

Abstract
Osimertinib is a third‐generation epidermal growth factor receptor tyrosine kinase inhibitor (EGFR‐TKI) for lung adenocarcinoma (LUAD) harboring activating mutations, but patients ultimately develop acquired resistance. Circular RNAs are involved in EGFR‐TKI resistance, while the role of hsa_circ_0005576 in the osimertinib resistance of LUAD remains unknown. In this study, we demonstrated that hsa_circ_0005576 could facilitate osimertinib‐resistant LUAD cells. Briefly, knockdown of hsa_circ_0005576 not only suppressed the proliferation and promoted the apoptosis of resistant LUAD cells, but also increased their sensitivity to osimertinib. Mechanistically, hsa_circ_0005576, serving as an miRNA sponge, could directly interact with miR‐512‐5p and subsequently upregulate the miR‐512‐5p‐targeted insulin‐like growth factor 1 receptor. Rescue assays indicated that miR‐512‐5p inhibition could reverse the effects of hsa_circ_0005576 knockdown in LUAD cells resistant to osimertinib. Overall, our study revealed that hsa_circ_0005576 regulates proliferation and apoptosis through miR‐512‐5p/IGF1R signaling, which contributes further to the resistance of LUAD cells to osimertinib. In addition, this study provides a novel insight into the mechanisms underlying osimertinib resistance of LUAD.
Keywords: hsa_circ_0005576, IGF1R, lung adenocarcinoma, miR‐512‐5p, osimertinib resistance
This study revealed that hsa_circ_0005576 regulates proliferation and apoptosis through miR‐512‐5p/IGF1R signaling, which contributes further to the resistance of LUAD cells to osimertinib. In addition, it provides a novel insight into the mechanism underlying osimertinib resistance of LUAD.

Abbreviations
- EGFR‐TKIs
epidermal growth factor receptor tyrosine kinase inhibitors
- IGF1R
insulin‐like growth factor 1 receptor
- IGFBPs
insulin‐like growth factor‐binding proteins
- IGFs
insulin‐like growth factors
- IPA
Ingenuity Pathway Analysis
- LUAD
lung adenocarcinoma
- NSCLC
non–small‐cell lung cancer
- RTK
receptor tyrosine kinase
1. INTRODUCTION
Osimertinib is a third‐generation, irreversible epidermal growth factor RTK inhibitor (EGFR‐TKI) that selectively suppresses both EGFR‐sensitizing mutations and the T790M resistance mutation. 1 A recent clinical trial demonstrated that, compared with first‐generation EGFR‐TKIs in first‐line clinical treatment of EGFR mutation‐positive NSCLC patients, osimertinib was associated with a longer progression‐free survival and promising overall survival. 1 Based on the encouraging results of the FLAURA trial, first‐line treatment with osimertinib has become a favorable standard in therapies for NSCLC patients who carry the EGFR‐positive mutation. Nevertheless, acquired resistance to osimertinib is also inevitable. Based on preclinical and clinical studies, various mechanisms of osimertinib resistance have been reported, these EGFR‐dependent mechanisms involved in osimertinib resistance include the acquisition of EGFR G719X, G796X, and C797S mutations and amplification of wild‐type EGFR. 2 Moreover, multiple EGFR‐independent mechanisms can be implicated in osimertinib resistance including histologic and phenotypic transformation, activation of bypass pathways such as amplification of c‐Met, HER2, mutations of NRAS, KRAS, and acquired oncogenic fusions in RET, ALK, BRAF. 3 However, 40%‐50% of the mechanisms of resistance to osimertinib are not comprehensively understood. 2 Therefore, it is extremely vital to clarify osimertinib resistance to compensate therapeutic strategies and improve prognosis.
Circular RNAs (circRNAs), a special category of non‐coding RNAs, are characterized as covalently closed, endogenous biomolecules in eukaryotes. 4 CircRNAs, serving as miRNAs sponges, can interfere with the transcriptional and translational processes of genes or delay the degradation of specific mRNAs. 4 , 5 , 6 The expression profile of circRNAs is complex and variable. 7 Furthermore, there is increasing evidence that circRNAs are closely implicated in human cancers and can act as stable biomarkers due to their asymmetric distribution in cancers, in which there is frequent cell type‐specific, tissue‐specific, or developmental‐stage‐specific expression. 6 , 8 , 9 Similarly, mounting evidence has confirmed that the dysregulation and aberrant expression patterns of circRNAs are associated with the malignant behavior of LUAD. 10 , 11 Recently, hsa_circ_0005576 has been demonstrated to promote malignant progression in colorectal cancer through the miR‐874/CDK8 axis. 12 Furthermore, upregulated hsa_circ_0005576 was associated with cervical cancer progression through the miR‐153/KIF20A axis. 13 Nevertheless, the expression patterns and functions of hsa_circ_0005576 involved in EGFR‐TKI resistance of LUAD are still almost unknown. In summary, these clues led us to investigate if the differential expression and functions of hsa_circ_0005576 made it a new osimertinib resistance driver in LUAD.
In our study, we confirmed the differential expression of hsa_circ_0005576 in parental osimertinib‐sensitive and paired osimertinib‐resistant LUAD cells. We provided new insight into the biological consequences of upregulated hsa_circ_0005576 participating in osimertinib resistance progression of LUAD. More importantly, we identified that hsa_circ_0005576 mediates resistance to osimertinib by sponging miR‐512‐5p and, simultaneously, contributes to the IGFR1 regulation associated with osimertinib resistance. These findings further extend the horizon of the mechanisms underlying acquired resistance to osimertinib and enabled us to recognize potential biomarkers of osimertinib resistance in patients with LUAD.
2. MATERIALS AND METHODS
2.1. Transfection of miRNA mimics, inhibitors, or plasmid vectors
Lentivirus packaging of plasmids psPAX2 and pMD2.G was performed as previously described. 14 Plasmids for hsa_circ_0005576‐specific shRNA, and Mock (sequences are shown in Table S1) were added into the pGLVH1/GFP+Puromycin vector, plasmids for hsa_circ_0005576 overexpression and control were added into the pGLV‐CMV‐MCS‐EF1‐circRNA+Puromycin vector (GenePharma). Lentivirus was infected into cells using 5 μg/mL of the enhancer polybrene (GenePharma), then stable cell lines were selected with 2 μg/mL puromycin (Gibco) for 14 d. In addition, miR‐512‐5p (miRbase accession:MI0000463) mimics, inhibitors, and miR‐NC (Thermo Fisher Scientific) were transfected into H1975OR cells using Lipofectamine 2000 (Invitrogen), according to the manufacturer's protocol.
2.2. Reverse transcription and quantitative real‐time PCR (qRT‐PCR)
Genomic DNA (gDNA) was isolated using the QIAamp DNA Mini Kit (Qiagen). Total RNA from LUAD cells was extracted using TRIzol reagent. For RNA samples requiring linear RNA depletion, 10 μg total RNA alone was incubated for 2‐3 h at 37°C with 4 U/μg RNase R (BioVision), then was purified using the RNeasy MinElute Cleaning Kit (Qiagen). For circRNA analysis, reverse transcription was performed using the Goscript™ Reverse Transcription System and GoTaq® qPCR Master Mix (Promega). For microRNAs analysis, total RNA was reverse transcribed and detected using the All‐in‐One™ miRNA qRT‐PCR Detection Kit (Genecopoeia) according to the manufacturer’s instructions. Quantitative RT‐PCR was performed using the Cobas Z480 Analyzer (Roche). circRNA and mRNA levels were normalized to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH), miRNA levels were normalized using small nuclear U6. The Ct value for each sample was calculated using the ΔΔCt method. All primers were synthesized by Tsingke Biological Technology (Beijing, China) and are listed in Tables S2 and S3. Sanger sequencing of hsa_circ_0005576 was performed by Tsingke Biological Technology (Beijing, China).
2.3. RNase R and Actinomycin D treatments
Adding actinomycin D (2 mg/mL, Selleck) or DMSO into the culture medium of H1975OR and HCC827OR suppressed transcription. Total RNA (10 μg) extracted from LUAD cells was incubated with 2 U/μg RNase R (BioVision) at 37°C for 30 min, and then RNA was purified using the RNeasy MinElute Cleaning Kit (Qiagen). Finally, RNA was reverse transcribed into cDNA, and the expressive abundance of CDC42 and hsa_circ_0005576 was detected by qRT‐PCR assay.
2.4. Cell proliferation assay
For the colony forming assay, LUAD cells were seeded into 6‐well plates at a concentration of 500 cells/well, divided into groups and treated with osimertinib (1 μM) or vehicle (DMSO). After 1‐2 wk of culture, colonies were fixed in 4% paraformaldehyde (Sangon Biotech), stained with crystal violet, and analyzed using Image‐Pro Plus 7.0 software. For the MTS assay, each cell line was seeded (2 × 103 cells/well) into a 96‐well plate, divided into an osimertinib group and a control group (4 wells/group), and treated with reagents as mentioned in the colony forming assay. After incubation for 0, 24, 48, or 72 h, MTS reagents (Promega) were added to assess cell viability according to the manufacturer's instructions.
2.5. Cell apoptosis analysis
LUAD cells were seeded into 6‐well plates and treated with osimertinib (1 μM) or vehicle (DMSO). After incubation for 48 h, the cell apoptosis rate was assessed using the Annexin‐V‐FITC/propidium iodide apoptosis kit (BD Bioscience) according to the manufacturer's instructions.
2.6. Western blot analysis
For western blot analysis, all samples were harvested separately and lysed in RIPA buffer (Cell Signaling Technology) containing 1× protease inhibitor and PhosSTOP (Roche). The protein concentrations of whole‐cell lysates were evaluated using the BCA Protein Assay Kit (Pierce Biotechnology). Immunoblot analysis was performed as previously described. 14 Antibodies are listed in Table S4.
2.7. Biotin‐labeled RNA pull‐down assay
A biotin‐labeled hsa_circ_0005576 probe and a negative control probe (shown in the Table S5) were synthesized by GenePharm. The probes were incubated with M280 streptavidin Dynabeads (Invitrogen) at 25°C for 2 h. The extraction was lysed and ultrasonicated from H1975OR cells and was incubated with the probe‐coated bead mixture at 4°C overnight. Biotin‐labeled RNA complexes were pulled down by streptavidin magnetic beads with the wash buffer and then were eluted and purified with TRIzol (Invitrogen) for further qRT‐PCR analysis of circRNA and miRNA expression abundance.
2.8. Dual‐luciferase reporter assay
The binding sites of miRNAs were predicted by TargetScan (http://www.targetscan.org/vert_71/). The different 3'‐UTR sequences of hsa_circ_0005576‐WT, hsa_circ_0005576‐mutant, IGF1R‐WT, and IGF1R‐mutant were synthesized by GenePharma and added into the pmirGLO vector (Promega) to generate reporter plasmids. All cells transfected with the indicated reporter plasmid were seeded into 96‐well plates and co‐transfected with 50 nM miR‐NC or miR‐512‐5p mimics using Lipofectamine 2000 reagent. Then, luciferase activity was assessed after post‐transfection for 48 h using the Dual‐Luciferase Assay System (Promega) and according to the manufacturer's instructions.
2.9. Animal experiments for tumor growth in vivo
The research protocol was approved and the mice were maintained according to the Institutional Guidelines of the Animal Ethics Committee of Central South University. Female BALB/c nude mice aged 4‐6 wk were randomized into 2 groups (10 mice/group) and inoculated separately in the left axilla with H1975OR‐sh‐Mock or H1975OR‐sh‐circ_0005576 cells (4 × 106 cells/100 μL). Tumor volumes and body weight of each mouse were measured at least weekly. When all tumors reached a mean volume of 50 mm3, the nude mice in each group were randomized into 2 different subgroups (5 mice/subgroup) and treated with osimertinib (20 mg/kg/d) or vehicle (equal volume DMSO) by oral gavage for 21 d. Tumor volumes were calculated using the following equation: V = length × width2/2 as in the previously described method. 14 All nude mice were sacrificed 21 d after the initial osimertinib treatment and tumor tissues were collected for western blot analysis.
3. RESULTS
3.1. Identification of overexpressed hsa_circ_0005576 in the osimertinib‐resistant LUAD cells
Based on our early circRNAs microarray analysis (detailed method is shown in the Document S1), it was confirmed that the expression of hsa_circ_0005576 in the osimertinib‐resistant H1975OR cells was almost 3 times that of the osimertinib‐sensitive H1975 cells (Figure 1A). We synthesized divergent primers specific for the backsplice junction of hsa_circ_0005576, and then investigated the expression of hsa_circ_0005576 in osimertinib‐resistant LUAD cells. qRT‐PCR results showed that hsa_circ_0005576 expression was significantly upregulated in H1975OR cells and HCC827OR cells compared with their respective parental cells H1975 and HCC827 (Figure 1B). Matching to the circBase database (http://www.circbase.org/) annotation found that hsa_circ_0005576 was transcribed from the cell division cycle 42 (CDC42) gene located on chr1:22404921‐22413359 and consisted of 4 exons with a length of 536 nt (Figure 1C). qRT‐PCR assay using divergent primers indicated that hsa_circ_0005576 was only amplified from cDNA templates compared with genomic DNA (gDNA) templates in H1975OR and HCC827OR cells. Next, Sanger sequencing of the qRT‐PCR products amplified by the specific divergent primers further directly confirmed the back‐splicing site of hsa_circ_0005576 (Figure 1D). Moreover, the RNase R treatment assay verified hsa_circ_0005576 expression in osimertinib‐resistant LUAD cells. Compared with CDC42 mRNA, resistance to RNase R digestion identified the typical circular form and loop structure of hsa_circ_0005576 (Figure 1E). Furthermore, actinomycin D assay was used to evaluate the stability of hsa_circ_0005576. The apparent half‐life of the circRNA transcript exceeded 24 h, whereas that of the linear isomer was less than 3 h, indicating that the circRNA isoform was much more stable (Figure 1F,G). In conclusion, these results revealed that hsa_circ_0005576 derived from CDC42 was upregulated in the osimertinib‐resistant LUAD cells, and may be associated with the development of LUAD cells resistant to osimertinib.
FIGURE 1.
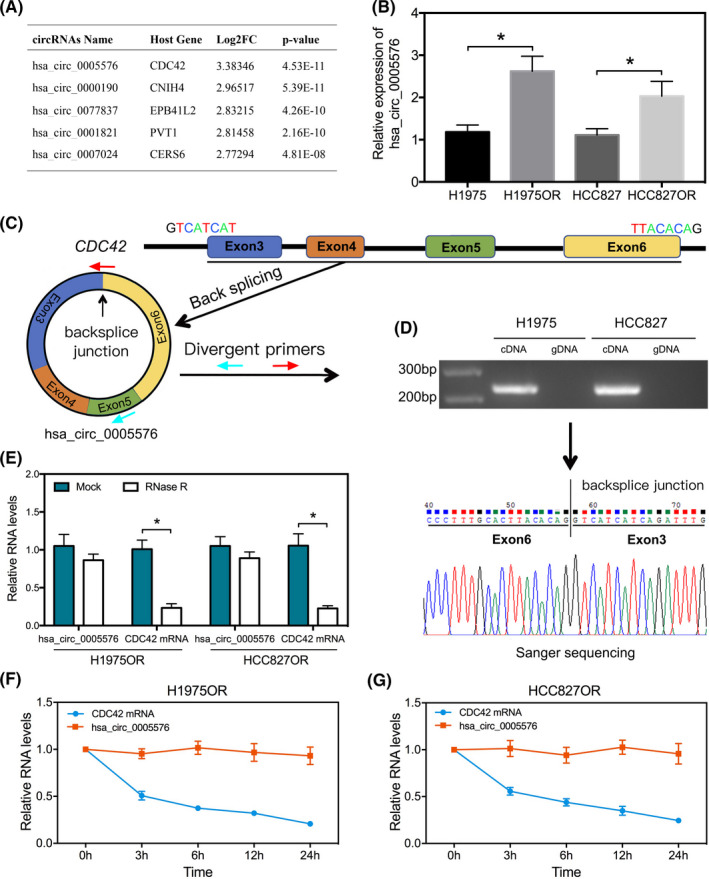
A, circRNA expression profiles between H1975OR and H1975 cells, the top 5 upregulated circRNAs were identified. B, qRT‐PCR analysis of hsa_circ_0005576 expression in H1975/H1975OR and HCC827/HCC827OR cells. C, Schematic diagram illustrates the genomic location (CDC42) and the backsplice conformation of hsa_circ_0005576. Red and blue arrows represent the fragment of divergent primers specific for backsplice junction of hsa_circ_0005576. D, Gel electrophoresis of hsa_circ_0005576 amplified by divergent primers in total cDNA and gDNA. Sanger sequencing validates the backsplice junction of hsa_circ_0005576. E, qRT‐PCR analysis of hsa_circ_0005576 and CDC42 mRNA expression in H1975OR and HCC827OR cells after treatment with RNase R. F, G, Actinomycin D assay for the abundance of CDC42 mRNA and hsa_circ_0005576 in H1975OR and HCC827OR cells. *P < .05
3.2. Knockdown of hsa_circ_0005576 reverses resistance of LUAD cells to osimertinib
To evaluate the potential functions of hsa_circ_0005576 in LUAD cells resistant to osimertinib, we constructed a specific shRNA vector for the back‐splicing site of hsa_circ_0005576. qRT‐PCR assay indicated that transfection of the specific shRNA vectors efficiently downregulated endogenous hsa_circ_0005576 expression abundance in osimertinib‐resistant LUAD cells (Figure 2A). Next, the function of hsa_circ_0005576 in osimertinib‐resistant‐LUAD cell viability, proliferation, and apoptosis was investigated. Knockdown of hsa_circ_0005576 expression significantly suppressed cell viability (Figure 2B) and colony forming ability (Figure 2C), which increased osimertinib sensitivity positively in resistant LUAD cells. Markedly, hsa_circ_0005576 knockdown stimulated apoptosis of resistant LUAD cells induced by osimertinib (Figure 2D). Furthermore, normal transformed cells (HBE cells) were used to perform a similar functional analysis by knockdown of hsa_circ_0005576; we found that normal HBE cells were not sensitive to osimertinib. In addition, knockdown of hsa_circ_0005576 had no significant effect on proliferation and apoptosis of HBE cells. In other words, knockdown of hsa_circ_0005576 in normal HBE cells did not significantly affect the sensitivity to osimertinib (Figure S1). In addition, the effect of hsa_circ_0005576 on osimertinib sensitivity in parental H1975 and HCC827 cells was investigated and showed that overexpression of hsa_circ_0005576 not only promoted cell viability and colony formation, but also decreased the apoptosis induced by osimertinib (Figure S2). In summary, the above results suggest that hsa_circ_0005576 not merely serves as a tumor promoter but also as an osimertinib resistance driver in LUAD cells.
FIGURE 2.
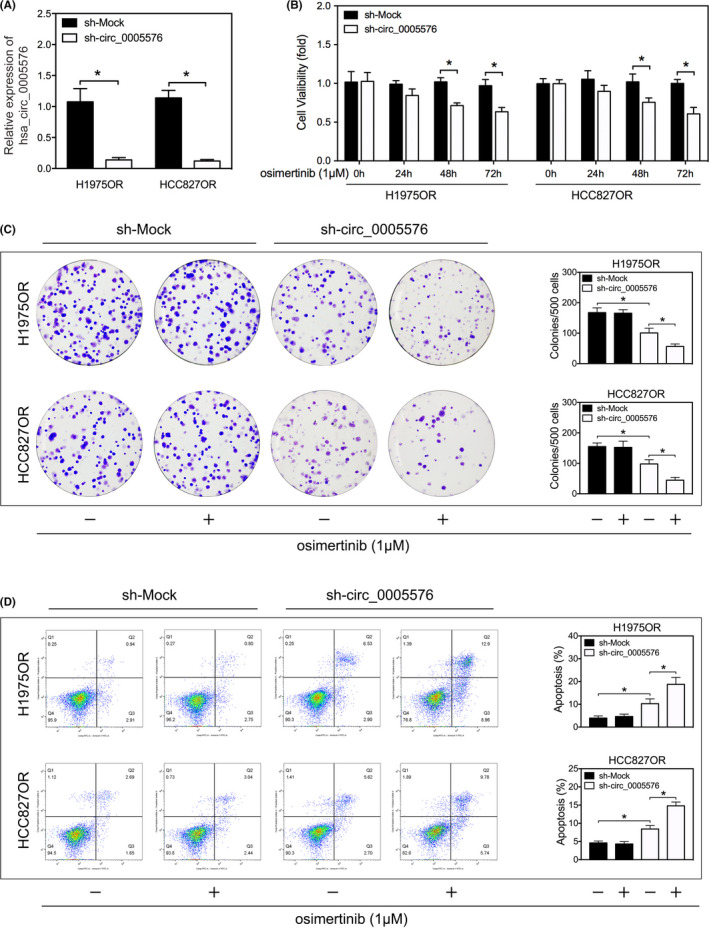
A, qRT‐PCR analysis for knockdown efficiency of sh‐circ_0005576 in H1975OR and HCC827OR cells. MTS assay (B), colony forming assay (C) and flow cytometry (D) were applied to measure the effects of hsa_circ_0005576 knockdown on osimertinib sensitivity in H1975OR and HCC827OR cells. *P < .05
3.3. Hsa_circ_0005576 serves as an efficient miRNA sponge for miR‐512‐5p
As circRNAs have been shown to have biological function as miRNA sponges, we next investigated the feasibility and capability of hsa_circ_0005576 to ‘sponge’ miRNAs. CircInteractome and regRNA2.0 (score > 130, free energy < −20) online tools were utilized to predict miRNA response elements (MREs) harbored by hsa_circ_0005576. Among them, miR‐146b‐3p, miR‐217, miR‐330‐5p, miR‐512‐5p, and miR‐593‐3p were the most highly ranked as candidate miRNAs by both databases (Figure 3A). Next, biotinylated probe binding of hsa_circ_0005576 was specifically performed by RNA pull down for purification and screening by qRT‐PCR to predict miRNA candidates. A distinct enrichment of hsa_circ_0005576 and miR‐512‐5p was found compared with other miRNAs (Figure 3A). Indeed, upregulation of miR‐512‐5p was the most obvious candidate when hsa_circ_0005576 was knocked down (Figure 3B). In addition, qRT‐PCR detected that miR‐512‐5p expression was downregulated in osimertinib‐resistant LUAD cells compared with the sensitive, parent LUAD cells (Figure S3). Subsequently, we generated dual‐luciferase reporter vectors encorporating the integral wild‐type (WT) hsa_circ_0005576 or the mutant hsa_circ_0005576, which contained the miR‐512‐5p binding sites (MUT) (Figure 3C). Luciferase activity was negative in H1975OR cells co‐transfected with miR‐512‐5p mimics or the WT hsa_circ_0005576 vector, but there was no change with the mutant hsa_circ_0005576 vector (Figure 3D). In summary, these results indicated that hsa_circ_0005576 serves as an efficient miRNA sponge for miR‐512‐5p.
FIGURE 3.
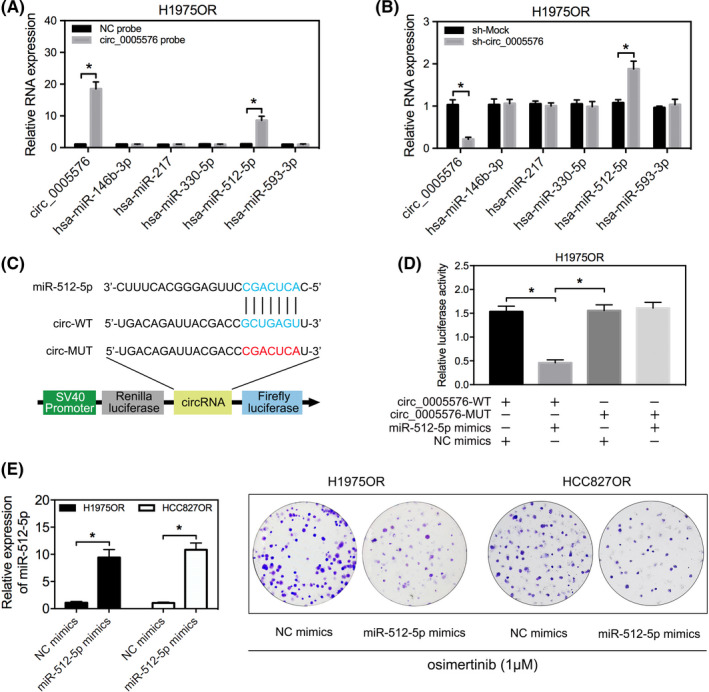
A, qRT‐PCR analysis for abundance of the candidate miRNAs pulled down by biotin‐labeled hsa_circ_0005576 in H1975OR cells. B, qRT‐PCR analysis measures the expression changes of candidate miRNAs in H1975OR cells transfected with or without sh‐circ_0005576. C, Predicted binding sites between miR‐512‐5p and hsa_circ_0005576. Construction of wild‐type (WT) and mutant (MUT) hsa_circ_0005576 dual‐luciferase reporter vectors. D, Dual‐luciferase activity of WT hsa_circ_0005576 3′UTR or mutant hsa_circ_0005576 3′UTR after transfection with miR‐512‐5p mimics in H1975OR cells. E, Colony forming assay evaluates cell proliferation in LUAD transfected with or without miR‐512‐5p mimics, then treated with osimertinib (1 μM). *P < .05
3.4. Hsa_circ_0005576 promotes osimertinib resistance of LUAD cells via miR‐512‐5p
Given the critical association between hsa_circ_0005576 with miR‐512‐5p, we next investigated the biological functions of miR‐512‐5p. We found that miR‐512‐5p truly acted as a tumor suppressor and that overexpression of miR‐512‐5p transfected with miR‐512‐5p mimics the increased osimertinib sensitivity of H1975OR cells, thereby inhibiting its colony forming ability (Figure 3E). Rescue experiments were carried out to confirm whether miR‐512‐5p was a direct mediator of the hsa_circ_0005576 effects of LUAD cells resistance to osimertinib. First, we showed that transfection with sh‐circ_0005576 can effectively reduce the expression of endogenous hsa_circ_0005576 (Figure S4A) and that miR‐512‐5p inhibitors can effectively decrease the expression of endogenous miR‐512‐5p (Figure S4B). Results from the proliferation assays demonstrated that transfection with miR‐512‐5p inhibitors significantly reversed the suppression effect by hsa_circ_0005576 knockdown on cell viability (Figure 4A,B) and the colony forming ability (Figure 4C) in H1975OR and HCC827OR cells. Similarly, flow cytometry analysis revealed that apoptotic stimulation of hsa_circ_0005576 knockdown was reversed by transfection with miR‐512‐5p inhibitors (Figure 4D). Taken together, the above results illustrated that hsa_circ_0005576 facilitated LUAD cell resistance to osimertinib by ‘sponging’ miR‐512‐5p, and thereby attenuated the tumor suppressor effect of miR‐512‐5p.
FIGURE 4.
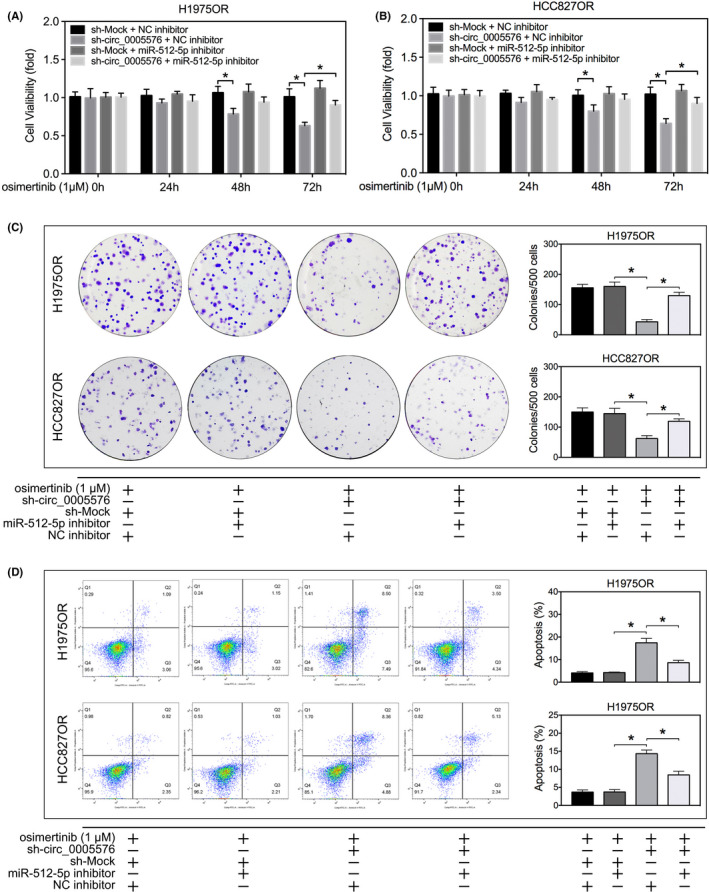
MTS assay (A, B), colony forming assay (C) and flow cytometry (D) were applied to measure the effects of hsa_circ_0005576 knockdown and miR‐512‐5p inhibition on osimertinib sensitivity in H1975OR and HCC827OR cells. *P < .05
3.5. IGF1R is a direct target gene of miR‐512‐5p and is regulated by hsa_circ_0005576
To evaluate the tumor‐promoting and osimertinib resistance‐assisting effects of hsa_circ_0005576, we knocked down hsa_circ_0005576 with a specific shRNA vector (sequences are shown in Table S1) against the back‐splicing site of hsa_circ_0005576 and used a transcriptome microarray (detailed method is shown in Document S1) to analyze the gene expression signature in H1975OR cells. We were able to identify 565 genes that changed in expression when hsa_circ_0005576 was knocked down in osimertinib‐resistant H1975OR cells (Figure 5A). Subsequently, IPA software further was applied to perform functional analysis of differentially expressed genes; the involvement of hsa_circ_0005576 knockdown in multiple cellular processes and pathways was found. More importantly, pathways enrichment analysis showed that many changes were observed in the pathway analysis. These genes were most enriched in the suppression of the signaling pathway for IGF1R (Figure 5B), a tyrosine protein kinase involved in downstream signaling of EGFR‐TKI resistance, which was most associated with knockdown of hsa_circ_0005576 in H1975OR cells. The differentially expressed genes related to the IGF1R signaling pathway were displayed to confirm downregulation of IGF1R expression after knockdown of hsa_circ_0005576 in H1975OR cells. Among them, differential downregulation of IGF1R by nearly 3 times was the most significant (Figure S5). Furthermore, qRT‐PCR assay also confirmed that IGF1R mRNA expression was upregulated in osimertinib‐resistant LUAD cells compared with their sensitive, parent LUAD cells (Figure S6). Next, we measured the comprehensive activation of IGF1R signaling by western blot analysis to clarify the regulatory effects of hsa_circ_0005576 sponging of miR‐512‐5p, the results revealed a positively regulated relationship between hsa_circ_0005576 and IGF1R expression in H1975OR cells. Phosphorylated AKT and the phosphorylated ERK1/2 were highly related to the expression and activation of phosphorylated IGF1R (Figure 5C). Nevertheless, regulation of miR‐512‐5p in IGF1R signaling revealed the opposite effect (Figure 5C). Moreover, we generated dual‐luciferase reporter vectors that included the integral WT IGF1R mRNA or the mutant IGF1R mRNA which contained miR‐512‐5p binding sites (MUT) (Figure 5D). Luciferase activity was reduced in H1975OR cells co‐transfected with miR‐512‐5p mimics and the WT IGF1R mRNA vector, but no change was observed with the mutant IGF1R mRNA vector (Figure 5E). Considering the above data, this study, therefore, presents the facilitating effects of hsa_circ_0005576 on osimertinib resistance through the miR‐512‐5p/IGF1R axis in LUAD cells.
FIGURE 5.
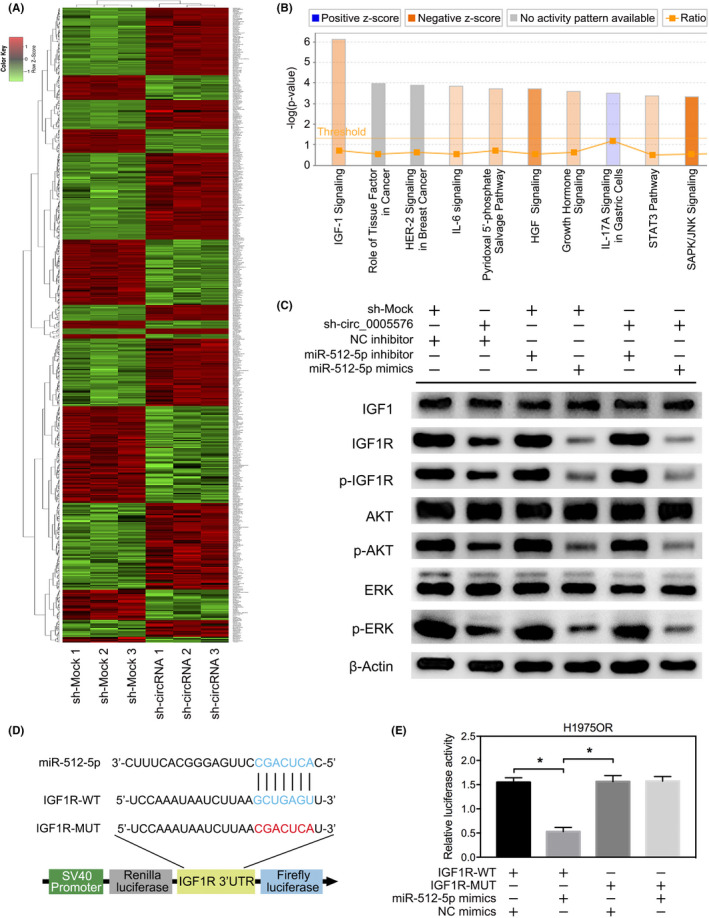
A, Heat map of the transcriptome microarray of differentially expressed genes in H1975OR cells transfected with or without sh‐circ_0005576. B, Signaling pathway enrichment analysis by transcriptome microarray for differentially expressed genes of H1975OR cells transfected with or without sh‐circ_0005576. C, Western blot analysis of the expression of EGFR and IGF1R signaling activation. D, Predicted binding sites of miR‐512‐5p with IGF1R mRNA 3′UTR sites, predicted binding sites between miR‐512‐5p and hsa_circ_0005576. Construction of wild‐type (WT) and mutant (MUT) IGF1R mRNA 3′UTR dual‐luciferase reporter vectors. E, Dual‐luciferase activity of WT IGF1R mRNA 3′UTR or mutant IGF1R mRNA 3′UTR after transfection with miR‐512‐5p mimics in H1975OR cells. *P < .05
3.6. Hsa_circ_0005576 facilitates osimertinib resistance of H1975OR xenograft tumor by the miR‐512‐5p/IGF1R pathway
To evaluate the tumor‐suppressing and osimertinib‐sensitizing effects of hsa_circ_0005576 in vivo, we established stable hsa_circ_0005576 knockdown cells H1975OR‐sh‐circ_0005576 and their corresponding control cells H1975OR‐sh‐Mock. In vivo, knockdown of hsa_circ_0005576 inhibited the tumorigenicity of H1975OR with no significant loss of mouse body weight (Figure S7). Compared with the sh‐Mock group, the mean xenograft tumor volume of the sh‐circ_0005576 group was significantly reduced, moreover treatment with osimertinib for 21 d further inhibited xenograft tumor growth in the sh‐circ_0005576 group compared with the group treated with DMSO (Figure 6A,B). Western blot analysis also showed that, in response to treatment with osimertinib, although EGFR was inactivated, phosphorylation activation downstream for AKT and ERK1/2 was not completely blocked (Figure 6C). When IGF1R was downregulated by hsa_circ_0005576 knockdown and then treated with osimertinib, its combined inhibition of AKT and ERK1/2 phosphorylation was significantly enhanced (Figure 6C). Briefly, our findings demonstrated that in vivo hsa_circ_0005576 promoted tumor growth and neutralized the tumor‐suppressing effect of osimertinib by the miR‐512‐5p/IGF1R pathway.
FIGURE 6.
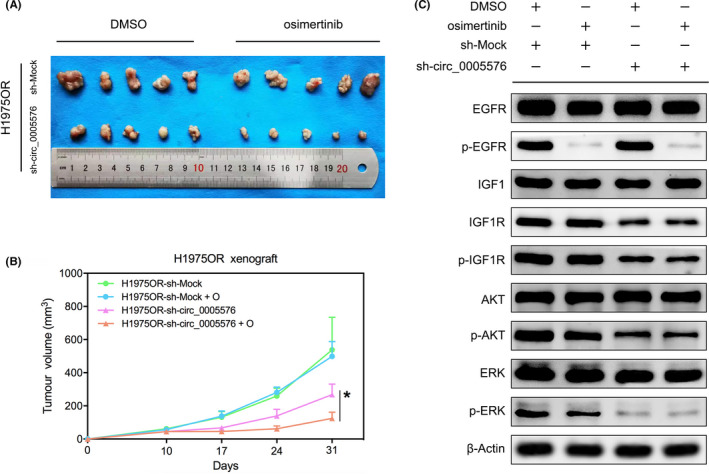
A, BALB/c nude mice were given H1975OR cells transfected with or without sh‐circ_0005576 and then treated with DMSO or osimertinib (20 mg/kg/d) for 21 d. B, Growth curve of xenograft tumors (n = 5). C, Western blot analysis of the protein expression of the largest xenograft tumor in each group. *P < .05
4. DISCUSSION
Biological functions and regulatory mechanisms, as well as circRNAs, sponge miRNAs, thereby potentially interfering with miRNA activity and levels available to bind to their target mRNAs. This effectively regulates target gene expression or activates signaling pathways, therefore facilitating tumorigenesis and stimulating cancer progression, which has been extensively validated. 5 , 6 , 15 circRNAs as modulators of miRNAs function immensely expand the diversity and complexity of transcriptomes in cancers. 5 Two previous studies pointed out that hsa_circ_0005576 is an oncogene that can mediate the progression of colorectal cancer and cervical cancer. 12 , 13 At this time, there have been no other reports on hsa_circ_0005576 affecting human cancer progression. Through circRNA microarray analysis, we found differential expression patterns for hsa_circ_0005576 between H1975 and H1975OR cells. Further investigation showed that hsa_circ_0005576 knockdown in vitro could reverse the sensitivity of LUAD cells to osimertinib. Hsa_circ_0005576 knockdown downregulated IGF1R expression, which decreased downstream activation of PI3K/AKT and MAPK/ERK1/2 signaling pathways, and therefore exerted the consequential anti‐proliferative and apoptotic effects. Similar results were found for animal experiments in vivo. These findings demonstrated, for the first time, that hsa_circ_0005576 is an oncogene in LUAD; it may not only serve as a diagnostic indicator for monitoring osimertinib resistance but also act as a potential therapeutic target for post resistance to osimertinib in LUAD.
In this study, we found a significant correlation between hsa_circ_0005576 and miR‐512‐5p. Moreover, miR‐512‐5p contains binding sites that almost perfectly complement hsa_circ_0005576 and IGF1R mRNA, as proved by dual‐luciferase reporter assay. When hsa_circ_0005576 was knocked down, the relative expression levels of miR‐512‐5p almost doubled in H1975OR cells. Importantly, effects of hsa_circ_0005576 on proliferation and anti‐apoptosis of LUAD cells can be abolished by overexpression of miR‐512‐5p. Sensitivity to osimertinib can be restored by overexpression of miR‐512‐5p, while suppression of miR‐512‐5p had the opposite effect. This finding suggests that hsa_circ_0005576 can interact with miR‐512‐5p and further regulate miR‐512‐5p levels and functional activity for IGF1R expression. More recently, the role of miR‐512‐5p as a tumor suppressor has been clarified in different types of human malignant tumors. 16 , 17 , 18 For lung cancer, specifically, the reactivation of epigenetically silenced miR‐512 sensitized lung cancer cells to cisplatin and restricted tumor growth. 19 Furthermore, overexpression of miR‐512‐5p induced apoptosis and inhibited glycolysis by targeting p21 in non–small‐cell lung cancer cells. 20 Findings from our study were consistent with those in previous studies and further revealed that miR‐512‐5p, acting as a tumor suppressor, was directly sponged and downregulated by hsa_circ_0005576, with upregulation of IGF1R and IGF1R signaling. The consequences of this chain were enhanced proliferative and anti‐apoptotic capabilities of LUAD cells and subsequent decreased susceptibility and sustainability of osimertinib.
Insulin‐like growth factor 1 receptor (IGF1R), a transmembrane RTK, plays an indispensable function in homeostasis and is responsible for cell survival, proliferation, and differentiation. 21 IGF1 ligand binding to IGF1R evokes dimerization of IGF1R and autophosphorylation of specific tyrosine residues, which activate the PI3K/AKT and MAPK/ERK1/2 signaling pathways and motivate strongly proliferative and anti‐apoptotic effects. 22 , 23 In particular, it has been reported previously that activation of IGF1R signaling is implicated in LUAD differentiation and survival in patients, 24 conferring acquired resistance to osimertinib in NSCLC containing the EGFR T790M mutation. 25 Many strategies to target IGF1R signaling have been developed, including siRNA and monoclonal antibodies for IGF1R and kinase inhibitors to inhibit the enzyme activity of RTK. 22 , 26 PI3K‐AKT and the MAPK/ERK1/2 pathway are 2 typical signal transduction pathways that are crucial for EGFR‐TKI resistance events in LUAD, 27 , 28 , 29 these 2 pathways are also defined as downstream of the IGF1R signaling pathways. 23 , 30 Although EGFR‐TKI resistance is more complex, much evidence has implied that activation of the PI3K/AKT pathway is required for resistance to EGFR‐TKIs in LUAD cells; activation of the MAPK/ERK1/2 pathway may also support a proliferative advantage to resistant cells. 31 Furthermore, multiple phospho‐RTK array analysis identified that AKT and ERK1/2 signaling was dependent on increased IGF1R phosphorylation activation, which was involved in resistance of LUAD cells to EGFR‐TKIs. 32 , 33 We therefore examined whether AKT and ERK1/2 were coordinators of IGF1R signaling using overexpression of hsa_circ_0005576 that facilitated acquired resistance to osimertinib in LUAD. We observed that, although expression of AKT and ERK1/2 was not significantly altered, their phosphorylation activation was consistent with that of IGF1R and was highly correlated. In addition, previous findings have highlighted the IGF family as biomarkers for response to advanced NSCLC through EGFR‐TKI treatment; 34 also IGF1R activated by IGF2 is relevant to osimertinib resistance in lung cancer. 35 We considered that the interaction of IGF1R with IGF1 would be required for optimal receptor activation for tyrosine kinase activity of IGF1R. 21 , 30 In the present study, transcriptome microarray combined with western blotting analysis showed that IGF1 and IGF2 expression levels were not significantly downregulated at the same time in osimertinib‐resistant H1975OR cells with knocked‐down hsa_circ_0005576. We confirmed that osimertinib resistance associated with hsa_circ_0005576 is directly related to IGF1R upregulation, leading to IGF1R signaling activation, rather than for IGF families.
It has already been illustrated that activation of IGF1R signaling associated with IGFBP3 loss of function plays a crucial role in the resistance to EGFR‐TKIs. 31 The IGFBP family, through binding to the extracellular portion of IGFs and preventing their activation, are negative regulators of IGF1R signal activation. 36 , 37 According to our transcriptome microarray analysis, IGFBP1 expression was upregulated (fold change: 1.32; P‐value: 1.65E−07) in osimertinib‐resistant H1975OR cells and knocked down hsa_circ_0005576. We thereby speculated that the slight downregulation effect of hsa_circ_0005576 on IGFBP1 expression may indirectly promote activation of the IGF1R signaling pathway.
In conclusion, for the first time we emphasized and demonstrated that hsa_circ_0005576 exerts an oncogenic role in proliferative stimulation and apoptotic suppression by sponging miR‐512‐5p to upregulate IGF1R expression, which promotes the resistance to osimertinib of LUAD cells.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Fig S1‐S7
Table S1‐S5
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the Hunan Province Science and Health Union Foundation (no. 2018JJ6135, no. 2018JJ6053) and the Hunan Province Natural Science Foundation (no. 2018JJ3777).
Liu S, Jiang Z, Xiao P, et al. Hsa_circ_0005576 promotes osimertinib resistance through the miR‐512‐5p/IGF1R axis in lung adenocarcinoma cells. Cancer Sci.2022;113:79–90. 10.1111/cas.15177
Contributor Information
Longyu Jin, Email: jinlongyu1123@163.com.
Wei Feng, Email: fweimail@163.com.
REFERENCES
- 1. Ramalingam SS, Vansteenkiste J, Planchard D, et al. Overall survival with osimertinib in untreated, EGFR‐mutated advanced NSCLC. N Engl J Med. 2020;382:41‐50. [DOI] [PubMed] [Google Scholar]
- 2. Leonetti A, Sharma S, Minari R, Perego P, Giovannetti E, Tiseo M. Resistance mechanisms to osimertinib in EGFR‐mutated non‐small cell lung cancer. Br J Cancer. 2019;121:725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Piper‐Vallillo AJ, Sequist LV, Piotrowska Z. Emerging treatment paradigms for EGFR‐mutant lung cancers progressing on osimertinib: a review. J Clin Oncol. 2020;38(25):2926‐2936. [DOI] [PubMed] [Google Scholar]
- 4. Kristensen LS, Andersen MS, Stagsted LVW, Ebbesen KK, Hansen TB, Kjems J. The biogenesis, biology and characterization of circular RNAs. Nat Rev Genet. 2019;20:675‐691. [DOI] [PubMed] [Google Scholar]
- 5. Chen LL. The biogenesis and emerging roles of circular RNAs. Nat Rev Mol Cell Biol. 2016;17:205‐211. [DOI] [PubMed] [Google Scholar]
- 6. Patop IL, Wust S, Kadener S. Past, present, and future of circRNAs. EMBO J. 2019;38:e100836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu Y‐C, Li J‐R, Sun C‐H, et al. CircNet: a database of circular RNAs derived from transcriptome sequencing data. Nucleic Acids Res. 2016;44:D209‐D215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333‐338. [DOI] [PubMed] [Google Scholar]
- 9. Ruan H, Xiang Y, Ko J, et al. Comprehensive characterization of circular RNAs in ~1000 human cancer cell lines. Genome Med. 2019;11:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang C, Tan S, Liu W‐R, et al. RNA‐Seq profiling of circular RNA in human lung adenocarcinoma and squamous cell carcinoma. Mol Cancer. 2019;18:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cheng X, Qiu J, Wang S, et al. Comprehensive circular RNA profiling identifies CircFAM120A as a new biomarker of hypoxic lung adenocarcinoma. Ann Transl Med. 2019;7:442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu C, Li S, Hu X. Circ_0005576 promotes malignant progression through miR‐874/CDK8 axis in colorectal cancer. Onco Targets Ther. 2020;13:7793‐7805. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13. Ma H, Tian T, Liu X, et al. Upregulated circ_0005576 facilitates cervical cancer progression via the miR‐153/KIF20A axis. Biomed Pharmacother. 2019;118:109311. [DOI] [PubMed] [Google Scholar]
- 14. Feng W, Xie Q, Liu S, et al. Kruppel‐like factor 4 promotes c‐Met amplification‐mediated gefitinib resistance in non‐small‐cell lung cancer. Cancer Sci. 2018;109:1775‐1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen LL. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat Rev Mol Cell Biol. 2020;21:475‐490. [DOI] [PubMed] [Google Scholar]
- 16. Saito Y, Suzuki H, Tsugawa H, et al. Chromatin remodeling at Alu repeats by epigenetic treatment activates silenced microRNA‐512‐5p with downregulation of Mcl‐1 in human gastric cancer cells. Oncogene. 2009;28:2738‐2744. [DOI] [PubMed] [Google Scholar]
- 17. Yang LU, Sun H, Liu X, et al. Circular RNA hsa_circ_0004277 contributes to malignant phenotype of colorectal cancer by sponging miR‐512‐5p to upregulate the expression of PTMA. J Cell Physiol. 2020. [DOI] [PubMed] [Google Scholar]
- 18. Zhang J, Wang L, Jiang J, Qiao Z. Elevation of microRNA‐512‐5p inhibits MUC1 to reduce radioresistance in cervical cancer. Cell Cycle. 2020;19:652‐665. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19. Adi Harel S, Bossel Ben‐Moshe N, Aylon Y, et al. Reactivation of epigenetically silenced miR‐512 and miR‐373 sensitizes lung cancer cells to cisplatin and restricts tumor growth. Cell Death Differ. 2015;22:1328‐1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chu K, Gao G, Yang X, et al. MiR‐512‐5p induces apoptosis and inhibits glycolysis by targeting p21 in non‐small cell lung cancer cells. Int J Oncol. 2016;48:577‐586. [DOI] [PubMed] [Google Scholar]
- 21. Li J, Choi E, Yu H, Bai XC. Structural basis of the activation of type 1 insulin‐like growth factor receptor. Nat Commun. 2019;10:4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fujita M, Ieguchi K, Cedano‐Prieto DM, et al. An integrin binding‐defective mutant of insulin‐like growth factor‐1 (R36E/R37E IGF1) acts as a dominant‐negative antagonist of the IGF1 receptor (IGF1R) and suppresses tumorigenesis but still binds to IGF1R. J Biol Chem. 2013;288:19593‐19603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li R, Pourpak A, Morris SW. Inhibition of the insulin‐like growth factor‐1 receptor (IGF1R) tyrosine kinase as a novel cancer therapy approach. J Med Chem. 2009;52:4981‐5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kikuchi R, Sonobe M, Kobayashi M, et al. Expression of IGF1R is associated with tumor differentiation and survival in patients with lung adenocarcinoma. Ann Surg Oncol. 2012;19(Suppl 3):S412‐S420. [DOI] [PubMed] [Google Scholar]
- 25. Hayakawa D, Takahashi F, Mitsuishi Y, et al. Activation of insulin‐like growth factor‐1 receptor confers acquired resistance to osimertinib in non‐small cell lung cancer with EGFR T790M mutation. Thorac Cancer. 2020;11:140‐149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yaktapour N, Übelhart R, Schüler J, et al. Insulin‐like growth factor‐1 receptor (IGF1R) as a novel target in chronic lymphocytic leukemia. Blood. 2013;122:1621‐1633. [DOI] [PubMed] [Google Scholar]
- 27. Sato H, Yamamoto H, Sakaguchi M, et al. Combined inhibition of MEK and PI3K pathways overcomes acquired resistance to EGFR‐TKIs in non‐small cell lung cancer. Cancer Sci. 2018;109:3183‐3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ercan D, Xu C, Yanagita M, et al. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2012;2:934‐947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Niederst MJ, Engelman JA. Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci Signal. 2013;6:re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chitnis MM, Yuen JS, Protheroe AS, Pollak M, Macaulay VM. The type 1 insulin‐like growth factor receptor pathway. Clin Cancer Res. 2008;14:6364‐6370. [DOI] [PubMed] [Google Scholar]
- 31. Cortot AB, Repellin CE, Shimamura T, et al. Resistance to irreversible EGF receptor tyrosine kinase inhibitors through a multistep mechanism involving the IGF1R pathway. Cancer Res. 2013;73:834‐843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Suda K, Mizuuchi H, Sato K, Takemoto T, Iwasaki T, Mitsudomi T. The insulin‐like growth factor 1 receptor causes acquired resistance to erlotinib in lung cancer cells with the wild‐type epidermal growth factor receptor. Int J Cancer. 2014;135:1002‐1006. [DOI] [PubMed] [Google Scholar]
- 33. Wang R, Yamada T, Kita K, et al. Transient IGF‐1R inhibition combined with osimertinib eradicates AXL‐low expressing EGFR mutated lung cancer. Nat Commun. 2020;11:4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vollebergh MA, Kappers I, Klomp HM, et al. Ligands of epidermal growth factor receptor and the insulin‐like growth factor family as serum biomarkers for response to epidermal growth factor receptor inhibitors in patients with advanced non‐small cell lung cancer. J Thorac Oncol. 2010;5:1939‐1948. [DOI] [PubMed] [Google Scholar]
- 35. Manabe T, Yasuda H, Terai H, et al. IGF2 autocrine‐mediated IGF1R activation is a clinically relevant mechanism of osimertinib resistance in lung cancer. Mol Cancer Res. 2020;18:549‐559. [DOI] [PubMed] [Google Scholar]
- 36. Evdokimova V, Tognon CE, Benatar T, et al. IGFBP7 binds to the IGF‐1 receptor and blocks its activation by insulin‐like growth factors. Sci Signal. 2012;5:ra92. [DOI] [PubMed] [Google Scholar]
- 37. Fürstenberger G, Senn HJ. Insulin‐like growth factors and cancer. Lancet Oncol. 2002;3:298‐302. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S7
Table S1‐S5
Supplementary Material