

Abstract
Precise quantification of copy‐number alterations (CNAs) in a tumor genome is difficult. We have applied a comprehensive copy‐number analysis method, digital multiplex ligation‐dependent probe amplification (digitalMLPA), for targeted gene copy‐number analysis in clear cell renal cell carcinoma (ccRCC). Copy‐number status of all chromosomal arms and 11 genes was determined in 60 ccRCC samples. Chromosome 3p loss and 5q gain, known as early changes in ccRCC development, as well as losses at 9p and 14q were detected in 56/60 (93.3%), 31/60 (51.7%), 11/60 (18.3%), and 33/60 (55%), respectively. Through gene expression analysis, a significant positive correlation was detected in terms of 14q loss determined using digitalMLPA and downregulation of mRNA expression ratios with HIF1A and L2HGDH (P = .0253 and .0117, respectively). Patients with early metastasis (<1 y) (n = 18) showed CNAs in 6 arms (in median), whereas metastasis‐free patients (n = 34) showed those in significantly less arms (3 arms in median) (P = .0289). In particular, biallelic deletion of CDKN2A/2B was associated with multiple CNAs (≥7 arms) in 3 tumors. Together with sequence‐level mutations in genes VHL, PBRM1, SETD2, and BAP1, we performed multiple correspondence analysis, which identified the association of 9p loss and 4q loss with early metastasis (both P < .05). This analysis indicated the association of 4p loss and 1p loss with poor survival (both, P < .05). These findings suggest that CNAs have essential roles in aggressiveness of ccRCC. We showed that our approach of measuring CNA through digitalMLPA will facilitate the selection of patients who may develop metastasis.
Keywords: ccRCC, copy‐number alteration, digitalMLPA, metastasis, prognosis
Our digitalMLPA approach of measuring copy‐number alterations for all chromosomal arms can distinguish patients with early metastasis from metastasis‐free patients, and will facilitate the selection of patients who may develop metastasis.
Abbreviations
- aCGH
array comparative genomic hybridization
- ccRCC
clear cell renal cell carcinoma
- CN
copy number
- CNA
copy‐number alteration
- digitalMLPA
digital multiplex ligation‐dependent probe amplification
- HEKn
human epidermal keratinocytes
- NGS
next‐generation sequencing
- PBL
peripheral blood lymphocytes
- TMB
tumor mutational burden
1. INTRODUCTION
NGS has revolutionized the analysis of nucleic acids, and has become an essential tool for the detection of somatic mutations in tumor tissues, for research and clinical use. 1 NGS is optimal for detection of single nucleotide variants and small insertions and deletions, but its application remains challenging for the detection of larger alterations such as single gene/exon CN variants. The precise quantification of CNAs in tumor genomes using NGS is difficult, especially when the tumor content of the tissue is low, and/or when target NGS approach is used.
Currently, tumor genome research with NGS is biased toward sequence‐level mutations that are easily detected and tends to overlook CNAs, which also have an essential role in tumorigenesis. 2 The standard tools for CN analysis are multiplex ligation‐dependent probe amplification (MLPA) and array comparative genomic hybridization (aCGH). 3 , 4 , 5 However, the former approach has a limited number of targets that can be assessed simultaneously, and the latter is labor intensive, and therefore suboptimal for clinical use.
digitalMLPA is a novel technique for CN detection that combines MLPA and NGS (Figure 1A), 6 in which amplicons are generated for each ligated digitalMLPA probe, and NGS sequencing is used to determine the read numbers of each amplicon. The CN status of the target regions is then determined by quantification of the relative read counts of the various amplicons.
FIGURE 1.
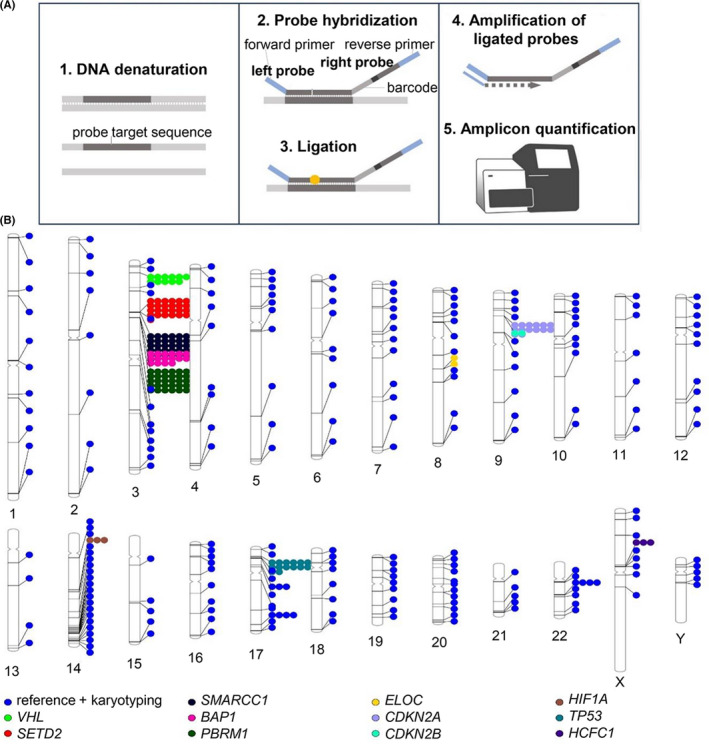
digitalMLPA. A, Assay principle. B, The probe regions of D017 (lot X1‐1018, RUO)
Among human carcinomas, clear cell renal cell carcinomas (ccRCCs) are known for substantial genetic heterogeneity, ie, involvement of CN losses and gains of large chromosomal regions, 7 , 8 , 9 , 10 , 11 and low rates of base‐substitution mutations. 12 In this study, we used a digitalMLPA assay containing 384 probes to determine the key CNAs in ccRCCs.
2. MATERIALS AND METHODS
2.1. Patient samples
Samples were obtained from patients at the Hospital of Hyogo College of Medicine diagnosed by a pathologist as having ccRCC. This study was approved by the Ethics Committee of Hyogo College of Medicine (Rinhi‐273), and performed in accordance with the Declaration of Helsinki (1995) of the World Medical Association (as revised in 2013 in Fortaleza, Brazil). All patients provided written informed consent.
Fresh ccRCC tissues were resected during surgery, and snap frozen at −80°C. Genomic DNA was extracted from frozen tissues using AllPrep DNA/RNA Mini kits (Qiagen, Hilden, Germany) and purified using NucleoSpin gDNA Clean‐up reagents (Macherey‐Nagel, Düren, Germany). Clinicopathological data for the patients included in study are described in Table 1.
TABLE 1.
Clinicopathological data for 60 ccRCC patients included in the study
| Sex | |
| Men | 44 |
| Women | 16 |
| Age, y, range (median) | 42‐85 (65.5) |
| TMN stage | |
| T1 | 32 |
| T2 | 7 |
| T3‐T4 | 21 |
| Metastasis | |
| Positive (at diagnosis or <1 y after tumorectomy) | 18 |
| Positive (≥1 y after tumorectomy) | 8 |
| Negative | 34 |
| Survival | |
| Dead | 10 a |
| Alive | 50 |
This number includes 1 death injury by external cause.
2.2. digitalMLPA
D017 probemix (lot X1‐1018, RUO) was developed for ccRCC and malignant mesothelioma. CN of the 11 target genes, von Hippel‐Lindau (VHL), SETD2, SMARCC1, BAP1, PBRM1, ELOC, CDKN2A, CDKN2B, HIF1A, TP53, and HCFC1, was evaluated using multiple probes. The D017 probemix included (i) 151 target probes for 11 genes known to have frequent genome alterations in ccRCC; (ii) 233 probes used for karyotyping (targeting each chromosomal arm), 141 of which target relatively CN stable regions in renal cancer and were used as reference probes for data normalization (Figure 1B and Table S1); (iii) 78 internal control probes were used for sample identification; and (iv) 53 probes were used for quality assessment.
The digitalMLPA protocol has previously been described in detail 6 and uses 20‐40 ng of genomic DNA. For each probe, at least 600 reads were generated. Data analysis was performed using in‐house software by MRC Holland. In each experiment batch, the read number generated for each probe was compared with those of the reference samples, which consisted of 20 genomic DNA samples of 20 healthy young Japanese men (each consist of Epstein‐Barr virus‐transformed B cells) obtained from the Riken DNA bank (Ibaraki, Japan) and then pooled. Samples with insufficient DNA quality or with suboptimal digitalMLPA reaction quality as indicated using quality control probes, were removed. In the same experiment batch for tumor analysis, 22 blood DNAs purified in the same procedure, from patients with ccRCCs were analyzed. The standard deviation (SD) values of CN ratios were calculated and the average SD value was 0.054 among all probes of autosomal genes (Figure S1A). It was also confirmed that blood DNAs from 16 Japanese healthy individuals showed the similar distribution of SD value for all probes of autosomal genes, indicating minimal variability.
The average ratios of 2 batches of experiments were calculated to determine CNA of each locus. A region containing at least 3 consecutive probes in the same chromosome, showing an average CN ratio outside the range 0.9 to 1.1, was regarded as a possible gene loss or gain, or chromosomal arm loss or gain. For continuing values surrounding the cut‐off value, that area was judged to be CNA if >60% of the probes in a chromosomal arm had CN ratios outside 0.9 to 1.1. We applied this particular cut‐off range for our present experiments because ccRCCs generally show CNAs of large chromosomal regions. However, it does not represent cut‐off value for other solid tumor tissues in general.
Using mesothelioma cell lines from HMMME or H2452 showing segmental CNAs as detected using single nucleotide polymorphism arrays and target NGS in our previous experiments, titration experiments were conducted by mixing DNA from HMMME or H2452 into DNA derived from normal human neonatal male epidermal keratinocytes, (HEKn) (Cascade Biologics, Carlsbad, CA) (n = 3). Samples of tumor cell line DNA and HEKn DNA were titrated in different proportions to produce 7 samples for further analysis, in the following tumor DNA contents: 0%, 5%, 10%, 20%, 30%, 50%, and 100%. For each digitalMLPA reaction, a total of 40 ng of DNA was used to identify the tumor cell DNA proportion at which monoallelic loss in the spiked cancer cell line DNA could be detected reproducibly.
2.3. Real‐time RT‐PCR
Total RNA was isolated from ccRCC tissues using an AllPrep DNA/RNA mini kit following the manufacturer's protocol. However, 15 isolated RNA samples degraded of the 60 ccRCCs. Subsequently, for the remaining 45 cases (Table S2), real‐time RT‐PCR was conducted using TaKaRa PrimeScript RT reagent kit and TB Green Premix Ex Taq II (or TB Green® Premix DimerEraser for L2HGDH) (TaKaRa, Shiga, Japan). The following primers were also used, after which normalization was conducted with glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH); HIF1A: 5′‐CACCACAGGACAGTACAGGAT‐3′ and 5′‐CGTGCTGAATAATACCACTCACA‐3′, L2HGDH: 5′‐TGTGCAGGACTTTACTCAGACC‐3′ and 5′‐GTAATCTCCCCGGAATGGTACA‐3′, KLHL33: 5′‐GCGTGGGACTGGTACGAAC‐3′ and 5′‐GGGTGTTGGAGTGACTGTAGA‐3′, GAPDH: 5′‐GCACCGTCAAGGCTGAGAAC‐3′ and 5′‐TGGTGAAGACGCCAGTGGA‐3′. Gene expression ratio was set to be 1.0 in the case of normal renal tissue (R06).
2.4. Target NGS
Libraries were prepared from 100 ng of genomic DNA from tumors, using Integrated DNA Technologies (IDT) Lotus™ DNA Library Prep Kits and xGen® Custom Target Capture Probes (Integrated DNA Technologies Inc., Coralville, IA, USA) according to the manufacturer's instructions. NGS was performed using an Illumina MiSeq (San Diego, CA) using paired‐end 150 bp runs. Illumina paired‐end reads were aligned to the human NCBI Build 37 reference sequence using bwa software (bio‐bwa.sourceforge.net/, version 0.6.0). The aligned sequence files were sorted and merged using SAMtools (samtools.sourceforge.net/, version 0.1.18). GATK (http://www.broadinstitute.org/gatk/) was used for realignment, base quality score recalibration, single nucleotide variant or indel (small insertions and deletions) variant calling, and variant quality recalibration. Germline or somatic mutations were confirmed by comparison between genomic DNA from tumors and that from the PBL of the patient, using Sanger sequencing. 13
2.5. Statistical analyses
Gene expression ratios were compared between, in ccRCCs, with 14q loss and the ones without 14q loss using the chi‐squared test. Multiple correspondence analysis was used to evaluate the associations among the variables and identify the risk factors associated with metastasis or poor survival. The variables used were: metastasis (metastasis‐free; late metastasis, more than 1 y after tumorectomy; early metastasis, <1 y including metastasis at diagnosis), survival excluding accidental death, 1p loss, 3p loss, 4p loss, 4q loss, 5p gain, 5q gain, 6q loss, 7 gain, 8p loss, 9p loss, 9q loss, 10q loss, 12 gain, 14q loss, 16p gain, 16q gain, 17p13 loss, 18 loss, 20 gain, 21q loss, 22 loss, and base‐substitution mutations in VHL, PBRM1, SETD2, and BAP1. Nonparametric pairwise multiple comparison analysis among 3 metastasis groups was carried out using Dunn multiple comparison test with the number of chromosome alterations. P‐values < .05 were considered to be statistically significant. JMP Pro v14.2 (SAS Institute, Cary, NC, USA) was used for the statistical analysis.
3. RESULTS
3.1. Sensitivity of digitalMLPA to detect CN loss
To estimate the sensitivity and reproducibility of CNA detection with digitalMLPA, titration DNA experiments were conducted. Tumor cell line DNA from HMMME or H2452, both with losses in 3p21 and 9p21 was mixed with DNA from normal cells DNA (HEKn) at proportions of 5%‐100%. Monoallelic losses were detected with high reproducibility at the proportion of 20% tumor cell line DNA (HMMME) in 80% of normal DNA in 3p21 (Figure 2A,B) and 17p13 regions (Figure 2C). The biallelic deletion of the CDKN2A/2B region could be detected at proportion of 10% of tumor cell line DNA (H2452) in 90% of normal DNA (Figure 2D).
FIGURE 2.
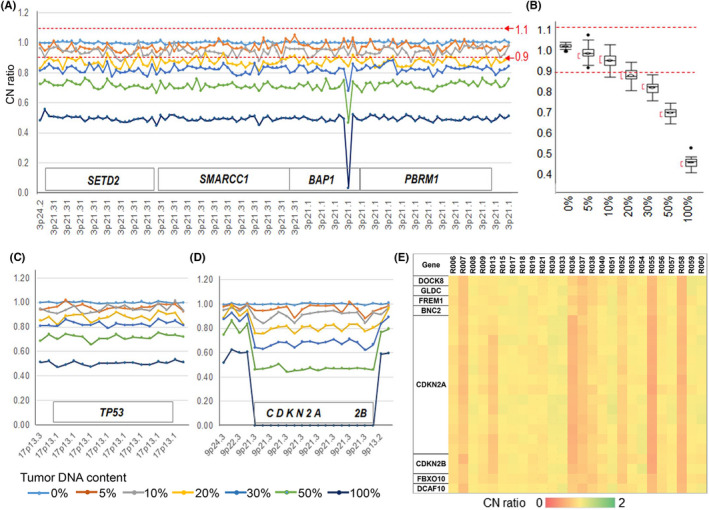
Titration experimental results of tumor cell line DNA into normal DNA from neonatal human epidermal keratinocytes (HEKn). A, CN ratio plot in the 3p21 region for DNA titration from tumor cell line HMMME. B, Box plot showing CN deviation of experiment A, omitting 1 probe showing biallelic deletion in exon 7 of BAP1; the average CN ratio was 1.002 at 0%, 0.972 at 5%, 0.941 at 10%, 0.874 at 20%, 0.824 at 30%, 0.717 at 50%, 0.495 at 100% of HMMME DNA. C, CN ratio plot in the 17p13 region for DNA titration from HMMME. D, CN ratio plot in the 9p region for DNA titration from tumor cell line H2452. In this experiment, 100% DNA from normal cells HEKn (n = 6) was used for normalization. E, CN ratio in the 9p region for ccRCCs with metastasis. Three cases, R13, R36, and R55, had biallelic CN losses in CDKN2A/2B
We confirmed precision of digitalMLPA assay by comparing CNA data at BAP1 with those CNA data obtained by conventional MLPA (P417‐B1 BAP1 probemix) which we reported for 34 ccRCCs previously. 14 The results of both technologies were essentially identical: 31 of 34 ccRCCs showed 1 allele loss, and the remaining 3 had no CNA (data not shown).
3.2. Detection of CNAs in ccRCCs using digitalMLPA
CN analysis was conducted using digitalMLPA on 60 ccRCCs, including 34 specimens used in our previous study. 14 Losses in 3p were detected in 56/60 tumors (93.3%) samples (Table 2). In most of these cases, large genomic regions between 3p26.2 and 3p21.1 carrying VHL, SETD2, SMARCC1, BAP1, and PBRM1 genes were deleted (Table 3 and Figure S2). The 5q gain was detected in 31/60 tumors (51.7%), of which 16 of 60 tumors (26.7%) showed gain of whole chromosome 5 (Table 2).
TABLE 2.
Summary of copy‐number alterations or nucleotide substitution mutations detected in ccRCCs
| Copy‐number alterations or nucleotide substitution mutations | The number of patients with each mutation | |||
|---|---|---|---|---|
| Metastasis | All (%, n = 60) | |||
| Metastasis‐free (n = 34) | Late (≥1 y) (n = 8) | Early (<1 y) (n = 18) | ||
| 1p loss | 1 | 2 | 3 | 6 (10.0%) |
| 3p loss | 31 | 8 | 17 | 56 (93.3%) |
| 4p loss | 1 | 0 | 2 | 3 (5.0%) |
| 4q loss | 1 | 2 | 6 | 9 (15.0%) |
| 5p gain | 10 | 5 | 1 | 16 (26.7%) |
| 5q gain | 18 | 6 | 7 | 31 (51.7%) |
| 6q loss | 9 | 2 | 3 | 14 (23.3%) |
| 7 gain | 12 | 4 | 13 | 29 (48.3%) |
| 8p loss | 8 | 2 | 8 | 18 (30.0%) |
| 9p loss | 3 | 1 | 7 | 11 (18.3%) |
| 9q loss | 3 | 2 | 7 | 12 (20.0%) |
| 10q loss | 8 | 3 | 1 | 12 (20.0%) |
| 12 gain | 8 | 5 | 5 | 18 (30.0%) |
| 14q loss | 17 | 3 | 13 | 33 (55.0%) |
| 16p gain | 3 | 0 | 4 | 7 (11.7%) |
| 16q gain | 3 | 0 | 1 | 4 (6.7%) |
| 17p13 loss | 1 | 0 | 2 | 3 (5.0%) |
| 18 loss | 2 | 3 | 2 | 7 (11.7%) |
| 20 gain | 4 | 3 | 4 | 11 (18.3%) |
| 21q loss | 1 | 2 | 1 | 4 (6.7%) |
| 22 loss | 2 | 0 | 2 | 4 (6.7%) |
| VHL | 28 | 8 | 14 | 50 (83.3%) |
| PBRM1 | 14 | 6 | 8 | 28 (46.7%) |
| SETD2 | 5 | 2 | 6 | 13 (21.7%) |
| BAP1 | 4 | 1 | 7 | 12 (20.0%) |
| The number of chromosome with CN alterations (median) | 3 | 5 | 6 | 4 |
TABLE 3.
The average CN ratios of 11 target genes, and chromosome X and Y in 60 ccRCCs
| Gene | Chr band | Sample ID | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R01 | R02 | R03 | R04 | R05 | R06 | R07 | R08 | R09 | R10 | R11 | R12 | R13 | R14 | R15 | R16 | R17 | R18 | R19 | R20 | ||
| VHL | 3p25.3 | 0.73 | 0.78 | 0.72 | 1.05 | 1.07 | 0.95 | 0.75 | 0.90 | 0.72 | 0.80 | 0.70 | 0.73 | 0.89 | 0.79 | 0.88 | 0.63 | 0.81 | 0.81 | 0.79 | 0.70 |
| SETD2 | 3p21.31 | 0.71 | 0.77 | 0.71 | 1.02 | 0.98 | 0.92 | 0.69 | 0.90 | 0.73 | 0.77 | 0.68 | 0.70 | 0.87 | 0.74 | 0.77 | 0.59 | 0.80 | 0.80 | 0.78 | 0.70 |
| SMARCC1 | 3p21.31 | 0.72 | 0.80 | 0.73 | 1.06 | 1.01 | 0.95 | 0.71 | 0.91 | 0.74 | 0.80 | 0.70 | 0.71 | 0.90 | 0.77 | 0.82 | 0.62 | 0.83 | 0.83 | 0.79 | 0.73 |
| BAP1 | 3p21.1 | 0.64 | 0.71 | 0.66 | 0.90 | 0.96 | 0.82 | 0.72 | 0.80 | 0.65 | 0.75 | 0.69 | 0.69 | 0.79 | 0.63 | 0.75 | 0.59 | 0.72 | 0.70 | 0.77 | 0.62 |
| PBRM1 | 3p21.1 | 0.71 | 0.77 | 0.71 | 1.03 | 0.99 | 0.94 | 0.68 | 0.90 | 0.72 | 0.77 | 0.69 | 0.70 | 0.89 | 0.76 | 0.80 | 0.60 | 0.81 | 0.81 | 0.80 | 0.70 |
| ELOC | 8q21.11 | 1.06 | 1.09 | 1.05 | 0.69 | 0.81 | 0.96 | 1.06 | 1.13 | 1.17 | 1.01 | 1.02 | 1.01 | 1.08 | 1.12 | 1.05 | 1.05 | 1.10 | 1.12 | 0.98 | 1.10 |
| CDKN2A | 9p21.3 | 1.01 | 1.00 | 1.00 | 1.00 | 1.01 | 0.91 | 0.73 | 1.02 | 0.93 | 0.99 | 0.68 | 0.93 | 0.77 | 1.00 | 1.01 | 1.02 | 1.01 | 1.02 | 0.96 | 1.04 |
| CDKN2B | 9p21.3 | 0.98 | 1.01 | 0.96 | 1.02 | 0.99 | 0.90 | 0.69 | 1.02 | 0.91 | 0.95 | 0.65 | 0.93 | 0.73 | 0.97 | 0.96 | 0.96 | 1.04 | 1.00 | 0.91 | 1.03 |
| HIF1A | 14q23.2 | 0.97 | 0.99 | 0.95 | 1.00 | 0.93 | 0.72 | 0.89 | 0.92 | 0.90 | 0.94 | 0.66 | 0.84 | 0.83 | 1.00 | 0.94 | 0.55 | 1.03 | 1.02 | 0.80 | 1.04 |
| TP53 | 17p13.1 | 1.07 | 1.06 | 1.07 | 1.09 | 1.09 | 0.98 | 1.09 | 1.06 | 1.07 | 1.05 | 1.05 | 1.04 | 0.88 | 1.08 | 1.11 | 1.10 | 1.04 | 1.08 | 0.99 | 1.08 |
| HCFC1 | Xq28 | 1.02 | 0.97 | 0.94 | 0.96 | 1.01 | 1.23 | 0.99 | 0.97 | 1.32 | 2.00 | 1.03 | 2.00 | 0.98 | 0.89 | 1.01 | 1.03 | 0.91 | 1.87 | 0.95 | 0.95 |
| chr X | 1.05 | 1.04 | 1.03 | 1.05 | 1.03 | 1.34 | 1.04 | 1.03 | 1.42 | 2.06 | 1.05 | 2.06 | 1.05 | 1.04 | 1.03 | 1.06 | 1.06 | 2.18 | 1.01 | 1.03 | |
| chr Y | 1.03 | 1.04 | 0.95 | 1.05 | 1.00 | 0.52 | 0.36 | 0.95 | 1.45 | 0.00 | 1.01 | 0.00 | 1.00 | 0.98 | 0.55 | 0.16 | 1.04 | 0.00 | 0.95 | 1.07 | |
| CN alterations a | 1 | 3 | 4 | 1 | 1 | 14 | 4 | 2 | 9 | 2 | 6 | 3 | 10 | 3 | 3 | 7 | 6 | 2 | 7 | 3 | |
| Gene | Chr band | Sample ID | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R21 | R22 | R23 | R24 | R25 | R26 | R27 | R28 | R29 | R30 | R31 | R32 | R33 | R34 | R35 | R36 | R37 | R38 | R39 | R40 | ||
| VHL | 3p25.3 | 0.69 | 0.78 | 0.81 | 0.87 | 0.69 | 1.00 | 0.68 | 0.77 | 0.77 | 0.72 | 0.68 | 0.75 | 0.70 | 0.71 | 0.82 | 0.83 | 0.73 | 0.73 | 0.81 | 0.85 |
| SETD2 | 3p21.31 | 0.68 | 0.72 | 0.77 | 0.85 | 0.67 | 1.00 | 0.66 | 0.77 | 0.75 | 0.69 | 0.60 | 0.71 | 0.68 | 0.69 | 0.80 | 0.81 | 0.68 | 0.70 | 0.83 | 0.83 |
| SMARCC1 | 3p21.31 | 0.69 | 0.74 | 0.80 | 0.87 | 0.69 | 1.02 | 0.67 | 0.79 | 0.77 | 0.72 | 0.62 | 0.75 | 0.71 | 0.71 | 0.82 | 0.82 | 0.69 | 0.73 | 0.84 | 0.85 |
| BAP1 | 3p21.1 | 0.65 | 0.76 | 0.76 | 0.78 | 0.62 | 0.88 | 0.64 | 0.70 | 0.69 | 0.66 | 0.67 | 0.65 | 0.60 | 0.62 | 0.66 | 0.76 | 0.76 | 0.64 | 0.68 | 0.80 |
| PBRM1 | 3p21.1 | 0.67 | 0.72 | 0.78 | 0.86 | 0.68 | 1.00 | 0.66 | 0.78 | 0.76 | 0.69 | 0.59 | 0.73 | 0.70 | 0.69 | 0.81 | 0.79 | 0.69 | 0.71 | 0.84 | 0.84 |
| ELOC | 8q21.11 | 1.03 | 1.02 | 1.03 | 1.07 | 1.08 | 1.03 | 1.08 | 1.11 | 1.06 | 1.10 | 1.03 | 1.11 | 1.11 | 1.09 | 1.11 | 1.04 | 1.05 | 0.94 | 0.89 | 1.08 |
| CDKN2A | 9p21.3 | 0.95 | 0.98 | 0.98 | 1.02 | 1.00 | 0.99 | 0.99 | 1.03 | 1.04 | 1.06 | 1.11 | 1.01 | 1.06 | 1.05 | 1.08 | 0.59 | 0.74 | 0.86 | 1.11 | 0.96 |
| CDKN2B | 9p21.3 | 0.95 | 0.92 | 0.95 | 1.00 | 0.98 | 0.97 | 0.94 | 1.01 | 1.00 | 1.02 | 1.02 | 1.01 | 1.03 | 0.99 | 1.09 | 0.62 | 0.72 | 0.81 | 1.12 | 0.91 |
| HIF1A | 14q23.2 | 1.08 | 0.87 | 0.74 | 0.88 | 0.97 | 0.59 | 0.97 | 0.75 | 0.99 | 0.71 | 0.93 | 1.00 | 0.96 | 1.02 | 0.90 | 0.79 | 0.66 | 0.91 | 1.07 | 0.82 |
| TP53 | 17p13.1 | 0.98 | 1.10 | 1.06 | 1.06 | 1.08 | 1.04 | 1.08 | 1.04 | 1.07 | 1.08 | 1.06 | 1.07 | 0.98 | 1.07 | 1.07 | 1.05 | 1.08 | 0.94 | 1.08 | 1.01 |
| HCFC1 | Xq28 | 1.14 | 1.09 | 2.11 | 2.23 | 0.91 | 0.90 | 1.03 | 0.95 | 1.98 | 2.03 | 1.29 | 0.94 | 1.87 | 0.95 | 0.89 | 1.00 | 1.15 | 1.23 | 0.87 | 0.96 |
| chr X | 1.18 | 1.05 | 2.12 | 2.30 | 1.03 | 1.03 | 1.07 | 1.06 | 2.12 | 2.15 | 1.13 | 1.03 | 2.12 | 1.06 | 1.06 | 1.09 | 1.08 | 1.39 | 1.10 | 1.03 | |
| chr Y | 1.14 | 0.45 | 0.00 | 0.00 | 1.05 | 0.08 | 0.96 | 0.51 | 0.00 | 0.00 | 1.04 | 1.01 | 0.00 | 1.08 | 0.95 | 1.07 | 1.02 | 0.47 | 0.60 | 0.61 | |
| CN alterations a | 6 | 6 | 3 | 2 | 2 | 6 | 3 | 2 | 2 | 6 | 2 | 4 | 3 | 3 | 3 | 9 | 8 | 12 | 4 | 7 | |
| Gene | Chr band | Sample ID | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R41 | R42 | R43 | R44 | R45 | R46 | R47 | R48 | R49 | R50 | R51 | R52 | R53 | R54 | R55 | R56 | R57 | R58 | R59 | R60 | ||
| VHL | 3p25.3 | 0.63 | 0.80 | 0.83 | 0.83 | 0.71 | 0.71 | 0.78 | 0.76 | 0.78 | 0.74 | 0.78 | 0.84 | 0.70 | 0.92 | 0.69 | 0.77 | 0.76 | 0.66 | 0.99 | 0.76 |
| SETD2 | 3p21.31 | 0.60 | 0.75 | 0.81 | 0.74 | 0.69 | 0.70 | 0.75 | 0.80 | 0.76 | 0.75 | 0.77 | 0.83 | 0.67 | 0.85 | 0.64 | 0.74 | 0.75 | 0.63 | 0.95 | 0.74 |
| SMARCC1 | 3p21.31 | 0.62 | 0.77 | 0.83 | 0.80 | 0.71 | 0.72 | 0.78 | 0.83 | 0.77 | 0.76 | 0.80 | 0.86 | 0.69 | 0.88 | 0.67 | 0.76 | 0.76 | 0.66 | 0.98 | 0.76 |
| BAP1 | 3p21.1 | 0.54 | 0.68 | 0.70 | 0.65 | 0.65 | 0.58 | 0.64 | 0.63 | 0.60 | 0.63 | 0.66 | 0.70 | 0.68 | 0.83 | 0.62 | 0.73 | 0.71 | 0.56 | 0.88 | 0.66 |
| PBRM1 | 3p21.1 | 0.61 | 0.75 | 0.80 | 0.77 | 0.70 | 0.70 | 0.77 | 0.80 | 0.75 | 0.74 | 0.77 | 0.85 | 0.68 | 0.85 | 0.64 | 0.74 | 0.74 | 0.65 | 0.95 | 0.75 |
| ELOC | 8q21.11 | 1.02 | 1.09 | 1.09 | 0.81 | 1.06 | 1.13 | 1.11 | 1.13 | 1.14 | 1.10 | 1.10 | 0.96 | 1.03 | 1.03 | 1.05 | 0.80 | 1.09 | 1.52 | 1.13 | 1.11 |
| CDKN2A | 9p21.3 | 0.61 | 0.77 | 1.03 | 1.24 | 1.05 | 1.05 | 1.07 | 1.03 | 1.04 | 1.03 | 1.05 | 0.82 | 0.99 | 1.03 | 0.53 | 0.95 | 1.02 | 0.63 | 1.00 | 1.04 |
| CDKN2B | 9p21.3 | 0.62 | 0.77 | 1.02 | 1.17 | 1.01 | 1.10 | 1.06 | 1.09 | 1.06 | 1.07 | 1.06 | 0.79 | 0.95 | 1.03 | 0.50 | 0.92 | 0.97 | 0.62 | 0.99 | 0.97 |
| HIF1A | 14q23.2 | 0.58 | 0.76 | 0.80 | 0.75 | 1.02 | 0.70 | 1.05 | 0.80 | 0.78 | 0.73 | 1.03 | 0.84 | 0.89 | 0.97 | 0.64 | 0.89 | 0.84 | 1.01 | 0.91 | 0.77 |
| TP53 | 17p13.1 | 0.98 | 0.97 | 1.04 | 0.82 | 1.05 | 1.05 | 1.03 | 1.01 | 1.05 | 1.05 | 1.03 | 0.81 | 1.06 | 1.06 | 1.13 | 1.01 | 1.08 | 1.08 | 1.03 | 1.07 |
| HCFC1 | Xq28 | 1.75 | 1.05 | 1.79 | 1.11 | 0.94 | 0.86 | 0.89 | 1.63 | 1.71 | 0.84 | 1.79 | 1.96 | 1.02 | 0.99 | 1.02 | 1.34 | 1.03 | 1.73 | 1.04 | 1.94 |
| chr X | 1.93 | 1.15 | 2.03 | 1.30 | 1.06 | 1.04 | 1.03 | 2.06 | 2.07 | 1.04 | 2.07 | 2.28 | 1.04 | 1.04 | 1.05 | 1.32 | 1.07 | 2.01 | 1.07 | 2.10 | |
| chr Y | 0.00 | 1.09 | 0.00 | 1.22 | 1.05 | 1.09 | 1.05 | 0.01 | 0.00 | 0.40 | 0.00 | 0.00 | 0.94 | 0.94 | 0.97 | 0.56 | 1.01 | 0.00 | 0.85 | 0.00 | |
| CN alterations a | 11 | 10 | 3 | 18 | 3 | 6 | 2 | 4 | 7 | 6 | 4 | 12 | 2 | 4 | 7 | 8 | 4 | 6 | 4 | 3 | |
The number of clinically significant chromosomes with CN alterations is shown.

The 9p loss and 14q loss, respectively, were detected in 11 (18.3%) and 33 tumors (55%), respectively (Table 2). Nine patients (15%) had both 9p and 14q losses. Of the 11 samples with 9p loss, 3 had biallelic CN loss encompassing CDKN2A/2B at 9p21.3 (cases R13, R36, and R55) (Table 3 and Figure 2E). Three samples (Cases R13, R44, and R52) showed monoallelic loss of 17p13, a region carrying the TP53 gene. Finally, 4 samples had monoallelic loss of a region carrying NF2 on chromosome 22; cases R36, R44, and R45 lost whole chromosome 22, and case R55 showed loss of 22q11.1‐22q12.2 (Tables 2, S2, and Figure S2).
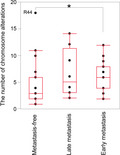
With regards to metastasis, median number of chromosome arm with CNA was 6 arms for 18 patients with early metastasis (<1 y). The number of altered arms was 5 for 8 patients with late metastasis (≥1 y). It was 3 arms for 34 patients without metastasis. Three tumors with biallelic deletion of CDKN2A/2B had multiple CNAs above 7 arms. Number of chromosome arms with CNA (median 6 arms) in 18 patients with early metastasis was significantly higher when compared with those (3 arms) in 34 patients without metastasis (P = .0289, Dunn test, Figure 3).
FIGURE 3.
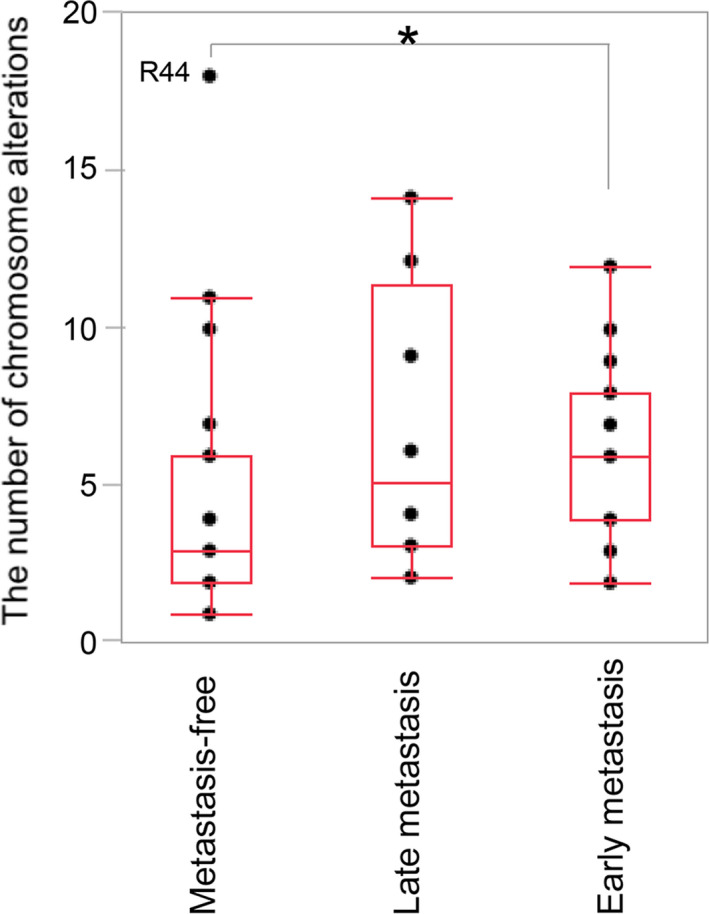
Association between metastasis status, ie, metastasis‐free, late metastasis (≥1 y), and early metastasis (<1 y) with the number of chromosome alterations (*P = .0289). R44 had the largest number of alterations, showing an extreme deviation from the mean. This patient, 1 y after tumorectomy of ccRCC, had suffered from primary lung cancer and mediastinum lymph node metastasis, which showed complete response to anti‐PD‐L1 antibody therapy
In male ccRCCs, loss of chromosome Y was often detected, but gain of chromosome X was frequently detected in ccRCCs with high complexity showing frequent CNAs: cases R06, R09, R38, R44, and R56 (Table 3).
3.3. Frequent 14q loss and the genes with downregulated expression
digitalMLPA results detected 14q losses at a higher frequency (55%). Target genes showing downregulated expression due to 14q loss have also remained largely unknown. Therefore, KLHL33 (14q11.2) or L2HGDH (14q21.3) has been reported to be a target of 14q losses, respectively, instead of HIF1A (14q23.2). 15 , 16 Therefore, to validate the significance of the frequent loss of 14q from different aspects, gene expression experiments for these 3 genes located on 14q were conducted in ccRCCs with and without 14q loss (22 and 23 cases, respectively). As observed, the gene expression of HIF1A and L2HGDH was significantly lower in ccRCCs with 14q loss compared with the ones without 14q loss (HIF1A; P = .0253, L2HGDH; P = .0117, chi‐squared test) (Figure 4). However, the expression of KLHL33 did not show a significant association with 14q loss (P = .0536). Markedly, for biallelic HIF1A deletions (case R06), low expression level of HIF1A (1/8 of normal tissue) was detected during RT‐PCR experiments compared with the normal renal tissue of this patient.
FIGURE 4.
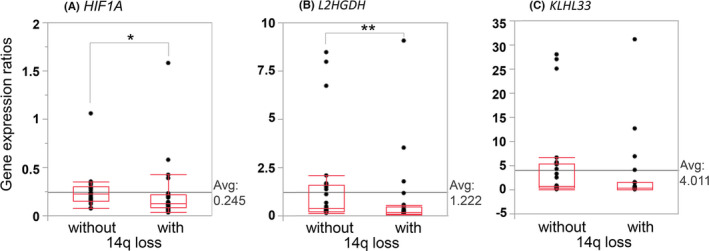
Association between 14q deletion and downregulated gene expression. The gene expression ratios of (A) HIF1A and (B) L2HGDH were significantly associated with 14q loss (HIF1A; *P = .0253, L2HGDH; **P = .0117, chi‐squared test). In contrast, (C) the gene expression ratios of KLHL33 did not show a significant association with 14q loss (P = .0536). The number of ccRCCs with and without this loss analyzed was 22 and 23, respectively. Gene expression ratios were normalized to GAPDH
3.4. Sequence‐level mutations and chromosomal complexity
The identification of genomic risk factors for metastasis should be as simple as possible for clinical use. We analyzed the sequences of only 4 genes: VHL, PBRM1, SETD2, and BAP1, located in 3p. In our samples, nucleotide substitution somatic mutations in the genes VHL, PBRM1, SETD2, and BAP1 were detected in 50/60 (83.3%), 28/60 (46.7%), 13/60 (21.7%), and 12/60 (20%), respectively (Tables 2, S2 and S3). Six of the 60 patients (10%) had mutations in both PBRM1 and SETD2 (cases R07, R15, R17, R21, R45 and R58), and 2/60 (3.3%) had mutations in both SETD2 and BAP1 (cases R57 and R59). A further 2 patients had concomitant PBRM1 and BAP1 mutations (cases R54 and R55).
3.5. Multiple correspondence analysis
We carried out multiple correspondence analysis to assess relationship between genome alterations, prognosis, and metastasis status (Figure 5). The early metastasis (<1 y) was associated with 9p loss and 4q loss (both P < .05). This analysis also indicated association of poor survival with 4p loss and 1p loss (both, P < .05).
FIGURE 5.
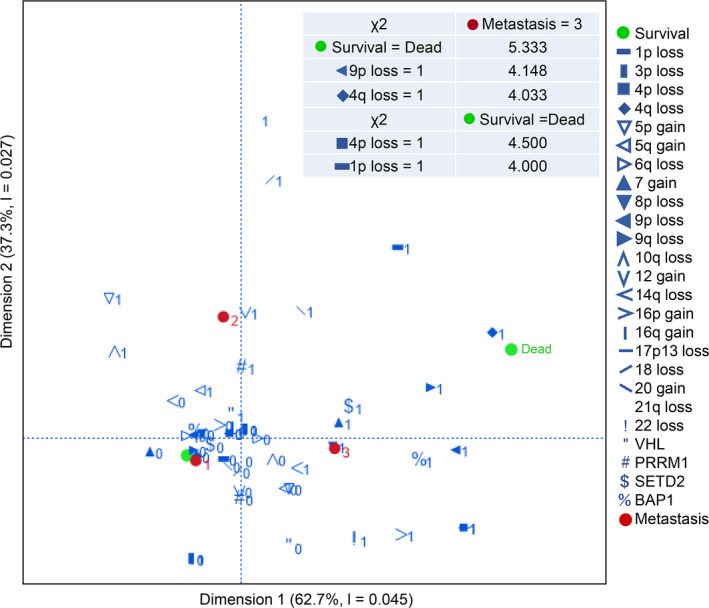
Multiple correspondence analysis among the metastatic status (1: metastasis‐free, 2: late metastasis, ≥1 y, 3: early metastasis, <1 y after tumorectomy) and each characteristic (1: CN alteration or nucleotide‐level mutation detected, 0: not detected). The chi‐squared test for independence on 2 categorical variables was conducted and χ 2 values associated with early metastasis or with poor survival are described in the figure. The critical value at the .05 level (P = .05) in 1 degree of freedom is 3.841
4. DISCUSSION
Our analysis indicated that CNAs had an essential role in aggressiveness of ccRCC. Detection of the subtle CN change observed in low tumor content experiments is challenging. To enable CN analysis in such situations, we used digitalMLPA, a high‐throughput NGS‐based MLPA tool. digitalMLPA allows detection of CNAs at 384 regions with as little as 20 ng of sample DNA in a single tube. It provides an easy preparation for NGS by skipping complicated purification steps in the preparation of libraries and yields compact volumes of data, which can be analyzed without extensive bioinformatic skills. We used this tool for copy‐number detection of multiple chromosomal regions using 384 digitalMLPA probes, of which 151 probes targeted 11 key genes in ccRCC and 233 probes were used for karyotyping of entire chromosomes. digitalMLPA could detect 1 allele loss with as low as 20% of tumor DNA mixed in 80% of normal DNA and 2 allele loss with as low as 10% of tumor DNA mixed in 90% of normal DNA.
We observed frameshift mutations in 2 cases; 1 was in exon 6 of SETD2 (R32) and exon 29 of PBRM1 (R21) (Table S3). In both cases, they mimicked apparent intergenic CN loss in the digitalMLPA assay by base mismatches located in the regions targeted by the digitalMLPA probe. The CN ratio showed a decrease of 25%‐30% in probes with low hybridization or ligation caused by base mismatches (Figure S2). BAP1 showed a slightly lower CN ratio than the other genes (VHL, SETD2, SMARCC1, PBRM1) located in chromosome 3p in blood DNAs used as the reference (Figure S1B). The reason why a decrease of c. 7% in the BAP1 CN ratio in comparison with the other genes in chromosome 3p is not clear.
The incidence of kidney cancer has been on the rise over the past decades, 17 and RCC is the most common type of kidney cancer. Early‐stage RCC is curable by clinical surgery, but 20%‐30% of patients have distant metastases at diagnosis and show resistance to standard chemotherapy and radiotherapy. 18 ccRCC accounts for c. 80% of all RCCs, and is characterized by allelic loss in chromosome 3p and loss‐of‐function mutations of the VHL gene on 3p25.3. 19 , 20 , 21 A 5q gain is known to initiate tumorigenesis. 7 , 22 High‐throughput NGS technologies have been widely used to detect specific genomic characteristics. For research use, comprehensive data, including sequencing data for the detection of somatic mutation, CN variation data, and the transcriptomic profiles underlying ccRCC have been reported by several collaborative global projects working on independent populations such as the Cancer Genome Atlas (TCGA)‐KIRC cohort (TCGA database, https://portal.gdc.cancer.gov/), a European Union cohort (ICGC database, https://dcc.icgc.org/), and a Tokyo cohort (cBioPortal database, https://www.cbioportal.org/). Low mutation rates have been observed, except for the genes VHL, PBRM1, SETD2, and BAP1, all of which are located on 3p. 8 , 10 ccRCC shows substantial genetic heterogeneity, with CN losses or gains of large chromosomal regions. CNAs are important biological characteristics, having the potential to predict the prognosis of ccRCC. 7 , 8 , 9 , 10 , 11 The Cancer Genomics Consortium Workgroup identified that losses on chromosomes 1p, 3p, 4p, 6q, 8p, 9p, 9q 10q, 14q, and 18, and gains on 5p, 5q, 7, 12, 16p, 16q, and 20 are frequently reported, and appear to have clinical significance. 23 More specifically, losses in 3p are important for diagnosis and losses in 9p or 14q are important for prognosis. The Cancer Genomics Consortium Workgroup also reported that the TP53 mutation had clinical significance for prognosis, and deletions on 17p13, carrying TP53, was added as a risk factor because this region is associated with an aggressive tumor phenotype. 24 , 25 Loss of chromosome 4 is not as well known as losses of chromosome 9 and 14q, but has been reported to be positively correlated with high tumor grade and candidate tumor suppressor genes. 26 , 27 , 28 In our ccRCCs, losses in 3p were detected in 56/60 (93.3%) and 5q gain was detected in 31/60 (51.7%). These alterations rates were similar to ones from previous reports, 22 , 23 showing the CN analysis using digitalMLPA worked well. Losses of chromosome 9p (18.3%), 14q (55.0%), and 17p13 (5%), known associate with poor prognosis, were detected. To detect genome alterations with clinical significance, we carried out multiple correspondence analysis (Figure 5). In the analysis, chromosome losses of 1p, 4p, 4q, and 9p were associated with poor survival or early metastasis (P < .05). In three of 13 patients having metastasis at diagnosis, biallelic deletion of the CDKN2A/2B genes was detected.
We detected loss of chromosome 14q at a higher frequency (55%) than the frequencies (~40%) previously described, whereas the frequencies of CNAs on other chromosomes and sequence‐level mutations were equivalent to those previously described. 23 Therefore, to validate the significance of high frequency 14q losses from different aspects, we conducted gene expression experiments for 3 representative genes located on 14q: HIF1A (14q23.2), L2HGDH (14q21.3), and KLHL33 (14q11.2). A significant positive correlation was detected with HIF1A and L2HGDH, but not with KLHL33, in terms of 14q loss determined using digitalMLPA and downregulation of mRNA expression ratios (Figure 4). Therefore, we showed through gene expression analysis, that digitalMLPA can detect fine CNAs and that HIF1A and L2HGDH were genes downregulated by 14q deletion.
Chromosomal complexity increases by chromosomal alterations accumulated over several decades before diagnosis, and is associated with a poor prognosis. 22 The number of chromosome alterations in the ccRCC tumors from the patients with metastasis several months to 3.5 y after tumorectomy showed a tendency to be higher than 6 (for 6/9 patients), but those from the 3 patients with metastasis occurring between 5 and 9 y were lower than 3. Frequent losses of the Y chromosome have been described in normal blood and bone marrow of elderly men, 29 , 30 and in many types of tumors, including ccRCC. 31 In our ccRCCs, loss of chromosome Y was detected independently of chromosomal complexity, but gain of chromosome X was frequently detected in ccRCCs with high complexity.
ccRCC has a low mutation rate of base substitution, 12 but mutations in several genes are risk factors for metastasis. Although PBRM1 mutations did not show a significant impact on overall survival or disease‐free survival in univariate analysis, 8 it has been reported that loss‐of‐function mutations in PBRM1 might alter the whole‐exome expression profiles of tumor cells, affecting their responsiveness to immune checkpoint blockade. 32 Nuclear BAP1‐negative tumors detected by immunohistochemistry (IHC) had larger tumors, higher Fuhrman grade, higher stage, and a higher incidence of metastases. 33 , 34 BAP1 mutations were mutually exclusive with PBRM1 mutations, and were significantly associated with shorter overall survival time. 35 SETD2 mutations were also associated with a high relapse rate. 8 In our samples, the BAP1 mutation carriers tend to have more frequent CNA (median 5 arms). There were 2 cases with mutations of both BAP1 and PBRM1, and 2 cases with mutations in both BAP1 and SETD2. All of these 4 cases belonged to a group of 13 patients who had metastasis at diagnosis. Our multiple correspondence analysis indicated that chromosome losses of 1p, 4p, 4q, and 9p had more important roles than sequence‐level mutations in judging malignant tumor behavior.
After the administration of immune checkpoint inhibitors and antiangiogenic targeted therapies, including anti‐vascular endothelial growth factor and multi‐target tyrosine kinase inhibitors to the patients with metastasis, overall survival improved significantly. 36 We hope that identification of novel characteristic CNAs and mutations using digitalMLPA and target NGS will lead to an understanding of the molecular mechanisms of ccRCCs and improvement in patient care.
DISCLOSURE
LA, JS, SS, KG are employed by MRC Holland, the manufacturer of MLPA and digitalMLPA products. SY has received research funding from Bayer Yakuhin, Ltd. and Ono Pharmaceutical Co., Ltd. The other authors have no conflicts of interest to declare.
Supporting information
Fig S1
Fig S2
Table S1‐S3
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by Bayer Academic Support from Bayer Yakuhin, Ltd., 2017, and by scholarship support from ONO Pharmaceutical Co., Ltd, 2020. We are grateful to Atsuko Iemoto for her technical assistance.
Yoshikawa Y, Yamada Y, Emi M, et al. Risk prediction for metastasis of clear cell renal cell carcinoma using digital multiplex ligation‐dependent probe amplification. Cancer Sci.2022;113:297–307. doi: 10.1111/cas.15170
Contributor Information
Yoshie Yoshikawa, Email: yoshiey@hyo-med.ac.jp.
Mitsuru Emi, Email: mitsuruemi@me.com.
REFERENCES
- 1. Salk JJ, Schmitt MW, Loeb LA. Enhancing the accuracy of next‐generation sequencing for detecting rare and subclonal mutations. Nat Rev Genet. 2018;19:269‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yoshikawa Y, Emi M, Hashimoto‐Tamaoki T, et al. High‐density array‐CGH with targeted NGS unmask multiple noncontiguous minute deletions on chromosome 3p21 in mesothelioma. Proc Natl Acad Sci U S A. 2016;113:13432‐13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation‐dependent probe amplification. Nucleic Acids Res. 2002;30:e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moreno‐Cabrera JM, Del Valle J, Castellanos E, et al. Evaluation of CNV detection tools for NGS panel data in genetic diagnostics. Eur J Hum Genet. 2020;28:1645‐1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zarrei M, MacDonald JR, Merico D, Scherer SW. A copy number variation map of the human genome. Nat Rev Genet. 2015;16:172‐183. [DOI] [PubMed] [Google Scholar]
- 6. Benard‐Slagter A, Zondervan I, de Groot K, et al. Digital Multiplex Ligation‐Dependent Probe Amplification for Detection of Key Copy Number Alterations in T‐ and B‐Cell Lymphoblastic Leukemia. J Mol Diagn. 2017;19:659‐672. [DOI] [PubMed] [Google Scholar]
- 7. Moore LE, Jaeger E, Nickerson ML, et al. Genomic copy number alterations in clear cell renal carcinoma: associations with case characteristics and mechanisms of VHL gene inactivation. Oncogenesis. 2012;1:e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sato Y, Yoshizato T, Shiraishi Y, et al. Integrated molecular analysis of clear‐cell renal cell carcinoma. Nat Genet. 2013;45:860‐867. [DOI] [PubMed] [Google Scholar]
- 9. Bi M, Zhao S, Said JW, et al. Genomic characterization of sarcomatoid transformation in clear cell renal cell carcinoma. Proc Natl Acad Sci U S A. 2016;113:2170‐2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ricketts CJ, De Cubas AA, Fan H, et al. The cancer genome atlas comprehensive molecular characterization of renal cell carcinoma. Cell Rep. 2018;23:3698. [DOI] [PubMed] [Google Scholar]
- 11. Maruschke M, Koczan D, Ziems B, Hakenberg OW. Copy Number Alterations with Prognostic Potential in Clear Cell Renal Cell Carcinoma. Urol Int. 2018;101:417‐424. [DOI] [PubMed] [Google Scholar]
- 12. Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Valencia CA, Pervaiz MA, Husami A, Qian Y & Zhang K Sanger sequencing principles, history, and landmarks. Next Generation Sequencing Technologies in Medical Genetics SpringerBriefs Genetics; 2013.
- 14. Togo Y, Yoshikawa Y, Suzuki T, et al. Genomic profiling of the genes on chromosome 3p in sporadic clear cell renal cell carcinoma. Int J Oncol. 2016;48:1571‐1580. [DOI] [PubMed] [Google Scholar]
- 15. Wang L, Li Y, Lyu Y, Wen H, Feng C. Association between copy‐number alteration of +20q, ‐14q and ‐18p and cross‐sensitivity to tyrosine kinase inhibitors in clear‐cell renal cell carcinoma. Cancer Cell Int. 2020;20:482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shenoy N. HIF1alpha is not a target of 14q deletion in clear cell renal cancer. Sci Rep. 2020;10:17642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Du Z, Chen W, Xia Q, Shi O, Chen Q. Trends and projections of kidney cancer incidence at the global and national levels, 1990–2030: a Bayesian age‐period‐cohort modeling study. Biomark Res. 2020;8:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Psutka SP, Master VA. Role of metastasis‐directed treatment in kidney cancer. Cancer. 2018;124:3641‐3655. [DOI] [PubMed] [Google Scholar]
- 19. Gnarra JR, Tory K, Weng Y, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet. 1994;7:85‐90. [DOI] [PubMed] [Google Scholar]
- 20. Shuin T, Kondo K, Torigoe S, et al. Frequent somatic mutations and loss of heterozygosity of the von Hippel‐Lindau tumor suppressor gene in primary human renal cell carcinomas. Cancer Res. 1994;54:2852‐2855. [PubMed] [Google Scholar]
- 21. Foster K, Prowse A, van den Berg A, et al. Somatic mutations of the von Hippel‐Lindau disease tumour suppressor gene in non‐familial clear cell renal carcinoma. Hum Mol Genet. 1994;3:2169‐2173. [DOI] [PubMed] [Google Scholar]
- 22. Mitchell TJ, Turajlic S, Rowan A, et al. Timing the landmark events in the evolution of clear cell renal cell cancer: TRACERx renal. Cell. 2018;173:611‐623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu YJ, Houldsworth J, Emmadi R, Dyer L, Wolff DJ. Assessing genomic copy number alterations as best practice for renal cell neoplasia: An evidence‐based review from the cancer genomics Consortium Workgroup. Cancer Genet. 2020;244:40‐54. [DOI] [PubMed] [Google Scholar]
- 24. Harlander S, Schonenberger D, Toussaint NC, et al. Combined mutation in Vhl, Trp53 and Rb1 causes clear cell renal cell carcinoma in mice. Nat Med. 2017;23:869‐877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eichenauer T, Shadanpour N, Kluth M, et al. Chromosome 17p13 deletion is associated with an aggressive tumor phenotype in clear cell renal cell carcinoma. World J Surg Oncol. 2020;18:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arai E, Ushijima S, Tsuda H, et al. Genetic clustering of clear cell renal cell carcinoma based on array‐comparative genomic hybridization: its association with DNA methylation alteration and patient outcome. Clin Cancer Res. 2008;14:5531‐5539. [DOI] [PubMed] [Google Scholar]
- 27. Kohn L, Svenson U, Ljungberg B, Roos G. Specific genomic aberrations predict survival, but low mutation rate in cancer hot spots, in clear cell renal cell carcinoma. Appl Immunohistochem Mol Morphol. 2015;23:334‐342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xiong Y, Qi Y, Bai Q, Xia Y, Liu L, Guo J. Relevance of arm somatic copy number alterations for oncologic outcomes and tumor immune microenvironment in clear cell renal cell carcinoma. Ann Transl Med. 2019;7:646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jacobs PA, Brunton M, Court Brown WM, Doll R, Goldstein H. Change of human chromosome count distribution with age: evidence for a sex differences. Nature. 1963;197:1080‐1081. [DOI] [PubMed] [Google Scholar]
- 30. Pierre RV, Hoagland HC. Age‐associated aneuploidy: loss of Y chromosome from human bone marrow cells with aging. Cancer. 1972;30:889‐894. [DOI] [PubMed] [Google Scholar]
- 31. Kovacs G, Frisch S. Clonal chromosome abnormalities in tumor cells from patients with sporadic renal cell carcinomas. Cancer Res. 1989;49:651‐659. [PubMed] [Google Scholar]
- 32. Miao D, Margolis CA, Gao W, et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science. 2018;359:801‐806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Joseph RW, Kapur P, Serie DJ, et al. Loss of BAP1 protein expression is an independent marker of poor prognosis in patients with low‐risk clear cell renal cell carcinoma. Cancer. 2014;120:1059‐1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Minardi D, Lucarini G, Milanese G, Di Primio R, Montironi R, Muzzonigro G. Loss of nuclear BAP1 protein expression is a marker of poor prognosis in patients with clear cell renal cell carcinoma. Urologic Oncol. 2016;34(338):e311‐338. [DOI] [PubMed] [Google Scholar]
- 35. Pena‐Llopis S, Vega‐Rubin‐de‐Celis S, Liao A, et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet. 2012;44:751‐759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Angulo JC, Shapiro O. The Changing Therapeutic Landscape of Metastatic Renal Cancer. Cancers (Basel). 2019;11(9):1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Table S1‐S3
Supplementary Material