

Abstract
Recent studies have reported that MLST8 is upregulated in many malignant tumors. Nevertheless, the underlying molecular mechanism is still unclear. The aim of this work was to investigate how MLST8 contributes to the development and progression of clear cell renal cell carcinoma (ccRCC). MLST8 is an oncogenic protein in the TCGA database and ccRCC clinical specimens. We also ascertain that MLST8 interacts with FBXW7, which was universally regarded as an E3 ubiquitin ligase. MLST8 can be degraded and ubiquitinated by tumor suppressor FBXW7. FBXW7 recognizes a consensus motif (T/S) PXX (S/T/D/E) of MLST8 and triggers MLST8 degradation via the ubiquitin‐proteasome pathway. Strikingly, the activated cyclin dependent kinase 1 (CDK1) kinase engages in the MLST8 phosphorylation required for FBXW7‐mediated degradation. In vitro, we further prove that MLST8 is an essential mediator of FBXW7 inactivation‐induced tumor growth, migration, and invasion. Furthermore, the MLST8 and FBXW7 proteins are negatively correlated in human renal cancer specimens. Our findings suggest that MLST8 is a putative oncogene that functions via interaction with FBXW7, and inhibition MLST8 could be a potential future target in ccRCC treatment.
Keywords: CDK1, FBXW7, protein degradation, renal cell carcinoma, ubiquitination
Schematic diagram of the CDK1‐FBXW7α signaling axis in the regulation of MLST8 protein degradation.
![]()
1. INTRODUCTION
Mammalian target of rapamycin (mTOR) associated protein (MLST8), also known as G protein beta subunit‐like (GBL), is a core component of mTORC1 and mTORC2, and contributes to the occurrence and progression of many tumors, including hepatocellular carcinoma (HCC) and colon cancer. 1 , 2 , 3 , 4 MLST8 has been reported to promote the proliferation and invasion of hepatocellular carcinoma cells through the AKT pathway. 5 Mouse embryo knockout experiments demonstrate an essential role of MLST8 in mTORC2 that directly affects the function of mTORC2. 6
F‐box and WD repeat domain‐containing 7 (FBXW7) is the substrate recognition component of the Skp1‐Cul1‐F‐box (SCF) ubiquitin ligase complex. It recognizes and binds to its substrates via the conserved Cdc4‐phosphodegron (CPD) motif (T/S‐P‐X‐X‐S/T/ D/E). 7 FBXW7 targets multiple oncoproteins for ubiquitination and degradation, including cyclin E, 8 c‐Myc, 9 c‐Jun, 10 Notch‐1, 11 and SREBP1. 12 Silencing of FBXW7 expression was found to render metabolic advantages in pancreatic cancer cells through the induction of aerobic glycolysis by downregulating the thioredoxin interacting protein (TXNIP) in a c‐Myc‐dependent manner. 13 , 14 , 15 Previous studies have demonstrated that decreased FBXW7 expression induces cancer antigen 125 (CA125) or Muc16 production in pancreatic cancer cells, which are both regarded as metastatic markers of pancreatic cancer cells. 16 , 17 , 18
In this study, we present data highlighting a crucial role of MLST8 towards tumor progression. We validate that FBXW7 physically interacts with MLST8 and mediates its ubiquitylation, which highlights potential future efforts towards developing therapeutic strategies against renal cancer (ccRCC).
2. MATERIAL AND METHODS
2.1. Cell culture and transfection
786‐O, A498, and 293T cells were obtained from the American Type Culture Collection (ATCC). 786‐O cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin, and incubated in a 5% CO2 humidified incubator at 37°C. A498 and 293T cells were cultured in DMEM medium supplemented with 10% FBS. All transient transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. To determine the contribution of cyclin B‐CDK1, cells were treated with a CDK inhibitor, such as butyrolactone I 18 , 19 or RO‐3306 (Calbiochem).
2.2. Expression constructs
Human cDNAs for c‐Myc, Fbxw7, and MLST8 were amplified from the human brain cDNA library (Clontech) by polymerase chain reaction (PCR). These cDNAs were inserted into the EcoRI and NotI restriction sites of pCMV‐HA vector, pCMV‐FLAG vector, and pCMV‐Myc. PCR amplified cDNAs for the phosphorylation site mutants of the mouse (T50A/S54A) and human (T50A) with primers corresponding to the mutant sequences and inserted into EcoRI/BamHI sites of pCMV‐HA vector to generate pCMV‐HA‐c‐Myc (T50A/S54A) and pCMV‐HA‐c‐Myc (T50A), respectively. All the constructs were confirmed by DNA sequencing.
2.3. Establish stable Cas‐9 knockout and overexpression 786‐O and A498 cell lines
The FBXW7 and MLST8 Cas‐9 gene knockout plasmid was constructed using the Lenti CRISPR V2 plasmid, then transfected with psPAX2 and pCMV‐VSV‐G plasmid at a ratio of 4:3:3 into 293 T cells. 786‐O cells in a 60 mm dish were infected with 1 mL of virus supernatant when the cell density was 60%‐80%. The positive cells were screened with puromycin for 2 weeks. The result was verified by Western blotting. The method is the same as the above: 10 μg of pCDH‐CD513B‐Flag‐FBXW7 plasmid and 10 μg of pCDH‐CD513B‐Myc‐MLST8 plasmid, respectively, and 10 μL of Lipofectamine 2000 were transfected into 786‐O and A498 cells, then screened with puromycin for 2 weeks. The primer sequences are listed in Table 1.
TABLE 1.
Sequences of primers for sgRNAs
| Gene name | Sequences of siRNAs |
|---|---|
| sgControl | Forward: 5′CACCGGGGCAGGTCCACAATACTAC3′ |
| Reverse: 5′AAACGTAGTATTGTGGACCTGCCCC3′ | |
| sgFBXW7#1 | Forward: 5′CACCGTTCGGCGTCGTTGTTGCCCT3′ |
| Reverse: 5′AAACAGGGCAACAACGACGCCGAAC3′ | |
| sgFBXW7#2 | Forward: 5′ CACCGAGTGGAAGTATGCCCATATA3′ |
| Reverse: 5′ AAACTATATGGGCATACTTCCACTC3′ | |
| sgMLST8#1 | Forward: 5′CACCGGACGGCCGCTGGATGTACAC3′ |
| Reverse: 5′ AAACGTGTACATCCAGCGGCCGTCC3′ | |
| sgMLST8#2 | Forward: 5′CACCGAGACGGCCGCTGGATGTACA3′ |
| Reverse: 5′ AAACTGTACATCCAGCGGCCGTCTC3′ | |
| siRBX1#1 | Forward: 5′GAAGCGCTTTGAAGTGAAA3′ |
| Reverse: 5′TTTCACTTCAAAGCGCTTC3′ | |
| siRBX1#2 | Forward: 5′GGGATATTGTGGTTGATAA3′ |
| Reverse: 5′TTATCAACCACAATATCCC3′ | |
| siRBX1#3 | Forward: 5′GGAACCACATTATGGATCT3′ |
| Reverse: 5′AGATCCATAATGTGGTTCC3′ | |
| siCUL1#1 | Forward: 5′CAACGAAGAGTTCAGGTTT3′ |
| Reverse: 5′AAACCTGAACTCTTCGTTG3′ | |
| siCUL1#2 | Forward: 5′CGAGGAAGACCGCAAACTA3′ |
| Reverse: 5′TAGTTTGCGGTCTTCCTCG3′ | |
| siCUL1#3 | Forward: 5′AGACAGTGCTTGATGTTCA3′ |
| Reverse: 5′TGAACATCAAGCACTGTCT3′ | |
| siCUL2#1 | Forward: 5′GGAAGTGCATGGTAAATTT3′ |
| Reverse: 5′AAATTTACCATGCACTTCC3′ | |
| siCUL2#2 | Forward: 5′CATCCAAGTTCATATACTA3′ |
| Reverse: 5′TAGTATATGAACTTGGATG3′ | |
| siCUL2#3 | Forward: 5′GCAGAAAGACACACCACAA3′ |
| Reverse: 5′TTGTGGTGTGTCTTTCTGC3′ | |
| siCUL3#1 | Forward: 5′GAGAAGATGTACTAAATTC3′ |
| Reverse: 5′GAATTTAGTACATCTTCTC3′ | |
| siCUL3#2 | Forward: 5′CGACAGAAAACATGAGATA3′ |
| Reverse: 5′TATCTCATGTTTTCTGTCG3′ | |
| siCUL3#3 | Forward: 5′GAAAGTAGACGACGACAGA3′ |
| Reverse: 5′TCTGTCGTCGTCTACTTTC3′ | |
| siCUL4A#1 | Forward: 5′GCACAGATCCTTCCGTTTA3′ |
| Reverse: 5′TAAACGGAAGGATCTGTGC3′ | |
| siCUL4A#2 | Forward: 5′GAACAGCGATCGTAATCAA3′ |
| Reverse: 5′TTGATTACGATCGCTGTTC3′ | |
| siCUL4A#3 | Forward: 5′GCATGTGGATTCAAAGTTA3′ |
| Reverse: 5′TAACTTTGAATCCACATGC3′ | |
| siCUL4B#1 | Forward: 5′TAAATAACCTCCTTGATGA3′ |
| Reverse: 5′TCATCAAGGAGGTTATTTA3′ | |
| siCUL4B#2 | Forward: 5′CAGAAGTCATTAATTGCTA3′ |
| Reverse: 5′TAGCAATTAATGACTTCTG3′ | |
| siCUL4B#3 | Forward: 5′CGGAAAGAGTGCATCTGTA3′ |
| Reverse: 5′TACAGATGCACTCTTTCCG3′ | |
| siCUL5#1 | Forward: 5′GACACGACGTCTTATATTA3′ |
| Reverse: 5′TAATATAAGACGTCGTGTC3′ | |
| siCUL5#2 | Forward: 5′GCAAATAGAGTGGCTAATA3′ |
| Reverse: 5′TATTAGCCACTCTATTTGC3′ | |
| siCUL5#3 | Forward: 5′TAAACAAGCTTGCTAGAAT3′ |
| Reverse: 5′ATTCTAGCAAGCTTGTTTA3′ |
2.4. Antibodies
Commercially available antibodies for Western blotting were as follows: FBXW7 (A5872, ABclonal), MLST8 (A13599, ABclonal), Myc (9E10; Sigma), FLAG (M2; Sigma), HA (MM5‐101R; Millipore), and Actin (M20011; Abmart). Anti‐human MLST8 phosphorylated Thr50 polyclonal antibody (anti‐p‐T50‐MLST8) was raised against keyhole limpet hemocyanin‐conjugated chemically synthesized phosphorylated Thr50 peptide, corresponding to the CPD region of MLST8 (amino acid residues 50‐61) (MBL). Antiserum obtained from an immunized guinea pig was bound to column chromatography conjugated with P‐T50 peptide. The affinity‐purified anti‐p‐T50‐MLST8 was then passed through a column conjugated to nonphosphorylated Thr50 peptide (amino acid residues 50‐61 of MLST8) to deplete antibodies against the nonphosphorylated antigen. ELISA confirmed the specific binding ability of the purified antibody to P‐T50 peptide.
2.5. Immunoprecipitation
To immunoprecipitate the ectopically expressed Flag‐tagged proteins, transfected cells were lysed 24 h post‐transfection in BC100 buffer. The whole‐cell lysates were immunoprecipitated with monoclonal anti‐Flag antibody‐conjugated M2 agarose beads (Sigma) at 4°C overnight. After three washes with Flag lysis buffer, followed by two washes with BC100 buffer, the bound proteins were eluted using Flag‐Peptide (Sigma)/BC100 for 3 h at 4°C. SDS‐PAGE resolved the eluted material. To immunoprecipitate the endogenous proteins, cells were lysed with 1× cell lysis buffer (Cell Signaling) and the lysate was centrifuged. The supernatant was precleared with protein A/G beads (Sigma) and incubated with indicated antibodies overnight. After that, protein A/G beads were applied, all at 4°C. After 2 h of incubation, pellets were washed five times with lysis buffer and resuspended in sample buffer and analyzed by SDS‐PAGE.
2.6. Western blot
Cell lysates or immunoprecipitates were subjected to SDS‐PAGE, and proteins were transferred to nitrocellulose membranes (GE Healthcare). The membranes were blocked in Tris‐buffered saline (TBS, pH 7.4) containing 5% nonfat milk and 0.1% Tween‐20, washed twice in TBS containing 0.1% Tween‐20, and incubated with primary antibody for 2 h followed by secondary antibody for 1 h at room temperature. The proteins of interest were visualized using an ECL chemiluminescence system (Santa Cruz). In vitro, phosphorylation was performed as described previously. 20 , 21 , 22
2.7. Quantitative RT‐PCR
Total RNA was isolated from transiently transfected cells using TRIzol reagent (Tiangen), and cDNA was reversed‐transcribed using a Superscript RT kit (TOYOBO), according to the manufacturer's instructions. PCR amplification was performed using a SYBR Green PCR master mix kit (TOYOBO). All quantization was normalized to the level of endogenous control GAPDH. The primer sequences are listed in Table 2.
TABLE 2.
Sequences of primers qT‐PCR
| Gene name | Forward (5′‐3′) | Reverse (5′‐3′) |
|---|---|---|
| FBXW7 | CCCACCTACCCCTCCTATGT | TGCCCATTCATCTTGTGGTA |
| MLST8 | TGATTGCTGCTGCAGGTTAC | GTTAATGGGTGCGTTCACCT |
| GAPDH | ACCAGAAGACTGTGGATGG | TGCTGTAGAAAAATTCGTTG |
2.8. CCK‐8 assay, cell migration, and invasion assay
The cell proliferation rate was determined using a Cell Counting Kit‐8 (CCK‐8) according to its protocol (Vazyme). Cell migration was assessed using a 24‐well transwell unit with polycarbonate membrane (pore size 8 μm) (Corning) according to the manufacturer's protocol. The membrane was coated with Matrigel basement membrane matrix (1 μg/μL) (BD Bioscience) for 24 h. Cells (0.5‐2.5 × 104) were then seeded into the upper chamber in a serum‐free medium. The lower chamber was filled with a medium containing 10% FBS. After 24 h of incubation, the cells in the upper chamber were removed and fixed in 4% paraformaldehyde, stained with crystal violet for 20 min. For invasion, the membranes utilized were Matrigel‐coated invasion chambers (BD Biosciences) that were prehydrated in a serum‐free medium.
2.9. Immunohistochemistry
Tissue microarrays (TMAs) were made using the above 79 paired tissues in Shanghai Outdo Biotech Company (Shanghai, China), including tumor and adjacent normal tissues. The immunohistochemistry (IHC) was performed by the streptavidin‐peroxidase method (Zymed Laboratories Inc). IHC scores were determined by the estimated proportion of positive tumor cells in percentage. To assess the average degree of staining within a tumor, multiple regions were analyzed and at least 100 tumor cells were assessed. The H‐score system assessed the cytoplasmic or nucleus expression. The H‐score formula is Histoscore = ∑(I × Pi), where I is the intensity of staining and Pi is the percentage of stained tumor cells, producing a cytoplasmic score ranging from 0 to 200. The scoring was independently assessed by two assessors (AWHC and JHMT) who were unaware of the clinical outcomes.
2.10. In vivo tumorigenesis and metastasis assay
Cells stably knockout MLST8 (2 × 106) or control cells were subcutaneously injected into the right flanks of 5 BALB/c (nu/nu) mice. Tumor size was measured weekly using a caliper. After 4 weeks, the mice were sacrificed for tumor burden analysis. The tumor volume was calculated as volume = (length × width2)/2. For intrahepatic metastasis assays, cells (3 × 106/mouse) were injected into the livers of nude mice. Tumor nodules formed on the liver surfaces were macroscopically determined and counted. The livers were excised and embedded in paraffin. Tissue sections (5 μm) were stained with H&E to visualize the structure. The Institutional Animal Care and Use Committee (IACUC) is from Ethics Committee of General Hospital of Shanghai Jiao Tong University.
2.11. Statistical analysis
Data were expressed as means ± SD. All experiments were carried out with three or more replicates. Statistical analyses were performed by Student's t‐test for most studies using GraphPad Prism v7.0 software. P < .05 was considered statistically significant.
3. RESULTS
3.1. Elevated expression of MLST8 in human renal cancer
We initially evaluated the expression of MLST8 mRNA in ccRCC from the cancer‐related databases GEPIA (http://gepia.cancer‐pku.cn/) and Oncomine (www.oncomine.org/). According to GEPIA database results, MLST8 is generally overexpressed in renal carcinoma (red) and downregulated in normal tissue (green) (Figure 1A‐B). We further explored the Oncomine database and collected the number of statistically significant datasets with MLST8 mRNA overexpression (red) or downregulation (blue) in Figure 1C. The expression level of MLST8 was significantly upregulated in ccRCC tissues in the Oncomine dataset (Jones Renal [Figure 1D], Cutcliffe Renal [Figure 1E[).
FIGURE 1.
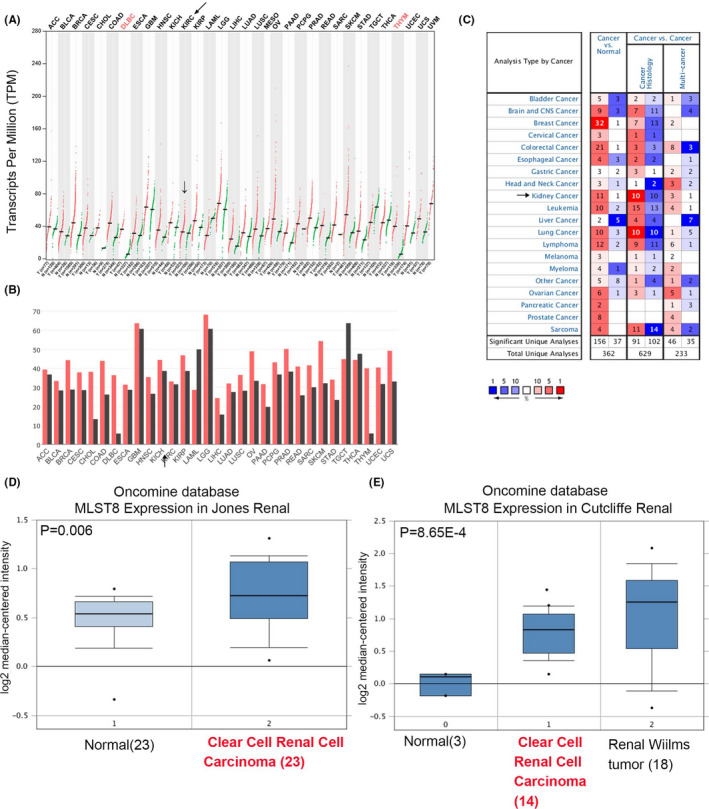
Analysis of MLST8 RNA expression on GEPIA and Oncomine database. A, MLST8 RNA expression in different cancers on GEPIA. B, Median TPM of MLST8 in different cancers on GEPIA. C, MLST8 RNA expression in different cancers on Oncomine database. D‐G, Exhibition of MLST8 expression (cancer vs normal) of other kidney cancers and ccRCC tissue in the Oncomine datasets
3.2. MLST8 is a potential biomarker for ccRCC according to TCGA
As shown in Figure 2A, MLST8 was significantly upregulated in ccRCC samples compared to normal tissues at the transcriptional level (P < .0001). We evaluated this association with clinical‐pathological characteristics. Although there was no difference between MLST8 expression and lymphatic metastasis, MLST8 was correlated to tumor stage M (Figure 2B‐E). Because the N1 stage samples from TCGA were small, this was insufficient to demonstrate a correlation between MLST8 and N metastasis. However, a strong correlation was identified between the MLST8 expression and the overall survival (OS) of ccRCC patients (Figure 2F, P = .0248). Therefore, we assumed that the nonsignificance between MLST8 and distant metastasis was potentially due to the limited sample size, necessitating future validation.
FIGURE 2.
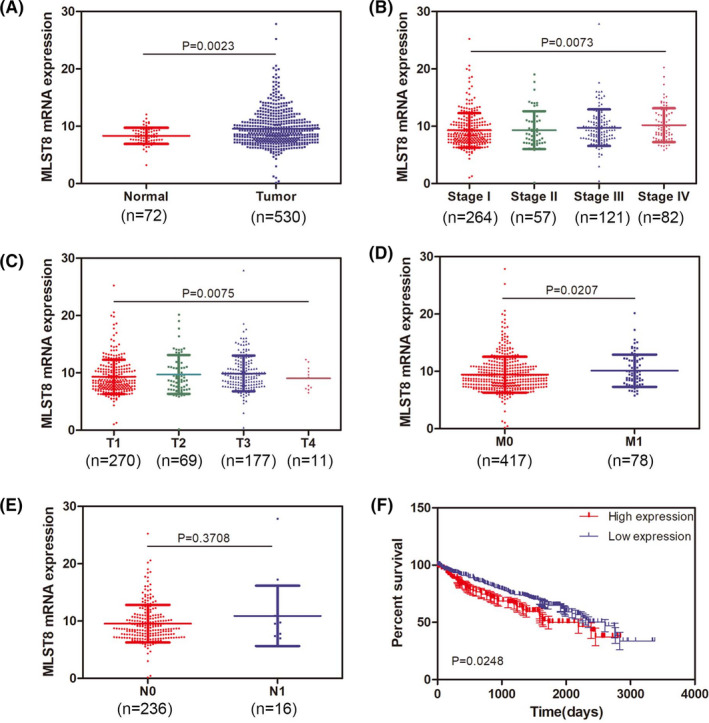
MLST8 is a potential biomarker for ccRCC according to TCGA. A, Expression level of MLST8 in ccRCC was elevated in tumor tissue compared to normal tissue. B, Expression of MLST8 was elevated gradually as ccRCC progressed to stage II and stage III. C, Expression of MLST8 presented as more upregulated in stages T2 and T3 compared to T1. D, Expression of MLST8 in ccRCC patients with stage M1 was remarkedly increased compared that in stage M0. E, There was no significant difference of MLST8 expression between stages N1 and N0 in ccRCC patients. F, OS curve of ccRCC patients based on MLST8 expression (P = .0248)
3.3. MLST8 is upregulated in ccRCC specimens and predicts poor clinical outcomes in ccRCC patients
Overexpressed MLST8 protein was found in ccRCC specimens compared to adjacent normal tissue (Table 3, P < .0001), and the results showed a significant increase in the expression levels of MLST8 as ccRCC progressed to an advanced stage (Figure 3A,B). The association between MLST8 expression and clinical pathological characteristics is shown in Table 3. The overexpression of MLST8 was strikingly correlated to the T stage (P = .009) and Furman grade (Figure 3D, P < .0001). However, there was no association between MLST8 expression levels and a patient's age, gender, and size. We developed Kaplan‐Meier curves on 75 patients with a log‐rank test for OS to elucidate the relationship between MLST8 expression and patient survival in ccRCC. Our results reveal that a high expression of MLST8 is associated with poor OS (Figure 3E, P = .0183). These results suggest that MLST8 is a potential prognostic marker in ccRCC.
TABLE 3.
Patient characteristics and clinicopathological factors by FBXW7 and MLST8 expression
| FBXW7 high | FBXW7 low | P value | MLST8 high | MLST8 low | P value | |
|---|---|---|---|---|---|---|
| No. of patients | 25 (31.6%) | 54 (68.4%) | 45 (60.0%) | 34 (40.0%) | ||
| Age | ||||||
| >60 | 11 (44.0%) | 31 (57.4%) | .267 | 27 (60.0%) | 15 (44.1%) | .161 |
| ≦60 | 14 (56.0%) | 23 (42.6%) | 18 (40.0%) | 19 (55.9%) | ||
| Gender | ||||||
| Male | 15 (60%) | 21 (38.9%) | .08 | 17 (37.8%) | 19 (55.9%) | .110 |
| Female | 10 (40%) | 33 (61.1%) | 28 (62.2%) | 15 (44.1%) | ||
| Size | ||||||
| >4 cm | 21 (84.0%) | 40 (74.1%) | .398 | 36 (80.0%) | 25 (73.5%) | .497 |
| ≦4 cm | 4 (16.0%) | 14 (25.9%) | 9 (20.0%) | 9 (26.5%) | ||
| Primary T stage | ||||||
| Low (pT1/pT2) | 21 (84.0%) | 43 (79.6%) | .449 | 32 (71.1%) | 32 (94.1%) | .009 |
| High (pT3/pT4) | 4 (16.0%) | 11 (20.4%) | 13 (28.9%) | 2 (5.9%) | ||
| TNM stage | ||||||
| Low (pT1/pT2) | 21 (84.0%) | 43 (79.6%) | .449 | 32 (71.1%) | 30 (88.2%) | .058 |
| High (pT3/pT4) | 4 (16.0%) | 11 (20.4%) | 13 (28.9%) | 4 (11.8%) | ||
| Fuhrman grade | ||||||
| Low (G1/G2) | 22 (88.0%) | 37 (68.5%) | .054 | 25 (55.6%) | 32 (94.1%) | .000 |
| High (G3/G4) | 3 (12.0%) | 17 (31.5%) | 20 (44.4%) | 2 (5.9%) | ||
FIGURE 3.
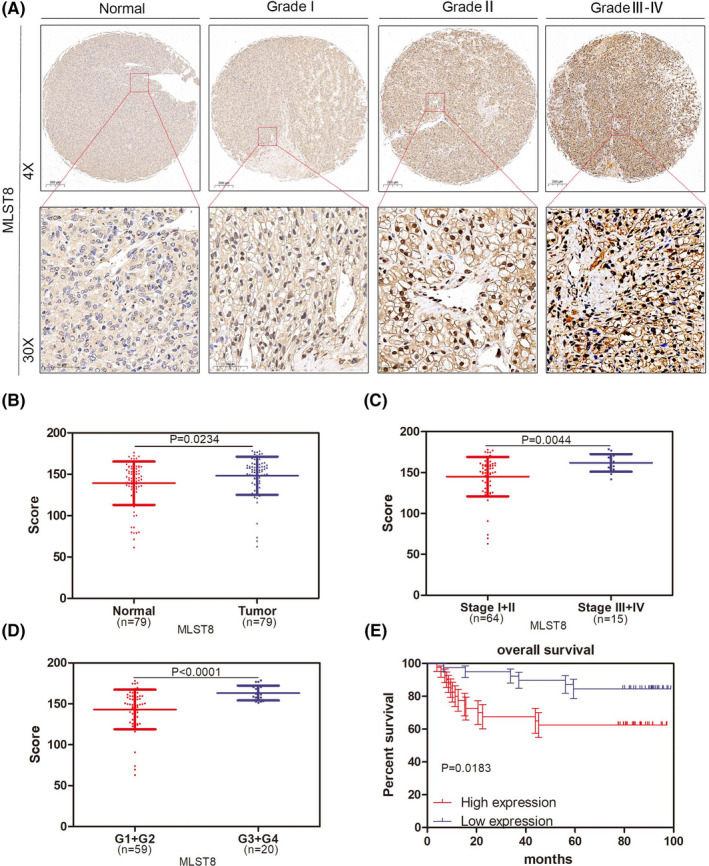
MLST8 is upregulated in ccRCC specimens and predicts poor clinical outcomes in ccRCC patients. A, Expression of MLST8 increased as ccRCC progressed to more advanced stages. MLST8 protein expression was accessed by IHC analysis in 79 paired ccRCC specimens. B, Box plots of MLST8 protein expression based on their staining index in normal renal normal tissues and ccRCC specimens. C, Box plots of MLST8 protein expression based on different clinical stages. D, Box plots of MLST8 protein expression based on IS at different clinical stages. G1, G2, G3, and G4 represent well‐differentiated, moderately differentiated, and poorly differentiated tumors, respectively. E, OS curve of ccRCC patients based on MLST8 expression according to Kaplan‐Meier analysis. Patients with high levels of MLST8 were associated with poor survival (P = .0183)
3.4. Downexpression of MLST8 inhibits ccRCC proliferation and metastasis
To confirm the clinical significance of MLST8, we were prompted to determine its biological role in the progression of ccRCC. We therefore performed a subcutaneous tumor formation test, and the results showed that MLST8 knockout could impair tumor formation in vivo (Figure 4A), suggesting that MLST8 promotes the proliferation and growth of ccRCC cells in vivo (Figure 4C‐D). Immunohistochemical analysis also showed that MLST8 expression was positively correlated with Ki67 expression, which is a recognized marker of cell proliferation (Figure 4B). According to the biological role of MLST8 mentioned above, further proof is necessary to demonstrate the in vivo role of MLST8 overexpression on metastasis. MLST8‐knocking out or empty vector‐transfected cells were subcutaneously injected into nude mice to mimic cell liver metastasis. Fewer liver metastatic nodules were observed in mice injected with MLST8‐knocking out cells compared with mice injected with control cells (Figure 4E). Histopathological studies confirmed that the liver disease was caused by ccRCC cell invasion and subsequent tumor growth (Figure 4F). We therefore conclude that MLST8 knockout inhibits renal cancer growth and metastasis.
FIGURE 4.
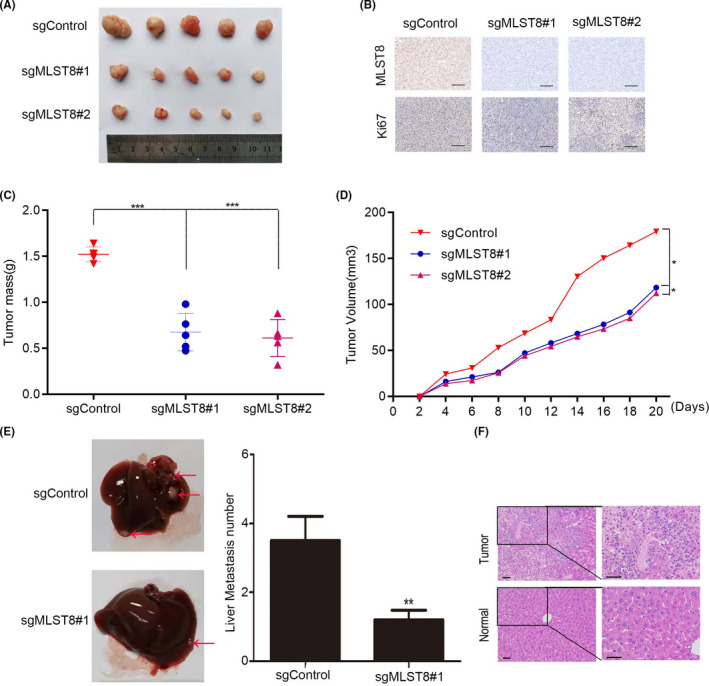
Downexpression of MLST8 inhibits ccRCC metastasis. A, Xenograft tumor formation assays show that downexpression of MLST8 inhibits subcutaneous tumor formation capacity. B, Immunohistochemical analysis of MLST8 and Ki67 expression in subcutaneous tumors. C and D, Tumor weight and tumor growth of xenografts of MLST8‐knocking out 786‐O stable cells in nude mice, respectively. E and F, Images showing intrahepatic metastasis and morphological characters by HE staining, respectively. **P < .01. All assays were conducted in triplicate
3.5. FBXW7 interacts with MLST8
A previous study performed quantitative mass spectrometry on FBXW7 knockout (KO) HCT116 cells. 23 Enrichment of MLST8 protein in the nuclear fraction of the HCT116 FBXW7‐KO cells could be a binding partner for FBXW7 and, most likely, the alpha isoform that resides in the nucleus (Figure 5A). To verify that FBXW7 is a bona fide MLST8 interactor, we first examined whether FBXW7 can interact with MLST8 cells. FLAG‐FBXW7 and Myc‐MLST8 constructs were co‐expressed in 293T cells. Cells were subsequently harvested for co‐immunoprecipitation (Co‐IP) with the anti‐FLAG antibody. As shown in Figure 5B, Myc‐MLST8 was immunoprecipitated by FLAG‐FBXW7, suggesting an exogenous interaction between these two proteins. In addition, a reciprocal Co‐IP assay was performed using lysates of 293T cells that were co‐transfected with FLAG‐MLST8 and Myc‐FBXW7 constructs. The results indicate that FLAG‐MLST8 is also able to immuneprecipitate Myc‐FBXW7 (Figure 5C). Further experiments showed that FLAG‐FBXW7 is capable of immunoprecipitating endogenous MLST8 in 786‐O cells (Figure 5D).
FIGURE 5.
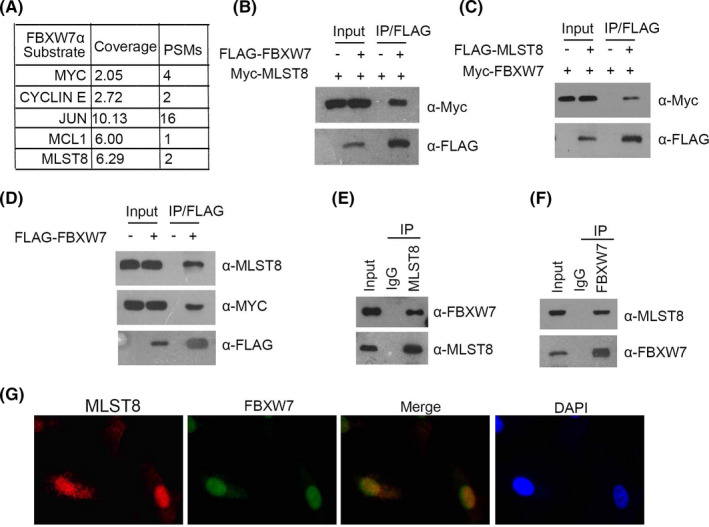
FBXW7 directly interacts with MLST8. A, Quantitative mass spectrometry using FBXW7 knockout (KO) HCT116 cells. B and C, 293T cells were co‐transfected with Myc‐MLST8 and FLAG‐FBXW7 constructs. D, Western blot and co‐IP samples of anti‐FLAG antibody obtained from 293T cells infected with expressing FLAG‐FBXW7 or control. E and F, After treatment with 20 μm MG132 for 4 h, 786‐O cell lysates were prepared for Co‐IP with anti‐MLST8 antibody and WB analyses with indicated antibodies. G, Confocal microscopy of 786‐O cells stained with MLST8 and FBXW7 antibodies. DAPI represents nuclear staining. Red, green, and blue channel images were captured by Olympus. Maximum projection images are shown, original magnification ×40
Immunoprecipitation using the anti‐MLST8 antibody was performed using cell lysates prepared from 786‐O cells. As shown in Figure 5E, endogenous FBXW7 was immunoprecipitated by MLST8, suggesting an endogenous interaction between these two proteins. Similarly, endogenous MLST8 was immunoprecipitated by FBXW7 (Figure 5F). To further confirm the endogenous interaction between MLST8 and FBXW7, we tested whether these two proteins are co‐localized to the same subcellular compartments. 786‐O Cells were immunostained with FBXW7 or MLST8 antibodies and observed by confocal microscopy. As shown in Figure 5G, FBXW7 and MLST8 were co‐located in the cell nucleus. Taken together, these results indicated that FBXW7 is associated with MLST8 in 786‐O cells.
3.6. MLST8 is degraded and ubiquitinated by tumor suppressor FBXW7
Myc‐FBXW7α and Flag‐MLST8 were co‐transfected in 293T cells, followed by treatment with DMSO and MG132, respectively. Although Myc‐FBXW7α promoted the degradation of MLST8, this effect was inhibited by treatment with the proteasome inhibitor MG132 (Figure 6A). According to previous reports, FBXW7, as the adaptor protein of the substrate, can interact with Cullin1 and RBX1 to form a ubiquitination complex, so whether Cullin1 or other members of the Culling family, for example CUL1, CUL2, CUL4A, CUL4B, and CUL5 are also involved in FBXW7's regulation of MLST8 protein stability. We therefore eliminated endogenous Culling 1, Culling 2, Culling 3, Culling 4A, Culling 4B, Culling 5, and RBX1 elements in the complex in 786‐O cells by transient transfection of siRNA. The change in MLST8 protein expression was detected by WB experiment. Interestingly, the results showed that, compared with the blank control group, the expression level of MLS8 protein was obviously increased except after CUL1 and RBX1 elimination, while the expression level of MLST8 protein was not changed by culling family protein elimination (Figure 6B). Similarly, we detected the expression changes of MLST8 protein after overexpressing Culling family negative effector mutants in 786‐O cells, and the results were consistent with the previous results. Only the overexpressing RBX1 and Cul1 negative effector mutants led to significant downregulation of MLST8 protein expression. This allowed us to determine that Cul1 specifically mediates degradation of MLST8 ubiquitin proteasome pathways in the Culling family (Figure 6C). Therefore, we conclude that Cul1 and RBX1 elements are essential for FBXW7 ubiquitination of MLST8, while other Culling family members do not appear to influence MLST8.
FIGURE 6.
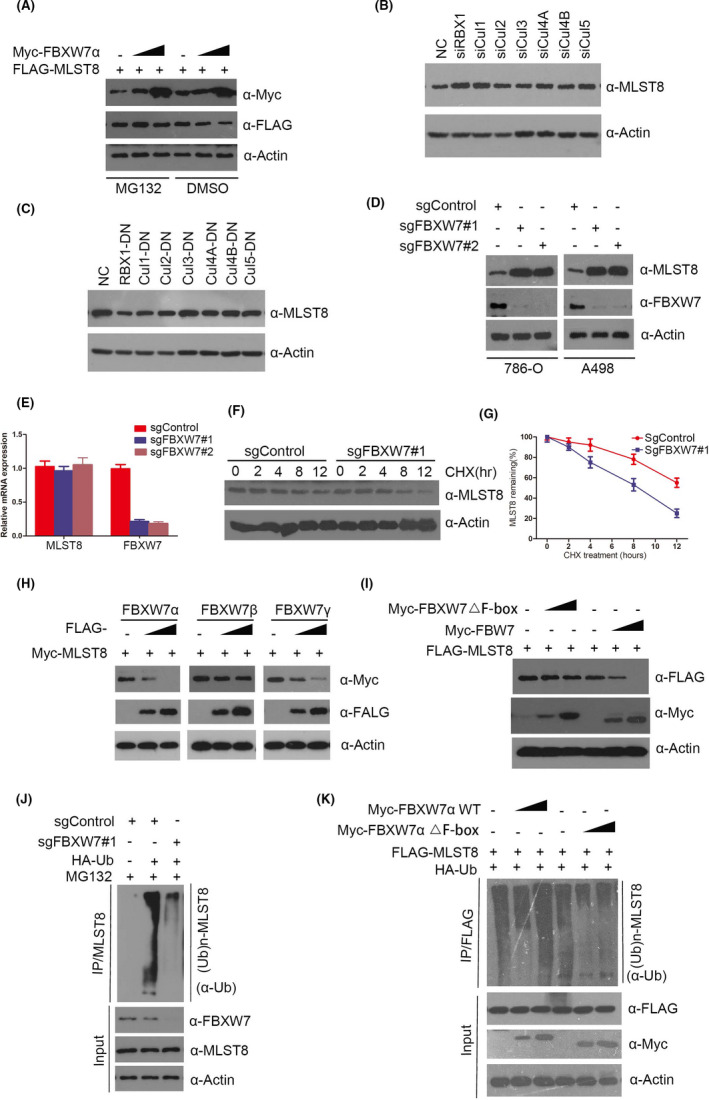
MLST8 is degraded and ubiquitination by tumor suppressor FBXW7. A, Western blotting analysis of WCLs of 293T cells transfected with the indicated constructs. B, The expressions of Cul1, Cul2, Cul3, Cul4A, Cul4B, Cul5, and RBX1 were reduced by transient transfection siRNA in 786‐O cells. Protein samples were collected 48 h after transfection, and the reduction effect and expression of MLST8 were detected by Western blot. C, In 786‐O cells, Cul1, Cul2, Cul3, Cul4A, Cul4B, and Cul5 mutants with the fLag label were overexpressed. Protein samples were collected 48 h after transfection, and the overexpression effect and expression level of MLST8 were detected by Western blot. D, Western blot of indicated proteins in WCLs from 786‐O and A498 cells with FBXW7 knockout through CRISPR/Cas9 methods. E, RT‐qPCR measurement of MLST8 mRNA expression in 786‐O cells infected with lentivirus expressing FBXW7‐specific sgRNA or NC; RT‐qPCR measurement of MLST8 mRNA expression in parental and FBXW7‐KO 786‐O cells. Data are shown as means ± SD (n = 3). F and G, Western blot of indicated proteins in WCLs of 786‐O cells infected with lentivirus expressing FBXW7‐specific sgRNA or NC for 48 h and then treated with 50 μg/mL cycloheximide (CHX) and harvested at different time points. H, 786‐O cells were transfected with the indicated plasmids for 24 h, followed by Western blotting analysis. I, 786‐O cells were transfected with the indicated plasmids for 24 h, followed by Western blotting analysis. J, Western blot of indicated proteins in WCLs from 786‐O with FBXW7 knockout through CRISPR/Cas9 methods, followed by treatment with 20 μM MG132 for 8 h. K, 786‐O cells were transfected with the indicated plasmids for 16 h, followed by treatment with 20 μM MG132 for 8 h
We used CRISPR‐Cas9 to knockout endogenous copies of FBXW7 in 786‐O and A498 cells with two FBXW7‐specific sgRNAs, resulting in an increase in MLST8 protein levels (Figure 6D) but no effect on the mRNA expression (Figure 6E). Further results showed that the depletion of FBXW7 significantly extends the half‐life of endogenous MLST8 protein and stabilizes the levels of MLST8 protein (Figure 6F,G). Furthermore, when 786‐O cells were transfected with three isoforms (α, β, and γ) of FBXW7, this resulted in a marked reduction in the protein levels of MLST8 in a dose‐dependent manner (Figure 6H). Since FBXW7 is an ubiquitinating enzyme, we wondered whether FBXW7 can ubiquitinate and downregulate MLST8 protein levels. As shown in Figure 6I, ectopic expression of FBXW7‐WT, but not the FBXW7‐△F‐box mutant (catalytic inactive), decreased the protein levels of co‐expressed MLST8 in a dose‐dependent manner, suggesting that the ubiquitination activity of FBXW7 promotes MLST8 destabilization. Conversely, the depletion of FBXW7 using CRISPR‐Cas9 knockout decreased MLST8 ubiquitination levels (Figure 6J), indicating that FBXW7 is responsible for MLST8 ubiquitination. Additionally, MLST8 protein was robustly polyubiquitinated in a dose‐dependent manner when FBXW7‐WT was co‐expressed. By contrast, minimal or no MLST8 polyubiquitination was observed in FBXW7‐△F‐box expressing cells (Figure 6K). In summary, these data demonstrate that the FBXW7 E3 ubiquitin ligase complex regulates MLST8 protein stability through the ubiquitin‐dependent proteasomal degradation pathway in ccRCC cell lines.
3.7. Mutants of FBXW7 are defective in promoting MLST8 degradation and ubiquitination
In the TCGA database, we did not find the high‐frequency mutation of FBXW7 in ccRCC, but did find it in pancreatic cancer, lung cancer, colon cancer, and breast cancer. We therefore selected the high‐frequency mutation sites of FBXW7, including R505C, W406A, T416A, L443F, W425R, S462P, H468R, D510E, and D527G, in other cancers to clarify the relationship between MLST8 and protein levels and inactive mutations of FBXW7 (Figure 7A). To test this, nine Myc‐tagged mutants of FBXW7 were generated: R505C, W406A, T416A, L443F, W425R, S462P, H468R, D510E, and D527G. We initially examined their interactions with MLST8 by co‐IP assay. The results showed that mutations of the residues at the corresponding domains substantially decreased the capacity of FBXW7 to interact with MLST8 in vivo, suggesting that FBXW7 mutants are defective in binding to MLST8 (Figure 7B). We then investigated the half‐life of additional targets of FBXW7 in cells transfected with wild type or W425R FBXW7 mutant. Thirty‐six hours following transfection cells were treated with cycloheximide (CHX) to prevent de novo protein synthesis. Western blot analyses confirmed an extended half‐life and reduced turnover of MLST8 in cells carrying W425R (Figure 7C). FBXW7 wild type, the FBXW7 R505C reportedly dead‐mutant, and additional FBXW7 mutants (S462P, H468R, D510E, D527G, W605R, T416A, W425R, and L443F) previously isolated from 786‐O cells were transiently transfected along with tagged‐expression vectors for MLST8. The ability of transfected FBXW7 and mutants to degrade targets wase assessed by Western blot analyses, As expected, wild‐type FBXW7 was able to induce degradation of MLST8 while R505C was not. Additional FBXW7 mutants (L443F, W425R, S462P, and H468R) present a phenotype similar to that of R505C and could not induce degradation of any targets (Figure 7D). Furthermore, in vivo ubiquitination assays indicated that some FBXW7 mutants (R505C, L443F, W425R, S462P, and H468R) lost the ability to promote MLST8 polyubiquitination compared to wild‐type FBXW7 (Figure 7E‐F). Taken together, our findings suggest that mutants of FBXW7 are defective in promoting MLST8 degradation and ubiquitination.
FIGURE 7.
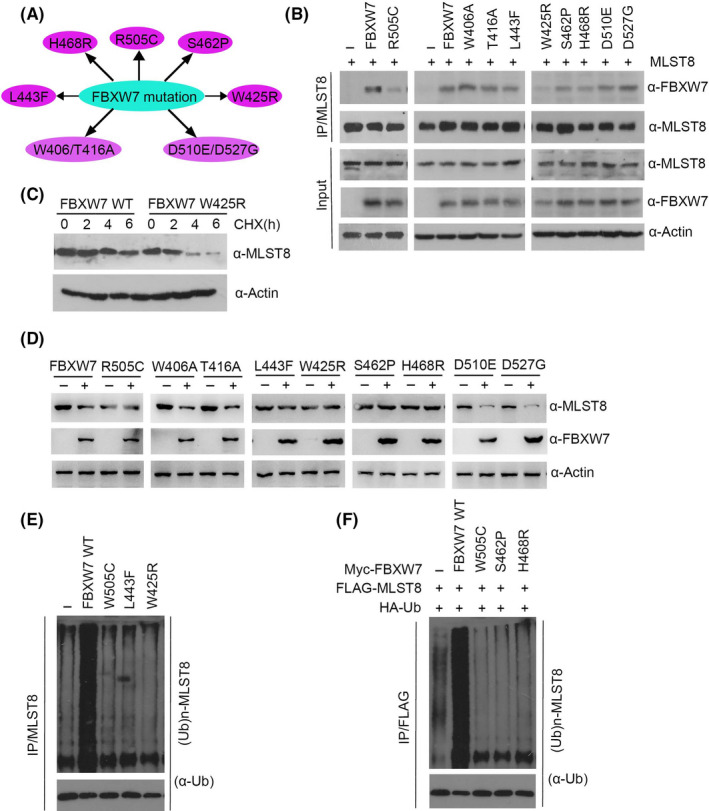
Mutants of FBXW7 are defective in promoting MLST8 degradation and ubiquitination. A, Distribution of the point mutations on the FBXW7 gene found in TCGA pan‐cancer samples. B, 293T cells were transfected with the indicated constructs. After 24 h, cell lysates were prepared for co‐IP assay with anti‐FLAG antibody and WB analyzes. C, FBXW7 W425R increased the half‐life of MLST. The effect of FBXW7 on the half‐life of oncoproteins was determined by Western blot after cells treated with 100 μg/mL cycloheximide (CHX) for 0, 2, 4, and 6 h. D, 293T cells were transfected with wild‐type or mutated FBXW7 constructs as indicated. After 24 h, cell lysates were prepared for WB analysis. E, 293T cells were transfected with the indicated constructs. After 24 h, cells were treated with 20 μmol/L MG132 for 4 h and cell lysates were prepared for immunoprecipitation and WB analysis. F, 293 T cells were cotransfected with indicated Myc‐ FBXW7, HA‐Ub, and Flag‐MLST8. Cells were treated with MG132 for 4 h before harvest. Immunoprecipitated FLAG and Western blot Ub showed the ubiquitination level of MLST8
3.8. Thr50‐dependence is required for FBXW7‐mediated degradation of MLST8
The FBXW7‐binding consensus motif (T/S)PXX(S/T/D/E) has been identified in several FBXW7 substrates, including cyclin E, c‐Myc, c‐ Jun, 9 , 10 and OASIS. 23 Interestingly, when we examined the amino acid sequence of MLST8, we discovered that MLST8 harbors one perfectly matched (50‐TPDRS‐54) FBXW7‐binding motif (Figure 8A,B). FBXW7 often recognizes phosphorylated Ser/Thr residues in the CPD motif. Therefore, we speculated that the regulation of MLST8 by FBXW7 might be mediated by the phosphorylation of Thr50 in its CPD. To examine whether this region is required for the FBXW7‐MLST8 interaction, we generated MLST8 mutants in which the acidic residues T50 and S54 were mutated to alanine. 293T cells were co‐transfected with FBXW7α and MLST8‐WT or MLST8 mutant. Although FBXW7 coimmunoprecipitated MLST8‐WT at a similar level, both MLST8 mutants completely lost their FBXW7‐binding capability (Figure 8C). These data indicate that the 50‐TPDRS‐54 motif is necessary for the FBXW7‐MLST8 interaction. Next, we sought to identify whether the 50‐TPDRS‐54 motif is required for the FBXW7‐mediated degradation of MLST8. As shown in Figure 8D, while FBXW7 efficiently targets MLST8‐WT for degradation, the MLST8‐T50A/S54A mutant displayed resistance to FBXW7‐mediated degradation, indicating that the 50‐TPDRS‐54 motif is crucial to FBXW7‐mediated MLST8 degradation. Furthermore, the T50A/S54A mutation prolonged the half‐life of MLST8 protein (Figure 8E,F). In vivo ubiquitination assays suggest that FBXW7 robustly enhances the polyubiquitination of MLST8‐WT, but not the MLST8‐T50A/S54A mutant (Figure 8G). Consistent with the protein degradation, the T50A/S54A mutation largely diminished FBXW7‐induced MLST8 ubiquitination. These data therefore demonstrate that the 50‐TPDRS‐54 motif functions as an MLST8 degron and is essential for FBXW7 binding and degradation through the ubiquitin‐proteasome pathway.
FIGURE 8.
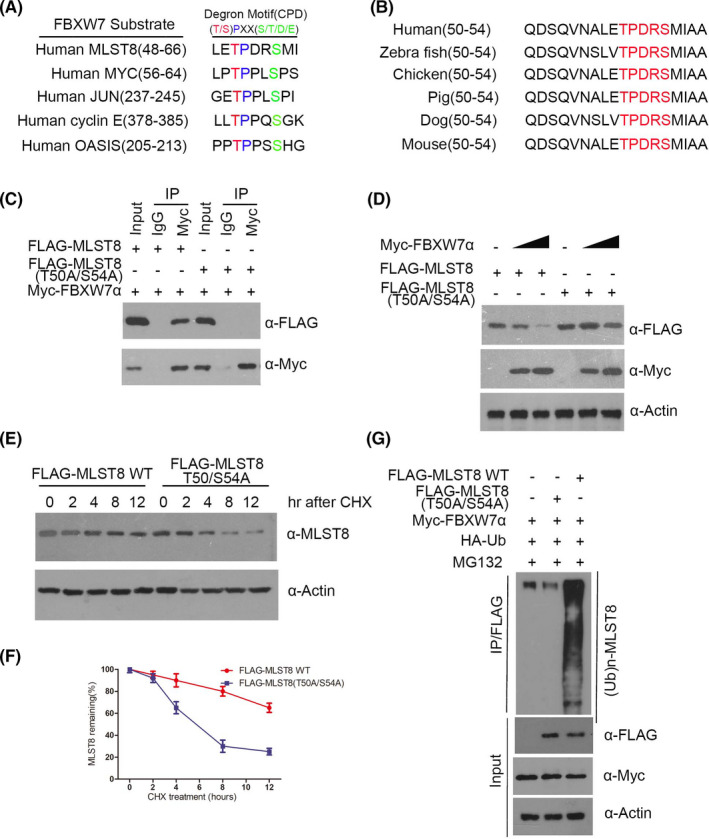
Thr50‐dependent is required for FBXW7‐mediated degradation of MLST8. A, Comparison of FBXW7 binding sites in MLST8 with the FBXW7‐binding consensus motif defined in the known FBXW7 substrates. B, Conserved CPD motifs in MLST8 in different species. C, Western blotting analysis of co‐immunoprecipitation of ectopically expressed FLAG‐MLST8 WT, FLAG‐MLST8 T50A/S54A, and Myc‐FBW7α in 293T cells. D, 786‐O cells were transfected with the indicated plasmids for 24 h, followed by Western blotting analysis. E and F, 786‐O cells were transfected with the indicated plasmids. After 24 h, cells were treated with 50 μg/mL CHX. G, 786‐O cells were transfected with the indicated plasmids for 16 h, followed by treatment with 20 μm MG132 for 8 h
3.9. Activated CDK1 induces degradation of MLST8 and decreases binding with FBXW7 and MLST8
FBXW7‐mediated degradation requires the phosphorylation of the first Ser/Thr in the CPD motif of FBXW7 target proteins by a variety of kinases, including GSK3β, 24 CDK1/2, 22 CDK4, and CDK5. 25 To identify the kinase involved in the phosphorylation of T50 in MLST8, we first analyzed the surrounding amino acid sequence and found that Thr50 in MLST8 perfectly matches the general CDK1/2 consensus motif (S/TPXK/R).26 WT‐MLST8 expressed in 293T cells treated with phosphatase and proteasome inhibitors was detected by an anti‐p‐T50‐MLST8 antibody, but this was not detected when using the T50A‐MLST8 mutant (Figure 9A). Thus, the Thr50 of exogenous MLST8 is phosphorylated in vivo. Furthermore, we detected endogenous MLST8 and its phosphorylation on Thr50 in 786‐O cells transfected with MLST8 control (Figure 9B), suggesting that endogenous MLST8 was phosphorylated at Thr50 in 786‐O cells. The CPD motif in MLST8 (Thr50‐Pro51‐Asp52‐Lys53‐Ser54) corresponds to the consensus motif for CDKs, which is Ser/Thr‐Pro‐X‐Lys/Arg (Figure 9C). To identify the kinase responsible for phosphorylation of MLST8 at residue Thr50, we performed an in vitro phosphorylation assay using purified GST‐MLST8 protein and recombinant cyclin‐CDK complexes. Phosphorylation of Thr50 in MLST8 by a variety of kinases, including CDK5, CDK4, and CDK2, was not detected, but phosphorylation on a control RB protein had the same efficiency (Figure 9D,E). CDK1 efficiently phosphorylated MLST8 at Thr50, while CDK2 slightly resulted in phosphorylation (Figure 9D). We also examined whether recombinant GSK3β phosphorylates GST‐MLST8 in vitro (Figure 9F). We determined that GSK3β did not phosphorylate Thr50 in MLST8 in vitro and is not the kinase responsible for the CPD motif of MLST8. Furthermore, we investigated whether phosphorylation of Thr50 in MLST8 was required for its recognition by FBXW7 using a GST pull‐down assay. Purified GST‐WT‐MLST8 or GST‐T50A‐MLST8 was phosphorylated by CDK1 and incubated with cell lysates expressing FLAG‐FBXW7. MLST8 in the presence of CDK1 migrated more slowly than the unphosphorylated form of MLST8, even when Thr50 was mutated to alanine (T50A). We also found that FBXW7 bound to GST‐WT‐MLST8 was detected by an anti‐p‐T50‐MLST8 antibody after treatment with CDK1, but did not bind to GST‐MLST8 without CDK1 (Figure 9G). Therefore, these results suggest that CDK1 phosphorylation of residue Thr50 in MLST8 is responsible for its recognition by FBXW7.
FIGURE 9.
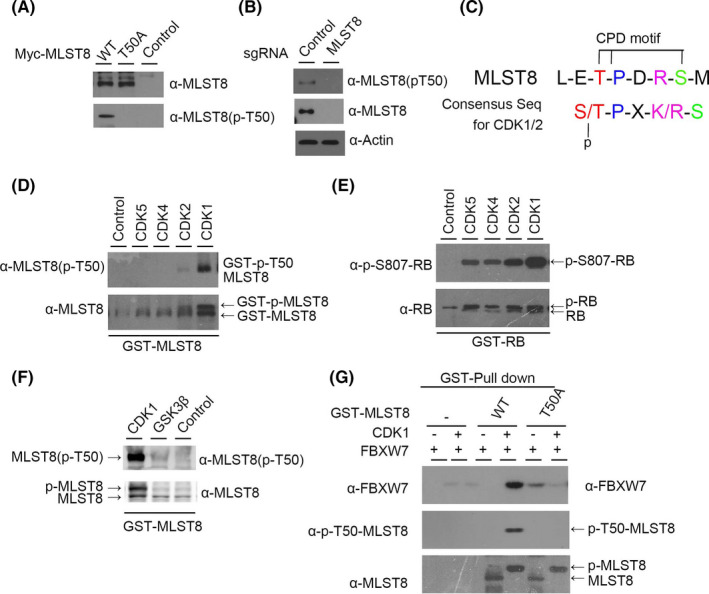
Activated CDK1 induces degradation of MLST8 and decreases binding with FBXW7 and MLST8. A, Evaluation of antibodies against phospho‐Thr50‐MLST8 (p‐T50‐MLST8). WT‐MLST8‐myc or T50A‐MLST8‐myc were transiently expressed in HEK293 cells. B, Thr50 of endogenous MLST8 protein is phosphorylated in 786‐O cells. Endogenous MLST8 protein was depleted by MLST8‐sgRNA, and lysates were subjected to immunoblotting with the indicated antibodies. C, Putative consensus motif for phosphorylation by CDK1 and CDK2 in the CPD motif containing Thr50 in MLST8. D and F, Thr50 in MLST8 is efficiently phosphorylated by CDK1 in vitro. G, Thr50‐phosphorylated MLST8 binds FBXW7 in vitro. Purified GST‐fused WT‐ or T50A‐MLST8 or GST protein using glutathione beads was incubated with or without recombinant CDK1 in reaction buffer containing 1 mM ATP for 30 min
3.10. MLST8 is an important mediator of FBXW7 inactivation‐induced cell proliferation, migration, and invasion in vitro
As determined by the CCK‐8 assay, the growth rate of MLST8‐depleted 786‐O and A498 cells was slower than that of the control cells. In contrast, the depletion of FBXW7 could significantly enhance cell proliferation, and the phenotypes could be rescued by MLST8 co‐depletion (Figure 10A,B). We observed similar effects for the cell proliferation assay, in which the increase of migration and invasion in 786‐O and A498 cells by FBXW7 depletion was partly diminished by MLST8 inactivation (Figure 10C,D). Thus, these results suggest that MLST8 is an essential mediator of FBXW7 inactivation‐induced cell proliferation, migration, and invasion.
FIGURE 10.
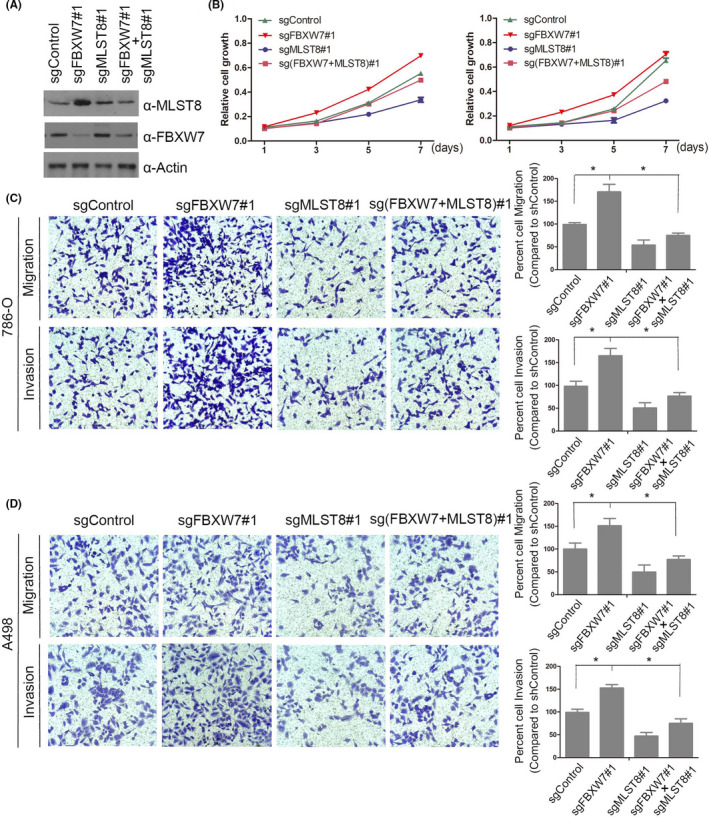
MLST8 is an important mediator of FBXW7 inactivation‐induced cell proliferation, migration, and invasion in vitro. A and B, Western blot and CCK8 cell proliferation assay of parental or FBXW7 and/or MLST8 knockout 786‐O and A498 cells. The standard deviation (SD) of at least three independent experiments is shown to indicate statistical significance. C and D, Cell migration and invasion assays of parental or FBXW7 and/or MLST8 knockout 786‐O and A498 cells. *P < .05
3.11. MLST8 and FBXW7 protein levels negatively correlate in human renal cancer specimens
Given the critical role of FBXW7 and MLST8 in ccRCC, we analyzed the expression of FBXW7 and MLST8 by IHC on a tissue microarray (TMA) in 79 pairs of renal cancer samples with clinical follow‐up information and observed that both are mainly expressed in the nucleus, which is consistent with the result of immunofluorescence. Statistical analysis revealed that MLST8 was remarkably overexpressed in renal cancer tissues compared to normal tissue. In contrast, FBXW7 was expressed at a lower level in tumor tissues (Figure 11A ). The clinical and pathologic characteristics of the patients are shown in Table. 2. In the 79 paired ccRCC cases, MLST8 protein was greatly overexpressed in 60.0% of tumor tissues and FBXW7 was found to be underexpressed in 68.4% of tumor tissues (Figure 11B ). Moreover, statistical analysis indicated that MLST8 was negatively correlated with FBXW7 expression in this cohort of patient samples (r = −.3124, P = .0051) (Figure 11C ). Furthermore, the univariate and multivariate analyses were used to determine the independent prognostic factors for ccRCC patients (Table 4). In univariate analysis, the MLST8 expression level instead of FBXW7, was found to be significantly associated with the OS (P = .006). TNM stage (P < .0001) and Fuhrman grade (P = .001) were also correlated significantly with OS, but other factors such as tumor size (P = .058) and age (P = .871) were uncorrelated significantly with OS. Moreover, multivariate analysis showed that the TNM stage (P < .0001) and tumor size (P = .050) were proven to be independent predictors of OS for ccRCC patients. Simultaneously, other variables, including age and Fuhrman grade, did not contribute to OS independently (P > .05).
FIGURE 11.
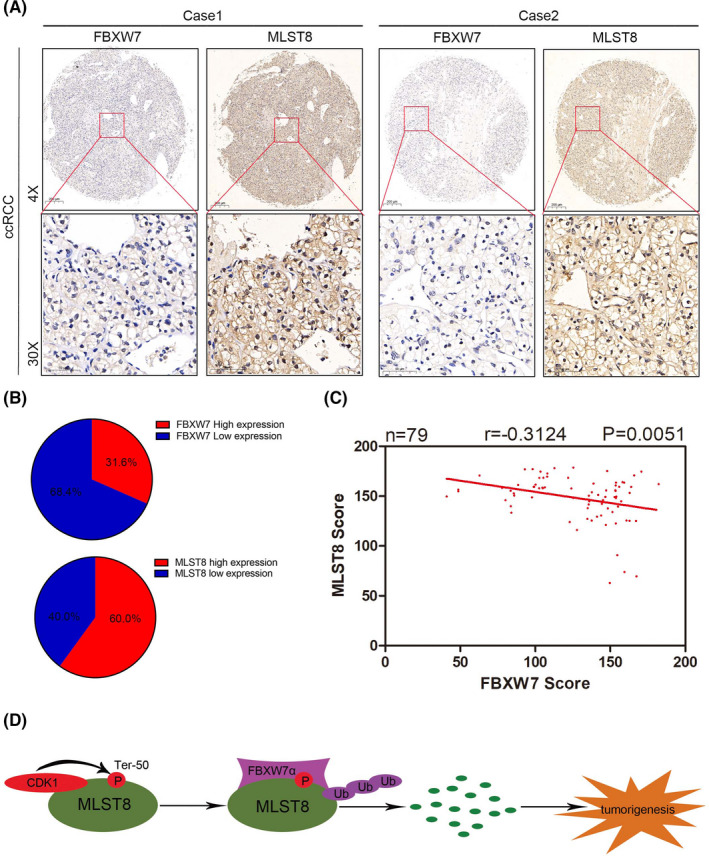
MLST8 and FBXW7 protein levels negatively correlate in human renal cancer specimens. A, Representative staining of IHC analysis of MLST8 and FBXW7 protein expression on tissue microarray. B, FBXW7 and MLST8 staining patterns in 79 paired ccRCC cases. C, Negative correlation of FBXW7 and MLST8 protein expression was observed in human renal cancer specimens (Pearson's test, P < .01). D, The CDK1 ‐FBXW7α signal axis in the regulation of MLST8 protein degradation. On phosphorylation of Ter‐50 by the CDK1 axis, MLST8 is recognized and ubiquitinated by FBXW7 and targeted for degradation, which in turn suppresses renal cancer progression
TABLE 4.
Summary of univariate and multivariate Cox regression analysis of OS duration in all ccRCCs
| Variables | Univariable analysis | Multivariable analysis | ||||
|---|---|---|---|---|---|---|
| HR | (95% CI) | P value | HR | (95% CI) | P value | |
| FBXW7 expression (low vs high) | 0.717 | 0.164‐1.452 | .197 | 0.628 | 0.161‐1.766 | .304 |
| MLST8 expression (low vs high) | 1.704 | 1.617‐18.684 | .006 | 1.274 | 0.888‐14.385 | .073 |
| Age (≦60 vs ﹥60) | 0.071 | 0.396‐2.194 | .871 | 0.561 | 0.231‐1.409 | .224 |
| TNM stage (III/IV vs Ⅰ/II) | 2.382 | 4.485‐26.135 | .000 | 2.235 | 3.310‐26.372 | .000 |
| Furman grade (III/IV vs Ⅰ/II) | 1.489 | 1.860‐10.571 | .001 | 0.039 | 0.315‐3.437 | .949 |
| Tumor size (≦4 vs >4) | 1.944 | 0.937‐52.070 | .058 | 2.043 | 1.004‐59.239 | .050 |
4. DISCUSSION
Over recent decades, aberrant activation of multiple signaling cascades, including the Hippo pathway and the PI3K/Akt/mTOR pathway, has been shown to be involved in the development and progression of ccRCC. 1 , 26 The mTORC2 complex consists of mTOR, Rictor, Sin1, and MLST8, 27 but the role of MLST8, as a core member of the mTOR pathway, is unsettled in cases of ccRCC.
In recent years there have been multiple reports suggesting the role of MLST8 in malignancies, including colon and prostate cancer. Studies have shown that the overexpression of MLST8 suppresses tumor growth and invasion. 4 , 26 Another study showed that MLST8 contributes to promoting cell proliferation and metastasis via the AKT pathway in HCC. 14 We observed that MLST8 upregulation was associated with the upregulation of other components of mTOR complexes, such as mTOR itself and RICTOR/RAPTOR. Therefore, MLST8 upregulation can be attributed to enhanced protein stabilization, which is in turn caused by the upregulation of binding partners that are modulated by other mechanisms, such as micro‐RNA silencing. 28
Our study first detected MLST8 expression and analyzed its correlation with clinical factors in ccRCC patients. We identified that the overexpression of MLST8 was a frequent event in ccRCC specimens, which is in line with TCGA. We also confirmed that increased MLST8 expression is predictive of an unfavorable prognosis for human renal cancer. The cellular functions and molecular mechanisms of MLST8 remain unknown in ccRCC.
Ubiquitin‐proteasome‐dependent protein degradation usually requires direct interaction between the substrate and an E3 ubiquitin ligase. 29 Fbw7 contains several conserved protein interaction domains. A stretch of eight WD40 repeats makes multiple contacts with the CPDs in the Fbw7 substrate. 29 Rapamycin binds to FK506 binding protein‐12 (FKBP12) to form the FKBP12‐rapamycin complex, which only partially inhibits mTOR kinase activity. 30 A recent study implied that concurrent targeting of mTORC1/2 by OSI‐027 potently inhibits the proliferation and the migration of keloid keratinocytes. 31 Other studies have implied that the concurrent targeting of mTORC1/2 by OSI‐027 potently inhibits the proliferation and migration of keloid keratinocytes. 31 Due to these limitations, direct mTOR kinase inhibitors have been developed and termed “second‐generation mTOR inhibitors”. 32
In this study, we demonstrate for the first time that the WD40 domain of Fbw7 indeed binds to the CPD of MLST8. We note that the depletion of FBXW7 shows impaired binding to MLST8 proteins, resulting in impaired proteasomal degradation and the accumulation of MLST8 in renal cancer cell lines and cancer specimens, which partly contributes to FBXW7 inactivation‐induced renal cancer cell migration and invasion. Interestingly, the exact mechanism by which CDK1, excluding the possibility of other protein kinases, phosphorylates MLST8 has also been determined (Figure 11D).
As a shared subunit between mTORC1 and mTORC2, MLST8 may play a divergent role in the activation of both mTORC1 and mTORC2. Genetic studies showed that the ablation of MLST8 in mice only influences mTORC2 function. 6 MLST8 protein inhibitors are emerging as promising anticancer therapies. The gene encoding the E3 ubiquitin ligase FBXW7 was the most frequently mutated gene in 20 CCA patients. 6 FBXW7, W406R, and T416A are similar in activity to wild‐type FBXW7, while the mutants D510E and D527G degrade all targets except for NOTCH1. 33 Hotspot mutations in FBXW7 have been reported for R505, R465, and R479, and represent 25.41%, 9.29%, and 13.40%, respectively, of all FBXW7 mutations in cancer. 34 Mutations in FBXW7 that prevent BRAF degradation were associated with resistance to BET inhibitors. 35 FBXW7 mutant expression confers BET inhibitor resistance.
In conclusion, our results show that the upregulation of MLST8 exerts a decisive role in ccRCC progression. MLST8 is an independent predictive indicator for OS and provides novel insights into the aberrant regulation of MLST8 in renal cancer, suggesting that MLST8 is an important mediator of FBXW7 inactivation‐induced cell proliferation, migration, and invasion. This understanding will contribute to the elucidation of new strategies in developing substrate‐specific FBXW7 inhibitors for therapeutic use.
CONFLICT OF INTEREST
The authors have no conflict of interest.
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (81570607).
Zhang E, Chen S, Tang H, et al. CDK1/FBXW7 facilitates degradation and ubiquitination of MLST8 to inhibit progression of renal cell carcinoma. Cancer Sci.2022;113:91–108. 10.1111/cas.15188
Encheng Zhang and Siteng Chen equal contributors and co‐first authors.
Contributor Information
Yu Fan, Email: fanyu1965@163.com.
Mingyue Tan, Email: tanmoon@163.com.
Xiang Wang, Email: seanw_hs@163.com.
REFERENCES
- 1. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960‐976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Janku F, Yap TA, Meric‐Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol. 2018;15:273‐291. [DOI] [PubMed] [Google Scholar]
- 3. Yu XN, Chen H, Liu TT, Wu J, Zhu JM, Shen XZ. Targeting the mTOR regulatory network in hepatocellular carcinoma: are we making headway? Biochim Biophys Acta Rev Cancer. 2019;1871:379‐391. [DOI] [PubMed] [Google Scholar]
- 4. Kakumoto K, Ikeda J, Okada M, Morii E, Oneyama C. mLST8 promotes mTOR‐mediated tumor progression. PLoS One. 2015;10:e0119015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yu XN, Zhang GC, Sun JL, et al. Enhanced mLST8 expression correlates with tumor progression in hepatocellular carcinoma. Ann Surg Oncol. 2020;27:1546‐1557. [DOI] [PubMed] [Google Scholar]
- 6. Guertin DA, Stevens DM, Thoreen CC, et al. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt‐FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11:859‐871. [DOI] [PubMed] [Google Scholar]
- 7. Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. Human F‐box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature. 2001;413:316‐322. [DOI] [PubMed] [Google Scholar]
- 8. Zhang W, Koepp DM. Fbw7 isoform interaction contributes to cyclin E proteolysis. Mol Cancer Res. 2006;4:935‐943. [DOI] [PubMed] [Google Scholar]
- 9. Yada M, Hatakeyama S, Kamura T, et al. Phosphorylation‐dependent degradation of c‐Myc is mediated by the F‐box protein Fbw7. Embo J. 2004;23:2116‐2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nateri AS, Riera‐Sans L, Da Costa C, Behrens A. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science. 2004;303:1374‐1378. [DOI] [PubMed] [Google Scholar]
- 11. Gupta‐Rossi N, Le Bail O, Gonen H, et al. Functional interaction between SEL‐10, an F‐box protein, and the nuclear form of activated Notch1 receptor. J Biol Chem. 2001;276:34371‐34378. [DOI] [PubMed] [Google Scholar]
- 12. Sundqvist A, Bengoechea‐Alonso MT, Ye X, et al. Control of lipid metabolism by phosphorylation‐dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab. 2005;1:379‐391. [DOI] [PubMed] [Google Scholar]
- 13. Akhoondi S, Sun D, von der Lehr N, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007;67:9006‐9012. [DOI] [PubMed] [Google Scholar]
- 14. Akhoondi S, Lindstrom L, Widschwendter M, et al. Inactivation of FBXW7/hCDC4‐beta expression by promoter hypermethylation is associated with favorable prognosis in primary breast cancer. Breast Cancer Res. 2010;12:R105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ji S, Qin Y, Liang C, et al. FBW7 (F‐box and WD repeat domain‐Containing 7) negatively regulates glucose metabolism by targeting the c‐Myc/TXNIP (Thioredoxin‐Binding protein) axis in pancreatic cancer. Clin Cancer Res. 2016;22:3950‐3960. [DOI] [PubMed] [Google Scholar]
- 16. Liang C, Qin Y, Zhang B, et al. Oncogenic KRAS targets MUC16/CA125 in pancreatic ductal adenocarcinoma. Mol Cancer Res. 2017;15:201‐512. [DOI] [PubMed] [Google Scholar]
- 17. Arabi A, Ullah K, Branca RM, et al. Proteomic screen reveals Fbw7 as a modulator of the NF‐kappaB pathway. Nat Commun. 2012;3:976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kitagawa M, Okabe T, Ogino H, et al. Butyrolactone I, a selective inhibitor of cdk2 and cdc2 kinase. Oncogene. 1993;8:2425‐2432. [PubMed] [Google Scholar]
- 19. Kitagawa M, Higashi H, Takahashi IS, et al. A cyclin‐dependent kinase inhibitor, butyrolactone I, inhibits phosphorylation of RB protein and cell cycle progression. Oncogene. 1994;9:2549‐2657. [PubMed] [Google Scholar]
- 20. Kitagawa K, Shibata K, Matsumoto A, et al. Fbw7 targets GATA3 through cyclin‐dependent kinase 2‐dependent proteolysis and contributes to regulation of T‐cell development. Mol Cell Biol. 2014;34:2732‐2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kitagawa M, Higashi H, Jung HK, et al. The consensus motif for phosphorylation by cyclin D1‐Cdk4 is different from that for phosphorylation by cyclin A/E‐Cdk2. EMBO J. 1996;15:7060‐7069. [PMC free article] [PubMed] [Google Scholar]
- 22. Nakajima T, Kitagawa K, Ohhata T, et al. Regulation of GATA‐binding protein 2 levels via ubiquitin‐dependent degradation by Fbw7: involvement of cyclin B‐cyclin‐dependent kinase 1‐mediated phosphorylation of THR176 in GATA‐binding protein 2. J Biol Chem. 2015;290:10368‐10381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yumimoto K, Matsumoto M, Onoyama I, Imaizumi K, Nakayama KI. F‐box and WD repeat domain‐containing‐7 (Fbxw7) protein targets endoplasmic reticulum‐anchored osteogenic and chondrogenic transcriptional factors for degradation. J Biol Chem. 2013;288:28488‐28502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koo J, Wu X, Mao Z, Khuri FR, Sun SY. Rictor undergoes glycogen synthase kinase 3 (GSK3)‐dependent, FBXW7‐mediated ubiquitination and proteasomal degradation. J Biol Chem. 2015;290:14120‐14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maskey D, Marlin MC, Kim S, et al. Cell cycle‐dependent ubiquitylation and destruction of NDE1 by CDK5‐FBW7 regulates ciliary length. EMBO J. 2015;34:2424‐2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shi X, Zhu HR, Liu TT, Shen XZ, Zhu JM. The Hippo pathway in hepatocellular carcinoma: non‐coding RNAs in action. Cancer Lett. 2017;400:175‐182. [DOI] [PubMed] [Google Scholar]
- 27. Sarbassov DD, Ali SM, Kim DH, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin‐insensitive and raptor‐independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296‐1302. [DOI] [PubMed] [Google Scholar]
- 28. Oneyama C, Ikeda J, Okuzaki D, et al. MicroRNA‐mediated downregulation of mTOR/FGFR3 controls tumor growth induced by Src‐related oncogenic pathways. Oncogene. 2011;30:3489‐3501. [DOI] [PubMed] [Google Scholar]
- 29. Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83‐93. [DOI] [PubMed] [Google Scholar]
- 30. Yu K, Toral‐Barza L. Biochemical and pharmacological inhibition of mTOR by rapamycin and an ATP‐competitive mTOR inhibitor. Methods Mol Biol. 2012;821:15‐28. [DOI] [PubMed] [Google Scholar]
- 31. Chen J, Liu K, Liu Y, Wang X, Zhang Z. Targeting mTORC1/2 with OSI‐027 inhibits proliferation and migration of keloid keratinocytes. Exp Dermatol. 2019;28:270‐275. [DOI] [PubMed] [Google Scholar]
- 32. Zhou HY, Huang SL. Current development of the second generation of mTOR inhibitors as anticancer agents. Chin J Cancer. 2012;31:8‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pancewicz J, Taylor JM, Datta A, et al. Notch signaling contributes to proliferation and tumor formation of human T‐cell leukemia virus type 1‐associated adult T‐cell leukemia. PNAS. 2010;107:16619‐16624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yeh CH, Bellon M, Nicot C. FBXW7: a critical tumor suppressor of human cancers. Mol Cancer. 2018;17:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yeh CH, Bellon M, Wang F, Zhang H, Fu L, Nicot C. Loss of FBXW7‐mediated degradation of BRAF elicits resistance to BET inhibitors in adult T cell leukemia cells. Mol Cancer. 2020;19:139. [DOI] [PMC free article] [PubMed] [Google Scholar]