St. Louis, MO, USA). SUM149 and SUM159 cells were maintained in Ham’s F-12 medium (Life Technologies Inc.) supplemented with 5% FBS, 1% antibiotic/antimycotic, 5 μg/mL insulin (Life Technologies Inc.), and 1 μg/mL hydrocortisone (Sigma-Aldrich Co. LLC). MCF10A cells were maintained in DMEM/F-12 medium supplemented with 10% horse serum (Thermo Fisher Scientific, Waltham, MA, USA), 1% antibiotic/antimycotic, 10 μg/mL insulin, 20 ng/mL epidermal growth factor (EGF; Sigma-Aldrich Co. LLC), 100 ng/mL cholera toxin (Sigma-Aldrich Co. LLC), and 500 μg/mL hydrocortisone. KTB6 cells were maintained in low-glucose DMEM and Ham’s F-12 medium (1:3) supplemented with 10% FBS, 1% penicillin/streptomycin, 5 μg/mL insulin, 20 ng/mL EGF, 0.4 μg/mL hydrocortisone, and 5 uM ROCK inhibitor Y-27632 (STEMCELL Technologies Inc., Vancouver, BC, Canada). All cell lines were validated by DNA typing at The University of Texas MD Anderson Cancer Center Cytogenetics and Cell Authentication Core (Houston, Texas, USA) and confirmed to be free of mycoplasma.
Gemcitabine hydrochloride was purchased from Sigma-Aldrich Co. LLC. Entinostat (class I histone deacetylase [HDAC] inhibitor), cisplatin, paclitaxel, voxtalisib (PI3K inhibitor), dasatinib (Src inhibitor), and erlotinib (EGF receptor inhibitor) were purchased from Selleckchem (Houston, TX, USA). Birinapant was purchased from Selleckchem and also provided by Medivir AB (Huddinge, Sweden). Ent-birinapant, a non-IAP-binding negative control, was provided by Medivir AB.
Cell viability assay
Cell viability was determined using a CellTiter-Blue viability assay (Promega Corporation, Madison, WI, USA) as described previously (33). Cells were seeded in 96-well plates and treated the next day with birinapant alone at 0-20 μM or with entinostat, cisplatin, paclitaxel, voxtalisib, dasatinib, or erlotinib at 0-20 μM alone or in combination with birinapant at a fixed concentration as indicated in Supplementary Tables 2–10. At 72 h after treatment, the CellTiter-Blue reagent was added into the plates, and optical density at 595 nm was determined using the VICTOR X3 plate reader (PerkinElmer, Waltham, MA, USA).
Soft agar colony formation assay
Anchorage-independent growth was determined using a soft agar colony formation assay as described previously (34). Cells were resuspended in 0.4% agarose growth medium in the presence of gemcitabine, birinapant, or gemcitabine plus birinapant and then plated in 6-well plates containing solidified 0.8% agarose in growth medium. Three weeks later, colonies greater than 80 μm in diameter were counted using the GelCount system (Oxford Optronix Ltd., Milton Park, Abingdon, UK).
Flow cytometry
For the cell cycle distribution analysis, cells (3 × 105 cells/3 mL) were seeded in 60-mm plates overnight and then, the next morning, treated with gemcitabine, birinapant, or gemcitabine plus birinapant. At 48 h or 72 h after treatment, the cells were harvested and fixed in 70% ethanol at −20 °C overnight. The next morning, the fixed cells were treated with RNase (10 μg/mL) at 37 °C for 15 min, stained with propidium iodide (20 μg/mL), and then subjected to flow cytometry analysis. For the apoptosis analysis, after 48 h or 72 h of treatment with gemcitabine, birinapant, or gemcitabine plus birinapant, cells were harvested, incubated at room temperature with annexin V–PE (BD Biosciences, Franklin Lakes, NJ, USA) and 7-AAD (BD Biosciences) for 15 min, and then subjected to flow cytometry analysis.
Western blotting
Cells (1 × 106 cells/10 mL) were seeded in 10-cm plates overnight and then, the next morning, treated with birinapant, gemcitabine, or birinapant plus gemcitabine. At 48 h following treatment, cells were harvested, and proteins were extracted for Western blotting analysis as described previously (34). Proteins of interest were probed using the following primary antibodies (1:1000 dilution) purchased from Cell Signaling Technology (Danvers, MA, USA) or other suppliers as indicated: anti-XIAP (#14334), anti-cIAP1 (#7065), anti-cIAP2 (#3130), anti-caspase 3 (#9662), anti-caspase 7 (#9492), anti-caspase 8 (#9746), anti-caspase 9 (#9502), anti-cleaved caspase 3 (#9661), anti-cleaved caspase 7 (#9491), anti-cleaved caspase 8 (#9748), anti-cleaved caspase 9 (#9501), anti-PARP (#9542), anti-cleaved PARP (#9542), and anti-α-tubulin (clone DM1A, #T9026, Sigma-Aldrich Co. LLC). The secondary antibodies used were horseradish peroxidaseconjugated IgG (Life Technologies Inc.) for chemiluminescence signal detection and Alexa Fluor-conjugated IgG (Life Technologies Inc.) for fluorescence signal detection. The intensity of target proteins on the blots was measured using ImageJ (National Institutes of Health, Bethesda, MD, USA).
Treatment with a pan-caspase inhibitor
Cells (2 × 103 cells/well) were seeded into a 96-well plate overnight and then pre-treated the next day with the pan-caspase inhibitor Z-VAD-FMK (10 μM; EMD Millipore, Burlington, MA, USA) for 2 h, followed by incubation with birinapant, gemcitabine, or birinapant plus gemcitabine for 5 days. On day 5 after treatment, the CellTiter-Blue reagent was added into the plate, and optical density at 595 nm was determined using a plate reader.
TNBC xenograft mouse model
All animals were maintained and handled in accordance with the guidelines of the MD Anderson Institutional Animal Care and Use Committee (00001429-RN01). SUM149 or MDA-MB-231 (4 × 106 cells/100 μL) cells were implanted into 1 of the mammary fat pads of 4- to 6-week-old female athymic BALB/c nude mice purchased from Envigo (Indianapolis, IN, USA). When tumors reached 75-150 mm3, mice were randomly divided into 4 groups (12 mice/group) and treated with vehicle (12.5% captisol), birinapant (15 mg/kg in 12.5% captisol), gemcitabine (15 mg/kg in 0.9% NaCl), or birinapant (15 mg/kg in 12.5% captisol) plus gemcitabine (15 mg/kg in 0.9% NaCl) via intraperitoneal injections twice per week for 21 days in the SUM149 xenograft model or 38 days in the MDA-MB-231 xenograft model. Birinapant and gemcitabine were given to the mice at 24-h intervals. At the end of the study, tumor samples were collected and processed for immunohistochemical (IHC) staining of target proteins.
IHC staining
Tumor tissues were fixed in 10% buffered formalin, embedded in paraffin, sectioned at 5 μm, and mounted on slides. The sections were deparaffinized in xylene, rehydrated in graded alcohols, and washed in distilled water. Antigens on sections were retrieved by steaming in 10 mM citric acid (pH 6.0) for 30 min. Endogenous peroxidases were quenched by incubation in 3% H2O2 for 10 min at room temperature. The slides were washed 3 times with phosphate-buffered saline and blocked for 30 min with 10% normal horse serum in 1% bovine serum albumin/phosphate-buffered saline. The slides were then incubated with the following antibodies: anti-Ki-67 (#RB-1510-RQ, Thermo Fisher Scientific), anti-XIAP (#ab21278; Abcam, Cambridge, UK), anti-cIAP2 (#ab137393, Abcam), anti-cleaved caspase 3 (#9664, Cell Signaling Technology), and anti-cleaved PARP (#5625, Cell Signaling Technology). Images were scanned using an Aperio ScanScope (Aperio, Vista, CA, USA) and captured at 20× magnification using ImageScope software (Leica Biosystems, Wetzlar, Germany). The intensity of IHC staining of target proteins on the tumor tissues was measured using ImageJ (National Institutes of Health).
Statistical analysis
All data are presented as mean ± standard deviation. Differences between 2 groups were analyzed using a 2-tailed Student t-test, and differences between more than 2 groups were analyzed using a 1-way analysis of variance. P values of < 0.05 were considered statistically significant.
The combination index (CI) and fraction affected (Fa) were determined using the CalcuSyn software (V2.1, Biosoft, Cambridge, UK) to evaluate the synergistic effect of birinapant and anti-cancer drugs. CI < 0.90 indicates synergistic, 0.91 ≤ CI < 1.10 indicates additive, and CI ≥ 1.11 indicates antagonistic effects of the 2-drug combination.
Results
Synergistic efficacy of birinapant and gemcitabine in TNBC cells in vitro
We first examined the in vitro anti-tumor efficacy of birinapant as a single agent using the CellTiter-Blue viability assay in a panel of 11 TNBC cancer cells. We found that birinapant was effective at inhibiting proliferation of HCC38 (IC50 = 0.63 μM), HCC70 (IC50 = 0.47 μM), MDA-MB-231 (IC50 = 0.71 μM), and HS578T cells (IC50 = 0.21 μM) but not other tested TNBC cells and normal breast epithelial cell lines MCF10A and KTB6 (IC50 > 20 μM; Supplementary Table S1). We also examined the targeting specificity of birinapant by comparing the anti-proliferation effect of birinapant with that of the control compound ent-birinapant in HCC70, SUM149, MDA-MB-157, MDA-MB-231, SUM159, and MDA-MB-468 cells, which were the major cells used in this study. While birinapant inhibited proliferation of HCC70 and MDA-MB-231 cells, ent-birinapant had no anti-proliferation effects on the tested TNBC cells (Supplementary Fig. S1A and Table S1). These studies indicate that birinapant inhibits proliferation of TNBC cells by specifically targeting IAPs.
We next examined whether birinapant had a synergistic effect with the commonly used anti-cancer drugs, including entinostat (class I HDAC inhibitor, Supplementary Tables S2 and S3), cisplatin (Supplementary Tables S4 and S5), and gemcitabine (Supplementary Tables S6 and S7) in TNBC cells, as well as paclitaxel, voxtalisib (PI3K inhibitor), dasatinib (Src inhibitor), and erlotinib (EGF receptor inhibitor) in SUM149 TNBC cells (Supplementary Tables S9 and S10). We treated cells with increasing doses of the drugs (0.005-20 μM) in combination with birinapant at IC20 or at 5 μM when IC20 was greater than 10 μM. Among the tested drugs, gemcitabine had the most synergy with birinapant in the tested TNBC cells. Among the tested cell lines (Supplementary Tables S6 and S7), MDA-MB-231, MDA-MB-157, SUM149, and HCC70 cells showed the most substantial reduction in cell viability following combination treatment with birinapant and gemcitabine compared to treatment with gemcitabine alone (Fig. 1A). In the presence of birinapant at IC20, the sensitivity to gemcitabine (0.05 μM) increased by 4.02-fold in MDA-MB-231 cells (Fa = 0.911, CI = 6.9 × 10−6), 1.47-fold in MDA-MB-157 cells (Fa = 0.904, CI = 9.87 × 10−6), 17.95-fold in SUM149 cells (Fa = 0.615, CI = 0.021), and 3.35-fold in HCC70 cells (Fa = 0.671, CI = 0.155). In contrast, the addition of birinapant did not enhance the sensitivity of normal breast epithelial cell lines MCF10A and KTB6 to gemcitabine (Fig. 1A and Supplementary Tables S6 and S8), suggesting the cancer-targeting specificity of the combination treatment. In addition, we examined the targeting specificity of birinapant by comparing the anti-proliferation effect of birinapant with that of the control compound ent-birinapant with or without gemcitabine. Birinapant alone suppressed proliferation of HCC70 (IC50 = 0.47) and MDA-MB-231 (IC50 = 0.71) cells, whereas ent-birinapant alone had no any anti-proliferation effect on these cells (Supplementary Fig. 1A and Table S1). While birinapant combined with gemcitabine enhanced the anti-proliferation effects of gemcitabine against MDA-MB-231, MDA-MB-157, SUM149, and HCC70 cells (Fig. 1A and Supplementary Tables S6 and S7), ent-birinapant did not show any such enhancement (Supplementary Fig. S1B), suggesting the targeting specificity of birinapant.
Figure 1. Birinapant increases sensitivity of TNBC cells to gemcitabine in vitro.

A, Cell viability was determined using CellTiter-Blue assay at 72 h following treatment with gemcitabine alone (0.0001-1 μM for SUM149 cells and 0.005-20 μM for other tested cells) or gemcitabine (at the same concentrations) plus birinapant at IC20 or 5 μM when IC20 was greater than 10 μM. B, Anchorage-independent growth was determined using soft agar colony formation assay at 3 weeks following treatment with birinapant alone, gemcitabine alone, or birinapant plus gemcitabine. *P < 0.05, **P < 0.01, ***P < 0.001 by 2-tailed Student t-test. G, gemcitabine; B, birinapant.
Birinapant also enhanced the sensitivity of TNBC cells to gemcitabine in a soft-agar assay, which reflects the in vivo tumorigenicity of cancer cells. The sensitivity to gemcitabine was increased 1.96-fold at 0.0001 μM and 71.33-fold at 0.001 μM in MDA-MB-231, 2.68-fold at 0.0001 μM and 4.21-fold at 0.001 μM in MDA-MB-157, 1.58-fold at 0.0001 μM and 2.97-fold at 0.001 μM in SUM149, and 1.77-fold at 0.0001 μM and 3.81-fold at 0.001 μM in HCC70 cells (Fig. 1B). No synergistic effect was found in MDA-MB-468 and SUM159 cells in either the CellTiter-Blue assay (Fig. 1A) or the soft-agar assay (Fig. 1B). Thus, based on the degree of sensitivity, we categorized MDA-MB-231, MDA-MB-157, SUM149, and HCC70 cells as sensitive cells and MDA-MB-468 and SUM159 as insensitive cells to study the underlying molecular mechanism of the synergy.
Birinapant enhances sensitivity of TNBC cells to gemcitabine through induction of apoptosis
Birinapant is designed to target IAP proteins to trigger an apoptotic response (26), while gemcitabine inhibits cell growth by suppressing DNA synthesis (35). Therefore, we examined the effects of combination treatment with birinapant and gemcitabine at 72 h on cell cycle progression and apoptosis induction by flow cytometry. Cell cycle analysis showed that compared with gemcitabine alone, the combination treatment increased the sub-G1 fraction (Fig. 2A) and reduced the S-phase fraction (Fig. 2B) in MDA-MB-231 (29.18%-38.37% increase in sub-G1 and 5.44%-31.63% reduction in S phase), MDA-MB-157 (5.14%-21.06% increase in sub-G1 and 11.41%-23.36% reduction in S phase), SUM149 (30.33%-40.66% increase in sub-G1 and 8.58%-14.76% reduction in S phase), and HCC70 (44.46%-53.51% increase in sub-G1 and 17.07%-51.45% reduction in S phase), which indicate induction of apoptosis and suppression of DNA synthesis, respectively. Apoptosis induction was further confirmed by annexin V–PE and 7-AAD staining. Compared with gemcitabine alone, the combination of birinapant and gemcitabine increased apoptotic populations by 18.86% at 0.001 μM gemcitabine and 28.91% at 0.01 μM gemcitabine in MDA-MB-231, 6.99% at 0.01 μM and 14.15% at 0.05 μM in MDA-MB-157, 16.18% at 0.001 μM and 45.29% at 0.01 μM in SUM149, and 18.96% at 0.01 μM and 30.08% at 0.05 μM in HCC70 (Fig. 2C). Enhanced apoptosis (Supplementary Figs. S2A and S2C) and suppression of DNA synthesis (Supplementary Fig. S2B) were also observed at 48 h following combination treatment with birinapant and gemcitabine compared with gemcitabine alone. These results demonstrate that birinapant enhances the sensitivity of TNBC cells to gemcitabine by inducing apoptosis.
Figure 2. Birinapant increases sensitivity of TNBC cells to gemcitabine by inducing apoptosis.
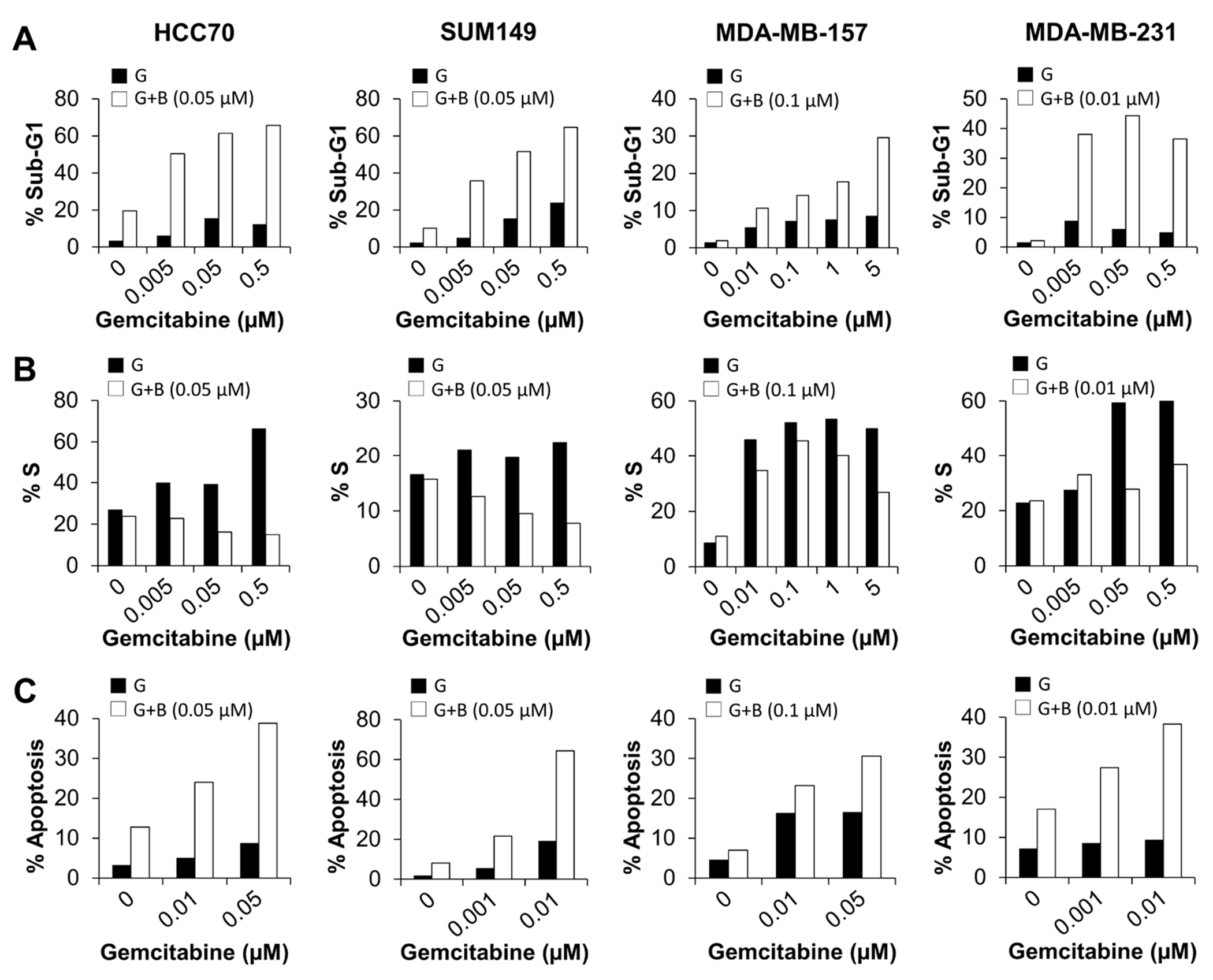
A-C, Cells were treated with birinapant alone, gemcitabine alone, or birinapant plus gemcitabine for 72 h and then subjected to cell cycle analysis (A) for sub-G1 fraction and (B) for S-phase fraction and (C) stained with annexin V and 7-AAD to assess apoptosis by flow cytometry analysis. G, gemcitabine; B, birinapant. The experiments were repeated twice, with no replicates for each treatment.
While birinapant alone was pro-apoptotic, birinapant plus gemcitabine was more pro-apoptotic than birinapant alone. Compared with birinapant alone, the combination treatment increased sub-G1 fraction by 30.87%-46.14% in HCC70, 25.7%-54.54% in SUM149, 8.73%-27.67% in MDA-MB-157, and 34.29%-42.18% in MDA-MB-231 cells (Fig. 2A). In addition, compared with birinapant alone, the combination treatment increased apoptotic populations by 11.25%-26.04% in HCC70, 13.56%-56.29% in SUM149, 16.19%-23.61% in MDA-MB-157, and 10.38%-21.31% in MDA-MB-231 cells (Fig. 2C). Similar results were observed at 48 h following the combination treatment (Supplementary Fig. S2). These results clearly show that birinapant sensitizes TNBC cells to gemcitabine by enhancing apoptotic cell death.
Birinapant enhances sensitivity of TNBC cells to gemcitabine through activation of the intrinsic apoptotic pathway
To identify the apoptosis pathway activated by the combination treatment with birinapant and gemcitabine, we analyzed cleavage of PARP and caspases by Western blotting. We found that combination treatment increased cleavage of PARP and caspases 3, 7, and 9 in a dose-dependent manner in HCC70, SUM149, MDA-MB-157, and MDA-MB-231 cells but had no effect on cleavage of caspase 8 (Fig. 3A). No significant induction of PARP and caspase cleavage was found in the insensitive cell lines MDA-MB-468 or SUM159 except for a slight induction of caspase 7 cleavage in MDA-MB-468 cells (Fig. 3A). Furthermore, the addition of Z-VAD-FMK (a pan-caspase inhibitor) partially inhibited the birinapant-mediated inhibition of cell proliferation by 44.3% (P < 0.0001) in SUM149 and 43% (P < 0.0001) in MDA-MB-231 cells treated with both birinapant and gemcitabine (Fig. 3B). These results indicate that the enhanced apoptosis resulting from the combination of birinapant and gemcitabine is a consequence of cleavage of caspase 9 and activation of the intrinsic pathway.
Figure 3. Birinapant increases sensitivity of TNBC cells to gemcitabine by activating the intrinsic apoptotic pathway.

A, Cells were treated with birinapant alone, gemcitabine alone, or birinapant plus gemcitabine for 72 h and then subjected to Western blot analysis for cleavage of PARP and caspases. B, Cells were pre-treated with Z-VAD-FMK and then with birinapant plus gemcitabine. On day 5 after treatment, cell viability was determined using CellTiter-Blue assay. *P < 0.001 by unpaired Student t-test. C, Cells were treated with birinapant alone, gemcitabine alone, or birinapant plus gemcitabine for 72 h and then subjected to Western blot analysis for IAP degradation.
Birinapant is designed to induce apoptosis by binding to and inducing degradation of IAP proteins (26). Thus, we examined IAP degradation following the combination treatment with birinapant and gemcitabine in HCC70, SUM149, MDA-MB-157, MDA-MB-231, SUM159, and MDA-MB-468 cells. The expression levels of cIAP2 and XIAP were significantly reduced in a dose-dependent manner in HCC70, SUM149, MDA-MB-157, and MDA-MB-231 cells (in which the combination treatment had shown synergistic effects) after 48 h of combination treatment (Fig. 3C). The expression levels of cIAP2 and XIAP were also slightly reduced in MDA-MB-468 cells but unaffected in SUM159 cells (Fig. 3C); these were the two cell lines, in which no synergistic anti-proliferation effect of the combination treatment had been observed. The reduced expression of XIAP and cIAP2 in MDA-MB-468 cells might have led to the observed cleavage of caspase 7 in those cells (Fig. 3A). However, the effect was not strong enough to trigger apoptotic cell death in MDA-MB-468 cells. No similar changes were seen for survivin (Fig. 3C). A dramatic reduction in cIAP1 was induced by birinapant alone in all tested cell lines (Fig. 3C), suggesting that birinapant preferentially targets cIAP1 over other IAP family members. This reduction in cIAP1 expression in both the sensitive cells and in the SUM159 and MDA-MB-468 cells, in which no synergy had been observed, suggests that reduced cIAP1 expression was not the cause of the synergistic effects of the combination treatment. Among the tested cells, MDA-MB-231 cells showed a dramatic reduction in expression levels of XIAP, cIAP1, and cIAP2 proteins following the combination treatment, which may be the reason we observed a greater induction of apoptosis by the combinational treatment in MDA-MB-231 cells compared with the other tested cells (Figs. 2A and 2C). Altogether, these results suggest that birinapant increases the sensitivity of TNBC cells to gemcitabine by inducing cleavage of cIAP2 and XIAP, leading to apoptotic cell death.
Birinapant enhances anti-tumor effectiveness of gemcitabine in TNBC xenograft mouse models by inducing apoptosis
Our in vitro results showed that birinapant and gemcitabine synergistically inhibited the growth of TNBC cells. We therefore examined the synergy of these 2 drugs using SUM149 and MDA-MB-231 xenograft mouse models. On the basis of the mouse studies by others (26,36–38), we administered birinapant at 15 mg/kg to test the combination treatment in vivo. Compared with the vehicle control, birinapant alone (P < 0.001, SUM149; P < 0.001, MDA-MB-231) and gemcitabine alone (P < 0.001, SUM149; P < 0.001, MDA-MB-231) significantly suppressed SUM149 (Fig. 4A) and MDA-MB-231 (Fig. 4B) tumor growth when applied at 15 mg/kg. More importantly, the combination of birinapant and gemcitabine resulted in significantly greater suppression of overall growth of SUM149 xenografts (Fig. 4A, P < 0.0001 vs. birinapant alone or gemcitabine alone) and MDA-MB-231 xenografts (Fig. 4B, P < 0.0001 vs. birinapant alone, P < 0.01 vs. gemcitabine alone). Gemcitabine alone more effectively inhibited tumor growth in the MDA-MB-231 xenograft model (Fig. 4B) than in the SUM149 model (Fig. 4A). However, even though gemcitabine alone markedly inhibited tumor growth in the MDA-MB-231 xenograft model, the addition of birinapant led to a significantly greater reduction in tumor volumes compared with gemcitabine alone at all time points assessed (Fig. 4B, P < 0.01). In both xenograft mouse models, no signs of toxicity and no reduction in mouse body weights were observed (Supplementary Fig. S3), indicating that both birinapant and gemcitabine at the tested doses are well tolerated. These results strongly suggest that birinapant potentiates gemcitabine in both mouse models.
Figure 4. Birinapant increases sensitivity of SUM149 and MDA-MB-231 xenografts to gemcitabine in vivo by inducing apoptosis.

A and B, Birinapant synergizes with gemcitabine in (A) SUM149 and (B) MDA-MB-231 xenograft mouse models. SUM149 or MDA-MB-231 cells (4 × 106) were injected into 1 of the mammary fat pads of female nude mice. When the tumors were about 75-150 mm3, the mice were treated with vehicle, birinapant alone, gemcitabine alone, or birinapant plus gemcitabine for 21 days in the SUM149 model or 38 days in the MDA-MB-231 model. **P < 0.01, ***P < 0.001, ****P < 0.0001 for gemcitabine alone vs. combination; #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 for birinapant alone vs. combination by a 2-tailed Student t-test. C and D, Immunohistochemical staining showing expression levels of Ki-67, XIAP, cIAP2, cleaved caspase 3, and cleaved PARP in tumors from mice implanted with (C) SUM149 cells or (D) MDA-MB-231 cells and treated with vehicle, birinapant alone, gemcitabine alone, or birinapant plus gemcitabine. Images were taken at 20× magnification. Scale bars, 200 μm. E and F, Measurement of the intensity of immunohistochemical staining for Ki-67, XIAP, cIAP2, cleaved caspase 3, and cleaved PARP in tumors from mice implanted with (E) SUM149 cells or (F) MDA-MB-231 cells and treated with vehicle, birinapant alone, gemcitabine alone, or birinapant plus gemcitabine. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by 2-tailed Student t-test.
Our in vitro results showed that birinapant enhanced the sensitivity of TNBC cells to gemcitabine by inducing cleavage of cIAP2 and XIAP, leading to apoptotic cell death. We therefore collected tumor samples on day 21 for SUM149 and on day 38 for MDA-MB-231 mouse models following treatment and analyzed the expression levels of Ki-67, XIAP, cIAP2, cleaved caspase 3, and cleaved PARP by IHC staining (Figs. 4C–4F). As expected, expression levels of Ki-67 in both models were significantly reduced in tumors from mice treated with birinapant alone (P < 0.05, SUM149 and MDA-MB-231) or gemcitabine alone (P < 0.01, SUM149; P < 0.05, MDA-MB-231) at 15 mg/kg compared with tumors from the vehicle-treated controls. The expression levels of Ki-67 were further reduced in tumors from mice treated with both birinapant and gemcitabine in both SUM149 xenografts (Figs. 4C and 4E, P < 0.01 vs. birinapant alone; P < 0.01 vs. gemcitabine alone) and MDA-MB-231 xenografts (Figs. 4D and 4F, P < 0.01 vs. birinapant alone; P < 0.0001 vs. gemcitabine alone). This result suggests that combination treatment more effectively suppresses tumor growth than monotreatment.
To confirm that birinapant enhanced sensitivity of TNBC cells to gemcitabine by inducing degradation of XIAP and cIAP2, we examined the effect of treatments on expression levels of XIAP and cIAP in tumors. As expected, the expression levels of XIAP and cIAP2 were significantly reduced in both SUM149 tumors from combination-treated mice (Figs. 4C and 4E, XIAP, P < 0.01 vs. birinapant alone, P < 0.001 vs. gemcitabine alone; cIAP2, P < 0.001 vs. birinapant alone, P < 0.0001 vs. gemcitabine alone) and MDA-MB-231 tumors from combination-treated mice (Figs. 4D and 4F, XIAP, P < 0.05 vs. birinapant alone, P < 0.05 vs. gemcitabine alone; cIAP2, P < 0.01 vs. birinapant alone, P < 0.001 vs. gemcitabine alone) compared with tumors from mice treated with birinapant alone or gemcitabine alone. This result demonstrates that combination treatment leads to degradation of both XIAP and cIAP2 to a significantly greater extent than monotreatment does.
To further confirm that the enhanced anti-tumor effect of combination treatment was a result of apoptosis induction, we examined the effect of the treatments on the expression levels of cleaved caspase 3 and cleaved PARP. As expected, the expression levels of cleaved caspase 3 and cleaved PARP were significantly increased in both SUM149 tumors from combination-treated mice (Figs. 4C and 4E, cleaved caspase 3, P < 0.001 vs. birinapant alone, P < 0.001 vs. gemcitabine alone; cleaved PARP, P < 0.05 vs. birinapant alone, P < 0.05 vs. gemcitabine alone) and MDA-MB-231 tumors from combination-treated mice (Figs. 4D and 4F, cleaved caspase 3, P < 0.0001 vs. birinapant alone, P < 0.01 vs. gemcitabine alone; cleaved PARP, P < 0.01 vs. birinapant alone, P < 0.05 vs. gemcitabine alone) compared with tumors from mice treated with birinapant alone or gemcitabine alone.
These results suggest that birinapant synergizes with gemcitabine by inducing apoptosis in tumors and that the combination of birinapant and gemcitabine is a potential therapeutic option for TNBC.
Discussion
Apoptosis resistance is a hallmark of cancer cells, and targeting regulators of apoptosis such as the IAP family of proteins to overcome apoptosis resistance has become an attractive strategy for cancer treatment. Here, we show that birinapant, a biindole-based bivalent SMAC mimetic, had differential effects at inhibiting the growth of TNBC cells in vitro when applied alone but significantly enhanced anti-tumor effectiveness of gemcitabine in TNBC cells both in vitro and in vivo. In contrast, birinapant did not enhance the anti-proliferation effect of gemcitabine against normal breast epithelial cells MCF10A and KTB6 (Fig. 1A and Supplementary Tables S6 and S8), suggesting the cancer-targeting specificity of the combination treatment. Furthermore, we found that birinapant sensitized TNBC cells to gemcitabine by inducing degradation of cIAP2 and XIAP, leading to activation of the intrinsic apoptosis pathway and eventually apoptotic cell death and tumor growth inhibition. Our findings demonstrate the therapeutic potential of birinapant for overcoming gemcitabine resistance in TNBC.
One of the mechanisms of cancer cells to evade apoptosis is the dysregulation of IAP proteins (4,5,14,19), which has been linked with resistance to chemotherapy. IAPs’ activity is regulated by endogenous IAP antagonists such as SMAC (25). Therefore, small-molecule inhibitors that mimic SMAC have been developed to overcome IAP-associated resistance in cancer cells (6,14,17,26). Similar to the endogenous SMAC, SMAC mimetics compete with caspases for binding to IAPs, which leads to the release of caspases from IAPs and subsequent caspase activation (6,14,17,26,39). In addition, SMAC mimetics induce the proteasomal degradation of cIAP proteins (17,19,20), which promotes the release of receptor-interacting protein 1 (RIP1) from the TNFR1 complex, leading to the formation of the RIP1-dependent caspase 8 activation complex (20,21) and eventually apoptotic cell death. Indeed, various studies have reported the ability of birinapant to bind IAPs, leading to their degradation, which results in cell death in a variety of cancer cell lines (26). Consistent with these findings, our results showed that birinapant induced complete degradation of cIAP1 in all tested cells and partial degradation of cIAP2 and XIAP in sensitive cells HCC70, SUM149, MDA-MB-231, and MDA-MB-157. No changes in survivin expression were seen following birinapant treatment, suggesting that the binding preferences and selective effects of birinapant on IAP proteins differ between cell lines and treatment conditions. In addition, birinapant-induced degradation of XIAP and cIAP1 at the tested concentrations had no significant effects on the proliferation of the tested TNBC cells. Furthermore, treatment with birinapant alone did not induce activation of caspase pathways in tested cells, which might account for birinapant’s lack of effect on cell proliferation. This ineffectiveness might be due to the compensatory effects of cIAP2 and survivin on apoptosis despite the degradation of cIAP1 and XIAP (5,40).
Our study showed that birinapant did not synergize with other commonly used anti-cancer drugs, including a class I HDAC inhibitor, cisplatin, a PI3K inhibitor, a Src inhibitor, and an mTOR inhibitor, in the tested TNBC cells. In contrast, we observed a strong synergistic effect when birinapant was combined with gemcitabine. Birinapant has been reported to potentiate the activity of chemotherapeutic drugs in both a TNF-dependent and a TNF-independent manner in a variety of cancer cell lines (26). In accordance with these findings, our results showed that birinapant sensitized TNBC cells to gemcitabine. Furthermore, birinapant alone induced complete degradation of cIAP1 in all tested cell lines and partial degradation of cIAP2 and XIAP in sensitive cell lines; when birinapant was applied together with gemcitabine, it induced degradation of both cIAP2 and XIAP in a dose-dependent manner in sensitive cell lines. These findings demonstrate that birinapant preferentially targets cIAP1 when applied alone and both cIAP2 and XIAP when combined with gemcitabine.
Furthermore, Western blot analysis showed that birinapant in combination with gemcitabine induced cleavage of caspases 3, 7, and 9 but not caspase 8. These results indicate that birinapant enhances the sensitivity of TNBC cells to gemcitabine through degradation of cIAP2 and XIAP and subsequent activation of the intrinsic pathway by inducing caspase 9 cleavage. This notion was confirmed by our in vivo study showing that the combination treatment significantly increased expression of cleaved caspase 3 in tumors. Studies by others suggest that antagonizing both cIAP1 and cIAP2 is required for inducing TNF-dependent cell death by SMAC mimetics and that birinapant promotes caspase 8/RIPK1 complex formation in response to TNF stimulation, activating downstream caspases (26). However, in this study, TNF neutralization with an anti-TNF antibody had no effects on birinapant-induced sensitization of gemcitabine, indicating the TNF independence of TNBC and multiple pro-apoptotic mechanisms of birinapant.
Currently, gemcitabine is used to treat metastatic TNBC in combination with other chemotherapeutic agents, such as platinum agents and paclitaxel. In a randomized open-label phase III trial, gemcitabine combined with cisplatin showed a median progression-free survival (PFS) of 7.7 months (95% confidence interval [CI] 6.2-9.3 months) with a median follow-up of 16.3 months, while gemcitabine combined with paclitaxel showed a median PFS of 6.5 months (95% CI 5.8-7.2 months) with a median follow-up of 15.9 months. This study suggests that gemcitabine plus cisplatin could be an alternative or even the preferred first-line chemotherapy for patients with metastatic TNBC (41). Gemcitabine has also been combined with targeted therapies, such as panitumumab and the PARP inhibitor iniparib. In a phase II trial of patients with metastatic TNBC, combination treatment with gemcitabine, panitumumab, and carboplatin showed an overall response rate of 42% and a median PFS of 4.4 months (95% CI 3.2-5.5 months) with a median follow-up of 11 months (42). In a single-arm phase II clinical trial of patients with triple-negative and BRCA1/2 mutation-associated breast cancer, combination treatment with gemcitabine, carboplatin, and iniparib yielded a promising pathologic complete response of 36% (43). Although iniparib is no longer considered a true PARP inhibitor, these results are compelling. These clinical trials demonstrate gemcitabine as an important agent for improved management of TNBC, both for anti-tumor activity and clinical benefit.
Given the importance of gemcitabine for treatment of TNBC, further studies considering the combination of gemcitabine with other anti-cancer drugs, especially targeted therapies, deserve high priority. These studies may well benefit from thorough knowledge of the metabolism, action mechanisms, and resistance profile of such combinations and the mode of interaction of gemcitabine with these other drugs. Our results here demonstrate the therapeutic potential of birinapant for improving the anti-tumor efficacy of gemcitabine in TNBC by targeting the IAP family of proteins. Our findings also provide strong evidence for continuing the preclinical and clinical development of a combinational treatment of birinapant with gemcitabine for patients with TNBC.
The observed roles of IAPs in cancer progression through regulation of apoptosis, proliferation, cell survival, and migration highlights IAPs as an essential target for cancer treatment. As described here, birinapant potentiated the anti-tumor efficacy of gemcitabine in preclinical TNBC models. Our study demonstrates that birinapant can effectively activate apoptotic signaling by targeting IAP proteins and has potential applications for TNBC treatment. Our findings, along with the findings of others, strongly suggest that the combination of anti-IAP therapy with other chemotherapies has tremendous promise for the future care of cancer patients.
Supplementary Material
Acknowledgments
The authors acknowledge Seayoung Lee at Duke University for discussion, Sarah Bronson of the Research Medical Library at MD Anderson Cancer Center for editorial assistance, and Wendy Schober and Nalini Patel of the Flow Cytometry and Cellular Imaging Facility at MD Anderson Cancer Center for assistance with cell cycle distribution and apoptosis analyses.
Financial support:
This work was supported by The University of Texas MD Anderson Cancer Center Morgan Welch Inflammatory Breast Cancer Research Program and the State of Texas Rare and Aggressive Breast Cancer Research Program (to Naoto T. Ueno); by MD Anderson’s Cancer Center Support Grant (P30CA016672; used the institutionally funded Flow Cytometry and Cellular Imaging Facility); by Cancer Center Support Grant (2P30CA016672-43 to The University of Texas MD Anderson Cancer Center); and by U.S. Department of Defense Grant (W81XWH-13-1-0047I to Gayathri R. Devi).
Footnotes
Disclosure of potential conflicts of interest
The authors declare no potential conflicts of interest.
References
- 1.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 2011;121(7):2750–67 doi 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carey L, Winer E, Viale G, Cameron D, Gianni L. Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol 2010;7(12):683–92 doi 10.1038/nrclinonc.2010.154. [DOI] [PubMed] [Google Scholar]
- 3.Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006;25:4798 doi 10.1038/sj.onc.1209608. [DOI] [PubMed] [Google Scholar]
- 4.Wong RSY. Apoptosis in cancer: from pathogenesis to treatment. Journal of Experimental & Clinical Cancer Research 2011;30(1):87 doi 10.1186/1756-9966-30-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Owens TW, Gilmore AP, Streuli CH, Foster FM. Inhibitor of Apoptosis Proteins: Promising Targets for Cancer Therapy. J Carcinog Mutagen 2013;Suppl 14 doi 10.4172/2157-2518.S14-004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rathore R, McCallum JE, Varghese E, Florea AM, Busselberg D. Overcoming chemotherapy drug resistance by targeting inhibitors of apoptosis proteins (IAPs). Apoptosis 2017;22(7):898–919 doi 10.1007/s10495-017-1375-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hassan M, Watari H, AbuAlmaaty A, Ohba Y, Sakuragi N. Apoptosis and molecular targeting therapy in cancer. Biomed Res Int 2014;2014:150845 doi 10.1155/2014/150845. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Baig S, Seevasant I, Mohamad J, Mukheem A, Huri HZ, Kamarul T. Potential of apoptotic pathway-targeted cancer therapeutic research: Where do we stand? Cell Death Dis 2016;7:e2058 doi 10.1038/cddis.2015.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falkenhorst J, Grunewald S, Muhlenberg T, Marino-Enriquez A, Reis AC, Corless C, et al. Inhibitor of Apoptosis Proteins (IAPs) are commonly dysregulated in GIST and can be pharmacologically targeted to enhance the pro-apoptotic activity of imatinib. Oncotarget 2016;7(27):41390–403 doi 10.18632/oncotarget.9159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krajewska M, Krajewski S, Banares S, Huang X, Turner B, Bubendorf L, et al. Elevated expression of inhibitor of apoptosis proteins in prostate cancer. Clin Cancer Res 2003;9(13):4914–25. [PubMed] [Google Scholar]
- 11.Lopes RB, Gangeswaran R, McNeish IA, Wang Y, Lemoine NR. Expression of the IAP protein family is dysregulated in pancreatic cancer cells and is important for resistance to chemotherapy. Int J Cancer 2007;120(11):2344–52 doi 10.1002/ijc.22554. [DOI] [PubMed] [Google Scholar]
- 12.LaCasse EC, Cherton-Horvat GG, Hewitt KE, Jerome LJ, Morris SJ, Kandimalla ER, et al. Preclinical Characterization of AEG35156/GEM 640, a Second-Generation Antisense Oligonucleotide Targeting X-Linked Inhibitor of Apoptosis. 2006;12(17):5231–41 doi 10.1158/1078-0432.CCR-06-0608 %J Clinical Cancer Research. [DOI] [PubMed] [Google Scholar]
- 13.Shaw TJ, Lacasse EC, Durkin JP, Vanderhyden BC. Downregulation of XIAP expression in ovarian cancer cells induces cell death in vitro and in vivo. Int J Cancer 2008;122(6):1430–4 doi 10.1002/ijc.23278. [DOI] [PubMed] [Google Scholar]
- 14.Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention in cancer. Nature Reviews Drug Discovery 2012;11:109 doi 10.1038/nrd3627 https://www.nature.com/articles/nrd3627#supplementary-information. [DOI] [PubMed] [Google Scholar]
- 15.LaCasse EC, Mahoney DJ, Cheung HH, Plenchette S, Baird S, Korneluk RG. IAP-targeted therapies for cancer. Oncogene 2008;27(48):6252–75 doi 10.1038/onc.2008.302. [DOI] [PubMed] [Google Scholar]
- 16.Cheng JQ, Jiang X, Fraser M, Li M, Dan HC, Sun M, et al. Role of X-linked inhibitor of apoptosis protein in chemoresistance in ovarian cancer: possible involvement of the phosphoinositide-3 kinase/Akt pathway. Drug Resist Updat 2002;5(3-4):131–46. [DOI] [PubMed] [Google Scholar]
- 17.Eschenburg G, Eggert A, Schramm A, Lode HN, Hundsdoerfer P. Smac mimetic LBW242 sensitizes XIAP-overexpressing neuroblastoma cells for TNF-alpha-independent apoptosis. Cancer Res 2012;72(10):2645–56 doi 10.1158/0008-5472.CAN-11-4072. [DOI] [PubMed] [Google Scholar]
- 18.Evans MK, Brown MC, Geradts J, Bao X, Robinson TJ, Jolly MK, et al. XIAP Regulation by MNK Links MAPK and NFkappaB Signaling to Determine an Aggressive Breast Cancer Phenotype. Cancer Res 2018;78(7):1726–38 doi 10.1158/0008-5472.CAN-17-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hunter AM, LaCasse EC, Korneluk RG. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis 2007;12(9):1543–68 doi 10.1007/s10495-007-0087-3. [DOI] [PubMed] [Google Scholar]
- 20.Dai Y, Lawrence TS, Xu L. Overcoming cancer therapy resistance by targeting inhibitors of apoptosis proteins and nuclear factor-kappa B. Am J Transl Res 2009;1(1):1–15. [PMC free article] [PubMed] [Google Scholar]
- 21.Holt SV, Brookes KE, Dive C, Makin GW. Down-regulation of XIAP by AEG35156 in paediatric tumour cells induces apoptosis and sensitises cells to cytotoxic agents. Oncol Rep 2011;25(4):1177–81 doi 10.3892/or.2011.1167. [DOI] [PubMed] [Google Scholar]
- 22.Cao C, Mu Y, Hallahan DE, Lu B. XIAP and survivin as therapeutic targets for radiation sensitization in preclinical models of lung cancer. Oncogene 2004;23(42):7047–52 doi 10.1038/sj.onc.1207929. [DOI] [PubMed] [Google Scholar]
- 23.Hu Y, Cherton-Horvat G, Dragowska V, Baird S, Korneluk RG, Durkin JP, et al. Antisense oligonucleotides targeting XIAP induce apoptosis and enhance chemotherapeutic activity against human lung cancer cells in vitro and in vivo. Clin Cancer Res 2003;9(7):2826–36. [PubMed] [Google Scholar]
- 24.Amantana A, London CA, Iversen PL, Devi GR. X-linked inhibitor of apoptosis protein inhibition induces apoptosis and enhances chemotherapy sensitivity in human prostate cancer cells. Mol Cancer Ther 2004;3(6):699–707. [PubMed] [Google Scholar]
- 25.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000;102(1):33–42. [DOI] [PubMed] [Google Scholar]
- 26.Benetatos CA, Mitsuuchi Y, Burns JM, Neiman EM, Condon SM, Yu G, et al. Birinapant (TL32711), a bivalent SMAC mimetic, targets TRAF2-associated cIAPs, abrogates TNF-induced NF-kappaB activation, and is active in patient-derived xenograft models. Mol Cancer Ther 2014;13(4):867–79 doi 10.1158/1535-7163.MCT-13-0798. [DOI] [PubMed] [Google Scholar]
- 27.Allensworth JL, Sauer SJ, Lyerly HK, Morse MA, Devi GR. Smac mimetic Birinapant induces apoptosis and enhances TRAIL potency in inflammatory breast cancer cells in an IAP-dependent and TNF-alpha-independent mechanism. Breast Cancer Research and Treatment 2013;137(2):359–71 doi 10.1007/s10549-012-2352-6. [DOI] [PubMed] [Google Scholar]
- 28.La V, Fujikawa R, Janzen DM, Nunez M, Bainvoll L, Hwang L, et al. Birinapant sensitizes platinum-resistant carcinomas with high levels of cIAP to carboplatin therapy. NPJ Precis Oncol 2017;1 doi 10.1038/s41698-017-0008-z. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Eytan DF, Snow GE, Carlson SG, Schiltz S, Chen Z, Van Waes C. Combination effects of SMAC mimetic birinapant with TNFalpha, TRAIL, and docetaxel in preclinical models of HNSCC. Laryngoscope 2015;125(3):E118–24 doi 10.1002/lary.25056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Promises Fulda S. and Challenges of Smac Mimetics as Cancer Therapeutics. Clin Cancer Res 2015;21(22):5030–6 doi 10.1158/1078-0432.CCR-15-0365. [DOI] [PubMed] [Google Scholar]
- 31.de Sousa Cavalcante L, Monteiro G. Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur J Pharmacol 2014;741:8–16 doi 10.1016/j.ejphar.2014.07.041. [DOI] [PubMed] [Google Scholar]
- 32.van Moorsel CJ, Peters GJ, Pinedo HM. Gemcitabine: Future Prospects of Single-Agent and Combination Studies. Oncologist 1997;2(3):127–34. [PubMed] [Google Scholar]
- 33.Gloeckner H, Jonuleit T, Lemke HD. Monitoring of cell viability and cell growth in a hollow-fiber bioreactor by use of the dye Alamar Blue. Journal of immunological methods 2001;252(1-2):131–8. [DOI] [PubMed] [Google Scholar]
- 34.Bartholomeusz C, Gonzalez-Angulo AM, Kazansky A, Krishnamurthy S, Liu P, Yuan LX, et al. PEA-15 inhibits tumorigenesis in an MDA-MB-468 triple-negative breast cancer xenograft model through increased cytoplasmic localization of activated extracellular signal-regulated kinase. Clinical cancer research : an official journal of the American Association for Cancer Research 2010;16(6):1802–11 doi 10.1158/1078-0432.CCR-09-1456. [DOI] [PubMed] [Google Scholar]
- 35.Schafer A, Schomacher L, Barreto G, Doderlein G, Niehrs C. Gemcitabine functions epigenetically by inhibiting repair mediated DNA demethylation. PLoS One 2010;5(11):e14060 doi 10.1371/journal.pone.0014060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou L, Zhang Y, Leng Y, Dai Y, Kmieciak M, Kramer L, et al. The IAP antagonist birinapant potentiates bortezomib anti-myeloma activity in vitro and in vivo. J Hematol Oncol 2019;12(1):25 doi 10.1186/s13045-019-0713-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eytan DF, Snow GE, Carlson S, Derakhshan A, Saleh A, Schiltz S, et al. SMAC Mimetic Birinapant plus Radiation Eradicates Human Head and Neck Cancers with Genomic Amplifications of Cell Death Genes FADD and BIRC2. Cancer Res 2016;76(18):5442–54 doi 10.1158/0008-5472.CAN-15-3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Srivastava AK, Jaganathan S, Stephen L, Hollingshead MG, Layhee A, Damour E, et al. Effect of a Smac Mimetic (TL32711, Birinapant) on the Apoptotic Program and Apoptosis Biomarkers Examined with Validated Multiplex Immunoassays Fit for Clinical Use. Clin Cancer Res 2016;22(4):1000–10 doi 10.1158/1078-0432.CCR-14-3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cossu F, Mastrangelo E, Milani M, Sorrentino G, Lecis D, Delia D, et al. Designing Smac-mimetics as antagonists of XIAP, cIAP1, and cIAP2. Biochem Biophys Res Commun 2009;378(2):162–7 doi 10.1016/j.bbrc.2008.10.139. [DOI] [PubMed] [Google Scholar]
- 40.Vaux DL, Silke J. Mammalian mitochondrial IAP binding proteins. Biochem Biophys Res Commun 2003;304(3):499–504. [DOI] [PubMed] [Google Scholar]
- 41.Hu XC, Zhang J, Xu BH, Cai L, Ragaz J, Wang ZH, et al. Cisplatin plus gemcitabine versus paclitaxel plus gemcitabine as first-line therapy for metastatic triple-negative breast cancer (CBCSG006): a randomised, open-label, multicentre, phase 3 trial. Lancet Oncol 2015;16(4):436–46 doi 10.1016/S1470-2045(15)70064-1. [DOI] [PubMed] [Google Scholar]
- 42.Yardley DA, Ward PJ, Daniel BR, Eakle JF, Lamar RE, Lane CM, et al. Panitumumab, Gemcitabine, and Carboplatin as Treatment for Women With Metastatic Triple-Negative Breast Cancer: A Sarah Cannon Research Institute Phase II Trial. Clin Breast Cancer 2016;16(5):349–55 doi 10.1016/j.clbc.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 43.O’Shaughnessy J, Osborne C, Pippen JE, Yoffe M, Patt D, Rocha C, et al. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N Engl J Med 2011;364(3):205–14 doi 10.1056/NEJMoa1011418. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.