

Abstract
We conducted a single-center, single-arm, open-label, dose-escalation phase 1 clinical trial to evaluate the tolerability of a single intravenous injection of ciprofol emulsion for the induction of short-term general anesthesia. Four doses of ciprofol (0.15 mg/kg, n = 2; 0.4 mg/kg, n = 10; 0.6 mg/kg, n = 6; 0.9 mg/kg, n = 6) were administered. Twenty-four subjects were enrolled, with 18 subjects in the 0.4 to 0.9 mg/kg dosage groups included in the data analysis. In total, 37 mild and 4 moderate adverse events (AEs), including 9 abnormal limb movements (3 moderate cases), 8 cases of sinus bradycardia, 11 cases of prolonged QTcF interval (including 1 moderate case), and 1 case of hypotension, were found, but no serious AEs were reported. The Modified Observer’s Assessment of Alertness/Sedation (MOAA/S) scores rapidly decreased after ciprofol administration. The duration of recovery of the verbal response, loss of verbal response duration, the duration of MOAA/S ≤1 and the duration until the return of responsiveness were all increased in a dose-dependent manner. The durations of bispectral index values <60 (6, 8 and 12 min) were similar to the durations of loss of verbal response (6, 8 and 14 min) and MOAA/S ≤1 (5, 5.5 and 13.5 min) in the 0.4, 0.6 and 0.9 mg/kg dose groups, respectively. The plasma concentration reached a peak value approximately 2 min after injection in the 0.4-0.9 mg/kg groups and all subjects fully recovered after ciprofol administration, with the shortest time being 9.2 min in the 0.4 mg/kg group. A ciprofol dosing regimen of 0.4-0.9 mg/kg was well-tolerated and exhibited rapid onset and recovery properties.
Keywords: Anesthetic, ciprofol, pharmacokinetics, pharmacodynamics, sedation
Introduction
Since propofol was first approved for clinical use in 1986, it has become one of the most commonly used intravenous general anesthetic agents, mainly because of its rapid onset and fast recovery characteristics [1-3]. However, a recent trend in pharmacology is to improve the pharmacokinetic (PK), pharmacodynamic (PD) and side effects of similar molecules by modifying the chemical structures of existing drugs [4]. Ciprofol (HSK3486) is a newly developed structural analog of propofol and a short-acting intravenous drug for induction and maintenance anesthesia in adults. Its application includes anesthesia/sedation during invasive endoscopy and for adult intensive care unit (ICU) patients. The active ingredient of ciprofol is similar to propofol, but with single R-configured diastereoisomers [5]. Besides, in preclinical experiments, the half-maximal effective inhibitory concentration (EC50) of ciprofol on γ-aminobutyric acid type A (GABAA) receptor-mediated currents was 5 times less than that of propofol (5.3 vs. 1.1 µM, respectively) [5]. In addition, the EC50 of the loss of the righting reflex in rat, dog and pig was also 5 times less than that of propofol (unpublished data). Phase 1a, 1b clinical trials of ciprofol were completed in Australia and revealed median terminal half-life times ranging from 68.8 to 245.7 minutes for ciprofol doses of 128 to 810 μg/kg with median central nervous system equilibrium half-life time of 1.3 minutes in males and 1.8 minutes in females. Otherwise ciprofol doses of 432 and 540 μg/kg had comparable clinical effects without different adverse event outcomes as 2,500 μg/kg propofol, which is commonly used for anesthesia in clinical practice [6]. Due to potential differences including genetic factors, lifestyle and environment [7-9], we carried out a phase 1 study on Chinese healthy subjects, with the primary endpoint of evaluating anesthetic dose monitoring of ciprofol.
Materials and methods
Conduct of the study
The trial was carried out in healthy adult subjects as a single-center, open-label, single-arm, dose-escalation phase 1 study in the West China Hospital of Sichuan University (Chengdu, China). The ethical committee of our institution approved the trial (approval No. 2016-25). Written informed consent was obtained from all subjects before they were enrolled. The study protocol was registered at ClinicalTrial.gov (registration number: NCT03773835).
Endpoints
The primary endpoint was the evaluation of anesthetic dose monitoring and the secondary endpoint was the evaluation of PD and PK parameters.
Subjects
Briefly, the inclusion criteria for this study were healthy adult male or female subjects, aged 18-49 years, weighing more than 45 kg, with a body mass index (BMI) between 19 and 24 kg/m2. Additional inclusion and exclusion criteria are listed in Supplementary File 1.
Procedures
After signing informed consent, subjects who met the inclusion criteria were assigned to the trial in a random sequence and all completed screening tests 21 days prior to commencement of the study. Subjects were initially assigned to 4 ciprofol dose groups (0.15, 0.4, 0.6 and 0.9 mg/kg). The day before the trial started, subjects were accommodated in a standardized phase 1 test room for good clinical practice (GCP). They were required to fast (no food or liquids of any kind) for at least 8 h before drug administration. PK/PD values were measured and safety items were recorded before and after injection of ciprofol. At the beginning of the trial, 2 subjects in the 0.15 mg/kg group and 4 subjects in the 0.4 mg/kg group were administered medication using infusion pumps. During the administration, it was found that the actual infusion volume of the syringe pump was significantly different from the planned infusion volume, revealing that the infusion volume of the syringe pump was inaccurate. In order to ensure the accuracy of the dosing volume, we decided to change the method of administration to manual injection, which was completed within 1 min. Considering that changing the drug administration method may have had an impact on PK analysis, the subjects who received a drug with the infusion pump were excluded from the PK and PD analysis (Figure 1). An anesthetist injected the study drug as a single bolus within 1 min and remained in the test room until a subject’s responsiveness returned with normal cardiovascular and respiratory functions. After drug administration, oxygen was administered through an oxygen mask at a flow rate of 10 L/min until the subject was awake. Epinephrine, atropine and metaraminol were prepared but were not administered unless absolutely necessary.
Figure 1.
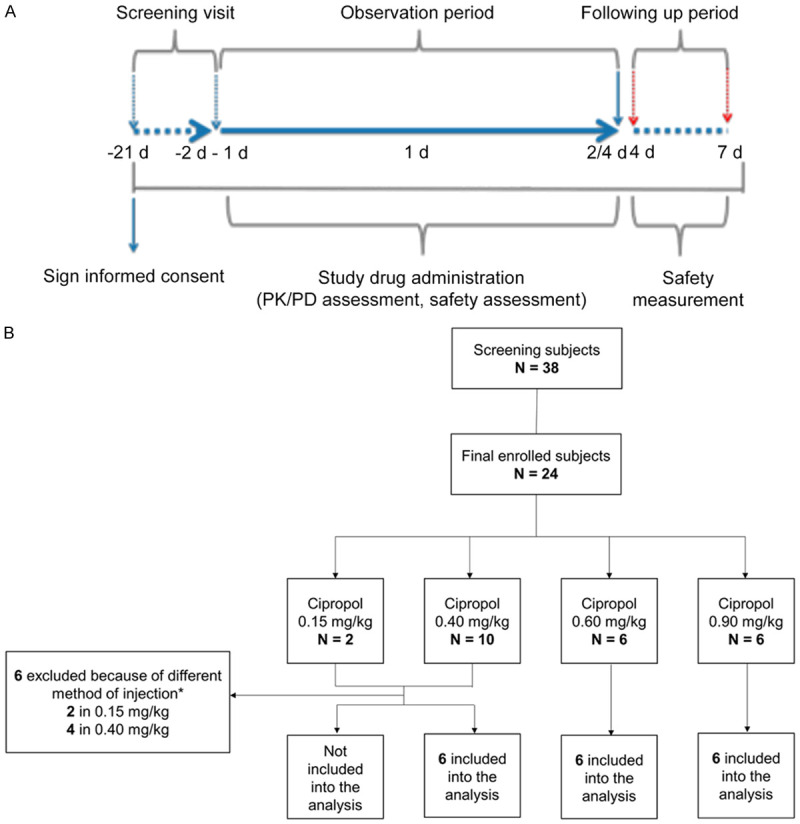
Flow diagrams of the study. A. Scheme of the study periods. B. Scheme of the study subjects. *At the beginning of the experiment, 2 subjects in the 0.15 mg/kg group and 4 subjects in the 0.4 mg/kg group received ciprofol by infusion pump, but the subsequent administrations were changed to manual injections, which were completed within 1 min. Subjects administered medication with an infusion pump were excluded from the PK and PD analyses.
Modified Observer’s Assessment of Alertness/Sedation (MOAA/S) [10] scores were assessed once 5 min before drug administration was initiated, which were taken as the baseline value, and then every 1 min for 30 min after administration, and once every 5 min for the next 30 min. If MOAA/S = 5 occurred consecutively 3 times during MOAA/Sverbal, stimuli ceased and recovery parameters were carefully recorded. The time to a subject becoming fully alert was determined from the time of the first record for MOAA/S <5 after drug administration until the first measurement of MOAA/S = 5 for 3 consecutive measurements. Heart rate (HR), blood pressure (BP) and a 12-lead real-time electrocardiogram (ECG) were monitored continuously during the trial. After intensive safety, PD and PK assessments in the morning of the second day, subjects left the trial unit and were followed up at 4-7 days intervals (up to 7 days), except for subjects in the 0.6 mg/kg group. Subjects in the 0.6 mg/kg group were required to stay in the hospital to perform 24-48 h urine and 24-72 h fecal tests, and complete the follow-up examination in the morning of day 4. The flow of subjects throughout testing, the study protocol and follow-ups are shown schematically in Figure 1.
Safety measurements
Adverse events (AEs) were monitored according to the MedDRA Preferred Term (version 13.0), for the observation period from the administration of the first dose to the last follow-up visit. Investigators monitored vital signs (HR, BP and respiratory parameters), laboratory measurements and a 12-lead ECG during the trial. During drug administration, at least 5 min after completion of anesthesia monitoring placements and establishment of venous access, a 12-lead ECG test was conducted every 5 min for 3 times, and the mean measurement was taken as the baseline value. Subsequently, data (QT interval, QTcF interval, PR interval, QRS interval, RR interval, ST segment changes and U waves occurrence) were recorded at 0.5 min, 1 min, 2 min, 3 min, 5 min, 8 min, 15 min, 30 min (±1 min), 1 h (±2 min), 1.5 h (±2 min), 2 h (±2 min), 3 h (±2 min), 4 h (±2 min), 6 h (±3 min), 8 h (±4 min) and 24 h (±5 min) after drug administration. If abnormal clinically significant data occurred at any time point, it was carefully recorded. All ECGs were evaluated by a qualified physician for the presence of abnormalities and any clinically significant findings were documented as potential AEs.
When chest wall motion disappeared for longer than 30 s or there was an absence of expired CO2 waveform for >15 s, it was recorded as apnea and positive airway pressure ventilation was applied. Injection pain was evaluated 0-2 min after trial drug dosing. Mild injection pain was defined as subjects showing small finger movements of the injection hand without obvious withdrawal. Moderate injection pain was defined as subjects showing obvious injection hand movements. Severe injection pain was defined as obvious injection arm withdrawal accompanied by increases in HR and BP. Definitions of AEs and serious adverse events (SAEs) are given in detail in Supplementary File 2 and Supplementary Table 1.
Plasma sampling and PK measurements
All venous blood samples were collected from a venous access in the contralateral arm of the administration site every 30 min before study drug injection and 0.5 min, 1 min, 2 min, 3 min, 5 min, 8 min, 15 min, 30 min, 1 h, 1.5 h, 2 h, 3 h, 4 h, 6 h, 8 h and 24 h after IV injection. Four mL of blood samples were centrifuged at 2-8°C, 3,000 RPM for 10 min to separate out the plasma. These plasma samples were then used to detect PK parameters by Wuxi AppTec (Shanghai, China). Further processing of the plasma samples is described in Supplementary File 3.
Population PK/PD modeling
Clinical PK (plasma concentration), PD (MOAA/S scores and bispectral index (BIS)) data were modeled by Nonlinear Mixed Effects Modelling (NONMEM), and evaluated by bootstrap and graphic methods. Finally, the parameters of the population PK/PD model effect-site elimination rate constant (Keo), the effect-site equilibration half-time (t1/2Keo) and half-effective target effect-site concentration (Ce50) as the main parameters) were obtained. For the population PK model, a one-compartment model, two-compartment model, three-compartment model and a saturable PK model with/without time lag (Tlag) were assessed. Since the objective function value (OFV) of the three-compartment model with Tlag is the minimum, it was chosen as the basic model of PK. The additive error, proportional error and proportional-additive error models were investigated as statistical models. Among them, the OFV of the proportion-additive error model was the minimum, but the covariance step failed in fitting, so a more stable proportional error model was chosen. In the final models, intra-individual variation was described by the proportional model and inter-individual variation by the exponential model. After evaluation using the bootstrap method and visual predictive checks (VPC), the results indicated that the values of parameters in the original samples were stable and reliable, and that this population PK model could be used to describe the PK characteristics of ciprofol. For the population PK/PD model, the sigmoid Emax model with baseline was selected for PD models (BIS and MOAA/S); PK and PD models were linked using an effect-compartment with time lag (Telag). As the OFV in the additive error model was least, the additive error model was selected to describe population PK/PD models. In the final models, intra-individual variation was described by the proportional model and the inter-individual variation was described by the exponential model. Finally, the evaluation results of the bootstrap method and VPC indicated that the values of parameters in the original samples were stable and reliable, and this population PK/PD model could be used to describe the changes of BIS and MOAA/S after administration of ciprofol.
Dose selection
The dose selection (0.15, 0.4, 0.6 and 0.9 mg/kg) for this phase 1 trial was based on the previous published Australian phase 1a and phase 1b clinical trials [6]. Two subjects did not exhibit sedation in the 0.15 mg/kg group (MOAA/S = 5 and BIS value between 96-98). Thus, the study team decided to stop further enrollments for this dose and move to the next highest dose. When escalated to the 0.9 mg/kg dosage group, all subjects reached deep sedation/anesthesia (loss of verbal response (LORverbal, defined as MOAA/S <3/MOAA/S ≤1).
After each dose assessment, the study team evaluated all available safety and PD data for ciprofol and collectively agreed on the next escalating dose to be administered.
PD measurements
The depth of sedation/anesthesia was assessed using a MOAA/S score and the BIS value. Loss of eyelash reflexes and orientation recovery were used to assess the onset and offset of sedation/anesthesia. Modified quality of recovery 9 (QoR-9) [11] (Supplementary Table 2) was used to evaluate the quality of subject recovery from anesthesia after 30±5 min when MOAA/S ≥4, measured before injection and 24 h±30 min after injection.
Data acquisition and management
The data acquisition and management methods are explained in detail in Supplementary File 4.
Statistical analysis
The sample size was based on other dose-escalation phase 1 studies [12,13] to ensure adequate subject safety and robust PK/PD data to achieve the objectives of the study. In addition, in order to expose as few subjects as possible to the study medication and procedures, the sample size for each dose group was 6. Data were summarized using descriptive statistics by Statistical Analysis System (SAS, ver. 9.3). Continuous variables were presented as the mean ± standard deviation (SD) or medians (quartile range). The bootstrap method was used to evaluate the stability of the population PK/PD models and a total of 1000 bootstrap replications were used to provide the parameter median and confidence intervals (CIs).
Results
A total of 24 subjects were enrolled in the study and 18 were included in the final analysis (Figure 1). The demographic characteristics were similar among three cohorts (Table 1).
Table 1.
Demographics
| 0.4 mg/kg (N = 6) | 0.6 mg/kg (N = 6) | 0.9 mg/kg (N = 6) | |
|---|---|---|---|
| Age* | 26.0 (25.0, 27.0) | 26.0 (25.0, 27.0) | 26.0 (25.0, 27.0) |
| Height* | 168.0 (164.0, 177.0) | 160.0 (155.0, 163.0) | 164.0 (163.0, 167.0) |
| Weight* | 65.5 (60.1, 74.0) | 52.5 (48.0, 58.0) | 57.5 (55.5, 65.0) |
| BMI (kg/m2)* | 23.1 (22.3, 23.6) | 21.1 (20.4, 21.8) | 21.4 (20.4, 23.6) |
| Gender, n (%) | |||
| Male | 3 (50.0) | 3 (50.0) | 3 (50.0) |
| Female | 3 (50.0) | 3 (50.0) | 3 (50.0) |
data are presented as the median (1st to 3rd) quartile.
Safety data
A total of 41 AEs in 18 subjects were reported during the trial. The incidence of AEs was 83.3% (15 of 18 subjects) (Table 2). Most of the AEs were considered mild in intensity and unrelated to the trial medication. No cases of SAEs were reported in any subject and no subjects withdrew from the trial because of AEs. The AEs considered probably or definitely related to the trial drug were mainly mild abnormal limb movements, sinus bradycardia and prolonged QTcF intervals.
Table 2.
The incidence of AEs according to SOC and PT
| SOC, PT | 0.4 mg/kg (N = 6) | 0.6 mg/kg (N = 6) | 0.9 mg/kg (N = 6) | |||
|---|---|---|---|---|---|---|
|
|
|
|
||||
| Number of subjects (%) | Number of events | Number of subjects (%) | Number of events | Number of subjects (%) | Number of events | |
| Total | 5 (83.3) | 13 | 5 (83.3) | 7 | 5 (83.3) | 21 |
| Nervous system disorders | 2 (33.3) | 2 | 2 (33.3) | 2 | 5 (83.3) | 5 |
| Abnormal limb movements | 2 (33.3) | 2 | 2 (33.3) | 2 | 5 (83.3) | 5 |
| Cardiac disorders | 4 (66.7) | 5 | 2 (33.3) | 2 | 2 (33.3) | 2 |
| Sinus bradycardia | 4 (66.7) | 4 | 2 (33.3) | 2 | 2 (33.3) | 2 |
| Atrioventricular block | 1 (16.7) | 1 | 0 (0) | 0 | 0 (0) | 0 |
| Investigations | 1 (16.7) | 4 | 1 (16.7) | 1 | 4 (66.7) | 7 |
| Prolonged QT interval | 1 (16.7) | 4 | 1 (16.7) | 1 | 3 (50.0) | 6 |
| Elevated urobilinogen | 0 (0) | 0 | 0 (0) | 0 | 1 (16.7) | 1 |
| Gastrointestinal disorders | 0 (0) | 0 | 1 (16.7) | 1 | 2 (33.3) | 2 |
| Thirst | 0 (0) | 0 | 0 (0) | 0 | 1 (16.7) | 1 |
| Canker sore | 0 (0) | 0 | 1 (16.7) | 1 | 1 (16.7) | 1 |
| General disorders and administration site conditions | 1 (16.7) | 1 | 0 (0) | 0 | 2 (33.3) | 2 |
| Medical devices related pain | 1 (16.7) | 1 | 0 (0) | 0 | 2 (33.3) | 2 |
| Infections and infestations | 0 (0) | 0 | 0 (0) | 0 | 1 (16.7) | 1 |
| Influenza | 0 (0) | 0 | 0 (0) | 0 | 1 (16.7) | 1 |
| Musculoskeletal and connective tissue disorders | 1 (16.7) | 1 | 0 (0) | 0 | 0 (0) | 0 |
| Musculoskeletal discomfort | 1 (16.7) | 1 | 0 (0) | 0 | 0 (0) | 0 |
| Psychiatric disorders | 0 (0) | 0 | 0 (0) | 0 | 1 (16.7) | 1 |
| Dysphoria | 0 (0) | 0 | 0 (0) | 0 | 1 (16.7) | 1 |
| Vascular disorders | 0 (0) | 0 | 1 (16.7) | 1 | 0 (0) | 0 |
| Hypotension | 0 (0) | 0 | 1 (16.7) | 1 | 0 (0) | 0 |
| Eye disorders | 0 (0) | 0 | 0 (0) | 0 | 1 (16.7) | 1 |
| Blurred vision | 0 (0) | 0 | 0 (0) | 0 | 1 (16.7) | 1 |
Note: PT, preferred terms; SOC, systematic organ classification.
The subjects in the ciprofol 0.4, 0.6 and 0.9 mg/kg groups exhibited a dose-dependent trend in body movements, limb twitches, body twitches, right upper limb twitches, muscle fibrillation and other limb movements within 1-6 min of administration (1 case occurred in the maintenance period, the others in the induction period), with an incidence of 33.3% (2/6), 33.3% (2/6) and 83.3% (5/6). Three AEs, probably related to the trial medication, were moderate, which included 2 cases of abnormal limb movements in the 0.4 mg/kg group and 1 case in the 0.9 mg/kg group. No other dose-related AEs occurred except for abnormal limb movements. There were no changes in hemoglobin saturation levels measured by pulse oximeter (desaturation was defined as SpO2 ≤95%), temperature or the respiration rate during the trial. All subjects had stable vital signs over the entire trial period except for 1 case with hypotension who presented with a low BP before the trial (90/57 mmHg) had begun, and 2 cases of sinus bradycardia (50-54 beats/min) after ciprofol injections, respectively (Figure 2). One subject in the 0.6 mg/kg group had the lowest BP (89/51 mmHg) before ciprofol administration, with a baseline value of 90/57 mmHg. The systolic BP fluctuated in the range of 82-89 mmHg in the 5-19 min period after ciprofol administration, with a minimum value of 82/50 mmHg. The subject recovered without any other medication being administered, so it was believed that the hypotension was likely related to ciprofol. Another subject in the 0.9 mg/kg group had a baseline HR of 67 beats/min before ciprofol administration, and developed a mild sinus bradycardia (50-54 beats/min, lasting 5 min) 38 min after administration, which was relieved after brief treatment with atropine, followed by repeated minor fluctuations (around 54 beats/min). No hypotension or other ECG abnormalities were observed during this period and the HR returned to the baseline value at approximately 90 min after ciprofol administration. Due to the normal HR before ciprofol administration and the development of an abnormal rate after administration, it was assumed that the occurrence of this sinus bradycardia was most likely related to ciprofol. A second subject in the 0.9 mg/kg group had a baseline HR of 57 beats/min before ciprofol administration, and developed a mild sinus bradycardia (<60 beats/min, lasting 5 min) 2 min after administration. The treatment regimen and HR fluctuation of this subject was similar to the previous one, but the recovery time of this subject took more than 4 h. Since this subject had a low HR before ciprofol administration and a slight HR fluctuation after administration, it was deemed that the occurrence of this sinus bradycardia was possibly related to ciprofol.
Figure 2.
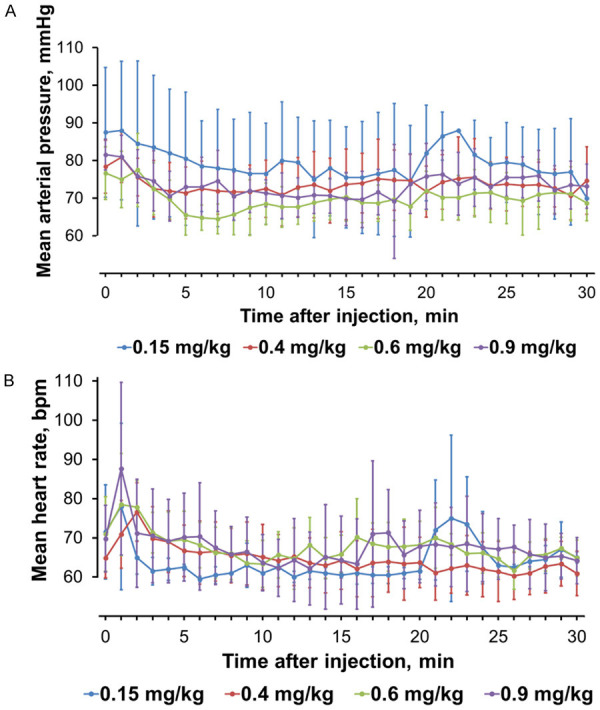
Changes in (A) mean MAP (mmHg), (B) mean HR (beats/min) for each ciprofol group.
Drug-related QTcF intervals were prolonged in 3 subjects (1 in the 0.6 mg/kg group and 2 in the 0.9 mg/kg group). No dose-dependent relationship between the ciprofol doses administered and prolonged QTcF intervals was detected. Injection pain, nausea or vomiting was not experienced by any subject who took part in the study. Laboratory blood and urine tests revealed no abnormalities before and 24 h after injections of ciprofol (Supplementary Table 3).
PK parameters
The non-compartmental PK parameters are shown in Table 3 and plasma concentration profiles are presented in Figure 3A. Plasma concentrations reached peak levels at 2.0 (2.0, 3.0) min, 2.0 (2.0, 3.0) min, and 2.0 (1.0, 2.0) min in the 0.4, 0.6 and 0.9 mg/kg groups, respectively. Median concentrations at LORverbal (Cp LORverbal) were 809.5, 768.5 and 1,750.0 ng/mL in the 0.4, 0.6 and 0.9 mg/kg groups, respectively. Recovery of verbal response (RORverbal) concentrations (Cp RORverbal) were not different among the 3 groups (median: 233.0, 261.0 and 315.0 ng/mL), which suggested that subjects would regain responsiveness when the plasma concentration was about 300 ng/mL (Table 3). A power function model was used to analyze the proportional relationship between dose and Cmax, AUC0-∞ and AUC0-t after drug administration. If the 90% confidence intervals (CI) of β fell in the range of 0.80-1.25 (linear interval after dose conversion was 0.725-1.275), it was assumed to be linear. The 90% CIs of the β value for Cmax, AUC0-t and AUC0-∞ were 0.257-1.393, 0.257-1.482 and 0.065-1.612, which were all beyond the linear range (0.725-1.275). Due to the small sample size and variations between individual subjects, the present results indicated that ciprofol did not exhibit a linear PK profile in the 0.4-0.9 mg/kg dosage range.
Table 3.
PK parameters and plasma concentration during loss/recovery of the verbal response after ciprofol injection
| 0.4 mg/kg (N = 6) | 0.6 mg/kg (N = 6) | 0.9 mg/kg (N = 6) | |
|---|---|---|---|
| Cmax (ng/mL) | 1,330.0 (985.0, 1,710.0) | 1,170.0 (839.0, 1,440.0) | 3,060.0 (1,750.0, 3,560.0) |
| AUC0-30 min (×104 min·ng/mL) | 0.59 (0.55, 0.71) | 0.83 (0.82, 0.85) | 1.59 (1.16, 1.78) |
| AUC0-1 h (×104 min ng/mL) | 0.82 (0.74, 1.28) | 1.19 (1.14, 1.27) | 2.07 (1.55, 2.14) |
| AUC0-t (×104 min·ng/mL) | 1.52 (1.36, 2.54) | 2.48 (2.26, 2.63) | 3.85 (3.25, 4.70) |
| AUC0-∞ (×104 min·ng/mL) | 1.63 (1.47, 3.03) | 2.54 (2.44, 2.75) | 4.24 (3.37, 5.15) |
| Tmax (min) | 2.00 (2.00, 3.00) | 2.00 (2.00, 3.00) | 2.00 (1.00, 2.00) |
| T1/2z (min) | 125.3 (111.8, 148.0) | 105.6 (100.1, 125.1) | 116.3 (107.9, 602.2) |
| Vz (×103 mL/kg) | 4.3 (2.1, 5.0) | 3.6 (3.5, 4.0) | 4.3 (4.3, 15.2) |
| CL (mL/min/kg) | 24.6 (13.2, 27.2) | 23.7 (21.8, 24.6) | 21.7 (17.5, 26.7) |
| λz (×10-3 L/min) | 5.6 (4.7, 6.2) | 6.6 (5.5, 6.9) | 6.0 (1.2, 6.4) |
| MRT0-∞ (min) | 141.7 (113.1, 142.3) | 126.1 (122.0, 132.8) | 156.4 (108.6, 404.5) |
| AUC%extrap in % | 7.7 (6.7, 12.4) | 4.8 (4.4, 7.3) | 7.3 (3.5, 9.2) |
| Cp LORverbal * | 809.5 (149.0, 2,030.0) | 768.5 (53.5, 2,960.0) | 1,750.0 (386.0, 7,900.0) |
| Cp RORverbal * | 233.0 (197.0, 242.0) | 261.0 (224.0, 362.0) | 315.0 (255.0, 361.0) |
Data are presented as the median and range; the remaining data are presented as medians (1st to 3rd) quartiles.
Cmax, maximum plasma concentration; AUC, area under the concentration; Tmax, time of maximum plasma concentration; T1/2z, terminal elimination half-life; CL, clearance; LORverbal, loss of verbal response; RORverbal, recovery of verbal response.
Figure 3.
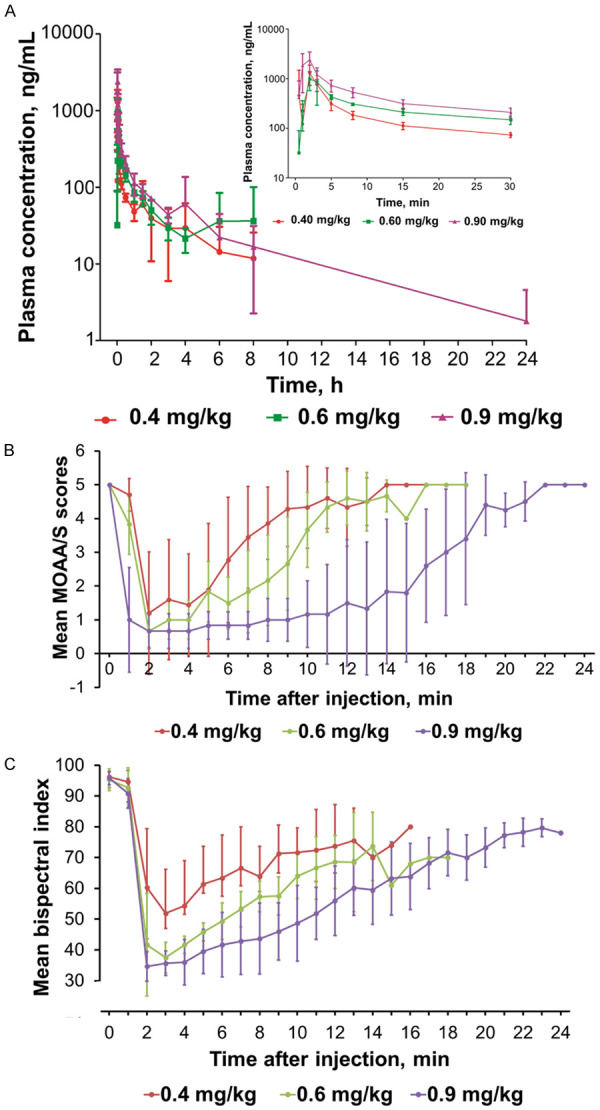
Changes in (A) ciprofol plasma concentrations, (B) MOAA/S scores, and (C) BIS values for each ciprofol group.
PD parameters
The MOAA/S scores rapidly decreased in all groups after administration of 0.4, 0.6 and 0.9 mg/kg ciprofol. LORverbal occurred in 83.3%, 100% and 100% of subjects in the 0.4, 0.6 and 0.9 mg/kg groups, respectively. The duration of recover of the verbal response (TRORverbal), LORverbal duration, the duration of MOAA/S ≤1 and the duration until return of responsiveness (duration of first record of MOAA/S <5 after drug administration to the first record of 3 consecutive times MOAA/S = 5) (Talert) were all increased in a dose-dependent manner (Figure 3B; Table 4). The durations of BIS values <60 were 6, 8 and 12 min in the 0.4, 0.6 and 0.9 mg/kg groups, respectively, which were similar to the durations of LORverbal and MOAA/S ≤1 (Figure 3C; Table 4). In addition, the median (Q1, Q3) time to loss of the eyelash reflex was 105.0 (90.0, 120.0), 90.0 (75.0, 90.0) and 60.0 (60.0, 60.0) s in the 0.4, 0.6 and 0.9 mg/kg groups, respectively. The median (Q1, Q3) orientation recovery time exhibited a dose-dependent increase when ciprofol was administered in doses of 0.4, 0.6 and 0.9 mg/kg [9.0 (7.0, 11.0), 11.0 (9.0, 12.0), and 19.5 (17.0, 21.0) min, respectively]. All modified QoR-9 summary scores were >17 points and recovered to the baseline values after ciprofol administration ceased.
Table 4.
MOAA/S and BIS values assessment
| 0.4 mg/kg (N = 6) | 0.6 mg/kg (N = 6) | 0.9 mg/kg (N = 6) | |
|---|---|---|---|
| LORverbal, n (%) | 5 (83.3) | 6 (100) | 6 (100) |
| RORverbal, n (%) | 5 (100) | 6 (100) | 6 (100) |
| TLORverbal (min) | 2.0 (2.0, 2.0) | 2.0 (2.0, 2.0) | 1.0 (1.0, 1.0) |
| TRORverbal (min) | 8.0 (7.0, 8.0) | 10.0 (7.0, 10.0) | 15.0 (14.0, 17.0) |
| LORverbal duration (min) | 6.0 (5.0, 6.0) | 8.0 (5.0, 8.0) | 14.0 (13.0, 16.0) |
| Talert (min) | 7.0 (6.0, 8.0) | 9.5 (9.0, 11.0) | 19.0 (16.0, 21.0) |
| MOAA/S ≤1, n (%) | 5 (83.3) | 6 (100) | 6 (100) |
| MOAA/S ≤1, duration (min) | 5.0 (4.0, 5.0) | 5.5 (3.0, 7.0) | 13.5 (12.0, 15.0) |
| MOAA/S = 0, n (%) | 5 (83.3) | 2 (33.3) | 4 (66.7) |
| MOAA/S = 0, duration (min) | 2.0 (1.0, 4.0) | 2.0 (1.0, 3.0) | 6.0 (3.5, 11.5) |
| BISpeak | 41.0 (38.0, 48.0) | 34.5 (28.0, 37.0) | 29.0 (26.0, 33.0) |
| TBISpeak (min) | 3.0 (3.0, 4.0) | 2.5 (2.0, 3.0) | 2.5 (2.0, 4.0) |
| BIS at LORverbal | 56.0 (44.0, 57.0) | 38.0 (32.0, 44.0) | 89.0 (85.0, 96.0) |
| BIS at RORverbal | 60.0 (60.0, 63.0) | 52.5 (47.0, 57.0) | 61.0 (60.0, 64.0) |
Note: Data are presented as medians (1st to 3rd quartile) and numbers with percentages. LORverbal: loss of the verbal response when MOAA/S <3; RORverbal: recovery of the verbal response when MOAA/S ≥3; TLORverbal (min): duration of loss of the verbal response; TRORverbal (min): duration of recovery of the verbal response; LORverbal duration (min): duration of LORverbal and RORverbal; Talert (min): duration of return of responsiveness (duration of first record of MOAA/S <5 after drug administration to the first record of 3 consecutive MOAA/S = 5); BISpeak: peak BIS value; TBISpeak: the duration when the peak BIS value was reached; BIS at LORverbal: BIS value at loss of the verbal response; BIS at RORverbal: BIS value when recovery of the verbal response.
Population PK/PD analyses
Population PK/PD analysis revealed that the population PK model conformed to a time delay three-compartment model (Tlag = 0.37 min). The estimated compartmental volumes (V1, V2 and V3) were 20.5 L, 176.0 L and 61.7 L, and the estimated compartmental clearances (CL1, CL2 and CL3) were 1.07 L/min, 1.27 L/min and 3.27 L/min, respectively (Table 5). BIS followed a sigmoid Emax model according to baseline values. BIS and the PK model were associated with a time delay at the effect-site concentration (Ce) (Telag = 0.455 min). The estimated median values of model parameters such as Ce50, Keo and t1/2Keo were 284.0 ng/mL, 2.2 min-1 and 0.315 min, respectively. A final MOAA/S PD model, established according to the baseline of the sigmoid Emax model and median Ce50, was estimated to be 326.0 ng/mL. There was no time delay between Ce connection and the PK model. The estimated median values for Keo and t1/2Keo were 0.626 min-1 and 1.11 min, respectively.
Table 5.
Population PK/PD parameters after ciprofol injection
| Parameters | Estimated value | 1,000 Bootstrapped | |
|---|---|---|---|
|
| |||
| Median | 95% CI | ||
| Final PK model | |||
| V1 (L) | 20.5 | 20.6 | 13.1-26.4 |
| V2 (L) | 176 | 182.7 | 116.4-290.2 |
| V3 (L) | 61.7 | 60.9 | 16.4-90.4 |
| CL1 (L/min) | 1.1 | 1.1 | 0.7-1.3 |
| CL2 (L/min) | 1.3 | 1.3 | 0.7-2.9 |
| CL3 (L/min) | 3.3 | 3.3 | 1.9-4.4 |
| Tlag (min) | 0.4 | 0.4 | 0.2-0.5 |
| Final BIS PD model (sigmoid Emax model) | |||
| E0, BIS | 94.5 | 94.6 | 93.5-95.8 |
| Emax, BIS | 37.1 | 36.4 | 31.7-40.9 |
| Ce50 (ng/mL) | 284.0 | 285.6 | 253.2-332.8 |
| Keo (min-1) | 2.2 | 2.3 | 1.1-5.5 |
| Telag (min) | 0.5 | 0.5 | 0.4-0.6 |
| Final MOAA/S PD model (sigmoid Emax model) | |||
| E0, MOAA/S | 4.7 | 4.7 | 4.4-5.0 |
| Emax, MOAA/S | 0.8 | 1.0 | 0.5-1.1 |
| Ce50 (ng/mL) | 326.0 | 325.0 | 272.0-365.0 |
| Keo (min-1) | 0.6 | 0.7 | 0.5-1.2 |
Discussion
In Australian phase 1a and 1b trials, subjects tolerated ciprofol at a dosage range of 0.128-0.81 mg/kg, and the proportions of subjects who reached LOR after receiving 0.128 mg/kg, 0.192 mg/kg and ≥0.288 mg/kg were 20%, 40% and ≥70%, respectively [6]. Considering that the initial dosage for this phase 1 trial should be the dosage below the median effective dose (ED50, 50% subjects reached LOR), the median dose of 0.15 mg/kg between 0.128 and 0.192 mg/kg was selected as the initial dose. Taking into account the comparable effect of cipropofol-0.54 mg/kg and propofol-2.0 mg/kg, and the fact that the ciprofol-0.81 mg/kg did not achieve the maximum tolerable dose (MTD) in the previous phase 1a and phase 1b trials [6], doses of ciprofol at 4.0, 6.0 and 9.0 mg/kg were selected for our phase 1 study.
Our results showed that ciprofol in a dose range between 0.4 mg/kg and 0.9 mg/kg was well-tolerated. All AEs occurred were mild and only 4 cases of AEs were moderate. The 0.9 mg/kg group of subjects exhibited the highest incidence of abnormal limb movements. The BIS values of the 3 moderate limb abnormal movement cases were all <60. The frequency of occurrence of abnormal limb movements appeared to increase in a dose-dependent manner, but due to the small sample size, statistical significance was not reached. However, these symptoms may not be indicative of true seizures and other intravenous anesthetics such as etomidate [14] and propofol [15] are known to elicit seizure-like phenomena, similar to those induced by ciprofol in the present study. These abnormal movements may be related to GABAergic inhibition in the central nervous system, which may sensitize the cortex to enter oscillatory excitation states in response to small initial stimuli [16]. Most studies indicate that propofol-induced dystonias and abnormal movements occur in the induction phase [17], which was also the case in the present study after ciprofol administration.
Though QTcF intervals were prolonged in 3 subjects, ciprofol did not induce dose-dependent prolonged QTcF intervals. However, it is noteworthy that QTcF intervals were also prolonged by other general anesthetics including sevoflurane [18,19], an action attributed to a reduction in the intracellular free Ca2+ concentration of cardiac muscle through actions on protein kinase C, thereby inhibiting myocardial contractility [20]. However, for a conclusive assessment on the effects of ciprofol on QTcF intervals, a larger sample size trial will need to be conducted.
BP was slightly decreased (SBP decreased by <10%) for 5 min after injections, but became stable thereafter (Figure 2). In the 0.6 mg/kg group, 1 subject had hypotension (82/50 mmHg), but this may have been due to the subject’s initial low baseline value (90/57 mmHg). In addition, 2 cases of sinus bradycardia occurred, when subjects already had low pulse rates before administration of ciprofol. It should be noted, however, that hypotension and bradycardia have been reported as side effects of propofol [21,22].
In the present trial, we found that ciprofol did not elicit pain in any of the subjects. Injection pain commonly occurs during an IV propofol injection [23]. It has been documented that propofol caused pain or discomfort on injection in 28% to 90% of subjects studied [24-26], while Tan et al. reported that several factors can lead to injection pain, including the injection site, vein size, rate of injection, propofol concentration in the aqueous phase and the buffering effect of blood [23].
The PD results showed that the onset and recovery from ciprofol were rapid and produced good quality clinical effects. The MOAA/S scores and BIS results revealed that ciprofol produced sedation in 1-2 min. Loss of the verbal response occurred within 2 min in the 0.4, 0.6 and 0.9 mg/kg groups, which indicated that ciprofol produced a rapid anesthetic effect. TRORverbal, LORverbal duration, a duration of MOAA/S ≤1 and Talert all exhibited dose-dependent increases in effects, while the peak BIS values exhibited a decreasing dose-dependent relationship. The duration of BIS values <60 was also extended with increasing ciprofol dosage.
In addition, the eyelash reflex disappeared within 2 min in the 0.4, 0.6 and 0.9 mg/kg groups. Orientation recovery time increased when the doses of ciprofol ranged from 0.4 to 0.9 mg/kg. The 0.4 and 0.6 mg/kg groups recovered in about 10 min while the 0.9 mg/kg group required about 18 min. All modified QoR-9 summary scores were >17 points and there was no change in the modified QoR-9 scores compared to the subjects’ baseline after recovery from ciprofol. These results indicated that ciprofol had a rapid onset of action and a fast recovery time.
PK parameters for ciprofol were calculated using a non-compartment model. The results revealed that T1/2z for single IV administrations for different doses in healthy subjects was between 2-5 h and the average T1/2z was about 3 h. T1/2z in the 0.4 and 0.6 mg/kg groups was about 2 h and approximately 5 h in the 0.9 mg/kg group. The plasma concentrations of ciprofol at loss of the verbal response (Cp LORverbal) exhibited a large variability in the different dose groups probably due to the rapid distribution of ciprofol into central and peripheral compartments, while the plasma concentration of ciprofol at recovery of the verbal response (Cp RORverbal) was most likely determined by drug redistribution. Therefore, it is difficult to accurately use Cp LORverbal determined by drug plasma concentration parameters. These results suggested that subjects regained responsiveness when the plasma concentration fell to about 300 ng/mL.
Limitations of the present study were the small sample size and that the injection mode of the infusion pump was unfortunately difficult to control to inject the drug at a constant volume within 1 min. The reason was probably related to individual vascular resistance patterns, but this conjecture requires further analysis.
In conclusion, a ciprofol dosage regimen of 0.4 to 0.9 mg/kg was well-tolerated and had a rapid onset and fast recovery. A dose of ciprofol >0.4 mg/kg, which induced deep sedation, is recommended for phase 2 clinical studies. On the basis of these data, further research on the potential usefulness of ciprofol for sedation and/or general anesthesia in endoscopy examinations and sedation of adults in ICUs will be necessary.
Acknowledgements
The study was supported by the National Key R&D Program of China (grant number 2018YFC2001800) and Sichuan Haisco Pharmaceutical Group Co., Ltd.
Disclosure of conflict of interest
Xiao Liu and Yong Liang were employees of Sichuan Haisco Pharmaceutical Group Co., Ltd. The remaining authors declared no conflicts of interest.
Supporting Information
References
- 1.White PF. Clinical uses of intravenous anesthetic and analgesic infusions. Anesth Analg. 1989;68:161–171. doi: 10.1213/00000539-198902000-00017. [DOI] [PubMed] [Google Scholar]
- 2.Cockshott ID. Propofol (‘diprivan’) pharmacokinetics and metabolism--an overview. Postgrad Med J. 1985;61(Suppl 3):45–50. [PubMed] [Google Scholar]
- 3.Smith I, White PF, Nathanson M, Gouldson R. Propofol. An update on its clinical use. Anesthesiology. 1994;81:1005–1043. [PubMed] [Google Scholar]
- 4.Mahmoud M, Mason KP. Recent advances in intravenous anesthesia and anesthetics. F1000Res. 2018;7:F1000 Faculty Rev-470. [Google Scholar]
- 5.Qin L, Ren L, Wan S, Liu G, Luo X, Liu Z, Li F, Yu Y, Liu J, Wei Y. Design, synthesis, and evaluation of novel 2,6-disubstituted phenol derivatives as general anesthetics. J Med Chem. 2017;60:3606–3617. doi: 10.1021/acs.jmedchem.7b00254. [DOI] [PubMed] [Google Scholar]
- 6.Ludbrook G, Li F, Sleigh J, Liang Y. Assessments of onset and duration of drug effects and pharmacokinetics by dose level of HSK3486, a new sedative-hypnotic agent, in healthy female/male subjects: a phase I multiarm randomized controlled clinical trial. Anesth Analg. 2021 doi: 10.1213/ANE.0000000000005343. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 7.Dahaba AA, Zhong T, Lu HS, Bornemann H, Liebmann M, Wilfinger G, Reibnegger G, Metzler H. Geographic differences in the target-controlled infusion estimated concentration of propofol: bispectral index response curves. Can J Anaesth. 2011;58:364–370. doi: 10.1007/s12630-011-9453-2. [DOI] [PubMed] [Google Scholar]
- 8.Lampotang S, Lizdas DE, Derendorf H, Gravenstein N, Lok B, Quarles JP. Race-specific pharmacodynamic model of propofol-induced loss of consciousness. J Clin Pharmacol. 2016;56:1141–1150. doi: 10.1002/jcph.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ortolani O, Conti A, Ngumi ZW, Texeira L, Olang P, Amani I, Medrado VC. Ethnic differences in propofol and fentanyl response: a comparison among Caucasians, Kenyan Africans and Brazilians. Eur J Anaesthesiol. 2004;21:314–319. doi: 10.1017/s0265021504004119. [DOI] [PubMed] [Google Scholar]
- 10.Chernik DA, Gillings D, Laine H, Hendler J, Silver JM, Davidson AB, Schwam EM, Siegel JL. Validity and reliability of the observer’s assessment of alertness/sedation scale: study with intravenous midazolam. J Clin Psychopharmacol. 1990;10:244–251. [PubMed] [Google Scholar]
- 11.Myles PS, Hunt JO, Nightingale CE, Fletcher H, Beh T, Tanil D, Nagy A, Rubinstein A, Ponsford JL. Development and psychometric testing of a quality of recovery score after general anesthesia and surgery in adults. Anesth Analg. 1999;88:83–90. doi: 10.1097/00000539-199901000-00016. [DOI] [PubMed] [Google Scholar]
- 12.Antonik LJ, Goldwater DR, Kilpatrick GJ, Tilbrook GS, Borkett KM. A placebo- and midazolam-controlled phase I single ascending-dose study evaluating the safety, pharmacokinetics, and pharmacodynamics of remimazolam (CNS 7056): part I. safety, efficacy, and basic pharmacokinetics. Anesth Analg. 2012;115:274–283. doi: 10.1213/ANE.0b013e31823f0c28. [DOI] [PubMed] [Google Scholar]
- 13.Kalman S, Koch P, Ahlen K, Kanes SJ, Barassin S, Bjornsson MA, Norberg A. First human study of the investigational sedative and anesthetic drug AZD3043: a dose-escalation trial to assess the safety, pharmacokinetics, and efficacy of a 30-minute infusion in healthy male volunteers. Anesth Analg. 2015;121:885–893. doi: 10.1213/ANE.0000000000000831. [DOI] [PubMed] [Google Scholar]
- 14.Miner JR, Danahy M, Moch A, Biros M. Randomized clinical trial of etomidate versus propofol for procedural sedation in the emergency department. Ann Emerg Med. 2007;49:15–22. doi: 10.1016/j.annemergmed.2006.06.042. [DOI] [PubMed] [Google Scholar]
- 15.Walder B, Seeck M, Tramer MR. Propofol [correction of propfol] versus methohexital for electroconvulsive therapy: a meta-analysis. J Neurosurg Anesthesiol. 2001;13:93–98. doi: 10.1097/00008506-200104000-00005. [DOI] [PubMed] [Google Scholar]
- 16.Voss LJ, Sleigh JW, Barnard JP, Kirsch HE. The howling cortex: seizures and general anesthetic drugs. Anesth Analg. 2008;107:1689–1703. doi: 10.1213/ane.0b013e3181852595. [DOI] [PubMed] [Google Scholar]
- 17.Al-Humaidan A, Hussein G, Lewis M. Can propofol cause dystonia and movement disorders? Clin Res Regul Aff. 2000;17:19–26. [Google Scholar]
- 18.Kuenszberg E, Loeckinger A, Kleinsasser A, Lindner KH, Puehringer F, Hoermann C. Sevoflurane progressively prolongs the QT interval in unpremedicated female adults. Eur J Anaesthesiol. 2000;17:662–664. doi: 10.1046/j.1365-2346.2000.00739.x. [DOI] [PubMed] [Google Scholar]
- 19.Nagele P, Pal S, Brown F, Blood J, Miller JP, Johnston J. Postoperative QT interval prolongation in patients undergoing noncardiac surgery under general anesthesia. Anesthesiology. 2012;117:321–328. doi: 10.1097/ALN.0b013e31825e6eb3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanaya N, Gable B, Wickley PJ, Murray PA, Damron DS. Experimental conditions are important determinants of cardiac inotropic effects of propofol. Anesthesiology. 2005;103:1026–1034. doi: 10.1097/00000542-200511000-00017. [DOI] [PubMed] [Google Scholar]
- 21.Farhan M, Hoda MQ, Ullah H. Prevention of hypotension associated with the induction dose of propofol: a randomized controlled trial comparing equipotent doses of phenylephrine and ephedrine. J Anaesthesiol Clin Pharmacol. 2015;31:526–530. doi: 10.4103/0970-9185.169083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tramer MR, Moore RA, McQuay HJ. Propofol and bradycardia: causation, frequency and severity. Br J Anaesth. 1997;78:642–651. doi: 10.1093/bja/78.6.642. [DOI] [PubMed] [Google Scholar]
- 23.Tan CH, Onsiong MK. Pain on injection of propofol. Anaesthesia. 1998;53:468–476. doi: 10.1046/j.1365-2044.1998.00405.x. [DOI] [PubMed] [Google Scholar]
- 24.Stark RD, Binks SM, Dutka VN, O’Connor KM, Arnstein MJ, Glen JB. A review of the safety and tolerance of propofol (‘Diprivan’) Postgrad Med J. 1985;61(Suppl 3):152–156. [PubMed] [Google Scholar]
- 25.Jalota L, Kalira V, George E, Shi YY, Hornuss C, Radke O, Pace NL, Apfel CC Perioperative Clinical Research Core. Prevention of pain on injection of propofol: systematic review and meta-analysis. BMJ. 2011;342:d1110. doi: 10.1136/bmj.d1110. [DOI] [PubMed] [Google Scholar]
- 26.Picard P, Tramer MR. Prevention of pain on injection with propofol: a quantitative systematic review. Anesth Analg. 2000;90:963–969. doi: 10.1097/00000539-200004000-00035. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.