

Abstract
Host genetic factors have been shown to play a role in SARs-CoV-2 infection in diverse populations. However, the genetic landscape differs among various ethnicities; therefore, we explored the host genetic factors associated with COVID-19 disease susceptivity and disease severity in a Thai population. We recruited and genotyped 212 unrelated COVID-19 Thai patients and 36 controls using AxiomTM Human Genotyping SARs-COV-2 array, including 847,384 single nucleotide polymorphisms related to SARs-COV-2 pathogenesis, immune response, and related comorbidity No SNPs passed the genome-wide significance threshold of p value <1 × 10−8. However, with a threshold of p value <1 × 10−5, a locus on chromosome 5q32 was found to have a suggestive association with COVID-19 disease susceptibility (p value 6.9 × 10−6; Q–Q plot λ = 0.805, odds ratio 0.02). Notably, IL17B is a gene located in this linkage disequilibrium block and is previously shown to play a part in inflammation and pneumonia. Additionally, a suggestive locus on chromosome 12q22, harboring EEA1 and LOC643339, was associated with COVID-19 disease severity (p value 1.3 × 10−6 – 4.4 × 10−6, Q–Q plot λ = 0.997, odds ratio 0.28–0.31). EEA1 is involved in viral entry into cells, while LOC643339 is a long non-coding RNA. In summary, our study suggested loci on chromosomes 5q32 and 12q22 to be linked to COVID-19 disease susceptibility and disease severity, respectively. The small sample size of this study may lessen the likelihood that the association found is real, but it could still be true. Further study with a larger cohort is required to confirm these findings.
Subject terms: Genetics research, Genome-wide association studies
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARs-CoV-2) was first reported in 2019 in Wuhan, China. Since then, the coronavirus disease 2019 (COVID-19) resulting from SARs-CoV-2 infection has become a global pandemic. Up to the first week of June 2021, more than 173 million individuals worldwide were confirmed cases with nearly 4 million deaths [1]. Thailand had the first outside-China confirmed case in January 2020 [2, 3] and is currently (June-July 2021) dealing with another outbreak of COVID-19 in the country [4]. COVID-19 disease had a wide range of manifestations from asymptomatic and mild to very severe respiratory failure leading to death. Several clinical factors were identified as risk factors for severe COVID-19 symptoms, including ageing, male gender, comorbidity such as hypertension, diabetes, obesity, and other cardiovascular diseases [5–12].
Undoubtedly, host genetic factors also play a role in SARs-CoV-2 pathophysiology, influencing an individual’s susceptibility to infection, disease severity, and disease progression, as shown by several studies with genome-wide association analysis (GWAS) [13–24]. The early works suggested the role of human leukocyte antigen (HLA) and renin-angiotensin pathway genes (ACE1 and ACE2) in the pathophysiology of COVID-19 disease [13–15]. A large GWAS study in Italian and Spanish populations with COVID-19 patients with respiratory failure [17] reported associations on chromosome 3p21.31 (rs11385942) corresponded with a cluster of genes including SLC6A20, LZtFL1, CCR9, FYCO1, and XCR1 and chromosome 9q34.2 (rs657152) coincided with the ABO-blood group system. Another GWAS study in the UK [20], focusing on critically ill patients with COVID-19, showed associations on chromosome 12q24.13 (rs10735079), chromosome 19p13.2 (rs74956615), chromosome 19p13.3 (rs2109069), and chromosome 21q22.1 (rs2236757), which were corresponded to antiviral restriction enzymes genes cluster (OAS1, OAS2, and OAS3), tyrosine kinase 2 (TYK2), dipeptidyl peptidase 9 (DPP9) and the interferon receptor gene (IFNAR2), respectively [20]. In Europeans, chromosome 21 was also explored as an in-dept genetic analysis showing that five single nucleotide polymorphisms (SNPs) within TMPRSS2 and MX1 were correlated with severe COVID-19 [19]. Recently, a global GWAS study including data of 46 studies from 19 countries known as COVID-19 Host Genetic Initiative (COVID-19 HGI) [24] showed several genome-wide significant loci which were associated with SARs-CoV-2 infection, including chromosome 3 (RPL24), 5 (DNAH5), 9 (ABO), and 19 (PLEKHA4) and COVID-19 disease severity including chromosome 1 (THBS3), 2 (SCN1A), 3 (LZTFL1), 6 (FOXP4), 8 (TMEM65), 12 (OAS1), 17 (KANSL1), 19 (DPP9 and RAVER1), and 21 (IFNAR2).
Data regarding host genetic factors in COVID-19 infection and disease progression in Asia are limited. However, one report from a Chinese group suggested the association of chromosome 21q22.11 (IFNAR2 and IL10RB) to COVID-19 susceptibility [18]. As the genetic landscape differs among various ethnicities, we aimed to explore the host genetic factors associated with COVID-19 disease susceptivity and disease severity, specifically in Thai. The obtained information may benefit from identifying risk groups that need special care or guiding the vaccination programs based on the future genetic risks of COVID-19 infection.
Materials and methods
This study used biobank samples and clinical data of the Faculty of Medicine, Chulalongkorn University and King Chulalongkorn Memorial Hospital, Bangkok, Thailand, from the project entitled: Collection and management of COVID-19-related clinical data and biological specimens for researches (COA No. 464/2020). The study was also approved by the Institutional Review Board of the Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand (COA No. 691/2021).
Study participants
The total of 248 participants (cases/controls = 212/36; males/females = 91/157) was recruited into the study. Cases (n = 212) were COVID-19 patients diagnosed with polymerase-chain-reaction (PCR) test from nasopharyngeal swabs and were admitted to King Chulalongkorn Memorial Hospital from February 2020 to March 2021. Controls were exposed individuals with negative PCR tests for SARs-CoV-2 viral infection (n = 36).
The severity of COVID-19 disease was assessed by the attending medical staffs of the Thai Red Cross Emerging Infectious Diseases Clinical Centre, King Chulalongkorn Memorial Hospital, Bangkok, Thailand, using the following criteria and was classified into four conditions: (1) mild: asymptomatic patients (2) moderate: symptomatic patients without pneumonia, comorbidity and risk factors for severe disease; (3) severe: symptomatic patients with mild pneumonia, comorbidity or risk factors for severe disease (any of these factors including age >60 years, obesity, chronic obstructive pulmonary disease, chronic kidney disease, cardiovascular disease, cerebrovascular disease, uncontrolled diabetes mellitus, liver cirrhosis, immunocompromised, or lymphocyte count <1000 cell/m3); and (4) critical: symptomatic patients with pneumonia together with resting O2 saturation <96% or exercise-induced hypoxemia.
Genomic DNA preparation and bioanalysis
Genomic DNA was extracted from 200 μL of peripheral ethylenediaminetetraacetic acid-anticoagulated blood using QIAamp® DNA Blood Mini Kit (Qiagen, Germany) and was adjusted to the concentration of 15 ng/µL with the total volume of 80 uL. DNA purity was evaluated by OD260/OD280 and OD260/OD230 ratios (NanoDropTM One/One Microvolume UV-Vis Spectrophotometers, Thermo Fisher Scientific, Wilmington, DE, USA). The acceptance criteria of DNA purity were OD260/OD280 ratio of 1.8–2.0 and OD260/OD230 ratio >1.5. DNA degradation was assessed on a 1% agarose gel using an appropriate size standard control. DNA concentration was also quantitated using Qubit™ dsDNA HS (High Sensitivity) Assay Kit (Thermo Fisher Scientific) before bioanalysis with AxiomTM Human Genotyping SARs-COV-2 Array (Thermo Fisher Scientific), which includes >800,000 SNPs of COVID-19 susceptibility, severity and immune response variants.
Genotype calling, quality control, and imputation
Genotype calling from intensity data file was performed with Axiom Analysis Suite (AxAS) version 5.1.1 software [25] using default parameters yielding 847,384 SNPs on 248 participants. Quality control (QC) was carried out following Ricopili pipeline [26] using criteria as follow: - SNPs were removed if call rate <0.98, call rate difference between cases and controls >0.02 or Hardy–Weinberg equilibrium p value <10−6 in controls or <10−10 in cases. Samples were also removed if call rate <0.98, |inbreeding coefficient | >0.2, or sex discordant were detected.
There were 558,132 SNPs left post-QC (cases/controls = 207/33; males/females = 88/152). Principal component analysis (PCA, Supplementary Fig. 1) for remaining QCed cases and controls was conducted using the Ricopili pipeline [26] with the default parameters to assess relatedness between samples and population stratification. In brief, SNPs were pruned to minimize linkage disequilibrium (LD) between SNPs with the criteria of R2 < 0.2, and the number of SNPs in the window for pruning was 200 until there were less than 100,000 SNPs. The resulting pruned SNPs were used to assess recent common ancestry and population stratification with the threshold of identity by descent (IBD) equals 0.2. After this step, there were 240 samples left for further analysis. With our inspection of the PC1 vs PC2 (Supplementary Fig. 1A), we remove additional two samples, leaving the final 222 samples for analyses (Supplementary Fig. 1A–C, cases/controls = 191/31). Genotype imputation was done for chromosomes 1–22 using Michigan Imputation Server [27]. The reference panel used was Genome Asia Pilot (GAsP) with reference genome version GRCh37/hg19.
Association analysis
Scalable and Accurate Implementation of GEneralized mixed model (SAIGE, https://github.com/weizhouUMICH/SAIGE) [28], a software-implemented for efficiently controlling for unbalanced case-control ratios, sample relatedness, and population stratification for GWAS, was applied for association analysis. SAIGE uses saddle point approximation to control case-control imbalance in a logistic mixed-effects model and reports score test results. Three models were carried out with the same covariates, including age, age2, sex, age*sex, 20 PCs, and phases of the COVID-19 outbreak in Thailand. Samples without age information were removed.
Figure 1 provides information on three GWAS models analyzed in this study. In Model 1: Susceptibility (total n = 222; cases/controls = 191/31), cases were patients of any severity. Controls were individuals who were exposed and tested negative for COVID-19. Model 2: Severity I compared 66 patients with moderate, severe, or critical conditions versus 125 controls who were patients with mild symptoms. In Model 3: Severity II, severity levels were analyzed as quantitative traits, coded as 0 for exposed individuals with negative PCR results (n = 31), and 1 to 4 for patients with mild (n = 125), moderate (n = 30), severe (n = 25), and critical (n = 11), respectively.
Fig. 1.

Study flow chart. GWAS genome-wide association study, PCR polymerase-chain-reaction
Linkage disequilibrium pattern
LD blocks using LDBlockShow [29] were obtained, and genes residing in the blocks which contained SNPs with statistical significance were acquired using the University of California, Santa Cruz (UCSC) Genome Browser [30].
Results
Baseline characteristics
Table 1 showed baseline characteristics of COVID-19 patients and individuals with COVID-19 exposure but negative PCR test results recruited in this study.
Table 1.
Characteristics of participants
| Characteristics | Individuals with the negative PCR test result | Individuals with the positive PCR test result | |||
|---|---|---|---|---|---|
| Mild | Moderate | Severe | Critical | ||
| N | 36 | 139 | 34 | 27 | 12 |
| Gender (Male/Female), (n) | 15/21 | 44/95 | 10/24 | 13/14 | 8/4 |
| Age (years), (mean ± standard deviation) | 45 ± 20 | 34 ± 11 | 42 ± 12 | 49 ± 17 | 51 ± 12 |
| The onset of disease (days) | 4 (1–6) | 4 (2–6) | 5 (3–7) | 3 (2–6) | 6 (5–7) |
| Concomitant disease (n(%)) | |||||
| Diabetes mellitus | 4 (11.1) | 7 (5.0) | 5 (14.7) | 5 (18.5) | 6 (50.0) |
| Hypertension | 11 (30.6) | 8 (5.8) | 1 (2.9) | 7 (25.9) | 3 (25.0) |
| Dyslipidemia | 8 (22.2) | 1 (0.7) | 0 (0.0) | 3 (11.1) | 4 (33.3) |
| Chronic kidney disease | 1 (2.8) | 0 (0.0) | 1 (2.9) | 1 (3.7) | 0 (0.0) |
| Cardiovascular diseases | 13 (36.1) | 7 (5.0) | 3 (8.8) | 6 (22.2) | 5 (41.7) |
| Lung disease | 1 (2.8) | 2 (1.4) | 0 (0.0) | 4 (14.8) | 0 (0.0) |
| Liver disease | 0 (0.0) | 1 (0.7) | 0 (0.0) | 2 (7.4) | 2 (16.7) |
| Immunocompromise host | 3 (8.3) | 0 (0.0) | 1 (2.9) | 1 (3.7) | 2 (16.7) |
| Cancer | 1 (2.8) | 1 (0.7) | 2 (5.9) | 2 (7.4) | 1 (8.3) |
| Symptoms and signs at admission (n(%)) | |||||
| Fever | 28 (77.8) | 71 (51.1) | 30 (88.2) | 18 (66.7) | 9 (75.0) |
| Cough | 30 (83.3) | 82 (59.0) | 28 (82.4) | 18 (66.7) | 10 (83.3) |
| Sore throat | 11 (30.6) | 61 (43.9) | 20 (58.8) | 10 (37.0) | 5 (41.7) |
| Runny nose | 16 (44.4) | 56 (40.3) | 15 (44.1) | 6 (22.2) | 5 (41.7) |
| Dyspnea | 15 (41.7) | 31 (22.3) | 14 (41.2) | 14 (51.9) | 9 (75.0) |
| Anosmia | 1 (2.8) | 31 (22.3) | 8 (23.5) | 2 (7.4) | 1 (8.3) |
| Dysuria | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 2 (16.7) |
| Hematuria | 7 (19.4) | 4 (2.9) | 5 (14.7) | 4 (14.8) | 5 (41.7) |
| Proteinuria | 8 (22.2) | 4 (2.9) | 4 (11.8) | 8 (29.6) | 8 (66.7) |
| Lung involvement (n(%)) | |||||
| None | 17 (47.2) | 127 (91.4) | 2 (5.9) | 1 (3.7) | 0 (0.0) |
| Focal | 10 (27.8) | 7 (5.0) | 12 (35.3) | 5 (18.5) | 0 (0.0) |
| Bilateral | 9 (25.0) | 5 (3.6) | 20 (58.8) | 21 (77.8) | 12 (100.0) |
| Medications | |||||
| Favipiravir (n(%)) | – | 8 (5.6) | 17 (50.0) | 26 (96.3) | 11 (91.7) |
| Time to starting favipiravir since admission (days) | – | 0 (0–1) | 2 (0–5) | 1 (0–3) | 1 (0–3) |
| Duration of favipiravir treatment (days) | – | 5 (5–5) | 5 (5–5) | 10 (10–10) | 14 (10–14) |
| Remdesivir (n(%)) | – | – | – | 1 (3.7) | 1 (8.3) |
| Time to starting remdesivir since admission (days) | – | – | – | 3 | 5 |
| Duration of remdesivir treatment (days) | – | – | – | 10 | 10 |
| Outcome of disease | |||||
| Time to clinical response (days) | – | 3 (2–3) | 6 (5–8) | 8 (6–12) | 19 (11–44) |
| Length of hospital stay before transferring to hospitel or discharge (days) | 2 (1–2) | 5 (3–9) | 8 (5–10) | 13 (10–16) | 22 (16–41) |
| Type of discharge (n(%)) | |||||
| Home | 36 (100.0) | 139 (100.0) | 33 (97.1) | 27 (100.0) | 10 (83.3) |
| Refer back to other hospitals | 0 (0.0) | 0 (0.0) | 1 (2.9) | 0 (0.0) | 1 (8.3) |
| Still in hospital | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (8.3) |
Data were presented in the median (interquartile range) unless stated otherwise
Genome-wide association analysis
No SNPs passed the genome-wide significance threshold in Model 1: Susceptibility. However, at a threshold of a p value <1 × 10−5, loci on chromosome 5q32 (position 148710242–148768047, p value 6.8745 × 10−6 – 6.8755 × 10−6; odds ratio 0.02, Fig. 2, Table 2, and Supplementary Table 1) and chromosome 9q21.13 (position 77748151–77762449, p value 2.3197 × 10−6 – 9.5083 × 10−6; odds ratio 0.11–0.13, Fig. 2, Table 2, and Supplementary Table 2) were suggested to be associated with COVID-19 disease susceptibility. A quantile–quantile (Q–Q) plot showed significant associations in the tail of the distribution with deflation (λ = 0.805; Supplementary Fig. 2A). The LD block containing SNPs with statistical significance (significant block) contained four genes on chromosome 5 (AFAP1L1, GRPEL2, PCYOX1L, and IL17B), and one gene on chromosome 9 (OSTF1), (Table 2 and Supplementary Fig. 2B, C). We further extended a segment of 200 kilobase pairs (kbp) in both directions from the boundary of the significant block. ABLIM3, BX640700, CSNK1A1, L26953, and MIR143 were also identified on chromosome 5 and C9orf41, BC043649, and NMRK1 were identified on chromosome 9 (Table 2).
Fig. 2.
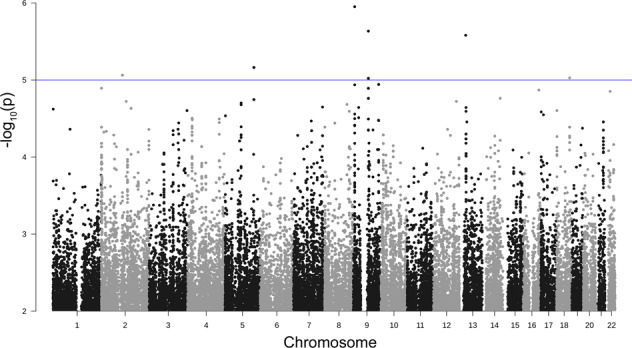
Manhattan plot of Model 1: susceptibility. Observed −log10 p values (y-axis) are shown for all SNPs on each autosomal chromosome (x-axis). The blue line indicates a suggestive line of p value <1 × 10−5
Table 2.
Summary of the loci and genes of the three models
| Model | Phenotypic impact | Chromosome | Position | p value | Odds ratio or Effect size | Genes in the significant block | Genes in extended of the significant block |
|---|---|---|---|---|---|---|---|
| Model 1 | Susceptibility | 5 | 148710242–148768047 | 6.8745 × 10−6 × 6.8755 × 10−6 | Odds ratio 0.02 | AFAP1L1 GRPEL2 PCYOX1L IL17B | ABLIM3, BX640700 CSNK1A1 L26953 MIR143 |
| 9 | 77748151–77762449 | 2.3197 × 10−6 – 9.5083 × 10−6 | Odds ratio 0.11–0.13 | OSTF1 | C9orf41 BC043649 NMRK1 | ||
| Model 2 | Severity I | 12 | 93426633–93446082 | 1.3490 × 10−6 – 4.3527 × 10−6 | Odds ratio 0.28–0.31 | LOC643339 | EEA1, LINC02412 |
| Model 3 | Severity II | 3 | 21141028–21235640 | 5.0649 × 10−7 – 2.5343 × 10−6 | Effect size 1.60–1.73 | – | VENTXP, ZNF385D |
Similar to Model 1, no SNPs passed the genome-wide significance threshold in Model 2: Severity I. Nevertheless, at a threshold of a p value <1 × 10−5, one locus on chromosome 12q22 (position 93456633–93446082) was plausibly associated with COVID-19 disease severity (p value 1.3490 × 10−6 – 4.3527 × 10−6; odds ratio 0.28–0.31; Fig. 3; Table 2; and Supplementary Table 3). A Q–Q plot was slightly deflated (λ = 0.997; Supplementary Fig. 3A). The associated region contains the LOC643339 gene (Table 2 and Supplementary Fig. 3B). No additional gene was found in the significant block. EEA1 and LINC02412 were found in a ±200 kbp segment from the significant block (Table 2).
Fig. 3.
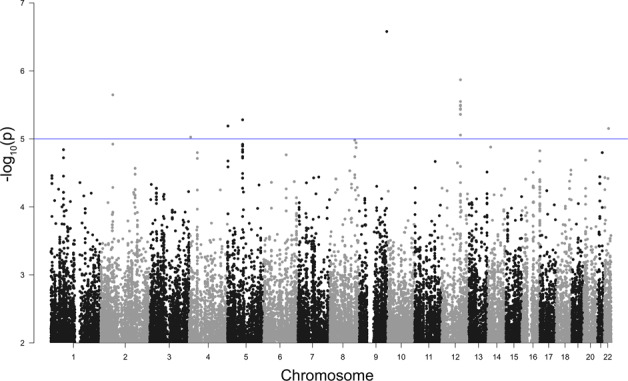
Manhattan plot of Model 2: severity I. Observed −log10 p values (y-axis) are shown for all SNPs on each autosomal chromosome (x-axis). The blue line indicates a suggestive line of p value <1 × 10−5
For Model 3: Severity II, no SNPs passed the genome-wide significance threshold; however, at a threshold of a p value <5 × 10−5, one locus on chromosome 3p24.3 (position 21141028–21235640) was identified to be associated with COVID-19 disease severity level (p value 5.0649 × 10−7 – 2.5343 × 10−6; Fig. 4; Table 2; and Supplementary Table 4). The Q–Q plot was slightly deflated in the middle but inflated at the tail of the distribution (λ = 0.931; Supplementary Fig. 4A). There was no gene in the significant region or the significant block (Table 2 and Supplementary Fig. 4B). However, in an extended segment (±200 kbp) from the boundary of the significant block, VENTXP7 and ZNF385D were identified (Table 2).
Fig. 4.
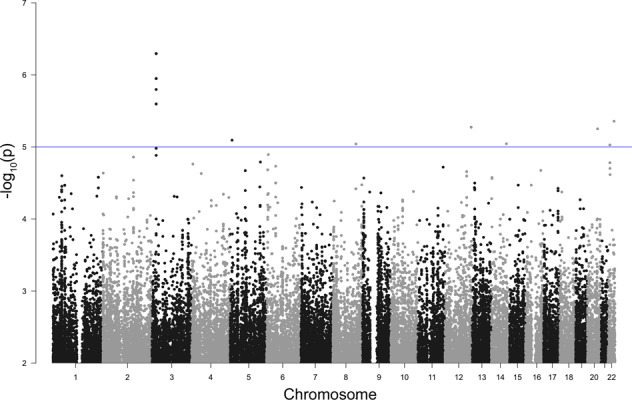
Manhattan plot of Model 3: severity II. Observed −log10 p values (y-axis) are shown for all SNPs on each autosomal chromosome (x-axis). The blue line indicates a suggestive line of p value <1 × 10−5
Discussion
Different ethnicity has different genetic composition. Therefore, studying various human populations could give better biological insights. We performed a GWAS in a Thai population to identify genetic loci associated with COVID-19 susceptibility and severity. As a result, two suggestive loci on chromosomes 5q32 and 9q21.13 and two suggestive loci on chromosomes 12q22 and 3p24.3 were associated with COVID-19 disease susceptibility and disease severity, respectively.
Three GWAS studies have shown associations of loci on chromosome 12 to the severity of COVID-19 disease [20, 24, 31]. Nelson et al. [31] reported three loci associated with plasma angiotensin-2 concentration in men. One was on chromosome 12 around the HNF1α gene, which encodes a transcription factor in regulating ACE2 expression. Results from the Genetics Of Mortality in Critical Care (GenOMICC) study in 2244 critically ill COVID-19 patients in intensive care units across the UK identified the associations on chromosomes 12q24.13, 19p13.2, 19p13.3, and 21q22.1, which were linked to two biological mechanisms including innate immunity (IFNAR2 and OAS) and host-driven inflammatory lung injury (DPP9, TYK2, and CCR2) [20]. Likewise, the most recent GWAS study from the COVID-19 HGI group also revealed the association of chromosome 12 on COVID-19 severity (OAS1) [24].
Here, we have suggested two plausible genome-wide significant associations on chromosomes 5q32 and 9q21.13 in our disease susceptibility model. Since the controls were all collected from the same hospital and same ethnics as cases, stratification bias is unlikely. Allele frequencies in Thai were 0.03 and 0.15 for chromosome 5 and 0.11 and 0.31–0.32 for chromosome 9, for cases and controls, respectively (Supplementary Tables 1, 2). The association signal on chromosome 5q32 coincided with IL17B. The protein encoded by IL17B is a T cell-derived cytokine known as interleukin-17B (IL-17B). Immunohistochemical analysis of several tissues indicated that IL-17B is primarily localized to chondrocytes and neurons [32, 33]. Furthermore, IL-17B was reported to play a role as a proinflammatory inducer in inflammatory disease, stimulating the release of tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β) from a monocytic cell line resulting in neutrophil infiltration [32, 33]. Evidence suggests that IL-17B may participate in the host defense mechanism of infection, but data were still conflicting [33–35]. IL-17B concentrations were increased in patients with community-acquired pneumonia, and the mechanism was linked to the evidence that IL-17B induces gene and protein expression of interleukin-8 in bronchial epithelial cells [34]. However, a study of colitis in mice infected with C. rodentium, a murine-specific model for human Gram-negative E. coli infection, reported IL-17B as a protective factor for infection [35]. Regarding COVID-19, cytokines are fundamental to the pathophysiology of SARs-CoV-2 viral infection. Some cytokines, including TNF-α and IL-1β, appear detrimental, particularly in the cytokine storm [36] and changes in the hemopoietic system, including neutrophilia and lymphopenia during infection, are significant prognostic factors [36]. Hence, further studies for the role of IL17B as a host factor related to the susceptibility of SARs-CoV-2 viral infection is warranted.
Our severity models have further suggested two plausible loci on chromosomes 12q22 and 3p24.3 linked to disease severity. The UK GWAS identified a gene cluster that encodes antiviral restriction enzyme activator on chromosome 12q24.13 (OAS1, OAS2, and OAS3) associated with severe COVID-19 disease [20]. Additionally, loci on chromosome 3 were observed to be associated with disease susceptibility (rs11919389: RPL24) and disease severity (rs10490770: LZTFL1) in the COVID-19 HGI report [24]. From our results, the extended LD block on chromosomes 12q22 contains EEA1, LOC643339, and LINC02412. Early endosomal antigen 1, encoded by EEA1, is a protein found in cytosol, endosome, and plasma membrane of cells in various organs, including the lungs [37]. Recently, a study reported the microscopic changes in the small airways and lung parenchyma of smokers and patients with COPD, showing an increased expression of SARs-CoV-2 receptor ACE2 and proteins involved in viral entry, including EEA1 [38]. Coronavirus is an enveloped virus that requires cellular ACE2 receptor binding and membrane fusion to enter and eject their RNA into the host cell. The viruses attack the host endocytosis pathway to enter via endosome, proceed to lysosomes, and fuse the viral and lysosomal membrane [39–41]. Our finding of EEA1 residing in an associated locus linked to COVID-19 disease severity suggests an overactive cellular response that may facilitate viral entry and processing contributing to COVID-19 severity. Additionally, LOC643339 encodes a long non-coding RNA (lncRNA) with little known functions. However, some lncRNAs have an essential role in pathogenic infection [42]. Therefore, further studies to determine the role of LOC643339 in regulating SARs-CoV-2 propagation seem justified.
The limitation of this study was the relatively small sample size. However, we believe that the mitigation of the widespread devastation caused by the COVID-19 pandemic requires scientific contributions from every corner of the world. The number of exposed individuals with negative PCR tests was smaller than the cases. However, as the use of the general population as a control group carried the potential for misclassification bias with an unknown extent of developing COVID-19 disease [43], we decided not to include the general population in our control group. The small sample size may lessen the likelihood that the association found is real (decreasing the chance of getting a true positive without increasing a false positive), but it could still be true [44]. Additionally, the reported effect estimates (e.g., odds ratio) can be inflated due to the increased sampling variability from a small sample size. Further studies with more samples are needed to verify the findings. In addition, we are participating in an international collaborative effort to uncover the host factors associated with COVID-19. Finally, our report will provide more details of the Thai population to complement the global initiative.
In conclusion, our GWAS study in Thai COVID-19 patients suggests plausible disease susceptibility loci on chromosome 5q32 containing IL17B and chromosome 9q21.13. In addition, the disease severity was suggestive of being linked to loci on chromosomes 12q22 containing LOC643339 and EEA1 and 3p24.3.
Supplementary information
Acknowledgements
This work was supported by Ratchadapiseksompotch Fund, Faculty of Medicine, Chulalongkorn University (RA (PO)003/63), Ratchadapiseksompotch Endowment Fund, Chulalongkorn University (764002-HE01), the e-ASIA Joint Research Program (e-ASIA JRP) as administered by the National Science and Technology Development Agency, TSRI Fund (CU_FRB640001_01_30_10), Thailand Research Fund (DPG6180001), and Health Systems Research Institute, Thailand (64–132). MC and TBS were supported by Ratchadaphiseksomphot Fund, Chulalongkorn University, for Postdoctoral Fellowship. We wish to thank Dr. Surachai Leksuwankun and Dr. Suwiwat Bunnag for their support in collecting the clinical data.
Author contributions
PC, NH, and VS conceived and designed the study; WJ, PT, and OP acquired all clinical data; PS coordinated for sample collection; MC and TBS performed all bioanalysis; MP performed all bioinformatics analysis; PC, MC, and MP interpreted results and wrote the manuscript; PC, NH, and VS acquired funding; CP, NH, and VS revised the manuscript; PC finalized the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s10038-021-01009-6.
References
- 1.WHO Coronavirus (COVID-19) Dashboard. https://covid19.who.int/ (2021).
- 2.Pongpirul WA, Pongpirul K, Ratnarathon AC, Prasithsirikul W. Journey of a Thai taxi driver and novel coronavirus. N Engl J Med. 2020;382:1067–8. doi: 10.1056/NEJMc2001621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wacharapluesadee S, Tan CW, Maneeorn P, Duengkae P, Zhu F, Joyjinda Y, et al. Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nat Commun. 2021;12:972. doi: 10.1038/s41467-021-21240-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Department of Disease Control MoPH, Thailand; Coronavirus disease (COVID-19): Thailand situation. https://ddc.moph.go.th/viralpneumonia/eng/index.php (2021).
- 5.Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395:1054–62.. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X, Xu S, Yu M, Wang K, Tao Y, Zhou Y, et al. Risk factors for severity and mortality in adult COVID-19 inpatients in Wuhan. J Allergy Clin Immunol. 2020;146:110–8. doi: 10.1016/j.jaci.2020.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen R, Liang W, Jiang M, Guan W, Zhan C, Wang T, et al. Risk factors of fatal outcome in hospitalized subjects with coronavirus disease 2019 from a nationwide analysis in China. Chest. 2020;158:97–105. doi: 10.1016/j.chest.2020.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Docherty AB, Harrison EM, Green CA, Hardwick HE, Pius R, Norman L, et al. Features of 20 133 UK patients in hospital with covid-19 using the ISARIC WHO Clinical Characterisation Protocol: prospective observational cohort study. BMJ. 2020;369:m1985. doi: 10.1136/bmj.m1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richardson S, Hirsch JS, Narasimhan M, Crawford JM, McGinn T, Davidson KW, et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA. 2020;323:2052–9. doi: 10.1001/jama.2020.6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leong A, Cole J, Brenner LN, Meigs JB, Florez JC, Mercader JM, et al. Cardiometabolic risk factors for COVID-19 susceptibility and severity: a Mendelian randomization analysis. PloS Med. 2021;18:e1003553. doi: 10.1371/journal.pmed.1003553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Freuer D, Linseisen J, Meisinger C. Impact of body composition on COVID-19 susceptibility and severity: a two-sample multivariable Mendelian randomization study. Metabolism. 2021;118:154732. doi: 10.1016/j.metabol.2021.154732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leong A, Cole JB, Brenner LN, Meigs JB, Florez JC, Mercader JM. Cardiometabolic risk factors for COVID-19 susceptibility and severity: a Mendelian randomization analysis. PLoS Med. 2021;18:e1003553. doi: 10.1371/journal.pmed.1003553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kachuri L, Francis SS, Morrison ML, Wendt GA, Bosse Y, Cavazos TB, et al. The landscape of host genetic factors involved in immune response to common viral infections. Genome Med. 2020;12:93. doi: 10.1186/s13073-020-00790-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang F, Huang S, Gao R, Zhou Y, Lai C, Li Z, et al. Initial whole-genome sequencing and analysis of the host genetic contribution to COVID-19 severity and susceptibility. Cell Discov. 2020;6:83. doi: 10.1038/s41421-020-00231-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gemmati D, Bramanti B, Serino ML, Secchiero P, Zauli G, Tisato V. COVID-19 and individual genetic susceptibility/receptivity: role of ACE1/ACE2 genes, immunity, inflammation and coagulation. Might the double X-chromosome in females be protective against SARS-CoV-2 compared to the single X-chromosome in males? Int J Mol Sci. 2020;21:3474. [DOI] [PMC free article] [PubMed]
- 16.Pathak GA, Singh K, Miller-Fleming TW, Wendt F, Ehsan N, Hou K, et al. Integrative analyses identify susceptibility genes underlying COVID-19 hospitalization. Nat Commun. 2021;12:4569. doi: 10.1038/s41467-021-24824-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Severe Covid GG, Ellinghaus D, Degenhardt F, Bujanda L, Buti M, Albillos A, et al. Genomewide association study of severe covid-19 with respiratory failure. N Engl J Med. 2020;383:1522–34. doi: 10.1056/NEJMoa2020283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma Y, Huang Y, Zhao S, Yao Y, Zhang Y, Qu J, et al. Integrative genomics analysis reveals a 21q22.11 locus contributing risk to COVID-19. Hum Mol Genet. 2021;30:1247–58. [DOI] [PMC free article] [PubMed]
- 19.Andolfo I, Russo R, Lasorsa VA, Cantalupo S, Rosato BE, Bonfiglio F, et al. Common variants at 21q22.3 locus influence MX1 and TMPRSS2 gene expression and susceptibility to severe COVID-19. iScience. 2021;24:102322. doi: 10.1016/j.isci.2021.102322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pairo-Castineira E, Clohisey S, Klaric L, Bretherick AD, Rawlik K, Pasko D, et al. Genetic mechanisms of critical illness in COVID-19. Nature. 2021;591:92–8. doi: 10.1038/s41586-020-03065-y. [DOI] [PubMed] [Google Scholar]
- 21.Chen HH, Shaw DM, Petty LE, Graff M, Bohlender RJ, Polikowsky HG, et al. Host genetic effects in pneumonia. Am J Hum Genet. 2021;108:194–201. doi: 10.1016/j.ajhg.2020.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu J, Li C, Wang S, Li T, Zhang H. Genetic variants are identified to increase risk of COVID-19 related mortality from UK Biobank data. Hum Genomics. 2021;15:10. doi: 10.1186/s40246-021-00306-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hernandez Cordero AI, Li X, Milne S, Yang CX, Bosse Y, Joubert P, et al. Multi-omics highlights ABO plasma protein as a causal risk factor for COVID-19. Hum Genet. 2021;140:969–79.. doi: 10.1007/s00439-021-02264-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Initiative C-HG. Mapping the human genetic architecture of COVID-19. Nature. 2021;600:472–77. [DOI] [PMC free article] [PubMed]
- 25.Thermo Fisher Scientific Inc. AxiomTM Analysis Suite (AxAS) v5.1. https://downloads.thermofisher.com/AffymetrixSoftwares/Axiom_Analysis_Suite_AxAS_v5.1_User_Guide.pdf (2018).
- 26.Lam M, Awasthi S, Watson HJ, Goldstein J, Panagiotaropoulou G, Trubetskoy V, et al. RICOPILI: rapid imputation for COnsortias PIpeLIne. Bioinformatics. 2020;36:930–3. doi: 10.1093/bioinformatics/btz633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michigan imputation server. https://imputationserver.sph.umich.edu/ (2021).
- 28.Zhou W, Nielsen JB, Fritsche LG, Dey R, Gabrielsen ME, Wolford BN, et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat Genet. 2018;50:1335–41.. doi: 10.1038/s41588-018-0184-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dong SS, He WM, Ji JJ, Zhang C, Guo Y, Yang TL. LDBlockShow: a fast and convenient tool for visualizing linkage disequilibrium and haplotype blocks based on variant call format files. Brief Bioinform. 2020;22:bbaa227. [DOI] [PubMed]
- 30.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nelson CP, Sama IE, Codd V, Webb TR, Ye S, Lang CC, et al. Genetic associations with plasma angiotensin converting enzyme 2 concentration: potential relevance to COVID-19 risk. Circulation. 2020;142:1117–9. doi: 10.1161/CIRCULATIONAHA.120.049007. [DOI] [PubMed] [Google Scholar]
- 32.Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity. 2011;34:149–62.. doi: 10.1016/j.immuni.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 33.Pappu R, Ramirez-Carrozzi V, Sambandam A. The interleukin-17 cytokine family: critical players in host defence and inflammatory diseases. Immunology. 2011;134:8–16. doi: 10.1111/j.1365-2567.2011.03465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou J, Ren L, Chen D, Lin X, Huang S, Yin Y, et al. IL-17B is elevated in patients with pneumonia and mediates IL-8 production in bronchial epithelial cells. Clin Immunol. 2017;175:91–8. doi: 10.1016/j.clim.2016.12.008. [DOI] [PubMed] [Google Scholar]
- 35.Reynolds JM, Lee YH, Shi Y, Wang X, Angkasekwinai P, Nallaparaju KC, et al. Interleukin-17B antagonizes interleukin-25-mediated mucosal inflammation. Immunity. 2015;42:692–703. doi: 10.1016/j.immuni.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agbuduwe C, Basu S. Haematological manifestations of COVID-19: from cytopenia to coagulopathy. Eur J Haematol. 2020;105:540–6. doi: 10.1111/ejh.13491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.EEA1 early endosome antigen 1 [Homo sapiens (human)]. https://www.ncbi.nlm.nih.gov/gene/8411 (2021).
- 38.Eapen MS, Lu W, Hackett TL, Singhera GK, Thompson IE, McAlinden KD, et al. Dysregulation of endocytic machinery and ACE2 in small airways of smokers and COPD patients can augment their susceptibility to SARS-CoV-2 (COVID-19) infections. Am J Physiol Lung Cell Mol Physiol. 2021;320:L158–L63.. doi: 10.1152/ajplung.00437.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burkard C, Verheije MH, Wicht O, van Kasteren SI, van Kuppeveld FJ, Haagmans BL, et al. Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog. 2014;10:e1004502. doi: 10.1371/journal.ppat.1004502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med. 2005;11:875–9. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang X, Zheng J, Yan Y, Ruan Z, Su Y, Wang J, et al. Angiotensin-converting enzyme 2 regulates autophagy in acute lung injury through AMPK/mTOR signaling. Arch Biochem Biophys. 2019;672:108061. doi: 10.1016/j.abb.2019.07.026. [DOI] [PubMed] [Google Scholar]
- 42.Shirahama S, Miki A, Kaburaki T, Akimitsu N. Long non-coding RNAs involved in pathogenic infection. Front Genet. 2020;11:454. doi: 10.3389/fgene.2020.00454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wellcome Trust Case Control C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–78. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Button KS, Ioannidis JP, Mokrysz C, Nosek BA, Flint J, Robinson ES, et al. Power failure: why small sample size undermines the reliability of neuroscience. Nat Rev Neurosci. 2013;14:365–76. doi: 10.1038/nrn3475. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.