

Abstract
Due to the considerable success of cancer immunotherapy for leukemia, the tumor immune environment has become a focus of intense research; however, there are few reports on the dynamics of the tumor immune environment in leukemia. Here, we analyzed the tumor immune environment in pediatric B cell precursor acute lymphoblastic leukemia by analyzing serial bone marrow samples from nine patients with primary and recurrent disease by mass cytometry using 39 immunophenotype markers, and transcriptome analysis. High‐dimensional single‐cell mass cytometry analysis elucidated a dynamic shift of T cells from naïve to effector subsets, and clarified that, during relapse, the tumor immune environment comprised a T helper 1‐polarized immune profile, together with an increased number of effector regulatory T cells. These results were confirmed in a validation cohort using conventional flow cytometry. Furthermore, RNA transcriptome analysis identified the upregulation of immune‐related pathways in B cell precursor acute lymphoblastic leukemia cells during relapse, suggesting interaction with the surrounding environment. In conclusion, a tumor immune environment characterized by a T helper 1‐polarized immune profile, with an increased number of effector regulatory T cells, could contribute to the pathophysiology of recurrent B cell precursor acute lymphoblastic leukemia. This information could contribute to the development of effective immunotherapeutic approaches against B cell precursor acute lymphoblastic leukemia relapse.
Keywords: B cell leukemia, immune response, regulatory T cell, relapse, Th1
Tumor immune environment characterized by a T helper 1‐polarized immune profile was observed in recurrent B cell precursor acute lymphoblastic leukemia. Regulatory T cells were activated in recurrent B cell precursor acute lymphoblastic leukemia.

Abbreviations
- BCP‐ALL
B cell precursor acute lymphoblastic leukemia
- BM
bone marrow
- CCR
C‐C chemokine receptor
- CSM
cell‐staining medium
- CTLA
CTL‐associated antigen
- CXCR
C‐X‐C motif chemokine receptor
- CyTOF
cytometry by time‐of‐flight
- GSEA
gene set enrichment analysis
- IFN‐γ
interferon‐γ
- IL
interleukin
- PD
programmed cell death
- PD‐L
programmed cell death‐ligand
- ROSE
recognition of outliers by sampling ends
- RT
room temperature
- Th
T helper
- TIE
tumor immune environment
- Treg
regulatory T cell
- UMAP
uniform manifold approximation and projection
- viSNE
visualization of t‐distributed stochastic neighbor embedding
1. INTRODUCTION
B cell precursor acute lymphoblastic leukemia is a hematopoietic cell malignancy derived from B‐lineage lymphoid precursors. 1 Despite intensive chemotherapy, including central nervous system prophylaxis, approximately 20% of children with BCP‐ALL will relapse, 2 and the outcome of recurrent BCP‐ALL remains unsatisfactory. In recent years, immunotherapy against leukemia has become a research focus, due to the success of chimeric antigen receptor T cell therapy 3 and other Ab‐related drugs, such as bispecific T cell engagers, 4 which can dramatically improve treatment outcomes. 5
Bone marrow is a hematopoietic tissue composed of various cell types, including mesenchymal stem cells, endothelial cells, osteoprogenitors, and immune cells, and provides an essential niche for hematopoietic stem cells; however, leukemic cells also take advantage of BM to aid their survival. 6 Recently, the immune environment of BM, the primary site of leukemia, has been the subject of intense research, 7 leading to elucidation of its role in relapse 8 , 9 ; however, the immunological background of BCP‐ALL in the BM remains unclear. Understanding the dynamics of the BM TIE in BCP‐ALL has potential to facilitate the identification of novel risk factors and the development of better treatment strategies, applicable to the different disease status of individual patients.
Immune responses involve concerted actions by several immune cell types, of which Th cells play a central, orchestrating role. Over recent decades, Th17, Th9, Th22, T follicular‐helper, and Tregs have been recognized, in addition to the classical biphasic model of Th1 and Th2 cell differentiation. 10 Of Th cell subgroups, Tregs have an immunosuppressive effect, which is indispensable for the maintenance of immune homeostasis. 11 FOXP3 (a master regulator of Tregs) and CD45RA can be used to classify Tregs into three phenotypically and functionally distinct populations: FOXP3loCD45RA+ (fraction I, naïve or resting Tregs), FOXP3hiCD45RA– (fraction II, effector Tregs), and FOXP3loCD45RA– (fraction III, non‐Tregs) cells. Of these Treg populations, only FOXP3hiCD45RA– cells truly have effective immunosuppressive capacity. 12 , 13 , 14 Regulatory T cells contribute to tumor development and progression by inhibiting antitumor immunity. 15 , 16 There have been several reports of increased Treg numbers and activity in BCP‐ALL 17 , 18 , 19 ; however, detailed analysis has not been carried out.
In this study, we focused on the immune environment of pediatric BCP‐ALL in the BM by analyzing serial samples of primary and recurrent phase leukemias, using high‐dimensional, single‐cell mass cytometry (also known as “cytometry by time‐of‐flight” or CyTOF) and transcriptome analysis. The use of mass cytometry with metal conjugated Abs for detecting cellular protein markers is a relatively new technology; however, the underlying principle is similar to that of conventional flow cytometry, and results from mass cytometry analyses are comparable with those from flow cytometry. 20 We also undertook a flow cytometry experiment to validate our mass cytometry findings. We comprehensively assessed the BM immune environment in BCP‐ALL, and closely investigated T cells, revealing that a Th1‐polarized inflammatory TIE, with an increase in effector Tregs, is characteristic of recurrent BCP‐ALL.
2. MATERIALS AND METHODS
2.1. Patient samples
Primary and recurrent BM samples were collected serially from nine pediatric patients with BCP‐ALL at Kyoto University Hospital from 2006 to 2013, with consent from patients or their guardians. Mononuclear cells were isolated by density centrifugation using Lymphoprep (Alere Technologies) and viably preserved in CELLBANKER 1 (ZENOAQ RESOURCE) until they were used. The proportion of BCP‐ALL cells in the samples was 87.3%‐97.8% (mean, 94.0%) at onset, and 81.3%‐98.8% (mean, 92.2%) at relapse. Flow cytometric analysis was carried out using serial samples from a validation cohort comprising seven patients collected at The University of Tokyo, Saitama Prefectural Children’s Medical Center, and our institution, with consent from the patients or their guardians.
This study was approved by the Kyoto University Hospital Ethical Board, approval number G‐1030.
2.2. Mass cytometry
Thawed BM mononuclear cells were resuspended in RPMI‐1640 medium (Sigma‐Aldrich) with 10% FCS and penicillin‐streptomycin (Meiji Seika Pharma), and then washed twice with PBS. For barcoding of viable samples, cells were stained with combined isotope‐tagged cisplatins in PBS (1 μM or 2 μM) at RT for 10 minutes. Details of the combinations of cisplatins used are provided in Figure S1. To evaluate batch effects, a healthy peripheral blood reference sample was included in each barcoding plate. After washing twice with PBS, all serial samples from the same patient and the reference sample were mixed together. Blocking was carried out using Fc receptor Binding Inhibitor Functional Grade Monoclonal Antibody (eBioscience), following the manufacturer’s instructions. Antibodies for chemokine markers were added to yield 100 μl final reaction volumes in RPMI, and samples were incubated at 37°C for 45 minutes, washed twice with CSM (PBS containing 0.1% BSA, 2 mM EDTA, and 0.01% sodium azide), and stained for surface markers in 100 µl CSM at RT for 45 minutes. After washing twice with PBS, cells were rested at RT for 10 minutes and stained for viability in PBS containing 500 nM dichloro‐(ethylenediamine)palladium (II) (Sigma‐Aldrich). 21 Intracellular staining was undertaken using the FOXP3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific), according to the manufacturer’s instructions. Finally, PBS containing 2% formaldehyde and 1:500 Cell‐ID Intercalator‐Rh (Fluidigm) was added, and samples were stored at 4°C overnight or for up to 48 hours. Before mass cytometry analysis, samples were washed once with CSM and twice with double‐distilled water, filtered to remove aggregates, and resuspended in Maxpar water containing 15% EQ Four Element Calibration Beads (Fluidigm). Throughout the analysis, cells were introduced at a constant rate of approximately 400 cells/s. The CyTOF Ab panel, including 39 Abs, is presented in Table S1.
Acquired data were normalized using bead normalization and uploaded into Cytobank software (https://www.beckman.jp/flow‐cytometry/software/cytobank‐premium). Following single‐cell gating, live cells were gated based on palladium staining, debarcoded, and assigned to each original sample.
2.3. Flow cytometry
Thawed BM mononuclear cells were resuspended as described above, and then washed twice with PBS. After blocking with Fc receptor binding inhibitor, fluorescent‐labeled Abs were added to yield 100 μl final reaction volumes in CSM. Samples were incubated at RT for 30 minutes and washed twice with PBS. Intracellular staining was carried out using the FOXP3/Transcription Factor Staining Buffer Set with an anti‐FOXP3 Ab, according to the manufacturer’s instructions. Before flow cytometry analysis, samples were resuspended in 500 μl CSM and measured using FACSVerse (Becton Dickinson). Acquired data were analyzed using Cytobank software (https://www.beckman.jp/flow‐cytometry/software/cytobank‐premium). The Ab panel for flow cytometry is presented in Table S2.
2.4. Nucleic acid preparation and RNA sequencing
RNA isolation from BM mononuclear cells was carried out using NucleoSpin TriPrep (MACHREY‐NAGEL), according to the manufacturer’s instructions. RNA integrity was measured using an Agilent 2200 TapeStation and RNA Screen Tapes (Agilent Technologies). Sequencing libraries were prepared using an NEBNext Ultra II RNA Library Kit for Illumina (New England Biolabs), according to the manufacturer’s protocol, and prepared libraries were run on an Illumina HiSeq × high‐throughput sequencing system. Paired‐end reads were aligned to the hg19 human genome assembly using STAR. 22 Fusion transcripts were detected using Genomon version 2.6.2 (https://github.com/Genomon‐Project/) and filtered by excluding fusions: (a) mapping to repetitive regions; (b) with fewer than four spanning reads; (c) that occurred out of frame; or (d) had junctions not located at known exon‐intron boundaries. The expression level of each RefSeq gene was calculated from mapped read counts using HTSeq and normalized using the Bioconductor package, DESeq2 version 1.28.1. 23 Supercomputing resources were provided by the Human Genome Center, Institute of Medical Science, The University of Tokyo. To detect Ph‐like ALL, we applied ROSE algorithm 24 and performed hierarchical clustering using Ward’s method for Euclidian distances, based on data from three in‐house BCR‐ABL1 fusion‐positive ALL cases and deposited in the DNA Data Bank of Japan (accession number PRJD8942). Cluster stability was ascertained by consensus clustering using the R package, ConsensusClusterPlus, with 1000 iterations. Gene set enrichment analysis was carried out using software (version 4.1.0) from the Broad Institute. 25 , 26 Differentially regulated MSigDB ontology gene sets with a false discovery rate q value of less than 0.10 were filtered and evaluated.
2.5. Statistical analysis
Data analysis was undertaken using Cytobank software (https://www.beckman.jp/flow‐cytometry/software/cytobank‐premium), Prism 8 (GraphPad), and R statistical software (http://www.r‐project.org) within the CATALYST (Cytometry dATA anALYSis Tools) pipeline, referring to the CyTOF workflow (version 4). 27 , 28 , 29 To test the statistical significance of differences between two groups, a two‐tailed paired Student’s test was applied in Prism 8. Using the CyTOF workflow, differential abundance analysis was assessed using a linear mixed model to calculate adjusted P values. 28
3. RESULTS
3.1. Patient characteristics
A detailed list of patient characteristics is shown in Table 1. We collected serial samples from nine relapsed patients by BM aspiration at initial diagnosis and relapse. None of the subjects had apparent or proven infection (which could influence the proportion and characteristics of immune cells), such as sepsis, viral disease, or mycosis, at the time of sample collection. Hierarchical clustering using the ROSE algorithm (Figure S2) and detection of fusion transcripts by RNA sequencing of samples from relapsed patients identified the following rearrangements in the cohort: one Ph‐like, three KMT2A, one MEF2D, and one ZNF384. Seven of the relapsed patients had died of the disease. The time course and sampling points for each patient are summarized in Figure 1. Patients R6 and R9 experienced early relapse; however, they had entered complete remission in response to induction therapy before they relapsed. For validation flow cytometric analysis, we used serial samples from another seven patients, whose characteristics are summarized in Table S3.
TABLE 1.
Characteristics of children with recurrent B cell precursor acute lymphoblastic leukemia included in the study cohort
| Patient no. | Relapse status | Sex | Age a (y) | WBC count a (109/L) | Subgroup | Treatment protocol | Time from initial diagnosis to relapse (mo) | Alive at last follow‐up |
|---|---|---|---|---|---|---|---|---|
| R1 | Yes | Female | 5.8 | 23.7 | B‐other | JACLS ALL‐02 SR | 33 | No |
| R2 | Yes | Male | 0.2 | 369.0 | KMT2A | In accordance with Interfant‐99 HR | 17 | No |
| R3 | Yes | Female | 3.2 | 12.9 | ZNF384 | JACLS ALL‐02 ER | 37 | Yes |
| R4 | Yes | Female | 10.2 | 0.4 | B‐other | JACLS ALL‐02 ER | 43 | No |
| R5 | Yes | Female | 11.8 | 309.6 | Ph‐like | JACLS ALL‐02 ER | 28 | No |
| R6 | Yes | Female | 8.1 | 0.9 | MEF2D | JACLS ALL‐02 HR | 3 | No |
| R7 | Yes | Female | 2.4 | 91.7 | KMT2A | JACLS ALL‐02 ER | 16 | Yes |
| R8 | Yes | Female | 18.8 | 37.3 | B‐other | JACLS ALL‐02 HR | 16 | No |
| R9 | Yes | Female | 0.1 | 441.9 | KMT2A | JPLSG MLL‐10 HR | 3 | No |
Abbreviations: ALL, acute lymphoblastic leukemia; ER, extremely high risk; HR, high risk; JACLS, Japan Association of Childhood Leukemia Study; JPLSG, Japanese Pediatric Leukemia/Lymphoma Study Group; MLL, mixed lineage leukemia; SR, standard risk; WBC, white blood cells.
At initial diagnosis.
FIGURE 1.
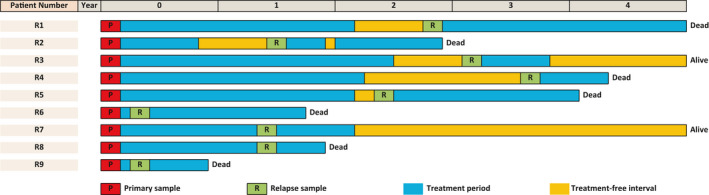
Time course and sample collection points for each patient with recurrent B cell precursor acute lymphoblastic leukemia. Samples analyzed in this study are indicated by red and green squares
3.2. Mononuclear cell landscape in BM samples from patients with primary and recurrent BCP‐ALL
To comprehensively understand the cell profiles in BCP‐ALL samples, data obtained from mass cytometric analysis were input into Cytobank software, and immune cell subsets were detected using viSNE. 30 First, a control sample was used to confirm the basic BM constitution; eight different cellular components were distinguished, as follows: CD4 T cells, CD3+CD4+CD8–; CD8 T cells, CD3+CD4–CD8+; Tregs (a subset of CD4 T cells), CD3+CD4+CD8–CD25+CD127–; monocyte/myeloid cells, CD3–CD19–CD56–CD11b+CD33+; natural killer cells, CD3–CD19–CD14–CD56+; B cells, CD10–CD19+CD20+CD22+; early B cells, CD10+CD19+CD20–CD22–; and stem/progenitor cells, CD34+CD3–CD19–CD14–CD56–CD33– (Figure 2A). Next, samples from patients with BCP‐ALL at initial diagnosis and relapse were analyzed. An example of visualization of cellular components using viSNE is shown in Figure 2B. The BCP‐ALL cells in the patient sample were clearly separated from immune cells (Figure 2B, left), and alterations in immune cell components could be serially tracked at onset and relapse (Figure 2B, middle and right). In this way, we analyzed the cellular composition of BM samples from all patients, and compared the transition between disease onset and relapse (Figure 2C). Patient samples were mostly comprised of BCP‐ALL cells, and there was no significant difference in the proportion of each component at relapse relative to onset. Furthermore, we did not detect any significant differences in the expression of immunomodulating markers (PD‐L1, PD‐L2, and CD86) on BCP‐ALL cells at onset and relapse (data not shown).
FIGURE 2.
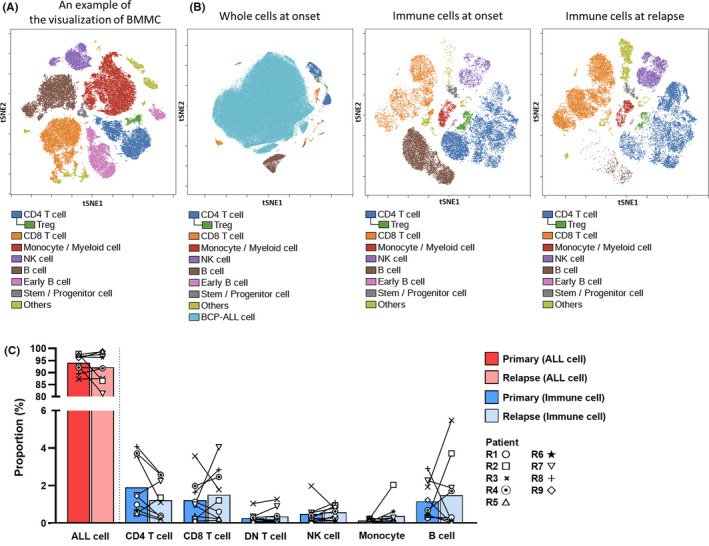
Classification of bone marrow component cells to understand the recurrent B cell precursor acute lymphoblastic leukemia (BCP‐ALL) tumor immune environment. A, Visualization of t‐distributed stochastic neighbor embedding (viSNE) projects each bone marrow (BM) cellular population onto a 2D surface (t‐distributed stochastic neighbor embedding 1 (tSNE1) and tSNE2). BM mononuclear cells (BMMC) from a remission sample are illustrated. B, A representative visualization of the cellular components of BM from a patient with BCP‐ALL. The onset sample comprises mostly ALL cells (left). Example serial visualizations of BM immune cells at disease onset (middle) and relapse (right). ALL cells were removed from the serial visualizations to facilitate understanding of the immune cell distribution (middle and right). C, Comparison of the BM composition of samples from nine patients with relapsed BCP‐ALL at diagnosis and relapse. DN, double negative; NK, natural killer; Treg, regulatory T cell
3.3. High‐dimensional single‐cell analysis of T cells in serial BM samples reveals dynamic immunological alteration toward a Th1‐dominant environment in recurrent BCP‐ALL
Next, we focused on T cell subtypes and their status in primary and recurrent BCP‐ALL. Samples from patient R9 did not contain sufficient T cells and were consequently excluded from this analysis. To evaluate T cells in detail, we adopted a two‐step approach, consistent with the CyTOF workflow. 27 , 28 , 29 First, we classified T cell‐related markers in the CyTOF panel into two groups: type markers and state markers. There were 10 type markers (CD3, CD4, CD8a, CD25, CD127, CD27, CD28, CCR7, CD45RA, and FOXP3) used to identify T cell subgroups and maturity, and eight state markers (CD38, HLA‐DR, PD‐1, CTLA‐4, CXCR3, CCR4, CCR6, and CXCR5) used to characterize the status of each T cell subgroup. Other markers expressed at low levels were excluded. In the next step, T cells from all serial samples were clustered using FlowSOM 31 and assigned to 13 optimal metaclusters, based on expression of type markers. Then, several similar metaclusters were merged manually by checking type marker expression patterns. Eventually, 10 distinct T cell subgroups were identified (Figure 3A). Dimensional reduction graphs of T cells colored by subgroup identity confirmed that the 10 subgroups were consistent with UMAP (arXiv:1802.03426) distribution, which represents cell similarity, indicating that the classification was performed appropriately (Figure 3B).
FIGURE 3.
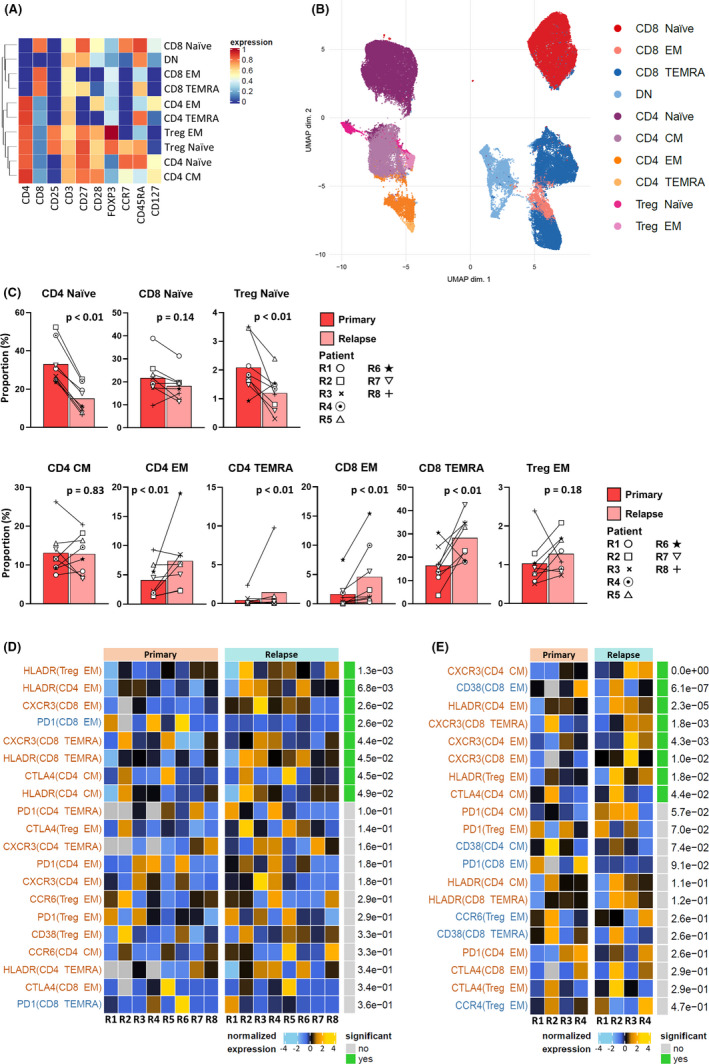
Serial mass cytometric analysis of T cells in bone marrow (BM) reveals dynamic immunological alteration to a T helper 1‐dominant tumor immune environment in recurrent B cell precursor acute lymphoblastic leukemia (BCP‐ALL). A, BM T cells from primary and recurrent BCP‐ALL samples classified into 10 subgroups using FlowSOM with manual merging. The type markers used in clustering are displayed as a heatmap. B, Dimensional reduction graph (UMAP) of T cells colored by subgroup identity. C, An increase in effector T cell subgroups was observed at relapse. Differential abundance analysis carried out using a generalized linear mixed model to calculate adjusted P values. D, Differential state test results and normalized expression of state markers by effector T cells in serial primary and relapse samples from eight patients. The top 20 state marker‐subgroup combinations are sorted according to adjusted P values calculated using a linear mixed model. Combinations upregulated at relapse are colored red; those downregulated are blue. E, Differential state test results and normalized expression of state markers in effector T cells, focusing on four patients without therapeutic effects at relapse. The top 20 state marker‐subgroup combinations are sorted according to adjusted P values calculated using a linear mixed model. Combinations upregulated at relapse are colored red; those downregulated are blue. CM, central memory; DN, double negative; EM, effector memory; TEMRA, terminally differentiated effector memory; Treg, regulatory T cell
To evaluate the transition of T cell subgroups in relapsed patients, differential abundance analysis was carried out, and adjusted P values were calculated using a generalized linear mixed model (Figure 3C). Interestingly, there was a tendency for naïve T cell subgroups to decrease, and effector T cell subgroups to increase, at relapse relative to onset. The significant increase of effector memory and terminally differentiated T cells indicated that the TIE of recurrent BCP‐ALL is in an immunologically stimulated state. Second, we compared normalized expression patterns of state markers in each effector T cell subgroup using a differential state test (Figure 3D). In this analysis, state marker‐subgroup combinations were examined by assessing the change in expression of state markers between primary and relapse samples. The 70 combinations were then ranked in ascending order of adjusted P values calculated using a linear mixed model. Both CXCR3 and HLA‐DR were significantly upregulated in the majority of effector T cell subgroups in relapse samples. Previous studies have reported roles for CXCR3 in trafficking of Th1 and CD8 T cells to peripheral sites of Th1‐type inflammation and establishment of a Th1 amplification loop mediated by IFN‐γ and INF‐γ‐inducible CXCR3 ligands. 32 These data indicate that T cells in relapsed BCP‐ALL BM had shifted to an effector phenotype with a Th1‐dominant state.
As the above analysis included patients who had relapsed during ALL therapy, we undertook further analyses focused on the four patients who relapsed more than 6 months after completion of treatments (patients R1‐R4), to rule out the influence of chemotherapeutic agents on the TIE. In this analysis, the upregulation of CXCR3 expression on effector T cells at relapse was more marked (Figure 3E). In summary, the BM microenvironment in recurrent BCP‐ALL was characterized by Th1 dominance.
3.4. Recurrent BCP‐ALL immune environment is enriched for effector Tregs
In the above analysis, effector memory Tregs showed a trend toward increasing at relapse (Figure 3C). To clarify the kinetics of Tregs in recurrent BCP‐ALL, we classified them according to previous research, 12 , 13 , 14 as follows: FOXP3loCD45RA+ cells (fraction I, naïve or resting Tregs), FOXP3hiCD45RA– cells (fraction II, effector Tregs), and FOXP3loCD45RA– cells (fraction III, non‐Tregs). An example of a consecutive gating to detect the FOXP3hi CD45RA– Treg subpopulation (fraction Ⅱ, effector Tregs) is shown in Figure 4A. According to this gating method, the effector Treg (CD25+CD127−FOXP3hiCD45RA–) subset was significantly increased in CD4 T cells at relapse (Figure 4B). Previously, it was reported that effector Tregs have a phenotype including PD‐1, CTLA‐4, TIM‐3, and CCR4 expression. 33 In our study, PD‐1, CTLA‐4, and CCR4 were significantly upregulated in CD25+CD127– Tregs at relapse, supporting an augmentation of effector characteristics (Figure 4C). An evaluation of Tregs in only the four patients who relapsed more than 6 months after completing treatments (patients R1‐R4) is shown in Figure S3, and the features were consistent with the results described above.
FIGURE 4.
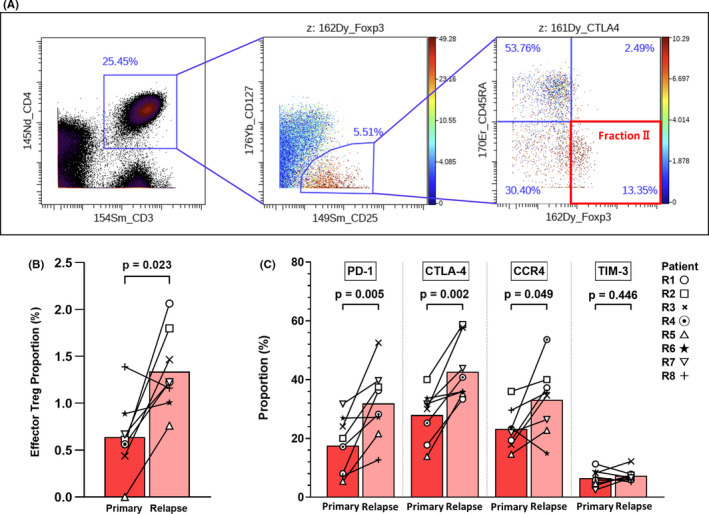
Enhancement of effector regulatory T cell (Treg) properties in recurrent B cell precursor acute lymphoblastic leukemia (BCP‐ALL). Results from 16 serial bone marrow samples from relapsed patients are shown. A, An example of consecutive gating for detection of FOXP3hi CD45RA– Tregs (effector Tregs, fraction II). B, Proportion of effector Tregs (CD25+CD127−FOXP3hiCD45RA–) among CD4 T cells. C, Expression levels of programmed cell death‐1 (PD‐1), CTL‐associated antigen‐4 (CTLA‐4), and C‐C chemokine receptor type 4 (CCR4), but not T cell immunoglobulin and mucin‐domain containing‐3 (TIM‐3), were significantly upregulated in CD25+CD127– Tregs at relapse
3.5. Validation of results of mass cytometric analysis by conventional flow cytometry in samples from a new patient cohort
To validate our mass cytometry findings, we undertook flow cytometric analysis of samples from a validation cohort, including seven patients (Table S3). Both CXCR3+ CD4 T cells and effector Tregs were detected by flow cytometry with the same Ab clones as those used in mass cytometry analysis (Table S2). Our findings confirmed that CXCR3+ CD4 T cells and effector Tregs showed a tendency to increase at the time of relapse in this cohort (Figure 5), consistent with the results of mass cytometry.
FIGURE 5.
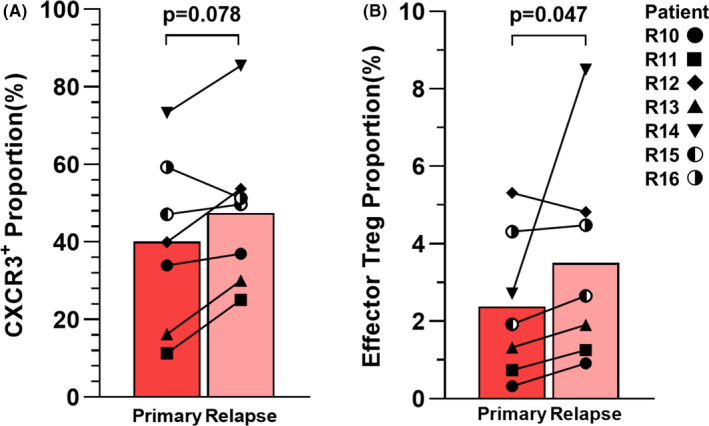
Validation of mass cytometric analysis by conventional flow cytometry. Serial bone marrow samples were obtained from seven additional pediatric patients with B cell precursor acute lymphoblastic leukemia. Proportions of (A) C‐X‐C motif chemokine receptor (CXCR3)+ and (B) effector regulatory T cells (Tregs) (CD25+CD127−FOXP3hi CD45RA–) among CD4 T cells. To test the statistical significance of differences between two groups, a two‐tailed Wilcoxon paired signed rank test was applied
3.6. Immune‐related pathways are broadly upregulated in BCP‐ALL cells at relapse
As described above, our data reveal that the immune environment in the BM during recurrent BCP‐ALL is dominated by Th1 cells and has increased levels of effector Tregs. To confirm the status of BCP‐ALL cells in this environment, we carried out whole RNA sequencing of primary and relapse samples from all nine patients. Average blast ratios were 94.0% and 92.2% at diagnosis and relapse, respectively, which was not a significant difference (P = .367, two‐tailed paired t test); therefore, the results of gene expression analysis mainly reflect the status of BCP‐ALL cells. The GSEA of RNA sequencing data using MSigDB ontology gene sets identified six immune‐related gene sets that were significantly upregulated in recurrent BCP‐ALL (false discovery rate <0.1). The six gene sets were as follows: chemokine activity, chemokine production, complement activation, positive regulation of cytokine production involved in immune response, positive regulation of lymphocyte chemotaxis, and positive regulation of lymphocyte migration. Enrichment plots for these significantly upregulated immune response gene sets are presented in Figure 6A.
FIGURE 6.
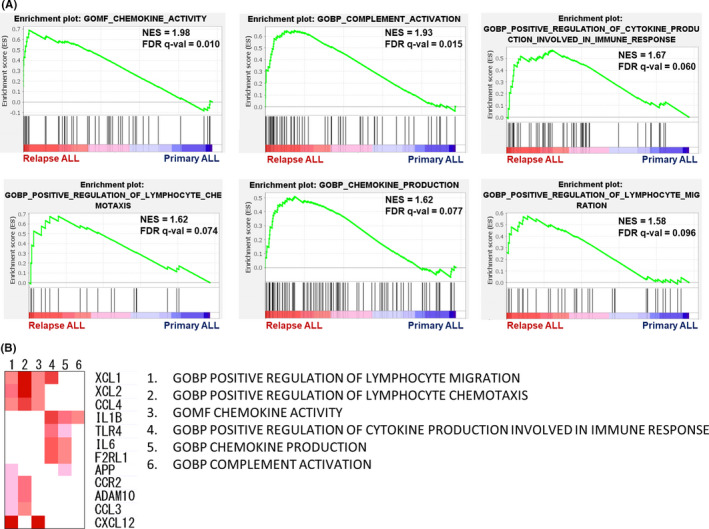
Transcriptional analysis identifies immune‐related pathways broadly upregulated in recurrent B cell precursor acute lymphoblastic leukemia cells. The result of gene set enrichment analysis of gene ontology gene sets are shown. A, Chemokine activity, chemokine production, complement activation, positive regulation of cytokine production involved in immune response, positive regulation of lymphocyte chemotaxis, and positive regulation of lymphocyte migration are illustrated as enrichment plots. B, Heatmap of the results of leading edge analysis using the six significantly upregulated immune response gene sets. Expression values of genes in each leading edge subset gene set are represented in red, and genes contained in at least two different gene sets are shown. NES, normalized enrichment score; FDR, false discovery rate; GOBP, gene ontology biological process; GOMF, gene ontology molecular function
Subsequently, leading edge analysis was carried out (Figure 6B) to determine which subsets of genes identified by GSEA mainly contributed to the enrichment signal of the leading edge or core enrichment, allowing detection of predominant genes in the selected gene sets. Various genes involved in inflammation and immune responses were detected as upregulated at relapse, including those encoding IL1B/CCL3/CCL4/CCR2 (Th1‐related immune response 34 , 35 , 36 ), XCL1/XCL2/CXCL12 (chemotaxis 37 , 38 ), and IL6/F2RL1 (B cell activation 39 and inflammation 40 ). Although the function of these genes in BCP‐ALL cells requires further investigation, these data show that BCP‐ALL cells have gene signatures that are predicted to attract lymphocytes to the inflammatory TIE in BM, and that these immune activities are upregulated at relapse.
4. DISCUSSION
Our comprehensive immune profiling data indicate that BCP‐ALL relapse is accompanied by a Th1‐polarized TIE and an increase of the effector Treg population. Generally, Th1 immune responses are important in anticancer immunity 41 ; however, several recent studies have suggested that BCP‐ALL could take advantage of the Th1‐dominant inflammatory environment. 42 , 43 , 44 In the clinical setting, children with BCP‐ALL often present with fever but no apparent evidence of infection. 45 , 46 A recent investigation showed that the concentrations of circulating pro‐inflammatory cytokines associated with fever (tumor necrosis factor‐α, IL‐6, IL‐8, and monocyte chemoattractant protein‐1) were elevated in patients with ALL, together with Th1 cytokines (IFN‐γ and IL‐12). 42 It has also been suggested that a highly inflammatory environment in the BM of patients with ALL could influence normal hematopoietic differentiation fates, creating favorable conditions for ALL proliferation. 43 Furthermore, an in vitro assay showed that BM Th1 cells can induce BCP‐ALL cell activation and proliferation through CD38 upregulation through IFN‐γ 44 ; interestingly, that study found that stimulated BCP‐ALL cells secreted IP‐10 (a CXCR3 ligand), which contributes to attraction of Th1 cells. As BCP‐ALL originates from precursor B cells, which require interactions with both Th2 and Th1 cells for proliferation, 47 unlike the situation in other malignancies, a Th1‐polarized TIE would not be an inhospitable environment for BCP‐ALL relapse.
T helper 1 cells are established supporters of CTL function, 41 and CTLs have the potential to eliminate ALL cells, threatening ALL progression and persistence. 48 The CTL suppression function is crucial in the context of tumor immune escape for relapse. Factors reported to be involved in suppressing CTL function are PD‐L1, 49 CD38, 50 and Tregs. 17 Expression of PD‐L1 on tumor cells is associated with poor prognosis in some hematologic malignancies; however, there is little information regarding the involvement of PD‐L1/L2 in BCP‐ALL, and PD‐L1/L2 expression is reported to be lower in BCP‐ALL than in other hematological malignancies, 51 although PD‐L1 expression has been detected in BCP‐ALL cells in some studies. 52 , 53 In our research, we did not detect distinct PD‐L1 expression (data not shown). Another possible mechanism by which BCP‐ALL cells suppress CTL function is overexpression of CD38 on tumor cells, leading to increased extracellular adenosine levels, which might contribute to resistance to CTLs. 50 In our analysis, CD38 was highly expressed on BCP‐ALL cells, regardless of the time at which BM was collected (Figure S4). Regulatory T cells are suppressors of antileukemic immune functions, including those of CTLs. Several studies reported that patients with BCP‐ALL had higher numbers of Tregs than healthy controls, 18 , 19 , 54 and that the immunosuppressive potential of Tregs increases with malignant progression in BCP‐ALL. 17 In the present study, we observed an increase in effector Tregs, which are reported to mediate suppression of antitumor immune responses. 16 Although we cannot definitively conclude whether the observed Th1‐polarized TIE and increase in the effector Treg population are causes or results of relapse, our GSEA results show that BCP‐ALL cells at relapse had gene signatures predicted to attract lymphocytes and enhance immunological responses, relative to the onset phase, indicating a close relationship between BCP‐ALL cells and the TIE.
There are some limitations to this study. We examined nine relapsed patients with mass cytometry and seven with flow cytometry; however, our sample size was insufficient for analysis of BCP‐ALL subgroups, which could have undiscovered differences in TIE. In addition, functional assessment could not be carried out due to the small quantities of samples available. These aspects may warrant further study.
In conclusion, a Th1‐polarized immune environment became dominant, and an increase of the effector Treg population was observed in BM samples from children with recurrent BCP‐ALL. Consistent with the context, immune‐related pathways were broadly upregulated in BCP‐ALL cells at relapse. Although the precise mechanisms involved in interactions between the TIE and BCP‐ALL cells are poorly understood, our data indicate that a Th1‐polarized immune environment with an increase of effector Tregs might be associated with BCP‐ALL relapse, and that targeted therapy modifying the TIE has potential to enhance the efficiency of existing immunotherapies.
DISCLOSURE
The authors declare no conflict of interest.
Supporting information
Fig S1
Fig S2
Fig S3
Fig S4
Table S1
Table S2
Table S3
ACKNOWLEDGMENTS
The mass cytometric data used in this research have been deposited in FlowRepository (Repository ID: FR‐FCM‐Z3EK). 55 Rika Ishii provided technical assistance with mass cytometry experiments. This investigation was partly supported by AMED under Grant Number JP21ck0106531, a grant from Bristol Myers Squibb, The Mochida Memorial Foundation for Medical and Pharmaceutical Research, and The Mother and Child Health Foundation.
Mikami T, Kato I, Wing JB, et al. Alteration of the immune environment in bone marrow from children with recurrent B cell precursor acute lymphoblastic leukemia. Cancer Sci.2022;113:41–52. 10.1111/cas.15186
REFERENCES
- 1. Malard F, Mohty M. Acute lymphoblastic leukaemia. The Lancet. 2020;395:1146‐1162. [DOI] [PubMed] [Google Scholar]
- 2. Kelly ME, Lu X, Devidas M, et al. Treatment of relapsed precursor‐B acute lymphoblastic leukemia with intensive chemotherapy: POG (Pediatric Oncology Group) study 9411 (SIMAL 9). J Pediatr Hematol Oncol. 2013;35:509‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B‐cell lymphoblastic leukemia. N Engl J Med. 2018;378:439‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kantarjian H, Stein A, Gokbuget N, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376:836‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Patrick AB, Lingyun J, Xinxin X, et al. A randomized phase 3 trial of Blinatumomab Vs. chemotherapy as post‐reinduction therapy in high and intermediate risk (HR/IR) first relapse of B‐acute lymphoblastic leukemia (B‐ALL) in children and adolescents/young adults (AYAs) demonstrates superior efficacy and tolerability of Blinatumomab: A report from children's oncology group study AALL1331. Blood. 2019;134(Supplement_2):LBA–1. [Google Scholar]
- 6. Bonomo A, Monteiro AC, Goncalves‐Silva T, Cordeiro‐Spinetti E, Galvani RG, Balduino A. A T cell view of the bone marrow. Front Immunol. 2016;7:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aoki T, Takami M, Takatani T, et al. Activated invariant natural killer T cells directly recognize leukemia cells in a CD1d‐independent manner. Cancer Sci. 2020;111:2223‐2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hohtari H, Bruck O, Blom S, et al. Immune cell constitution in bone marrow microenvironment predicts outcome in adult ALL. Leukemia. 2019;33:1570‐1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blaeschke F, Willier S, Stenger D, et al. Leukemia‐induced dysfunctional TIM‐3(+)CD4(+) bone marrow T cells increase risk of relapse in pediatric B‐precursor ALL patients. Leukemia. 2020;34(10):2607‐2620. [DOI] [PubMed] [Google Scholar]
- 10. Hirahara K, Nakayama T. CD4+ T‐cell subsets in inflammatory diseases: beyond the Th1/Th2 paradigm. Int Immunol. 2016;28:163‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775‐787. [DOI] [PubMed] [Google Scholar]
- 12. Miyara M, Yoshioka Y, Kitoh A, et al. Functional delineation and differentiation dynamics of human CD4+ T cells expressing the FoxP3 transcription factor. Immunity. 2009;30:899‐911. [DOI] [PubMed] [Google Scholar]
- 13. Ohkura N, Hamaguchi M, Morikawa H, et al. T cell receptor stimulation‐induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity. 2012;37:785‐799. [DOI] [PubMed] [Google Scholar]
- 14. Wing JB, Tanaka A, Sakaguchi S. Human FOXP3(+) regulatory T cell heterogeneity and function in autoimmunity and cancer. Immunity. 2019;50:302‐316. [DOI] [PubMed] [Google Scholar]
- 15. Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019;110:2080‐2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27:109‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bhattacharya K, Chandra S, Mandal C. Critical stoichiometric ratio of CD4+ CD25+ FoxP3+ regulatory T cells and CD4+ CD25‐ responder T cells influence immunosuppression in patients with B‐cell acute lymphoblastic leukaemia. Immunology. 2014;142(1):124‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Idris SZ, Hassan N, Lee LJ, et al. Increased regulatory T cells in acute lymphoblastic leukemia patients. Hematology. 2015;20:523‐529. [DOI] [PubMed] [Google Scholar]
- 19. Salem ML, El‐Shanshory MR, Abdou SH, et al. Chemotherapy alters the increased numbers of myeloid‐derived suppressor and regulatory T cells in children with acute lymphoblastic leukemia. Immunopharmacol Immunotoxicol. 2018;40:158‐167. [DOI] [PubMed] [Google Scholar]
- 20. Gadalla R, Noamani B, MacLeod BL, et al. Validation of CyTOF Against Flow Cytometry for Immunological Studies and Monitoring of Human Cancer Clinical Trials. Front Oncol. 2019;9:415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hartmann FJ, Simonds EF, Bendall SC. A universal live cell barcoding‐platform for multiplexed human single cell analysis. Sci Rep. 2018;8:10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29:15‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harvey RC, Mullighan CG, Wang X, et al. Identification of novel cluster groups in pediatric high‐risk B‐precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome‐wide DNA copy number alterations, clinical characteristics, and outcome. Blood. 2010;116:4874‐4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mootha VK, Lindgren CM, Eriksson KF, et al. PGC‐1alpha‐responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267‐273. [DOI] [PubMed] [Google Scholar]
- 26. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nowicka M, Krieg C, Crowell HL, et al. CyTOF workflow: differential discovery in high‐throughput high‐dimensional cytometry datasets. F1000Res. 2017;6:748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Weber LM, Nowicka M, Soneson C, Robinson MD. diffcyt: Differential discovery in high‐dimensional cytometry via high‐resolution clustering. Commun Biol. 2019;2:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chevrier S, Crowell HL, Zanotelli VRT, Engler S, Robinson MD, Bodenmiller B. Compensation of Signal Spillover in Suspension and Imaging Mass Cytometry. Cell Syst. 2018;6(5):612‐620.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. el Amir AD, Davis KL, Tadmor MD, et al. viSNE enables visualization of high dimensional single‐cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. 2013;31:545‐552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Van Gassen S, Callebaut B, Van Helden MJ, et al. FlowSOM: Using self‐organizing maps for visualization and interpretation of cytometry data. Cytometry A. 2015;87:636‐645. [DOI] [PubMed] [Google Scholar]
- 32. Groom JR, Luster AD. CXCR3 in T cell function. Exp Cell Res. 2011;317:620‐631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol. 2014;27:1‐7. [DOI] [PubMed] [Google Scholar]
- 34. Tominaga K, Yoshimoto T, Torigoe K, et al. IL‐12 synergizes with IL‐18 or IL‐1β for IFN‐γ production from human T cells. Int Immunol. 2000;12:151‐160. [DOI] [PubMed] [Google Scholar]
- 35. Vilgelm AE, Richmond A. Chemokines modulate immune surveillance in tumorigenesis, metastasis, and response to immunotherapy. Front Immunol. 2019;10:333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boring L, Gosling J, Chensue SW, et al. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C‐C chemokine receptor 2 knockout mice. J Clin Invest. 1997;100:2552‐2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fox JC, Nakayama T, Tyler RC, Sander TL, Yoshie O, Volkman BF. Structural and agonist properties of XCL2, the other member of the C‐chemokine subfamily. Cytokine. 2015;71:302‐311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Janssens R, Struyf S, Proost P. The unique structural and functional features of CXCL12. Cell Mol Immunol. 2018;15:299‐311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dienz O, Eaton SM, Bond JP, et al. The induction of antibody production by IL‐6 is indirectly mediated by IL‐21 produced by CD4+ T cells. J Exp Med. 2009;206:69‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rothmeier AS, Ruf W. Protease‐activated receptor 2 signaling in inflammation. Semin Immunopathol. 2012;34:133‐149. [DOI] [PubMed] [Google Scholar]
- 41. Haabeth OA, Lorvik KB, Hammarstrom C, et al. Inflammation driven by tumour‐specific Th1 cells protects against B‐cell cancer. Nat Commun. 2011;2:240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Perez‐Figueroa E, Sanchez‐Cuaxospa M, Martinez‐Soto KA, et al. Strong inflammatory response and Th1‐polarization profile in children with acute lymphoblastic leukemia without apparent infection. Oncol Rep. 2016;35:2699‐2706. [DOI] [PubMed] [Google Scholar]
- 43. Vilchis‐Ordonez A, Contreras‐Quiroz A, Vadillo E, et al. Bone marrow cells in acute lymphoblastic leukemia create a proinflammatory microenvironment influencing normal hematopoietic differentiation fates. Biomed Res Int. 2015;2015:386165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Traxel S, Schadt L, Eyer T, et al. Bone marrow T helper cells with a Th1 phenotype induce activation and proliferation of leukemic cells in precursor B acute lymphoblastic leukemia patients. Oncogene. 2019;38:2420‐2431. [DOI] [PubMed] [Google Scholar]
- 45. Agyeman P, Kontny U, Nadal D, et al. A prospective multicenter study of microbiologically defined infections in pediatric cancer patients with fever and neutropenia: Swiss Pediatric Oncology Group 2003 fever and neutropenia study. Pediatr Infect Dis J. 2014;33:e219‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Khurana M, Lee B, Feusner JH. Fever at diagnosis of pediatric acute lymphoblastic leukemia: Are antibiotics really necessary? J Pediatr Hematol Oncol. 2015;37:498‐501. [DOI] [PubMed] [Google Scholar]
- 47. Smith KM, Pottage L, Thomas ER, et al. Th1 and Th2 CD4+ T cells provide help for B cell clonal expansion and antibody synthesis in a similar manner in vivo. J Immunol. 2000;165:3136‐3144. [DOI] [PubMed] [Google Scholar]
- 48. Bollard CM, Barrett AJ. Cytotoxic T lymphocytes for leukemia and lymphoma. Hematology. 2014;2014:565‐569. [DOI] [PubMed] [Google Scholar]
- 49. Homet Moreno B, Ribas A. Anti‐programmed cell death protein‐1/ligand‐1 therapy in different cancers. Br J Cancer. 2015;112:1421‐1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mittal D, Vijayan D, Smyth MJ. Overcoming Acquired PD‐1/PD‐L1 Resistance with CD38 Blockade. Cancer Discov. 2018;8:1066‐1068. [DOI] [PubMed] [Google Scholar]
- 51. Dufva O, Polonen P, Bruck O, et al. Immunogenomic Landscape of Hematological Malignancies. Cancer Cell. 2020;38(3):380‐399.e13. [DOI] [PubMed] [Google Scholar]
- 52. Kohnke T, Krupka C, Tischer J, Knosel T, Subklewe M. Increase of PD‐L1 expressing B‐precursor ALL cells in a patient resistant to the CD19/CD3‐bispecific T cell engager antibody blinatumomab. J Hematol Oncol. 2015;8:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Feucht J, Kayser S, Gorodezki D, et al. T‐cell responses against CD19+ pediatric acute lymphoblastic leukemia mediated by bispecific T‐cell engager (BiTE) are regulated contrarily by PD‐L1 and CD80/CD86 on leukemic blasts. Oncotarget. 2016;7:76902‐76919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu CP, Qing X, Wu CY, Zhu H, Zhou HY. Immunophenotype and increased presence of CD4(+)CD25(+) regulatory T cells in patients with acute lymphoblastic leukemia. Oncol Lett. 2012;3:421‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Spidlen J, Breuer K, Rosenberg C, Kotecha N, Brinkman RR. FlowRepository: a resource of annotated flow cytometry datasets associated with peer‐reviewed publications. Cytometry A. 2012;81:727‐731. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Fig S4
Table S1
Table S2
Table S3