

Keywords: Lewis lung carcinoma, mitochondrial function, mitochondrial quality control, muscle atrophy, skeletal muscle contractility
Abstract
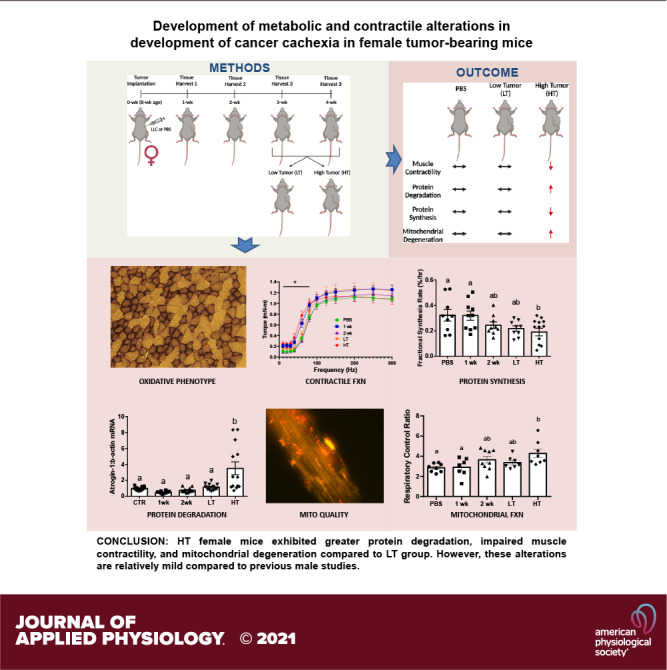
Cancer cachexia (CC) results in impaired muscle function and quality of life and is the primary cause of death for ∼20%–30% of patients with cancer. We demonstrated mitochondrial degeneration as a precursor to CC in male mice; however, whether such alterations occur in females is currently unknown. The purpose of this study was to elucidate muscle alterations in CC development in female tumor-bearing mice. Sixty female C57BL/6J mice were injected with PBS or Lewis lung carcinoma at 8 wk of age, and tumors developed for 1, 2, 3, or 4 wk to assess the time course of cachectic development. In vivo muscle contractile function, protein fractional synthetic rate (FSR), protein turnover, and mitochondrial health were assessed. Three- and four-week tumor-bearing mice displayed a dichotomy in tumor growth and were reassigned to high tumor (HT) and low tumor (LT) groups. HT mice exhibited lower soleus, tibialis anterior, and fat weights than PBS mice. HT mice showed lower peak isometric torque and slower one-half relaxation time than PBS mice. HT mice had lower FSR than PBS mice, whereas E3 ubiquitin ligases were greater in HT than in other groups. Bnip3 (mitophagy) and pMitoTimer red puncta (mitochondrial degeneration) were greater in HT mice, whereas Pgc1α1 and Tfam (mitochondrial biogenesis) were lower in HT mice than in PBS mice. We demonstrate alterations in female tumor-bearing mice where HT exhibited greater protein degradation, impaired muscle contractility, and mitochondrial degeneration compared with other groups. Our data provide novel evidence for a distinct cachectic development in tumor-bearing female mice compared with previous male studies.
NEW & NOTEWORTHY Our study demonstrates divergent tumor development and tissue wasting within 3- and 4-wk mice, where approximately half the mice developed large tumors and subsequent cachexia. Unlike previous male studies, where metabolic perturbations precede the onset of cachexia, females appear to exhibit protections from the metabolic perturbations and cachexia development. Our data provide novel evidence for divergent cachectic development in tumor-bearing female mice compared with previous male CC studies, suggesting different mechanisms of CC between sexes.
INTRODUCTION
Cancer is the second leading cause of death worldwide and ∼600,000 people die from cancer each year in the United States (1). Fifty to eighty percent of the patients with cancer experience the wasting syndrome known as cancer cachexia (CC), which is inversely associated with health-related quality of life (2, 3). CC is a multifactorial wasting syndrome characterized by significant skeletal muscle mass loss with or without loss of fat mass that may not be reversed by nutritional intervention alone and is responsible for 20%–30% of cancer-related deaths (2). Moreover, cachexia is often clinically defined by greater than 5% unintentional weight loss over the past 6 mo, which often is associated with progressive dysfunctions or degenerations in multiple tissues and organs (2). Unfortunately, there are limited treatments that mitigate the effects of CC. Therefore, appropriate early diagnosis and treatment of cancer-induced cachexia continues to be the most effective strategy to alleviate this condition. Furthermore, understanding the precachectic processes and the development of preventive approaches become crucial. However, although we have provided recent evidence for such precachectic processes in male tumor-bearing mice (4–6), to date there is limited understanding of precachectic processes in females.
Previous evidence has suggested that impaired skeletal muscle oxidative capacity may reduce muscle mass in the cachectic state (7), although the initial mechanisms remain elusive. Accordingly, we have focused on aspects of mitochondrial health including mitochondrial respiratory control ratio (RCR) and reactive oxygen species (ROS) emission during the development of CC in male mice, revealing that mitochondrial degeneration precedes the cachectic phenotypes in skeletal muscle of tumor-bearing male mice (4). Prior evidence denoted that elevated oxidative stress has been linked to altered protein turnover and skeletal muscle integrity (8). Oxidative stress to protein yields increases the hydrophobicity of both intracellular and cytoplasmic proteins, which are often toxic and can threaten cell viability, inducing protein aggregation, thereby undergoing selective proteolysis (9). Specifically, we found smaller cross-sectional area (CSA) of muscle fibers and lower protein fractional synthesis rates (FSRs), as well as altered cellular signaling associated with protein turnover in LLC (Lewis lung carcinoma)-induced cachectic male mice (5), in line with the previous works (7, 10).
There is a growing body of evidence suggesting distinct biological sex differences in different models of animal muscle atrophy (11), with phenotypical disparities between males and females having been found in different forms of human cancers including lung cancer (12). In fact, males appear more susceptible to inflammation-mediated muscle atrophies with greater ubiquitin-proteasome system (UPS) (4, 13), whereas females seem more susceptible to disuse-induced muscle atrophies with lower UPS compared with males (13, 14). Moreover, recent evidence from our laboratory suggests sex-based differences in the development of muscle disuse atrophy (11). Notably in cancer cachexia, male ApcMin/+ mice demonstrate a high IL-6 dependency for cachexia development, whereas female mice exhibit an IL-6 independence (7, 15, 16). Although the initial development of cancer cachexia in male mice is a relatively understudied topic, investigations of the same pathology in female mice are very few despite suggestions of differing atrophic mechanisms between biological sexes. Therefore, the purpose of this study was to assess the precachectic signature of skeletal muscle in the development of CC in female tumor-bearing mice. Specifically, herein, we have focused assessment on aspects of muscle contractile function, protein turnover, and mitochondrial health.
METHODS
Animal Interventions
All animal methods were approved by the Institutional Animal Care and Use Committee of the University of Arkansas. Sixty female C57BL/6J mice (n = 10–14/cohort) were purchased from Jackson Laboratories (000664, Bar Harbor, ME) and kept on a 12:12-h light-dark cycle, housed at 22°C, and given ad libitum access to normal rodent chow and water for the duration of the study. At 8 wk of age, mice were subcutaneously given an injection of either Lewis lung carcinoma (LLC) cells (1 × 106) suspended in 100 μL of sterile phosphate-buffered saline (PBS) or an equal volume of sterile PBS as a control to the left hind flank. Per our prior works (4, 6), tumors were allowed to develop for 1, 2, 3, or 4 wk. PBS control mice were age-matched with 4-wk tumor-bearing mice at the time of tissue collection. After 3 or 4 wk of tumor growth, we noted a clear dichotomy in tumor size, with some females having greater tumor development than others within 3- and 4-wk animals. Therefore, 3- and 4-wk mice were combined and regrouped by low tumor (LT; ≤1.2 g)- and high tumor (HT; ≥2 g)-bearing groups (Supplemental Fig. S1, A and B; all Supplemental material is available at https://doi.org/10.6084/m9.figshare.16623064). Tissue collection was performed at 12 wk of age for 4 wk mice as well as PBS mice and the rest of the mice were euthanized at the designated time points and tissue wet weight was measured. Plasma was separated from the blood by centrifugation to determine female sex hormones. Tissue and plasma samples were then snap-frozen in liquid nitrogen and stored at −80°C for further analysis. All tissue weights were normalized to their tibia length to account for different body sizes (5). Final groups for analysis included PBS, 1 wk, 2 wk, LT, and HT mice.
Lewis Lung Carcinoma Cell Culture
LLC cells (CRL-1642, ATCC, Manassas, VA) were plated in 250 mL of culture flasks in DMEM (11965092, Life Technologies, Carlsbad, CA) containing 10% fetal bovine serum (26140079, Life Technologies) supplemented with 1% penicillin/streptomycin (15140122, Life Technologies) and media was changed every 48 h. Upon ∼80% confluence, cells were trypsinized, suspended via centrifugation, and diluted in PBS before tumor implantation as previously described (4).
Plasmid DNA Amplification and Electroporation
DH5-α Escherichia coli containing pMitoTimer plasmid were amplified and plasmid DNA was isolated using PureLink HiPure Plasmid Filter Maxiprep Kit (K211017, Life Technologies). At 6 wk of age, pMitoTimer plasmid transfection into the right flexor digitorum brevis (FDB) muscle was performed as described previously (4, 17). Briefly, while mice were anesthetized (isoflurane), 10 μL of 0.36 mg/mL of hyaluronidase in saline was injected using an insulin syringe with a 30-gauge needle subcutaneously under footpad. The needle penetrates the skin at a point close to the heel of the foot and advances the needle subcutaneously toward the base of the toe for ¼, which is above the FDB muscle. After 60 min, mice were anesthetized again and a total of 20 μg of the plasmid DNA for MitoTimer was injected subcutaneously in the footpad with the same procedure described for the hyaluronidase injection. Anesthesia was disconnected after the plasmid DNA injection for 10 min and reconnected for electroporation. Two gold-plated acupuncture needles were connected to an electrical stimulator placed under the skin near the heel and a second one near the base of the toes. Electrodes were perpendicular to the long axis of the foot and parallel to each other. Ten electrical pulses with 20 ms in duration each at 1 Hz and 75 V were applied using an S88 electrical stimulator (Grass Telefactor, West Warwick, RI).
In Vivo Muscle Contractility Test
Approximately 48 h before tissue harvest, in vivo peak isometric torque, isometric torque frequencies, frequency at half maximum torque, 1/2 relaxation time, time to maximal contraction, and fatigability (susceptibility to fatigue) of the anterior crural (shin) muscles were performed as described (18). Briefly, while mice were anesthetized, the left hindlimb was shaved and an ethanol pad applied to clean the skin. The foot was placed in a footplate attached to a servomotor (Model 300B-LR, Aurora Scientific, Aurora, ON, Canada) using surgical tape while their body temperature was maintained at 37°C using a heating pad. Two platinum electrodes (Model E2–12, Grass Technologies) were then inserted percutaneously on either side of the peroneal nerve. Consistent instant stimulation was monitored to ensure the electrodes were neither too deep nor too proximal as to yield recruitment of the posterior crural muscles. Muscle contractions were induced via electrical stimulator and stimulus isolation unit (Model S48 and SIU5, respectively; Grass Technologies). After resting the hindlimb for 1 min, torque and M-wave as a function of stimulation frequency were measured during 12 isometric contractions with the duration of 150 ms at various stimulation frequencies (10, 20, 30, 40, 60, 80, 100, 125, 150, 200, 250, and 300 Hz) to determine hindlimb motor unit recruitment based on two different physiological conditions: 1) hindlimb motor unit recruitments during normal movement, which occurs between 60 and 100 Hz (19) and 2) during supramaximal stimulation frequencies (i.e., >200 Hz) that can reveal different contractile phenotypes such as low-frequency fatigue (20) followed by resting the hindlimb for 4 min. After measurement of submaximal and maximal isometric torques at 150 Hz, the anterior crural muscles underwent a fatigability test via applying 120 isometric contractions with duration of 150 ms per stimulation at 40 Hz for a total protocol time of 2 min. During the recovery following the fatigability test, the normal movement of experimental mice was closely monitored to determine any injuries during the in vivo muscle force production and fatigability test.
Deuterium Injection and 24-h Muscle Fractional Synthesis Rate
FSR was quantified using gas chromatography-mass spectrometry (GC-MS) (7890 A and 5977 A, Agilent, Santa Clara, CA) as described (5, 11). Briefly, mice were given an intraperitoneal (ip) injection of 99.9% deuterium (D2O; 151882-1 L, Millipore Sigma, Burlington, MA) at 20 μL/g body wt 24 h before tissue harvest. Drinking water was thereafter supplemented with 4% D2O to maintain the plasma pool of D2O until tissue harvest (21). Mice were not fasted at tissue collection. Fifteen milligrams of gastrocnemius and tibialis anterior (TA) muscles were powdered and homogenized in a 10% trichloroacetic acid (TCA) solution for FSR. Mixed protein fractions were then washed three times with 10% TCA solution by centrifugation to eliminate cytosolic amino acids. Proteins were then placed in a tube with 200 μL of 6 M HCl and heated at 100°C to hydrolyze proteins into amino acids for 24 h. An aliquot of the hydrolysate was dried down and derivatized with a 3:2:1 vol/vol solution of methyl-8, methanol, and acetonitrile to determine 2H labeling of alanine on its methyl-8 derivative. The solution was then placed in a GC-MS capillary column (7890 A GC HP-5 ms capillary column, Agilent) and positioned in the GC-MS; 1 μL of the solution was run on the GC-MS at a 20:1 split. GC-MS settings have previously been described (22). A ratio of deuterated alanine over alanine was used to assess protein synthesis. The precursor pool of 2H2O in the plasma was reacted with 10 M NaOH and a 5% solution of acetone in acetonitrile for 24 h to conjugate the free 2H2O to acetone. The solution was extracted by adding Na2SO4 and chloroform and placed in capillary columns to be analyzed on the GC-MS to detect acetone at a 20:1 split. FSR of mixed proteins was calculated using the equation EA × [EBW × 3.7 × t (h)] − 1 × 100, where EA represents the amount of protein-bound [2H] alanine (mole% excess), EBW is the quantity of 2H2O in body water (mole% excess), 3.7 represents the exchange of 2H between body water and alanine (3.7 of 4 carbon-bound hydrogens of alanine exchange with water), and t (h) represents the time the label was present in hours.
Fluorescence Microscopy for pMitoTimer
pMitoTimer was analyzed as previously described (4). Briefly, at the time of tissue harvesting, freshly collected FDB muscles were fixed with 4% paraformaldehyde (PFA) for 20 min followed by 5-min incubation with PBS at room temperature. Muscles were then spread out on a gelatin-coated glass slide using two forceps and mounted with a drop of 50% glycerol in PBS as mounting media. A coverslip was added on the top of the mounted sample and four drops of nail polish were applied at the corners to anchor the coverslip. pMitoTimer images were acquired at ×100 magnification using the fluorescein isothiocyanate (FITC; green; excitation/emission, 488/518 nm) and tetramethylrhodamine isothiocyanate (TRITC; red; excitation/emission, 543/572 nm) fluorescent channels on a Nikon Eclipse Ti-S inverted epi-fluorescent microscope (Nikon, Melville, NY) with LED-based light source. Standardized acquisition parameters were established and followed for all imaging to match across samples. All assessments were performed in congruence with Laker et al. (17). A specially generated MATLAB program, a generous gift from Dr. Zhen Yan (University of Virginia), was used to analyze pMitoTimer red-to-green ratio (a mitochondrial oxidative stress marker) and pure red puncta number (a marker for completely degenerated mitochondria) (17).
Cryosection and Succinate Dehydrogenase Histological Staining
Cryosections and all histological staining methods were performed as described (4). Plantaris muscle was embedded with optimal cutting temperature (OCT) and frozen in liquid nitrogen-cooled isopentane. Each section was cut at 10 μm using a Leica CM1860 cryostat (Leica Biosystems, Wetzlar, Germany). Sections were stained for succinate dehydrogenase (SDH). Sections were in incubation media (50 mM sodium succinate, 50 mM phosphate buffer, 0.12 M KH2PO4, and 0.88 M Na2PHO4, 0.5 mg/mL nitroblue tetrazolium) for 40 min in a 37°C water bath. Slides were washed for 3 min with dH2O and imaged. Images were collected with Nikon Sight DS-Vi1 camera mounted on an Olympus CKX41 inverted microscope. Approximately 25 individual fibers of SDH+ (purple or oxidative) and SDH− fibers (nonpurple or glycolytic) per each muscle were counted and both fibers were manually circled for cross-sectional area (CSA) using Nikon Basic Research Imaging Software (Melville, NY). All fiber measurements were performed by a blinded investigator. Plantaris muscle was initially chosen to compare current SDH data with our previous male CC study (4).
Preparation of Separated and Permeabilized Muscle Fibers
During tissue harvesting, ∼15 mg of fresh plantaris muscle was collected and placed in 100-mm rounded petri dish containing ice-cold buffer X (60 mM K-MES, 35 mM KCl, 7.23 mM K2EGTA, 2.77 mM CaK2EGTA, 20 mM imidazole, 0.5 mM DTT, 20 mM taurine, 5.7 mM ATP, 15 mM phosphocreatine, and 6.56 mM MgCl2 at pH 7.1). Connective tissues and blood clots were eliminated, and the fiber bundles were gently separated into a near-single fiber bundle to maximize their surface area under an upright stereoscopic microscope (Fisher Scientific, Waltham, MA) using a pair of extra-sharp forceps (Fisher Scientific). The finely separated fiber bundles were then permeabilized in a tube with saponin (50 μg/mL) in ice-cold buffer X on a rotator for 30 min at 4°C. The permeabilized fiber bundles were washed in ice-cold buffer Z (110 mM K-MES, 35 mM KCl, 1 mM EGTA, 5 mM K2HPO4, 3 mM MgCl2, 0.005 mM glutamate, 0.02 mM malate, and 0.5 mg/mL BSA at pH 7.1) for 5 min × 3 times as previously described (4, 23). The permeabilized fiber bundles were then used for both mitochondrial RCR and ROS emission (4).
Mitochondrial Respiratory Control Ratio
Mitochondrial oxygen consumption was measured by polarography in a respiration chamber maintained at 37°C (Oxygraph+, Hansatech Instruments, King’s Lynn, UK) as described (4) and this method was adapted from Min et al. (23). Briefly, permeabilized plantaris muscles were transferred to a respiratory chamber filled with 1 mL of respiration buffer Z containing creatine (20 mM) to saturate creatine kinase and allowed to equilibrate with the tissue for 5 to 10 min before adding any substrate. State 2 respiration was assessed by the addition of malate and pyruvate (5 mM) using 10-μL 701RN Syringes (Hamilton, Reno, NV) followed by maximal ADP-stimulated respiration (State 3) by adding ADP (0.25 mM) and then basal mitochondrial respiration (State 4) by the addition of oligomycin (10 μg/mL) to inhibit ATP synthesis. States 3 and 4 respiration rates were normalized to dry tissue weight. The respiratory control ratio (RCR) was calculated by dividing State 3 by State 4 respiration.
ROS Emission
ROS emission was determined using Amplex UltraRed Reagent (Molecular Probes, Eugene, OR) in permeabilized muscle fibers as previously described (4). This assay is based on the concept that amplex red reacts with H2O2 in a 1:1 stoichiometric ratio in the presence of horseradish peroxidase (HRP) to produce the red fluorescent oxidation product resorufin. Following the addition of the permeabilized plantaris muscle, the baseline H2O2 production was measured followed by induction of H2O2 production by the addition of supraphysiological concentrations of succinate. The rate of H2O2 was normalized to dry weight of the tissue and calculated based on the rate of change from the baseline to succinate and then concentrations were calculated using a standard curve. Plantaris muscle was used for both mitochondrial respiration and ROS emission analysis.
Immunoblotting
Frozen gastrocnemius muscle samples were powdered, and 20-mg tissue samples were suspended in 250-μL 2× protein sample buffer [50 mM Tris-HCl, 1% sodium dodecyl sulfate (SDS), 10% glycerol, dithiothreitol (20 mM), 2-mercaptoethanol (127 mM), and 0.01% bromophenol blue combined with protease inhibitors, and phosphatase inhibitors at pH 6.8] and homogenized. Protein concentration was determined using the RC/DC assay (5000122, Bio-Rad, Hercules, CA). Forty micrograms of total protein in protein sample buffer was loaded into 10% gel and resolved by SDS-PAGE at 100 V for 2 h, transferred to a PVDF membrane at 20 V for 1.5 h. Ponceau S stain was applied to membranes. The membranes were then incubated with 5% milk in Tris-buffered saline (TBS) at room temperature for 1 h, then the membranes were incubated with primary antibodies with 1:500–1,000 dilution rate in LI-COR blocking buffer with 0.2% Tween 20. The membranes were incubated overnight for primary antibodies specific to cytochrome c oxidase subunit 4 (COX-IV) (4844S, Cell Signaling), voltage-dependent anion channel (VDAC) (4866S, Cell Signaling), Deptor (ABS222, Millipore Sigma), Ubiquitin (3933, Cell Signaling), Beclin-1 (3737, Cell Signaling), 1A/1B-light chain 3 (LC3) (4108, Cell Signaling), p62-Sequestosome-1 (p62) (p0067, Sigma), p-p38 MAPK (9211, Cell Signaling; T180/182), p38 MAPK (9212, Cell Signaling), p-ERK 1/2 MAPK (4370, Cell Signaling; T202/Y204), ERK 1/2 MAPK (4695, Cell Signaling), PGC-1α (sc-13067, Santa Cruz), peroxisome proliferator-activated receptor α (PPARα; sc-9000, Santa Cruz), PPARδ (sc-7197, Santa Cruz), transcription factor of mitochondria A (TFAM) (7495, Cell Signaling), Mitofusin 1 (MFN1) (sc-50330, Santa Cruz), MFN2 (sc-50331, Santa Cruz), optic atrophy-1 (OPA1) (sc-367890, Santa Cruz), dynamin-related protein 1 (DRP1) (14647, Cell Signaling), mitochondrial fission 1 (FIS1) (NB100-56646, Novus), BCL2 interacting protein 3 (BNIP3) (3769, Cell Signaling), superoxide dismutase type 1 (SOD1) (GTX100554, Genetex), SOD2 (131945, Cell Signaling), SOD3 (AF4817, R & D Systems), and Catalase (140975, Cell Signaling). Following overnight incubation with primary antibodies, membranes washed with TBS with 0.1% Tween 20 (TBST) for 5 min × 3 times and conjugated with HRP secondary antibodies (LI-COR Biosciences, Lincoln, NE) with 1:10,000 dilution rate were used according to the manufacturer’s protocol for an hour at room temperature. Membranes were then washed with TBST for 5-min × 3 times followed by an image scan on Li-Cor Odyssey FC via immunoreactive (IR) detection. All bands were normalized to the 45-kDa actin band of Ponceau S stain as a loading control as previously described (4, 24, 25).
RNA Isolation, cDNA Synthesis, and Quantitative Real-Time PCR
Frozen gastrocnemius muscle samples were powdered and 20-mg tissue samples were suspended in 1 mL of TRIzol reagent and homogenized using Polytron for ∼5 s × 5 times before being transferred into a 1.5-mL microtube placed on ice. After 15 min at room temperature, 200 μL of chloroform was added and samples were centrifuged for 25 min. The clear portion of the solution from the top was transferred into a new tube. An equal amount of 70% of diethyl pyrocarbonate (DEPC)-treated ethanol was added, and the samples were loaded into an RNeasy column. RNA isolation was then processed using an RNA isolation kit (K145002; Invitrogen, Carlsbad, CA). Thirty microliters of DNase-treated RNA was collected and RNA concentration and purity were determined by fluorometry using 260/280 nm ratios read on a Bio-Tek Power Wave XC microplate reader (BioTek Instruments Inc., Winooski, VT) with Take3 microvolume plate using Gen5 software. Subsequently, 1 μg of RNA was reverse transcribed into cDNA using VILO Superscript reagent (11755050, Invitrogen) and cDNA was diluted to 1:100 (10 ng/μL). Quantitative real-time PCR was performed using TaqMan probe reagent and QuantSudio3 PCR instrument (Thermo Fisher Scientific, Waltham, MA), and cycle threshold (Ct) values were analyzed according to the manufacturer’s instructions. PCR was as follows: 4 min of incubations, 45 cycles of denaturation, annealing, and extension at 95°C, 60°C, and 72°C, respectively. Target TaqMan probes for β-actin (Mm00607939_g1), Cox4i1 (Mm01250094_m1), Igf1 (Mm00439560_m1), Redd1 (Mm00512504_g1), Deptor (Mm01195339_m1), Ubc (Mm02525934_g1), Gadd45a (Mm00432802_m1), Atrogin1 (Mm00499523_m1), Murf1 (Mm01185221_m1), IL-6 (Mm00446190_m1), Tnf (Mm00443258_m1), NF-κB (Mm00476361_m1), Lc3 (Mm00458724_m1), p62 (Mm01700766_M1), Cyclin-D1 (Mm00432359_m1), Pax7 (Mm01354484_m1), MyoD (Mm01203489_g1), Myogenin (Mm00446194_m1), Mki67 (Mm01278617_m1), Pgc1α1 (Mm01208835_m1), Pparγ (Mm01184322_m1), Nrf2 (Mm_00477784m1), Drp1 (Mm_00432881_m1), and Mtif2 (Mm00505356_m1) were purchased from Applied Biosystems (Life Technologies). Additional custom primer pairs specific for Vdac, Pgc1α4, Beclin-1, Bnip3, Pparα, Tfam, Opa1, Mfn1, Mfn2, Mff, Fis1, TuFM, Taco1, and Mtif3 were used in which primer information has been reported previously (4, 5, 26). Final quantification of mRNA level was calculated using the ΔΔCT method as described previously (24, 26). All targets were normalized to the β-actin Ct value, which did not differ between experimental groups. Genes are italicized and proteins are not.
Statistics
A one-way ANOVA with a factor of group was utilized as the global analysis for each dependent variable. Where significant F ratios were found, statistical differences among means were determined by Tukey’s post hoc test. The comparison-wise error rate, α, was set at 0.05 for all statistical tests. Only differences that reached significance by the global F test and significant differences by Tukey-adjusted pairwise comparison are presented. All data were analyzed using GraphPad Prism (La Jolla, CA) and figures were compiled using GraphPad Prism as well. Data were expressed as means ± standard error of the mean.
RESULTS
Lower Muscle and Fat Tissue Weights with HT Animals
Tissue weights from all animals are presented in Table 1. In soleus muscle, HT mice had ∼14% and ∼12% lower weights than PBS mice (P = 0.003) and LT mice (P = 0.024). In TA muscle, HT mice had ∼12% and ∼9% lower weights than PBS (P < 0.001) and LT mice (P = 0.017), respectively. In extensor digitorum longus (EDL) and plantaris muscles, no significant differences were observed. In addition, spleen weight, a surrogate marker for the inflammatory state, was ∼63% and ∼296% greater in LT (P = 0.010) and HT mice (P < 0.001) than in PBS mice. Similarly, liver weight was ∼16% and ∼32% greater in LT (P = 0.006) and HT mice (P < 0.001) than in PBS mice. Lung weight was ∼19% greater in HT mice than in PBS mice (P < 0.001). Gonadal fat mass was lower in HT compared with LT (∼34%; P = 0.030) and PBS (∼38%; P = 0.008). Tumor weight was ∼290% higher in HT mice than in LT mice (P < 0.001; Table 1). These data suggest that the HT group displayed mild cachexia compared with our prior male CC study (4).
Table 1.
Characteristics
| PBS | 1 wk | 2 wk | LT | HT | |
|---|---|---|---|---|---|
| n | 10 | 11 | 12 | 13 | 14 |
| Body weight, g/mm | 1.154 ± 0.014a,b | 1.075 ± 0.013c | 1.125 ± 0.011b,c | 1.197 ± 0.017a,d | 1.287 ± 0.026e |
| Tumor weight, mg/mm | N/A | 2.376 ± 0.29a | 14.86 ± 2.24a | 43.85 ± 6.37b | 171.2 ± 9.23c |
| Body weight–tumor, g/mm | 1.154 ± 0.014a | 1.073 ± 0.013b | 1.110 ± 0.011a,b | 1.153 ± 0.016a | 1.115 ± 0.025a,b |
| Gastrocnemius, mg/mm | 5.542 ± 0.068a | 5.211 ± 0.045b | 5.346 ± 0.06a,b | 5.602 ± 0.073a | 5.369 ± 0.062a,b |
| Soleus, mg/mm | 0.449 ± 0.01a | 0.403 ± 0.01a,b | 0.418 ± 0.016a,b | 0.43 ± 0.009a | 0.385 ± 0.014b |
| Plantaris, mg/mm | 0.789 ± 0.016 | 0.762 ± 0.019 | 0.753 ± 0.015 | 0.797 ± 0.02 | 0.736 ± 0.015 |
| EDL, mg/mm | 0.510 ± 0.017 | 0.488 ± 0.007 | 0.479 ± 0.013 | 0.488 ± 0.013 | 0.475 ± 0.016 |
| TA, mg/mm | 2.282 ± 0.057a | 2.174 ± 0.043a,b | 2.151 ± 0.039a,b | 2.213 ± 0.04a | 2.017 ± 0.042b |
| Fat, mg/mm | 19.23 ± 1.856a | 17.6 ± 1.516a,b | 16.77 ± 1.17a,b | 18.14 ± 1.526a | 12 ± 1.359b |
| Heart, mg/mm | 6.336 ± 0.18 | 6.275 ± 0.072 | 6.22 ± 0.163 | 6.346 ± 0.154 | 6.268 ± 0.165 |
| Liver, mg/mm | 47.11 ± 1.948a | 46.74 ± 1.994a | 48.85 ± 1.218a,b | 54.78 ± 1.863b,c | 62.3 ± 2.36d |
| Lungs, mg/mm | 7.695 ± 0.146a | 7.529 ± 0.211a | 7.607 ± 0.228a | 8.146 ± 0.238a | 9.131 ± 0.263b |
| Spleen, mg/mm | 4.967 ± 0.217a | 4.274 ± 0.186a | 5.471 ± 0.371a | 8.104 ± 0.531b | 19.46 ± 1.123c |
| Tibia length, mm | 16.57 ± 0.053a | 16.15 ± 0.043b | 16.30 ± 0.079b,c | 16.62 ± 0.057a,d | 16.64 ± 0.049a,d |
| Estrogen, pg/mL | 67.61 ± 14.85a | 22.43 ± 4.40b | 28.27 ± 4.78b | 32.09 ± 4.19b | 21.77 ± 4.36b |
| Progesterone, pg/mL | 9.58 ± 1.28a | 5.55 ± 1.55a,b | 3.66 ± 0.63b | 5.02 ± 0.75b | 3.59 ± 1.21b |
Tissue weights are recorded as wet weights. All tissue weights were normalized to their tibia length to account for different body sizes. All values are represented as means ± SE. Different letters represent statistical differences at Tukey-adjusted with α value set at P ≤ 0.05. n = 10–14/group was used. EDL, extensor digitorum longus; HT, high tumor; LT, low tumor; PBS, phosphate-buffered saline; TA, tibialis anterior.
Reduced Female Sex Hormones with LLC Injection
Female sex hormone levels in plasma were assessed to determine hormonal changes during CC. Both estrogen and progesterone levels in plasma were ∼67% and 42%, respectively, lower in 1 wk following LLC injection, which remained suppressed in all tumor-bearing groups (P = 0.05–0.038; Table 1).
Unchanged Oxidative Phenotype in Tumor-Bearing Female Mice
The mean percentage of SDH+ fibers in the plantaris muscle was not different among experimental groups (P = 0.350; Fig. 1, A and B). Moreover, fiber CSA distribution and mean fiber CSA of both SDH+ and SDH− tended to be greater in the HT group than in the other groups, although statistically nonsignificant (P = 0.089; Fig. 1, C–F). mRNA abundance of Vdac was ∼40%–54% lower in HT groups than in PBS (P < 0.001), 1 wk (P = 0.071), 2 wk (P = 0.003), and LT groups (P = 0.041; Fig. 1G), whereas protein contents of mitochondrial content (COX IV and VDAC) were not different across the groups (Supplemental Fig. S2A).
Figure 1.

Oxidative phenotype plantaris muscle used for SDH staining. A: SDH staining images are representative of the five experimental groups. B: representative of the development of cancer cachexia through % of SDH+ fibers. Distribution through relative frequency (%) of cross-sectional area for SDH+ fibers (C) and SDH− fibers (D). Average cross-sectional area for SDH+ (E) and SDH− fibers (F). G: mRNA abundance for mitochondrial specific markers Cox4i1 and Vdac by using the gastrocnemius muscle. Different letters represent statistical differences at Tukey adjusted with α value set at P ≤ 0.05. n = 4–9 animals/group was used (each n represents average value for each animal across all measured fibers). CSA, cross-sectional area; HT, high tumor; LT, low tumor; PBS, phosphate-buffered saline; SDH, succinate dehydrogenase.
Impaired Skeletal Muscle Contractility with HT Animals
In vivo isometric torque frequency data (unnormalized) demonstrated that HT mice had ∼77%–121% higher isometric torque during lower frequencies (10–60 Hz) than PBS mice (P = 0.002–0.040; Fig. 2A). Peak isometric torque (unnormalized) was not different between experimental groups (Fig. 2B). Stimulation frequency at 50% peak torque was ∼18% lower in HT group than in other groups (P = 0.001–0.013; Fig. 2C). One-half relaxation time was ∼71% slower in HT group than in PBS group (P = 0.054; Fig. 2D). Time to maximal contraction was ∼127% slower in HT group than in other experimental groups (P < 0.001; Fig. 2E). The fatigability test revealed no statistical difference between PBS and LT or HT animals, although 2 wk animals had ∼12%–42% greater fatigability compared with 1 wk, LT, and HT animals at various time points (10–30 s and 60–70 s; P = 0.005–0.046; Fig. 2F).
Figure 2.
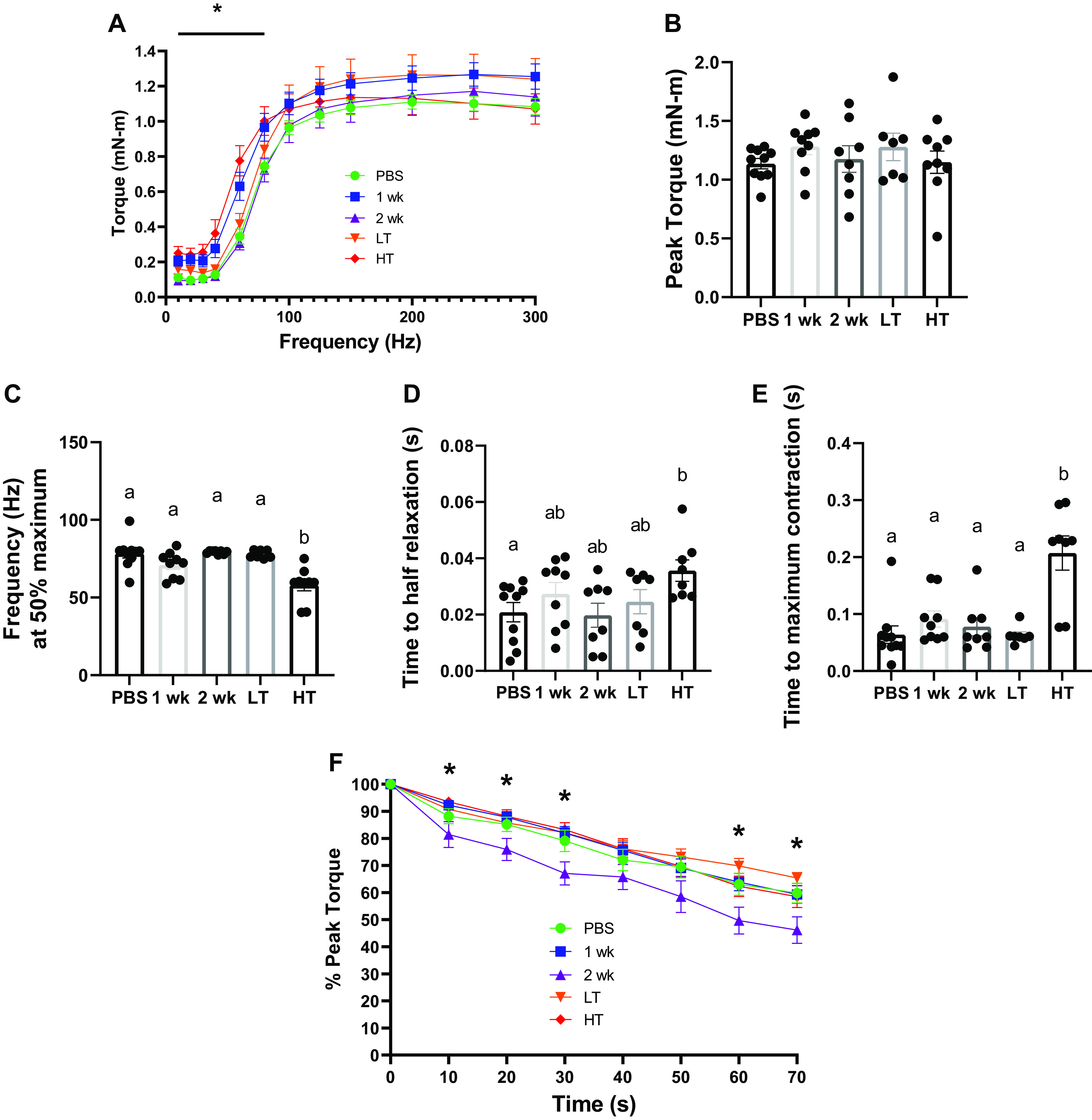
Skeletal muscle contractility. A: unnormalized isometric torque-frequency curve measured in mN·m for experimental animal groups. B: unnormalized average peak torque (mN·m) across experimental groups. C: frequency (Hz) delivered at 50% maximum torque across experimental groups. D: time (s) for muscle to achieve half relaxation across experimental groups. E: time to maximal contraction (s). F: % peak torque measured over time (s) across experimental groups during fatiguing protocol induced via continuous peroneal nerve stimulation. *Statistical differences observed between experimental groups at P< 0.05. Different letters represent statistical differences at Tukey adjusted with α value set at P ≤ 0.05. n = 7–10 animals/group was used. HT, high tumor; LT, low tumor; PBS, phosphate-buffered saline.
Inhibition of Muscle Protein Anabolism
To determine protein synthetic function, we assessed 24-h FSR determined by D2O labeling and cell signaling markers associated with protein synthesis. Mixed gastrocnemius muscle FSR was ∼40% lower in HT mice than in PBS mice (P = 0.043; Fig. 3A) although there were no statistical differences in TA (Supplemental Fig. S3A). mRNA content of Pgc1α4 was ∼62% lower in 1 wk group than in LT group (P = 0.015) and insulin growth factor-1 (Igf1) was greater in 2 wk group than in LT (∼66%, P = 0.009) and HT groups (∼57%, P = 0.016), respectively (Fig. 3B). Redd1 mRNA content was ∼17-fold greater in 1 wk group than in PBS group (P = 0.001), and approximately threefold to sixfold greater compared with 2 wk, LT, and HT groups (P = 0.004–0.014, Fig. 3C). Protein content of Deptor was greater in LT group than in PBS (approximately threefold; P = 0.067) and 2 wk groups (∼1.8-fold, P = 0.012; Fig. 3D) whereas mRNA content of Deptor was ∼45.1% greater in 2 wk group than in HT group (P = 0.046; Fig. 3E).
Figure 3.
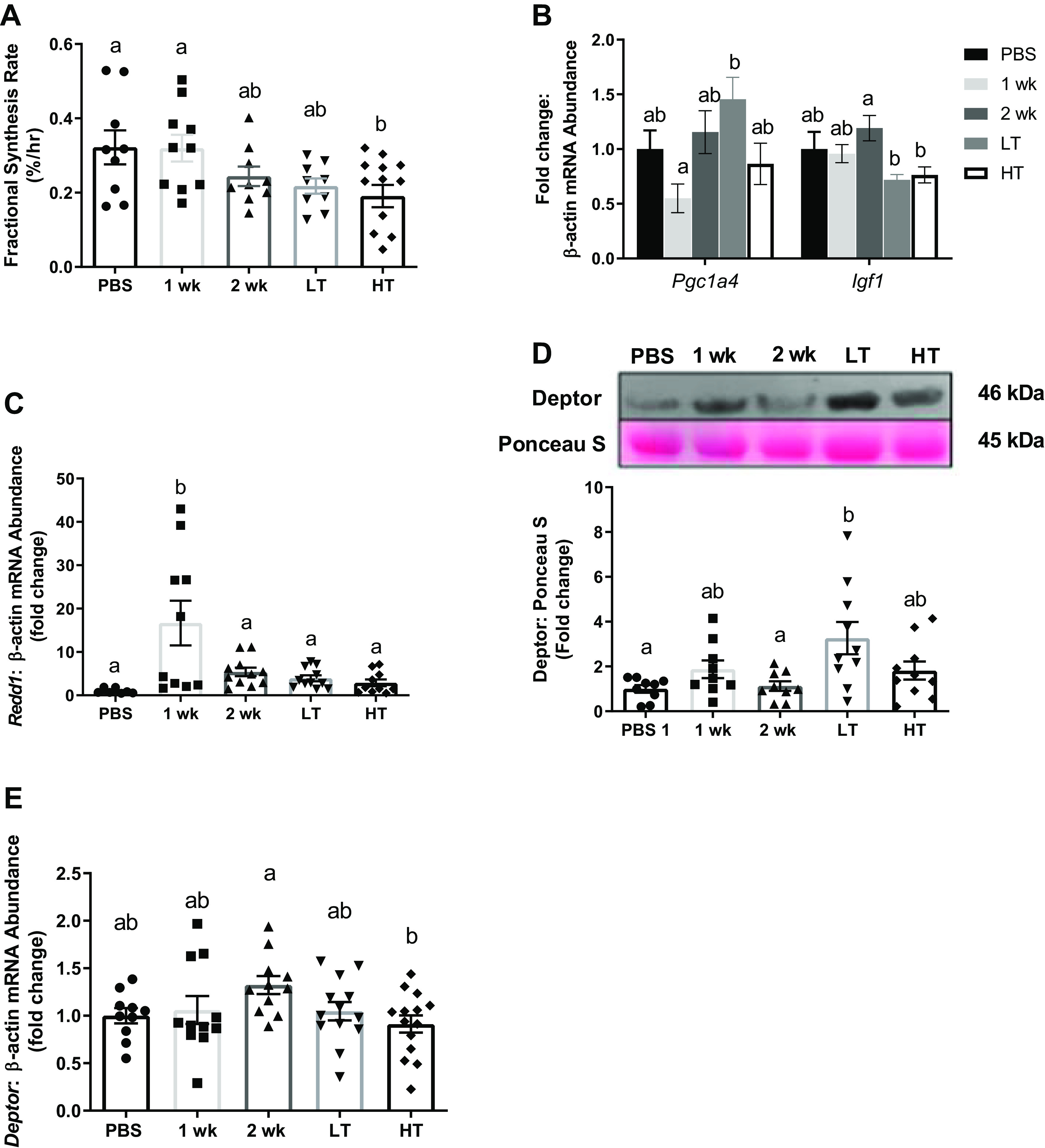
Protein synthesis 24-h mixed muscle fractional synthesis rates (A, %/h) across experimental groups. B: Pgc1α4 and Igf1 mRNA abundance measured among experimental groups. C: Redd1 mRNA abundance measured among experimental groups. D: protein content of Deptor relative to Ponceau S among experimental groups. E: Deptor mRNA abundance measured among experimental groups. Both mRNA and protein contents were analyzed from gastrocnemius muscle. Different letters represent statistical differences at Tukey adjusted with α value set at P ≤ 0.05. n = 9–14 animals/group was used. HT, high tumor; LT, low tumor; PBS, phosphate-buffered saline.
Atrophy-Associated Genes Are Upregulated with Tumor Development
We assessed mRNA content of genes from gastrocnemius within the UPS, autophagy-lysosomal pathway, and markers of inflammation to assess protein degradation systems during the development of CC (Fig. 4). First, mRNA content of Ubc (Ubiquitin C), Atrogin1, and Murf1 were ∼2.8-fold (P = 0.019), ∼3.5-fold (P < 0.001), and ∼4.5-fold (P = 0.004) greater in HT group than in other groups, respectively, whereas Gadd45a demonstrated no difference from PBS (Fig. 4A). Regarding autophagy, the protein content of Beclin1 (a central marker for autophagy, initiating autophagosome formation) was ∼54% higher in 1 wk group than in PBS group (P = 0.021) and LC3 II/I ratio (indication of autophagic activity) was ∼190% higher in HT group than in 2 wk group (P = 0.038), although there were no significant differences in total LC3 and p62 (a cargo protein that targets other proteins that bind to it for selective autophagy) protein content (Fig. 4, B–C). On the other hand, the mRNA content of Beclin1 was ∼44% lower in HT group than in PBS group (P = 0.035) and Lc3 was ∼39% higher in 2 wk group than in PBS and HT groups (P = 0.019–0.040). No significant differences in p62 content were observed (Fig. 4D). Regarding inflammatory markers, IL-6 mRNA content was ∼90% greater in HT compared with other groups (P < 0.001) and Tnfα was ∼185% greater in 2 wk and LT compared with 1 wk (P = 0.021–0.027; Fig. 4E). There was no statistical difference found in NF-κB mRNA content (Fig. 4E).
Figure 4.

Protein degradation and inflammation. A: protein degradation measured by mRNA abundance for Ubc, Atrogin-1, Murf1, and Gadd45a. B: protein content of autophagy-related markers; Beclin-1, LC3, LC3 II/I, and p62 normalized to Ponceau S. C: representative immunoblot image for autophagy-related markers from the same membrane. D: mRNA abundance of autophagy-related markers for Beclin-1, Lc3, and p62. E: mRNA abundance of inflammation markers; IL-6, Tnfα, and NF-κB. Both mRNA and protein contents were analyzed from gastrocnemius muscle. Different letters represent statistical differences at Tukey adjusted with α value set at P ≤ 0.05. n = 10–14 animals/group was used. HT, high tumor; LT, low tumor; PBS, phosphate-buffered saline; Ubc, Ubiquitin C.
Extrinsic Regulators of Skeletal Muscle Mass Are Altered during CC Development
To examine extrinsic regulators of skeletal muscle mass, we assessed mRNA contents from gastrocnemius for cell cycling and myogenesis. Cyclin-D1 (essential regulator of the G1-S transition) was ∼2.5-fold greater in 2 wk group than in other experimental groups (P < 0.001). Mki67 (cell proliferation marker) was ∼2.5-fold greater in HT group than in PBS (∼149%, P < 0.001), 2 wk (∼109%, P = 0.002), and LT groups (∼99%, P = 0.003). Acta2 (a marker of myofibroblast) was ∼57% higher in 1 and 2 wk groups than in PBS and HT groups (P = 0.001; Fig. 5A). Both Pax7 (the essential regulator for muscle precursor cell proliferation) and MyoD (regulating both skeletal muscle proliferations) were ∼31%–41% lower in HT group than in 1 and 2 wk groups (P = 0.001–0.045). No changes were observed in Myogenin (muscle differentiation marker) mRNA content (Fig. 5B).
Figure 5.
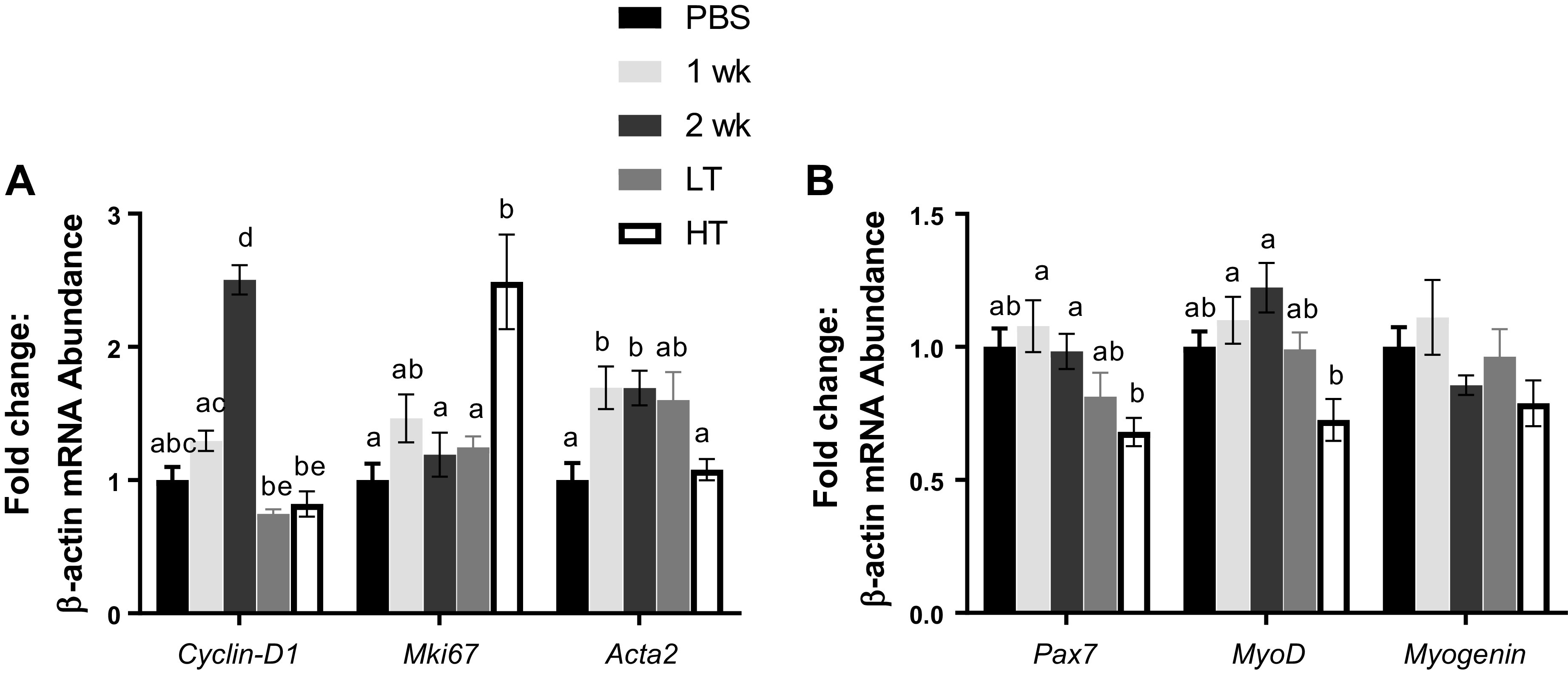
Myogenic and cell proliferation factors. Cell proliferation (A) and myogenic factors (B) measured for mRNA abundance. A: mRNA abundance measured for Cyclin-D1, Mki67, and Acta2. B: mRNA abundance measured for Pax7, MyoD, and Myogenin. mRNA contents were analyzed from gastrocnemius muscle. Different letters represent statistical differences at Tukey adjusted with α value set at P ≤ 0.05. n = 9–14 animals/group was used. HT, high tumor; LT, low tumor; PBS, phosphate-buffered saline.
Mitochondrial Degeneration in HT
Histological assessment of MitoTimer (redox-sensitive reporter gene) data demonstrated lower red-to-green ratio (oxidative stress) in 1 wk group than in LT (∼27%, P = 0.042) and HT groups (∼30%, P = 0.009; Fig. 6, A and B). Greater pure red puncta numbers (completely degenerated mitochondria) were observed in HT group compared with PBS and 1 wk groups (∼50%, P = 0.005–0.022, Fig. 6, A and C). In addition, mitochondrial respiratory function measured by the mitochondrial RCR was ∼49% higher in HT group than in PBS and 1 wk groups (P = 0.021–0.036, Fig. 6D), whereas mitochondrial ROS (mtROS) emission was not statistically different across experimental groups (Fig. 6E).
Figure 6.
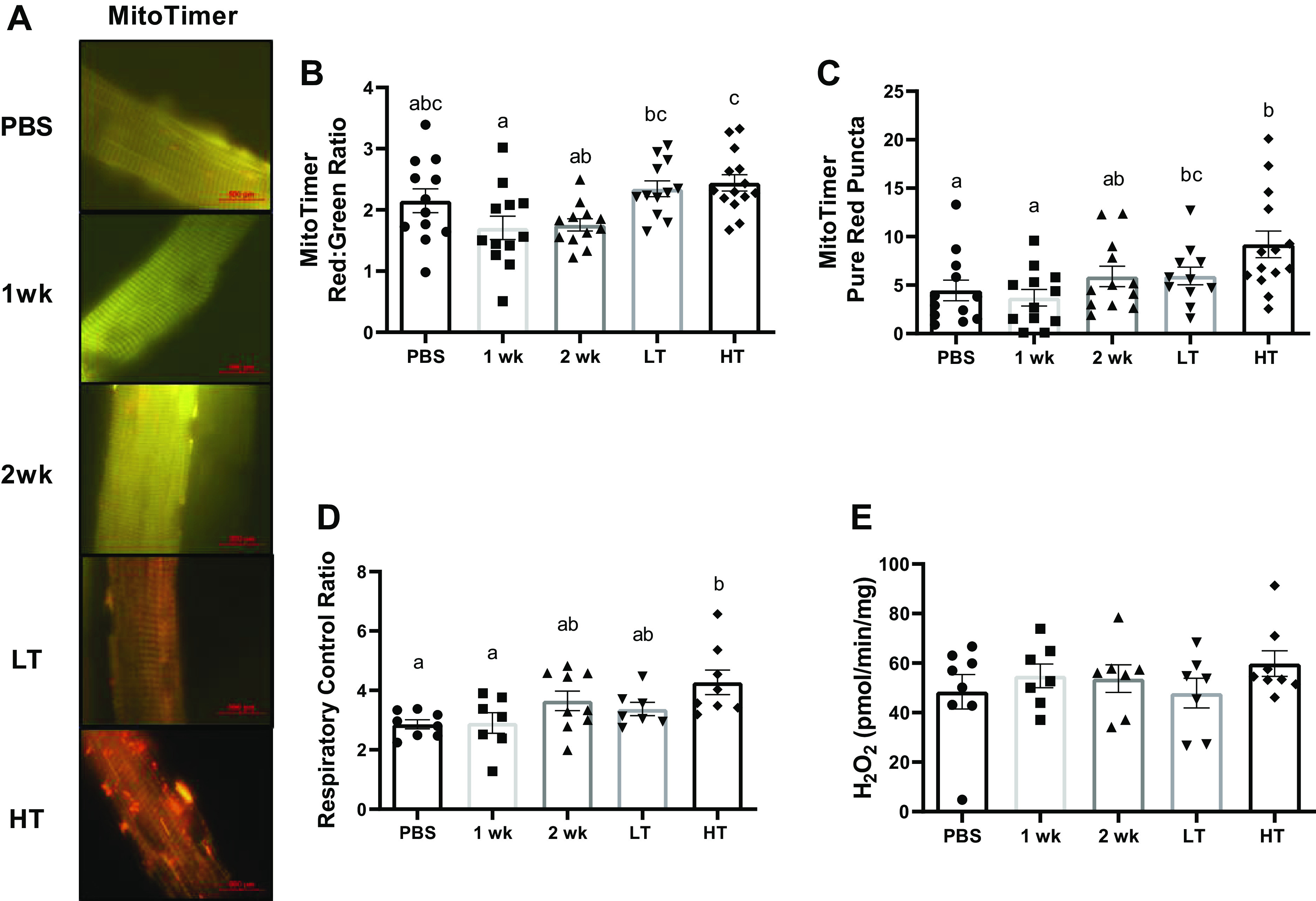
Mitochondrial function and network. A: representative images of MitoTimer in the FDB muscle taken at ×100 magnification. Scale bar set at 100 µm. B and C: quantifications of MitoTimer in FDB muscle. Quantification of red-to-green ratio (B) and pure red puncta numbers quantified for MitoTimer (C). Pure red puncta are representative of mitochondria that are marked for autophagy. D and E: mitochondrial function and mtROS emission measurements of the plantaris muscle. Respiratory control ratio (D; state 3:state 4 respiration) and H2O2 emission (E) of mitochondria measured with permeabilized plantaris. Different letters represent statistical differences at Tukey adjusted with α value set at P ≤ 0.05. n = 7–9 animals/group was used. FDB, flexor digitorum brevis; HT, high tumor; LT, low tumor; mtROS, mitochondrial reactive oxygen species; PBS, phosphate-buffered saline.
LLC Impairs Mitochondrial Quality Control and Mitophagy in HT, But Not in LT
Mitochondrial quality control was determined via assessing markers of mitochondrial biogenesis, dynamics, translation initiation factors, and mitophagy from gastrocnemius. In mitochondrial biogenesis, Pgc1α1 mRNA levels were ∼44% lower in HT group than in PBS, 1 wk, and 2 wk groups (P = 0.009–0.019; Fig. 7A). Tfam mRNA levels were ∼28% lower in HT group and ∼25% lower in 1 wk group than in PBS group (P < 0.001; Fig. 7A). Nrf2 mRNA content was ∼38% greater in HT group than in PBS (P = 0.026) and 1 wk groups (P = 0.029), whereas there were no statistical differences in Pparα or Pparδ (Fig. 7A). In mitochondrial dynamics, Drp1 and Mff were higher in 2 wk group and lower in HT group than in PBS group (∼32%; P = 0.024 and ∼43%; P = 0.002, respectively), whereas HT group was not different from PBS group (Fig. 7B). Mfn1 mRNA content was ∼34% lower in HT group than in PBS group (P = 0.014), whereas there were no statistical differences in Fis1 and Opa1 (Fig. 7B). In mitochondrial translation, TuFM, Taco1, and Mtif2 showed similar patterns that were gradually elevated in 2 wk group and reduced in HT group (P = 0.001–0.013), whereas no changes were observed in Mtif3 (Fig. 7C). In mitophagy, both protein and mRNA content of BNIP3 was ∼63% (P = 0.010) and ∼147% (P < 0.001) greater in HT groups than in other groups, respectively (Fig. 7, D–E). There were no statistical differences in protein contents for mitochondrial biogenesis and dynamics (Supplemental Fig. S2, A, B, and D).
Figure 7.
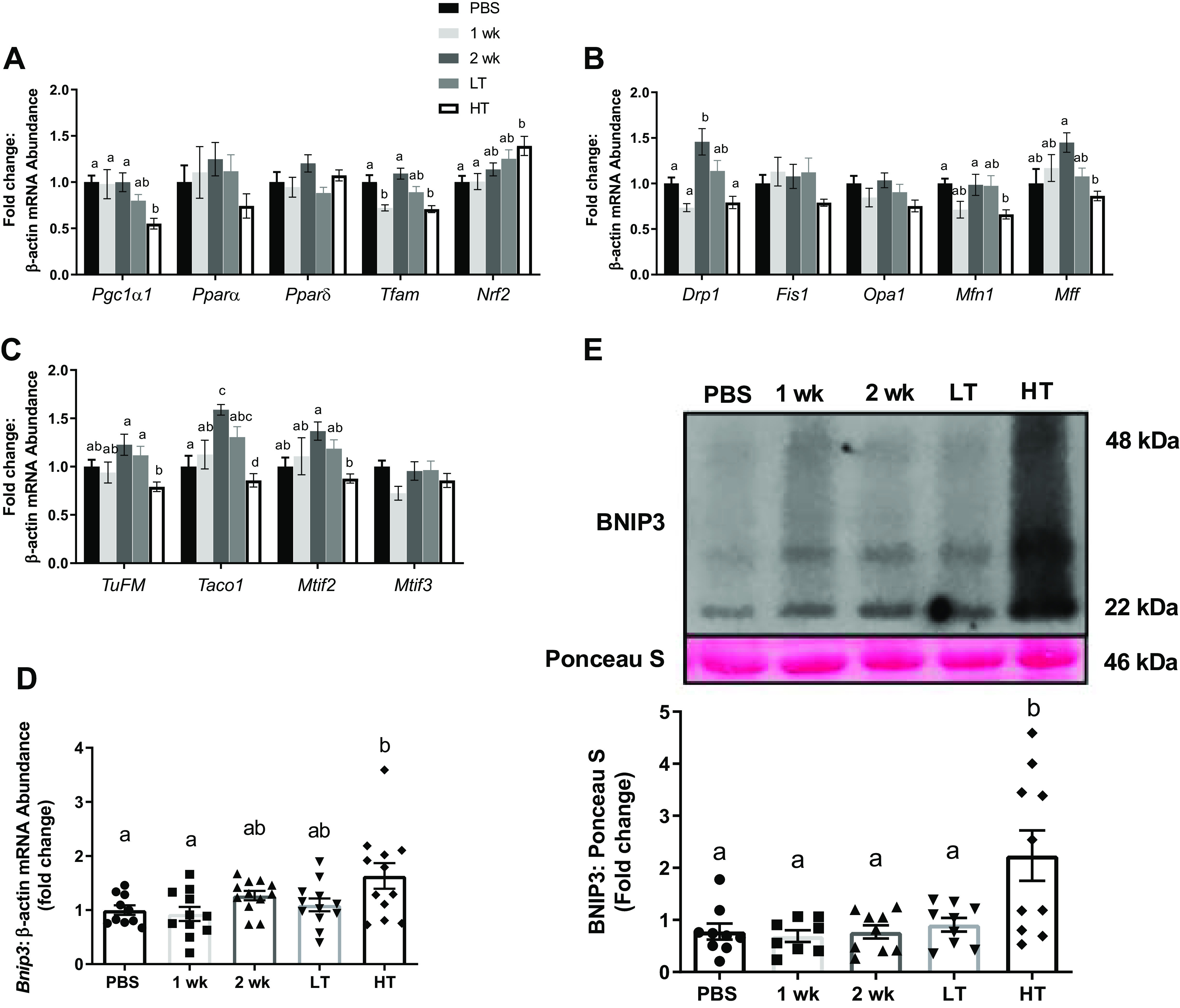
Mitochondrial quality control. A: mRNA abundance of mitochondrial biogenesis controllers for Pgc1α, Pparα, Pparδ, Tfam, and Nrf2 among experimental groups. B: mRNA abundance of mitochondrial dynamic controllers for Drp1, Fis1, Opa1, Mfn1, and Mff among experimental groups. C: mRNA abundance of mitochondrial translation factors for TuFM, Taco1, Mtif2, and Mtif3 among experimental groups. D and E: mitophagy regulator, BNIP3, measured for mRNA abundance (D) and protein content relative to Ponceau S with a representative image of immunoblot for Bnip3 and Ponceau S (E) among experimental groups. Both mRNA and protein contents were analyzed from gastrocnemius muscle. Different letters represent statistical differences at Tukey adjusted with α value set at P ≤ 0.05. n = 10–14 animals/group was used. HT, high tumor; LT, low tumor; PBS, phosphate-buffered saline.
Antioxidant Enzymes Are Unchanged During CC
The protein content of SODs −1, −2, and −3 and catalase from gastrocnemius was not statistically different among experimental groups (Supplemental Fig. S2C).
Altered MAPK Signaling in LT Group
MAPK signaling from gastrocnemius was determined via assessing the phosphorylation status of p38 and ERK. Although there were no statistical differences in protein content of p-p38T180/Y182/p38, p-ERK1/2T202/Y204/ERK was ∼7.3-fold higher in LT group than in PBS group (P = 0.034; Supplemental Fig. S4A).
DISCUSSION
In this study, we aimed to assess alterations in the development of CC in LLC tumor-bearing female mice. To do so, we assessed aspects of skeletal muscle contractile function and metabolism including protein turnover and mitochondrial health. In addition, we observed a distinct dichotomous effect in tissue wasting and cachectic phenotype within the 3- and 4-wk animals, where approximately half of the mice exhibited a relatively low tumor mass (<1.2 g), and the other half exhibited a substantially greater tumor mass (>2 g). This was a particularly interesting finding, as previous studies on male mice have shown gradual tumor development over time following tumor implantation (4, 27). Furthermore, animals with HT exhibited higher protein degradation, impaired skeletal muscle contractility, and accelerated mitochondrial degeneration, whereas LT mice did not exhibit such cachectic phenotypes, despite up to 4 wk of tumor growth. Our prior works have demonstrated impaired metabolic health in male mice before the onset of cachexia itself (4, 5), compared with those works, the current data suggest a relative protection of muscle contractile and metabolic functions in female tumor-bearing mice in these earlier stages of tumor development.
First, we observed a clear dichotomy in tumor burden among 3- and 4-wk tumor-bearing female mice, wherein mice with higher tumor burdens developed a cachectic phenotype. This dichotomy suggests a significant reliance upon tumor burden for the development of cancer cachexia in female mice whereby time frame of tumor development may not directly reflect increases to tumor burden and corresponding cachectic phenotype. Considering, these effects we regrouped 3- and 4-wk mice to low and high tumor-bearing conditions to better reflect the developmental process of cachexia in female mice. Our findings suggest LT mice may have a protective mechanism to prevent cancer-induced cachectic phenotypes including tumor development and muscle wasting although additional investigations are required to elucidate this mechanism. Despite marked muscle loss in HT mice, this atrophy was less overt compared with previous studies using tumor-bearing male mice (4, 28). Specifically, females revealed ∼3%–14% overall muscle loss with significant muscle wasting in two muscles (TA and soleus), whereas males underwent ∼11%–16% overall muscle loss with significant muscle wasting in four muscles (gastrocnemius, TA, plantaris, and soleus muscle) out of five (4). Furthermore, CSA distribution for oxidative and glycolytic muscle fibers as well as oxidative phenotype via SDH staining did not differ between experimental groups, although these markers were significantly lower in our previous study using tumor-bearing male mice (4). These findings imply that although female HT mice exhibit mild cachexia, female mice may have a protective effect on cancer-induced muscle wasting compared with their male counterparts consistent with suggestions that females may be less sensitive to inflammation-mediated muscle atrophies compared with males (13).
In addition to muscle wasting, impaired muscle contractile function is a strong indicator of lower survival rate and impaired functional outcomes in patients with advanced-stage cancer (29). CC is often associated with impaired muscle contractility including force production and fatigability (30). Here, we observed impaired muscle contractility in HT mice, in agreement with findings from a previous study (30). Indeed, we observed lower torque frequency at half maximum torque, slower muscle contraction, and relaxation times in HT mice, which was in line with prior works (31). Although speculative, these data suggest preferential protection of slower twitch fibers in early phases of cachexia itself, which aligns with preservation of oxidative fibers previously observed by our group (4). Prior evidence also demonstrated decreased isometric muscle force production and elevated muscle fatigue in male ApcMin/+ mice (colorectal cancer model) (30). However, another study demonstrated unaltered fatigability in tumor-bearing female mice (32), as observed here, which may be associated with the relatively mild cachectic phenotype in our study. Collectively, although fatigability may not be altered with early-stage CC, likely due to preferential survival of slow-twitch fibers, muscle contractile function does still appear to be impaired with high tumor development in female mice.
One of the major features of CC is the net loss of skeletal muscle protein as protein degradation exceeds protein synthesis. Therefore, we assessed muscle protein anabolism and catabolism across tumor development. In line with previous works (5, 22), we observed lower muscle protein FSR in gastrocnemius of female mice with greater tumor burdens. We postulate that this is due to altered function of the protein anabolic system during the development of CC, specifically the induction of anabolic/mTORC1 repressors, Deptor and Redd1, which were observed to be greater before the onset of the cachectic phenotype (Fig. 3, C and D). We previously observed a similar pattern in male tumor-bearing mice, whereby Deptor content was higher in precachectic male mice (5), suggesting that inductions of anabolic repressors precede cachexia and may account for diminished protein synthetic rates as early-stage cachexia develops. It is noteworthy to mention that although gastrocnemius FSR was lower in HT mice, this muscle mass was intact. We speculate that unlike males, female gastrocnemius muscle mass appears to be protected during the development of CC and may begin the wasting process at later stages of cancer, which may not be enough to detect the significant muscle loss at the time of tissue collection in the current study.
Protein degradation in skeletal muscle is largely mediated by two major processes, the UPS and the autophagy-lysosomal pathway. Of note, we observe induction of E3 ligases, Atrogin1 and Murf1, as well as UBC (11) in the HT mice (Fig. 4A), in accordance with previous male CC studies (5, 33). Moreover, we observed greater LC3II/I ratio in HT mice, whereas p62 level did not change, indicating an increase in autophagy initiation but not necessarily resolution in HT mice. In addition, cancer-induced protein degradation is often associated with elevated inflammation (7, 34) and inflammatory cytokines are well-known mediators of muscle wasting during CC (33–35). The elevated IL-6 mRNA content observed in the HT group has also been demonstrated previously in both clinical (35) and preclinical models of CC (7, 34). Furthermore, these effects are local induction of inflammatory cytokines at the level of the muscle, suggesting tumor-induced local inflammation in cachectic mice. Taken together, these upregulated E3 ligases and proinflammatory cytokines appear to coincide and associate with muscle wasting as well as tumor development in tumor-bearing female mice.
Adult skeletal muscle has a capacity to regenerate in response to proper stimuli (36) and is often altered in various myopathologies including CC (5). Of note, previous evidence suggests alteration to satellite cell function including the markers Pax7, Myogenin, MyoD, and Cyclin-D1 in tumor-bearing male mice (5, 37). However, here we observed lower Pax7 and MyoD in only the HT group (Fig. 5B), suggesting a relative protection of myogenic regulators in females compared with previous male CC study (5). Further in-depth research on the cell cycle and myogenic function is warranted to elucidate the regulation of muscle regeneration during CC, especially within female mice.
Prior evidence demonstrates cancer-induced muscle mitochondrial degeneration including diminished mitochondrial RCR, ATP production, and elevated mtROS emission (4, 28). In addition, we have demonstrated these mitochondrial degenerations occur before muscle loss in tumor-bearing male mice (4). Interestingly, here we report that female mice exhibited unchanged mtROS emission and greater mitochondrial RCR in HT group, in direct contrast to lowered RCR in cachectic male mice (4), suggesting specific protection of mitochondrial health during cancer cachexia in female mice. These data closely align with recent findings suggesting early mitochondrial degeneration in male, but not in female mice during disuse atrophy (38). Combined data suggest that a mechanism of mitochondrial degeneration to induce muscle atrophy is preserved across pathologies specifically in male mice; yet in females, these mitochondrial mechanisms may be more nuanced in relation to muscle atrophy. Impaired mitochondrial function is often associated with dysregulated mitochondrial autophagy (mitophagy) (4, 39), which contributes to exacerbated mitochondrial degeneration. We observed induction of BNIP3 and an increase in pMitoTimer pure red puncta in HT mice, reflecting the induction of mitophagy and accumulation of damaged mitochondria in female mice upon the onset of the cachectic phenotype (Figs. 6 and 7). Although similar data are observed in male mice by our group (4) as well as others (40), in our prior work these effects were observed well before the onset of the cachectic phenotype in males in contrast to later development in females. Further studies applying either pharmacological or transgenic animal models of BNIP3 are warranted to determine its precise role in CC. Considering data from the current study as well as other recent works (25, 41), we hypothesize that female mice specifically protect mitochondrial health during myopathologies. This specific mitochondrial protection may lead to attenuation of inflammatory forms of atrophy (i.e., CC) while prioritizing mitochondrial health at the expense of muscle mass in noninflammatory forms of atrophy (i.e., disuse).
Together with aforementioned mitophagy, mitochondrial quality control (MQC) is the accumulation of systems required for maintaining mitochondrial integrity and function and is an important defense mechanism for the cell’s survival from mitochondrial damage (42), often altered in cancer (4). Herein, we observed altered mitochondrial biogenesis markers in HT mice. Specifically, we found lower Pgc1α1 and Tfam, and greater Nrf2 contents in HT group, although these effects were absent in previous male CC studies (4). In addition, some mitochondrial fission markers (Drp1 and Mff) and translational factors (Taco1 and Mtif2) within females revealed a similar pattern of elevation at 2 wk of tumor-bearing state, which may reflect mechanisms to protect muscle mitochondrial health during early stages of tumor development. By contrast, several of these mitochondrial biogenesis and dynamic markers were suppressed during the development of CC in our previous male study (4) further exemplifying dichotomous effects between biological sexes relative to maintenance of mitochondrial health during atrophic pathologies.
Recent studies have addressed phenotypical differences between sexes in response to different types of muscle atrophy such as those induced by disuse and cancer (13, 43–45), with females possibly having exacerbated wasting compared with males in some myopathies such as disuse, with the opposite being true in males with cancer. However, prior preclinical CC studies have primarily focused on male animals, making it difficult to isolate the fundamental role of biological sex during CC. Previously, Hetzler et al. (16) demonstrated sex differences in IL-6 content in CC, whereby ApcMin/+ male mice had higher plasma IL-6 content than female counterparts. However, to our knowledge, the current study is one of the first studies to demonstrate various alterations in skeletal muscle during the initial development of CC in female tumor-bearing mice. Although some responses appear preserved between sexes, we observed distinct differences in response to tumor development in females compared with prior male CC studies (4, 5). Specifically, both sexes exhibit impaired protein anabolism, upregulated E3 ligase-mediated protein degradation, and mitophagy in mice with greater tumor burden. However, there were clear differences in oxidative phenotype, myogenic markers, MAPK signaling, mitochondrial RCR, mtROS emission, and mitochondrial dynamics between sexes, wherein many of these cellular alterations appear relatively protected in females, whereas that of males was significantly impaired before the onset of cachexia itself (4, 5). Among protected effects in females compared with previous male studies, mitochondrial protection during the development of CC appears to serve as a major contributor to preserve muscle mass, although further investigations regarding mitochondrial regulation in CC are warranted. Our findings suggest distinct biological sex differences in development of CC and these differentially regulated targets/pathways may be crucial in the protective mechanism in tissue wasting during CC in females. Therefore, a critical need remains to directly investigate comparative alterations in the skeletal muscle during the development of CC between males and females within a single study using various cancer types to better delineate the role of biological sex in CC.
It is worth mentioning that a limitation of this study is the use of younger mice (8- to 12-wk old) to compare outcomes with prior works in male mice (4, 5, 46–48). However, a majority of human cancers are mostly prevalent in older adults (49). Therefore, future studies should use aged animals to better recapitulate these clinical scenarios.
In conclusion, we set to investigate the initial development of CC in tumor-bearing female mice, revealing various metabolic and contractile impairments with tumor development. Specifically, although many metabolic perturbations precede the onset of cachexia in males, female tumor-bearing mice exhibit multiple protections in both metabolic and contractile functions in these earlier stages of tumor development. These data suggest that female mice may have a defense system protecting them from tumor-induced muscle degeneration via maintaining muscle regenerative processes, mitochondrial respiratory capacity, mitochondrial dynamics, and oxidative capacity during the development of CC. Our data strongly imply that females exhibit an enhanced capacity to mitigate aberrations associated with the onset of CC in the early stages of tumor development. However, further works are necessary to delineate these specific protections in female mice and their potential clinical use.
SUPPLEMENTAL DATA
Supplemental Figs. S1–S4: https://doi.org/10.6084/m9.figshare.16623064.
GRANTS
This study was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health Award Nos. R15AR069913/AR/NIAMS and R01AR075794-01A1/AR/NIAMS (to N.P.G.) and by the National Institute of General Medical Sciences of the National Institutes of Health under Award No. P20GM125503.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.A.W. and N.P.G. conceived and designed research; S.L., J.W.D., M.E.R-C., W.S.H., F.M.d.S, A.R.C., E.R.S., L.W.S., L.T.J., and K.R.D. performed experiments; S.L., J.W.D., M.E.R.-C, W.S.H., F.M.d.S, A.R.C., E.R.S., L.W.S., L.T.J., K.R.D., and N.P.G. analyzed data; S.L., J.W.D., M.E.R-C., M.P.W., T.A.W., and N.P.G. interpreted results of experiments; S.L. and W.S.H. prepared figures; S.L. drafted manuscript; S.L., J.W.D., M.E.R-C., F.M.d.S, A.R.C., E.R.S., L.W.S., L.T.J., K.R.D., M.P.W., T.A.W., and N.P.G. edited and revised manuscript; N.P.G. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank all the various faculty, staff, and students of the Exercise Science Research Center at the University of Arkansas for support and contributions herein and also thank Dr. Jarrod Call for consultation in the measure of muscle contractile function.
REFERENCES
- 1.Ryerson AB, Massetti GM. CDC’s public health surveillance of cancer. Prev Chronic Dis 14: E39, 2017. doi: 10.5888/pcd14.160480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, Jatoi A, Loprinzi C, MacDonald N, Mantovani G, Davis M, Muscaritoli M, Ottery F, Radbruch L, Ravasco P, Walsh D, Wilcock A, Kaasa S, Baracos VE. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 12: 489–495, 2011. doi: 10.1016/S1470-2045(10)70218-7. [DOI] [PubMed] [Google Scholar]
- 3.Kasvis P, Vigano M, Vigano A. Health-related quality of life across cancer cachexia stages. Ann Palliat Med 8: 33–42, 2019. doi: 10.21037/apm.2018.08.04. [DOI] [PubMed] [Google Scholar]
- 4.Brown JL, Rosa-Caldwell ME, Lee DE, Blackwell TA, Brown LA, Perry RA, Haynie WS, Hardee JP, Carson JA, Wiggs MP, Washington TA, Greene NP. Mitochondrial degeneration precedes the development of muscle atrophy in progression of cancer cachexia in tumour‐bearing mice. J Cachexia Sarcopenia Muscle 8: 926–938, 2017. doi: 10.1002/jcsm.12232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown JL, Lee DE, Rosa-Caldwell ME, Brown LA, Perry RA, Haynie WS, Huseman K, Sataranatarajan K, Van Remmen H, Washington TA, Wiggs MP, Greene NP. Protein imbalance in the development of skeletal muscle wasting in tumour-bearing mice. J Cachexia Sarcopenia Muscle 9: 987–1002, 2018. [Erratum in J Cachexia Sarcopenia Muscle 10: 712, 2019]. doi: 10.1002/jcsm.12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blackwell TA, Cervenka I, Khatri B, Brown JL, Rosa-Caldwell ME, Lee DE, Perry RA Jr, Brown LA, Haynie WS, Wiggs MP, Bottje WG, Washington TA, Kong BC, Ruas JL, Greene NP. Transcriptomic analysis of the development of skeletal muscle atrophy in cancer-cachexia in tumor-bearing mice. Physiol Genomics 50: 1071–1082, 2018. doi: 10.1152/physiolgenomics.00061.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.White JP, Baltgalvis KA, Puppa MJ, Sato S, Baynes JW, Carson JA. Muscle oxidative capacity during IL-6-dependent cancer cachexia. Am J Physiol Regul Integr Comp Physiol 300: R201–R211, 2011. doi: 10.1152/ajpregu.00300.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dasuri K, Zhang L, Keller JN. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic Biol Med 62: 170–185, 2013. doi: 10.1016/j.freeradbiomed.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 9.Shringarpure R, Grune T, Davies KJ. Protein oxidation and 20S proteasome-dependent proteolysis in mammalian cells. Cell Mol Life Sci 58: 1442–1450, 2001. doi: 10.1007/PL00000787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Norton JA, Shamberger R, Stein TP, Milne GWA, Brennan MF. The influence of tumor-bearing on protein metabolism in the rat. J Surg Res 30: 456–462, 1981. doi: 10.1016/0022-4804(81)90090-1. [DOI] [PubMed] [Google Scholar]
- 11.Rosa-Caldwell ME, Lim S, Haynie WA, Brown JL, Deaver JW, Morena DA Silva F, Jansen LT, Lee DE, Wiggs MP, Washington TA, Greene NP. Female mice may have exacerbated catabolic signalling response compared to male mice during development and progression of disuse atrophy. J Cachexia Sarcopenia Muscle 12: 717–730, 2021. doi: 10.1002/jcsm.12693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryberg D, Hewer A, Phillips DH, Haugen A. Different susceptibility to smoking-induced DNA damage among male and female lung cancer patients. Cancer Res 54: 5801–5803, 1994. [PubMed] [Google Scholar]
- 13.Rosa-Caldwell ME, Greene NP. Muscle metabolism and atrophy: let’s talk about sex. Biol Sex Differ 10: 1–14, 2019. doi: 10.1186/s13293-019-0257-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oliván S, Calvo AC, Manzano R, Zaragoza P, Osta R. Sex differences in constitutive autophagy. Biomed Res Int 2014: 652817, 2014. doi: 10.1155/2014/652817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.White JP, Puppa MJ, Gao S, Sato S, Welle SL, Carson JA. Muscle mTORC1 suppression by IL-6 during cancer cachexia: a role for AMPK. Am J Physiol Endocrinol Metab 304: E1042–E1052, 2013. doi: 10.1152/ajpendo.00410.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hetzler KL, Hardee JP, Puppa MJ, Narsale AA, Sato S, Davis JM, Carson JA. Sex differences in the relationship of IL-6 signaling to cancer cachexia progression. Biochim Biophys Acta 1852: 816–825, 2015. doi: 10.1016/j.bbadis.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laker RC, Xu P, Ryall KA, Sujkowski A, Kenwood BM, Chain KH, Zhang M, Royal MA, Hoehn KL, Driscoll M, Adler PN, Wessells RJ, Saucerman JJ, Yan Z. A novel MitoTimer reporter gene for mitochondrial content, structure, stress, and damage in vivo. J Biol Chem 289: 12005–12015, 2014. doi: 10.1074/jbc.M113.530527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Call JA, McKeehen JN, Novotny SA, Lowe DA. Progressive resistance voluntary wheel running in the mdx mouse. Muscle Nerve 42: 871–880, 2010. doi: 10.1002/mus.21764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorassini M, Eken T, Bennett DJ, Kiehn O, Hultborn H. Activity of hindlimb motor units during locomotion in the conscious rat. J Neurophysiol 83: 2002–2011, 2000. doi: 10.1152/jn.2000.83.4.2002. [DOI] [PubMed] [Google Scholar]
- 20.Jones LA, Hunter IW. Force sensation in isometric contractions: a relative force effect. Brain Res 244: 186–189, 1982. doi: 10.1016/0006-8993(82)90919-2. [DOI] [PubMed] [Google Scholar]
- 21.Gasier HG, Riechman SE, Wiggs MP, Previs SF, Fluckey JD. A comparison of 2H2O and phenylalanine flooding dose to investigate muscle protein synthesis with acute exercise in rats. Am J Physiol Endocrinol Metab 297: E252–E259, 2009. doi: 10.1152/ajpendo.90872.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith KL, Tisdale MJ. Increased protein degradation and decreased protein synthesis in skeletal muscle during cancer cachexia. Br J Cancer 67: 680–685, 1993. doi: 10.1038/bjc.1993.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Min K, Smuder AJ, Kwon O-S, Kavazis AN, Szeto HH, Powers SK. Mitochondrial-targeted antioxidants protect skeletal muscle against immobilization-induced muscle atrophy. J Appl Physiol (1985) 111: 1459–1466, 2011. doi: 10.1152/japplphysiol.00591.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosa-Caldwell ME, Jansen LT, Lim S, Dunlap KR, Haynie WS, Washington TA, Greene NP. Neither autophagy nor exercise training mode affect exercise-induced beneficial adaptations in high fat-fed mice. Sports Med Health Sci 2: 44–53, 2020. doi: 10.1016/j.smhs.2020.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosa-Caldwell ME, Lim S, Haynie WS, Jansen LT, Westervelt LC, Amos MG, Washington TA, Greene NP. Altering aspects of mitochondrial quality to improve musculoskeletal outcomes in disuse atrophy. J Appl Physiol (1985) 129: 1290–1303, 2020. doi: 10.1152/japplphysiol.00407.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greene NP, Lee DE, Brown JL, Rosa ME, Brown LA, Perry RA, Henry JN, Washington TA. Mitochondrial quality control, promoted by PGC‐1α, is dysregulated by Western diet‐induced obesity and partially restored by moderate physical activity in mice. Physiol Rep 3: e12470, 2015. doi: 10.14814/phy2.12470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cosper PF, Leinwand LA. Cancer causes cardiac atrophy and autophagy in a sexually dimorphic manner. Cancer Res 71: 1710–1720, 2011. doi: 10.1158/0008-5472.CAN-10-3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.VanderVeen BN, Fix DK, Carson JA. Disrupted skeletal muscle mitochondrial dynamics, mitophagy, and biogenesis during cancer cachexia: a role for inflammation. Oxid Med Cell Longev 2017: 3292087, 2017. doi: 10.1155/2017/3292087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kilgour RD, Vigano A, Trutschnigg B, Lucar E, Borod M, Morais JA. Handgrip strength predicts survival and is associated with markers of clinical and functional outcomes in advanced cancer patients. Support Care Cancer 21: 3261–3270, 2013. doi: 10.1007/s00520-013-1894-4. [DOI] [PubMed] [Google Scholar]
- 30.VanderVeen BN, Hardee JP, Fix DK, Carson JA. Skeletal muscle function during the progression of cancer cachexia in the male ApcMin/+ mouse. J Appl Physiol (1985) 124: 684–695, 2018. doi: 10.1152/japplphysiol.00897.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inaba S, Hinohara A, Tachibana M, Tsujikawa K, Fukada S-I. Muscle regeneration is disrupted by cancer cachexia without loss of muscle stem cell potential. PloS one 13: e0205467, 2018. doi: 10.1371/journal.pone.0205467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greenman AC, Albrecht DM, Halberg RB, Diffee GM. Sex differences in skeletal muscle alterations in a model of colorectal cancer. Physiol Rep 8: e14391, 2020. doi: 10.14814/phy2.14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baltgalvis KA, Berger FG, Peña MMO, Davis JM, White JP, Carson JA. Muscle wasting and interleukin-6-induced atrogin-I expression in the cachectic ApcMin/+ mouse. Pflugers Arch 457: 989–1001, 2009. doi: 10.1007/s00424-008-0574-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carson JA, Baltgalvis KA. Interleukin 6 as a key regulator of muscle mass during cachexia. Exerc Sport Sci Rev 38: 168–176, 2010. doi: 10.1097/JES.0b013e3181f44f11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Knüpfer H, Preiss R. Serum interleukin-6 levels in colorectal cancer patients—a summary of published results. Int J Colorectal Dis 25: 135–140, 2010. doi: 10.1007/s00384-009-0818-8. [DOI] [PubMed] [Google Scholar]
- 36.Forcina L, Cosentino M, Musarò A. Mechanisms regulating muscle regeneration: Insights into the interrelated and time-dependent phases of tissue healing. Cells 9: 1297, 2020. doi: 10.3390/cells9051297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas-Ahner J, Wang J, Bloomston M, Muscarella P, Nau P, Shah N, Butchbach MER, Ladner K, Adamo S, Rudnicki MA, Keller C, Coletti D, Montanaro F, Guttridge DC. NF-κB–mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest 123: 4821–4835, 2013. doi: 10.1172/JCI68523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosa‐Caldwell ME, Lim S, Haynie WS, Brown JL, Lee DE, Dunlap KR, Jansen LT, Washington TA, Wiggs MP, Greene NP. Mitochondrial aberrations during the progression of disuse atrophy differentially affect male and female mice. J Cachexia Sarcopenia Muscle, 2021. doi: 10.1002/jcsm.12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williams JA, Ni H-M, Ding Y, Ding W-X. Parkin regulates mitophagy and mitochondrial function to protect against alcohol-induced liver injury and steatosis in mice. Am J Physiol Gastrointest Liver Physiol 309: G324–G340, 2015. [Erratum in Am J Physiol Gastrointest Liver Physiol 310: G142, 2016]. doi: 10.1152/ajpgi.00108.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res 61: 6669–6673, 2001. [PubMed] [Google Scholar]
- 41.Ballarò R, Lopalco P, Audrito V, Beltrà M, Pin F, Angelini R, Costelli P, Corcelli A, Bonetto A, Szeto HH, O’Connell TM, Penna F. Targeting mitochondria by SS-31 ameliorates the whole body energy status in cancer-and chemotherapy-induced cachexia. Cancers (Basel) 13: 850, 2021. doi: 10.3390/cancers13040850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hurtley SM. Mitochondrial quality control. Science 350: 1052–1053, 2015. doi: 10.1126/science.350.6264.1052-f. [DOI] [Google Scholar]
- 43.Zhong X, Zimmers TA. Sex differences in cancer cachexia. Curr Osteoporos Rep 18: 646–654, 2020. doi: 10.1007/s11914-020-00628-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Montalvo RN, Counts BR, Carson JA. Understanding sex differences in the regulation of cancer-induced muscle wasting. Curr Opin Support Palliat Care 12: 394–403, 2018. doi: 10.1097/SPC.0000000000000380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lim S, Brown JL, Washington TA, Greene NP. Development and progression of cancer cachexia: perspectives from bench to bedside. Sports Med Health Sci 2: 177–185, 2020. doi: 10.1016/j.smhs.2020.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Puppa MJ, Gao S, Narsale AA, Carson JA. Skeletal muscle glycoprotein 130's role in Lewis lung carcinoma–induced cachexia. FASEB J 28: 998–1009, 2014. doi: 10.1096/fj.13-240580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Au ED, Desai AP, Koniaris LG, Zimmers TA. The MEK-inhibitor Selumetinib attenuates tumor growth and reduces IL-6 expression but does not protect against muscle wasting in Lewis lung cancer cachexia. Front Physiol 7: 682, 2017. doi: 10.3389/fphys.2016.00682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiappalupi S, Sorci G, Vukasinovic A, Salvadori L, Sagheddu R, Coletti D, Renga G, Romani L, Donato R, Riuzzi F. Targeting RAGE prevents muscle wasting and prolongs survival in cancer cachexia. J Cachexia Sarcopenia Muscle 11: 929–946, 2020. doi: 10.1002/jcsm.12561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cinar D, Tas D. Cancer in the elderly. North Clin Istanb 2: 73–80, 2015. doi: 10.14744/nci.2015.72691. [DOI] [PMC free article] [PubMed] [Google Scholar]