

Abstract
Nuclear-import receptors (NIRs) engage nuclear-localization signals (NLSs) of polypeptides in the cytoplasm and transport these cargo across the size-selective barrier of the nuclear-pore complex into the nucleoplasm. Beyond this canonical role in nuclear transport, NIRs operate in the cytoplasm to chaperone and disaggregate NLS-bearing clients. Indeed, NIRs can inhibit and reverse functional and deleterious phase transitions of their cargo, including several prominent neurodegenerative disease-linked RNA-binding proteins (RBPs) with prion-like domains (PrLDs), such as TDP-43, FUS, EWSR1, TAF15, hnRNPA1, and hnRNPA2. Importantly, elevated NIR expression can mitigate degenerative phenotypes connected to aberrant cytoplasmic aggregation of RBPs with PrLDs. Here, we review recent discoveries that NIRs can also antagonize aberrant interactions and toxicity of arginine-rich, dipeptide-repeat proteins that are associated with amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) caused by G4C2 hexanucleotide repeat expansions in the first intron of C9ORF72. We also highlight recent findings that multiple NIR family members can prevent and reverse liquid-liquid phase separation of specific clients bearing RGG motifs in an NLS-independent manner. Finally, we discuss strategies to enhance NIR activity or expression, which could have therapeutic utility for several neurodegenerative disorders, including ALS, FTD, multisystem proteinopathy, limbic-predominant age-related TDP-43 encephalopathy, tauopathies, and related diseases.
Graphical Abstract
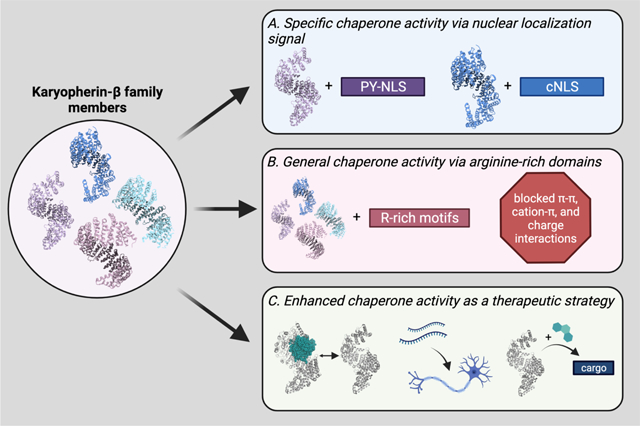
Kapβ family members as chaperones and dissolvases. Kapβ proteins are well-known for their roles in facilitating nucleocytoplasmic transport. However, they are also powerful chaperones, able to prevent and reverse aberrant material states of disease-relevant proteins, including those implicated in neurodegenerative disease. Kapβ family members can act as chaperones via a number of mechanisms. (A) The most well-understood manner in which Kapβ proteins interact with their cargo is through direct recognition of a NLS. For example, Kapβ2 recognizes proteins with a PY-NLS (purple), whereas Kapβ1 collaborates with a Kapα protein to recognize proteins with a classical NLS (blue). These interactions enable potent disaggregation of cargo. (B) A less specific means of recognition is via Kapβ proteins binding to arginine-rich motifs. Here, multiple Kapβ proteins can interact with cargo independent of any NLS, and in so doing prevent arginine residues from making π-π, cation-π, or electrostatic interactions that underlie the formation of higher-order assemblies. This mode of recognition also enables Kapβs to further regulate FUS self-assembly and shield R-DPRs from engaging in aberrant interactions. (C) As we learn more about how Kapβ proteins can act as chaperones, we may be able to design therapeutic techniques with the goal of enhancing their activity. This enhancement could be achieved by increasing the rate of cargo release in the nucleus, increasing Kapβ expression, or by using small molecules to enhance cargo binding. (PDBs used: 4XRK [Importin-β; dark blue]; 3ZKV [Importin 13; light blue]; 4C0P [Transportin 3; pink]; 4FDD [Karyopherin-β2; light purple, gray]; 1QBK [Karyopherin-β2 bound to Ran; gray and teal]). This figure was made with BioRender.
Introduction
A distinctive feature of several fatal and presently incurable neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), multisystem proteinopathy (MSP), and limbic-predominant age-related TDP-43 encephalopathy (LATE), is the depletion of specific RNA-binding proteins (RBPs) with prion-like domains (PrLDs) from the nucleus and their accumulation in cytoplasmic aggregates in degenerating neurons [1–8]. For example, TDP-43 exhibits this pathological phenotype in ~97% of ALS cases and ~50% of FTD cases, whereas another RBP with a PrLD, FUS, displays nuclear depletion and cytoplasmic aggregation in ~1% of ALS cases and ~9% of FTD cases [1, 8]. It is suggested that the loss of nuclear function of these RBPs coupled to a gain of toxic function due to cytoplasmic accumulation and aggregation may synergize to elicit neurodegeneration [1, 6, 8–10]. We have suggested that agents that reverse the cytoplasmic mislocalization and aggregation of these RBPs and restore their nuclear localization and function could be powerful therapeutics [6, 11–18]. Remarkably, nuclear-import receptors (NIRs) have emerged as agents capable of eliciting such a therapeutic effect [16, 19].
NIRs are members of the karyopherin family of proteins that bind tightly to nuclear-localization signals (NLSs) of polypeptide cargo in the cytoplasm [20–22]. NIRs are subdivided into a small family of karyopherin-α (Kapα) proteins, which engage classical NLSs and subsequently bind to a member of the larger karyopherin-β (Kapβ) family [22]. However, Kapβs can directly recognize non-classical NLSs (e.g. proline tyrosine [PY]-NLSs) independent of Kapαs [22]. Kapβs are flexible, superhelical proteins that typically comprise ~20 consecutive HEAT (Huntingtin, elongation factor 3 (EF3), protein phosphatase 2A (PP2A), and the yeast kinase TOR1) repeats, a type of protein tandem repeat structural motif composed of two alpha helices linked by a short loop (Figure 1A) [22]. Once bound to the NLS, NIRs can transport their cargo across the nuclear-pore complex (NPC) and into the nucleoplasm [22].
Figure 1. Specific and general interactions enable NIRs to chaperone cargo.
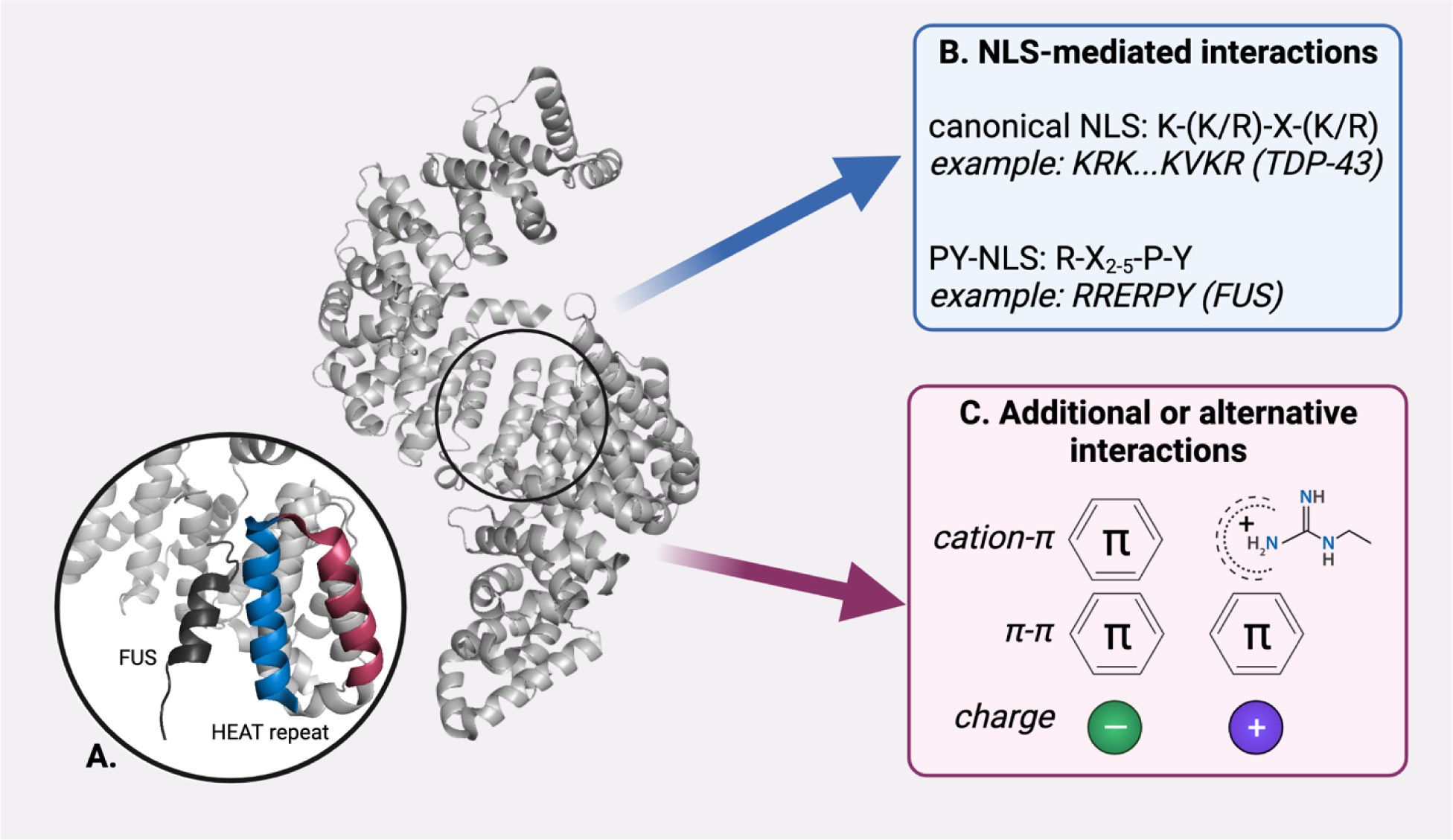
(A) NIRs like Kapβ2 (PDB: 4FDD) share a similar structure, with roughly 20 paired helical HEAT repeats coiled into an alpha solenoid structure. HEAT repeats comprise an outer helix (raspberry) and an inner, cargo-facing helix (blue; FUS-PY-NLS cargo, black). NIRs interact with cargo via two non-exclusive mechanisms. (B) Many NIRs will specifically interact with cargo bearing an NLS. For example, Kapβ2 (PDB: 4FDD) will interact with cargo bearing a PY-NLS, like FUS and hnRNPA1. (C) NIRs can also bind to cargo through additional or alternative interactions, including cation-π, π-π, and electrostatic interactions. Such interactions can occur with cargo that bears an NLS, and with cargo that has no known NLS sequence, such as the arginine-rich DPRs produced in c9ALS/FTD.
The NPC is an intricate structure [23] which operates as a size-selective barrier to prevent macromolecules with a molecular weight greater than ~30 kDa (or a Stokes radius greater than ~3 nm) from passively diffusing in and out of the nucleus [24]. This barrier is established by FG-rich nucleoporins, which form a phase-separated state inside the NPC channel [25–28]. NIRs can penetrate rapidly through this phase and transport cargo into the nucleus [29]. Once inside the nucleoplasm, the small GTPase Ran in its GTP-bound state binds to the incoming NIR, dissociating the NIR-cargo complex [22]. Cargo is thus released into the nucleus where a high concentration of RNA keeps the incoming RBP soluble so it can perform its regular function, and the NIR is recycled for further rounds of nuclear transport [22, 30–32]. By contrast, in the cytoplasm Ran is found in the GDP-bound state, which has a low affinity for NIRs, permitting NIR-cargo interactions [22].
Beyond this classical function in nuclear transport, NIRs are now understood to operate in the cytoplasm to chaperone and disaggregate NLS-bearing clients [19, 33–43]. In this context, NIRs engage cognate NLSs to inhibit and reverse physiological and deleterious phase transitions of their cargo (Figure 1B), which includes several prominent neurodegenerative disease-linked RBPs with PrLDs, including wild-type and disease-linked mutant forms of TDP-43, FUS, EWSR1, TAF15, hnRNPA1, and hnRNPA2 [19, 33–44]. For example, Karyopherin-β2 (Kapβ2; also known as Transportin 1) can prevent and reverse fibrillization of wild-type FUS, EWSR1, TAF15, hnRNPA1, hnRNPA2, and several disease-linked variants [19]. Kapβ2 also prevents and reverses FUS liquid-liquid phase separation (LLPS) [19, 33, 34, 36]. Moreover, Importin α (Impα) and Kapβ1 (also known as importin β) cooperate to prevent and reverse TDP-43 condensation and fibrillization [19, 37]. Importantly, elevated NIR expression can mitigate degenerative phenotypes connected with aberrant aggregation of RBPs with PrLDs in model systems [19, 34, 45]. Indeed, NIRs can disaggregate cytoplasmic inclusions formed by RBPs with PrLDs and return these proteins to the nucleus, thereby restoring their native function [19]. In this way, NIRs may simultaneously eliminate: (1) any gain of toxic function due to cytoplasmic mislocalization and aggregation of the RBP; and (2) any loss of nuclear RBP function, two facets of disease that likely combine to drive neurodegeneration [19]. Thus, NIRs join a growing class of ATP-independent protein disaggregases [14, 46–49]. These exciting advances have been reviewed in detail elsewhere [16–18, 20, 50–52].
In this review, we focus on recent developments concerning how NIRs can antagonize aberrant interactions and toxicity of dipeptide-repeat proteins (DPRs) that are produced via repeat-associated non-AUG (RAN) translation [53] of the G4C2 hexanucleotide repeat expansions (HRE) in C9ORF72 that cause ALS and FTD [37, 54]. We also highlight recent findings that multiple NIR family members can prevent and reverse the LLPS of specific cargo bearing RGG motifs [35]. In ALS/FTD and related degenerative disorders, NIRs can be mutated [55], expressed at lower levels [52, 56], sequestered in stress granules [57] and aggregated structures [52, 58–60], or fail to effectively recognize post-translationally modified cargo [61] or disease-linked mutant NLSs [19, 62–68]. Moreover, NIRs are critical for neuronal maintenance and function, and mutations in NIRs are associated with human developmental delays, neurologic deficits, and dysmorphic features [69]. Thus, we close the review by discussing strategies to enhance NIR activity or expression, which could have therapeutic utility for several presently untreatable disorders [16].
NIRs as safeguards against toxic DPRs
A large G4C2 HRE in the first intron of the C9ORF72 gene is the most common genetic cause of ALS and FTD (termed c9ALS/FTD) [10, 70–73]. Patients with c9ALS/FTD can have hundreds to thousands of G4C2 repeats in the first intron of C9ORF72, whereas healthy individuals typically harbor ~ 2–23 repeats [10, 70–74]. In c9ALS/FTD, the G4C2 HRE is bidirectionally transcribed into toxic repeat RNAs, which are RAN-translated to yield five different DPRs: poly(GA), poly(GP), poly(PR), poly(GR), and poly(PA) [71, 75–77]. In c9ALS/FTD models, arginine-rich DPRs (R-DPRs) are particularly toxic to neurons due to their positive charge and wide range of interacting partners [78–81]. More specifically, poly(GR) and poly(PR) can directly interact with the PrLD-containing RBP TDP-43, altering its phase-separation behavior and accelerating its aggregation both in vitro and in cells [37, 82]. In fact, poly(GR) and poly(PR) are notorious for their ability to disrupt the LLPS of multiple RBPs through interactions with low-complexity domains (LCDs), and their ability to disturb the dynamics of several membraneless organelles, including stress granules (SGs), nucleoli, nuclear speckles, Cajal bodies, and heterochromatin [71, 80, 81].
Given the high affinity of NIRs to arginine- or lysine-rich NLSs, it was postulated that NIRs might also target R-DPRs [37]. Several NIRs were in fact identified as modulators of R-DPR toxicity in c9ALS/FTD models, suggesting a mechanistic link between NIRs and R-DPRs [54, 83–85]. More recent studies found that R-DPRs directly interact with multiple NIRs, including Imp⍺, Kapβ1, and Kapβ2, causing an interruption in nucleocytoplasmic trafficking [37, 86]. Specifically, high concentrations of R-DPRs promote the insolubility of NIRs, disrupting their ability to bind and import their NLS-containing cargo [37, 86]. As such, this mechanism provides an explanation for why TDP-43 nuclear import deteriorates in c9ALS/FTD [37].
With the direct link between DPRs and NIRs now revealed, can we steer the chaperoning power of NIRs to combat R-DPR-associated toxicity? R-DPRs undergo RNA-stimulated phase separation [87], which likely leads to R-DPR accumulation in aggregated structures in c9ALS/FTD [76, 77]. Importantly, Kapβ1 and Kapβ2 suppress RNA-stimulated poly(GR) condensation, whereas Impα3 was ineffective [37]. Interestingly, while R-DPRs in molar excess can directly interact with NIRs and impair TDP-43 nuclear import, equimolar or elevated levels of Kapβ1 or Kapβ2 can shield R-DPRs, thereby suppressing their pathological interactions with TDP-43 [37]. Thus, increasing the concentration of NIRs prevents R-DPR phase separation, prevents R-DPRs from engaging in deleterious interactions, and also restores the nuclear localization of TDP-43 as it becomes available to interact with its own NIRs [37]. These findings suggest that the reported reduction in the endogenous concentrations of NIRs associated with neurodegenerative disease may contribute to the pathogenesis of c9ALS/FTD, and that interventions aiming to elevate NIR levels are a promising therapeutic strategy to combat R-DPR toxicity in c9ALS/FTD [52].
Besides R-DPRs, another c9ALS/FTD-linked DPR, poly(GA), as well as chimeric DPR species such as GA:GP can form cytoplasmic inclusions that may inhibit nuclear import of TDP-43 [88, 89]. Indeed, in hippocampal neurons, poly(GA) expression results in robust TDP-43 cytoplasmic mislocalization [88]. However, overexpression of Impα3 or Impα4, which may be involved in nuclear import of TDP-43 [56, 90], can likely restore TDP-43 to the nucleus [88]. Other repeat-expansion disorders can also produce DPRs, such as spinocerebellar ataxia 36 (SCA36), which presents with poly(PR) and poly(GP) [89, 91]. Thus, NIRs may also be promising therapeutics to mitigate DPR toxicity in SCA36.
Multiple NIRs exhibit chaperone and disaggregation activity
The family of Kapβ proteins is large, containing over a dozen subfamilies [92]. These proteins vary in their structure, directionality of transport (nuclear-export factors such as Crm1 are also members of the Kapβ family), and cargo repertoire [92]. And, whereas some cargo proteins show a clear preference for a single NIR, others can be ferried by multiple karyopherins, either individually or in concert with one another [93–99]. In addition to trafficking cargo proteins in and out of the nucleus, Kapβ family members like Kapβ2 can also act to prevent and reverse the self-assembly and aggregation of proteins, including those implicated in neurodegenerative disease [16]. However, it had been an unexplored question as to whether this disaggregation activity was a feature of karyopherin proteins in general, or an exclusive capability of only some. Focusing on the disease-associated RBP FUS, recent work from Baade et al. now establishes that multiple Kapβ family members can act as potent chaperones both in vitro and in cells [35].
To uncover the network of Kapβ proteins that FUS interacts with, Baade et al. performed pull-down assays using cell lysates or purified proteins and found that, in addition to Kapβ2, FUS also binds to Kapβ1, transportin-3, importin-7, importin-13, and exportin-4 [35]. Like Kapβ2, transportin-3, and importin-7 (either on its own or as a heterodimer with Kapβ1) interact stably with FUS, forming complexes that were sensitive to the addition of Ran-GTP [35]. Although these complexes were stable, of the NIRs tested, Kapβ2 demonstrated the strongest binding to FUS [35]. It is well established that Kapβ2 binds to FUS via its PY-NLS [62], but it was unclear if these other NIRs were interacting with FUS in the same way. Thus, Baade and colleagues assessed the binding of each NIR to truncated constructs of FUS [35]. They found that instead of interacting with the PY-NLS, Kapβ1, transportin-3, importin-7, and Kapβ1/importin-7 interact with FUS via its arginine- and glycine-rich RGG domains (Figure 1C) [35]. Interestingly, when the arginine residues of the RGG domains were mutated to lysine, the binding of Kapβ1, transportin-3, importin-7, and Kapβ1/importin-7 was impaired, underscoring the specific importance of arginine in mediating the interaction between Kapβ proteins and their cargo [35, 36, 40, 100].
Not only do multiple Kapβ proteins bind to FUS, but they can also chaperone its material state in vitro and in cells [35]. In the absence of any NIR, purified FUS protein will undergo LLPS [19, 33, 37, 101]. Addition of equimolar levels of Kapβ1, transportin-3, importin 7, or Kapβ1/importin-7 each prevented and reversed FUS LLPS, indicating that these NIRs both bind to and chaperone FUS [35]. Baade et al. also observed this chaperoning activity in cells, where addition of any of the NIRs tested was able to suppress the association of exogenous MBP-FUS with stress granules [35]. The work from Baade and colleagues illustrates that FUS self-assembly can be modulated by multiple Kapβ proteins and suggests that the mechanism by which this chaperone activity occurs may be a universal feature of Kapβ family members [35]. Although Kapβ1, transportin-3, importin 7, or Kapβ1/importin-7 can prevent and reverse FUS LLPS [35], it remains unclear whether Kapβ1, transportin-3, importin 7, or importin β/7 can reverse the formation of FUS fibrils like Kapβ2 [19]. Kapβ2 interacts more avidly with FUS than the other NIRs, which may confer stronger FUS chaperone and nuclear-import activity [35]. Further studies into the many Kapβ family members and their respective abilities to chaperone disease-related cargo proteins will therefore be an intriguing area for future research.
Of particular interest is Transportin-3. Intriguingly, mutations in transportin-3 have been connected to congenital limb-girdle myopathy [55, 102–107], indicating that transportin-3 is critical for human health. Transportin-3 binds to multiple proteins with RGG-motifs, including the cold-inducible RNA-binding protein which is involved in responding to cell stress [100]. Transportin-3 has also been shown to chaperone another disease-related cargo, the arginine-rich nuclear-speckle protein SRRM2 [108, 109]. In tauopathies, including FTD and Alzheimer’s disease, tau disrupts nuclear speckles and sequesters SRRM2 in cytoplasmic inclusions, which reduces SRRM2 splicing activity [110, 111]. Thus, upregulation of transportin-3 might enable extraction of SRRM2 from cytoplasmic tau aggregates and restore SRRM2 to the nucleus, which may mitigate degenerative phenotypes in tauopathies [112].
NIRs also affect phase transitions in physiological contexts. For example, Impα/Kapβ1 regulate the material state of Targeting Protein for XKlp2 (TPX2), a spindle-assembly factor whose activity must be tightly regulated for proper cell cycle progression [44, 113]. TPX2 condensation promotes its activity, whereas Impα and Kapβ1, either alone or combined, inhibit TPX2 condensation through a combination of specific binding and general low-affinity interactions [44, 113].
Interestingly, there are NIR family members that bind to cargo with no defined consensus NLS. For example, Transportin-3 and importin 13 can each bind dozens of cargo proteins with no annotated NLS [114]. Surprisingly, despite their close similarity, Transportin-3 and importin 13 have little overlap in which cargo they recognize, and for each NIR, the cargo vary in terms of sequence and structure [114]. Hence, there is yet much to uncover with respect to potential NIR:cargo interactions.
Leveraging NIRs as therapeutic agents
As newly appreciated dissolvases [18], Kapβ family members represent an exciting target to pursue in developing drugs for diseases related to the adoption of aberrant material states by various proteins [16]. There already exist several compounds that affect Kapβs, however these mainly work to prevent activity by occupying the cargo-binding surface of their target [115]. As such, there is an opening to develop compounds that can stimulate Kapβ activity (Figure 2A, i).
Figure 2. Therapeutic strategies to enhance NIR activity.
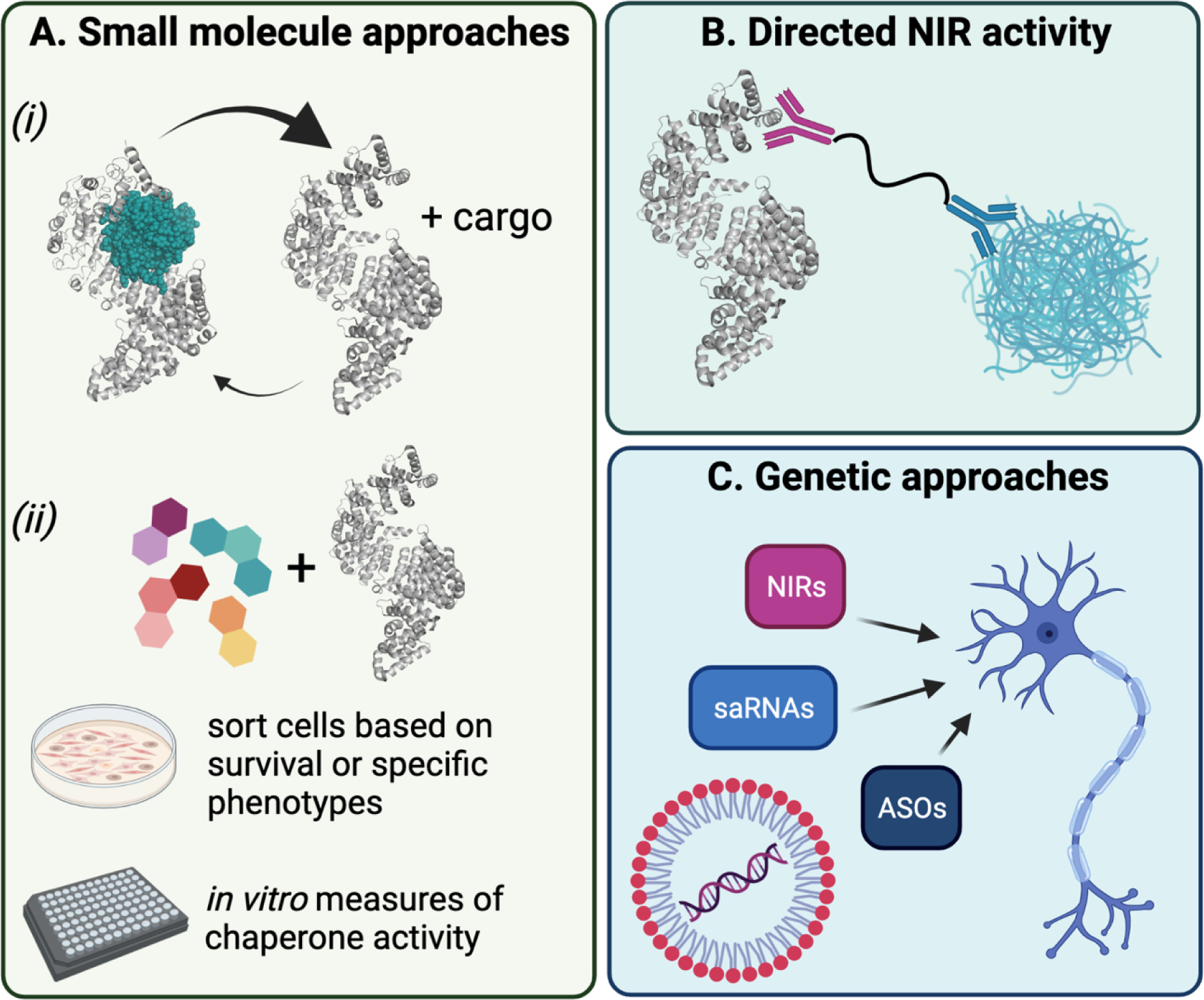
(A) One potential means by which NIR activity could be enhanced is through the use of small-molecule drugs. For example, a molecule that promotes the release of Ran-GTP (shown in teal) from NIRs in the nucleus would increase the pool of free NIRs available for binding to cargo (i). Alternatively, small molecule libraries or engineered compounds could be used to perform in vivo or in vitro screens for chaperone activity (ii). (B) Another approach could use a PROTAC-like molecule to direct NIRs to specified targets. (C) Finally, NIR activity could be augmented using genetic approaches to deliver saRNAs or ASOs to elevate NIR expression, or to deliver sequences via AAVs or lipid-containing nanoparticles to express NIRs directly.
What would such compounds look like? One approach could be to target disassembly of Ran-GTP:Kapβ complexes in the nucleus to promote Kapβ turnover [116]. Increasing the efficiency with which free Kapβ proteins are made available would increase the relative levels of free Kapβ, thereby potentially increasing their apparent activity.
There is also the opportunity to discover novel small molecules to enhance Kapβ activity directly (Figure 2A, ii). To this end, employing a high-throughput screening approach to identify compounds that stimulate Kapβ activity would be an exciting avenue to pursue. Such a screen could be done in vitro using purified proteins. For example, it will be of great interest to screen for drug-like compounds that enhance the ability of Kapβ2 to prevent or reverse FUS fibrillization or Impα/Kapβ1 to prevent and reverse TDP-43 fibrillization. Likewise, screening campaigns might also be considered in cell-based models to find drug-like compounds that enhance the ability of NIRs to restore nuclear localization of TDP-43 or FUS in response to stress or disease conditions.
Several structures of Kapβ2 bound to disease-linked cargo, including hnRNPA1, FUS, and ALS-linked FUS variants are available [40, 62, 117]. These structures could facilitate the design of small molecules that increase the affinity of Kapβ2 for these specific cargo. This approach may be particularly important for disease-linked hnRNPA1 and FUS variants with mutations in the PY-NLS that weaken the interaction with Kapβ2 [62, 67, 68]. Ideally, compounds could be uncovered that restore the affinity of Kapβ2 for disease-linked cargo to similar levels observed with wild-type cargo. Although structures of Impα/Kapβ1 bound to the TDP-43 NLS are not yet available, other structures of Impα family members bound to cargo have been solved [118–123], and these could also inform drug design. Here, it will be important to ensure that small-molecules do not make NIR-cargo interactions so tight that they cannot be dissociated by Ran-GTP, as release of cargo is essential for restoring nuclear activity and NIR recycling [16].
Small-molecule drugs that increase the expression of specific NIRs or even globally upregulate NIRs may also be an interesting therapeutic strategy. How NIR expression is regulated in response to nuclear-transport stress remains poorly understood and further studies in this area are likely to be informative. For example, it would be important to uncover compounds that globally upregulate nuclear-transport pathways akin to compounds that induce the heat-shock response to upregulate a battery of molecular chaperones [124–130]. Such compounds could provide a critical boost to nuclear transport to combat neurodegeneration.
Another strategy could resemble recent work with proteolysis-targeting chimeras (PROTACs) [131]. PROTACs are small molecules that comprise a moiety that targets an E3-ubiquitin ligase tethered to a recruiting compound that binds to a protein of interest [131]. Thus, PROTACs enable specific ubiquitination and subsequent degradation of a target protein, and have been successfully used in a wide range of applications [131]. However, in ALS/FTD, the disease-relevant proteins TDP-43 and FUS serve vital roles in biology, including in RNA metabolism, axonal transport, and responding to DNA damage and other stressors [6, 8, 34, 132–136]. A PROTAC-type model that disaggregates but does not degrade its target protein is therefore a potential adaptation well suited to Kapβ activity. Here, instead of an E3 ligase, Kapβ proteins could be utilized to target phase-separated and aggregated proteins, including those with a mutated NLS or no NLS at all (Figure 2B). In promoting a locally high concentration of NIRs, this strategy would liberate the target protein from phase-separated assemblies, allowing it to resume its normal activities.
In addition to a compound-based treatment, genetic approaches are also becoming an increasingly tractable strategy for preventing or reversing disease states (Figure 2C). For example, delivery of specific NIRs to neurons could be achieved using adeno-associated viruses (AAVs) [137, 138]. After a single administration of AAV, neurons can be successfully transduced, enabling stable expression of a therapeutic gene of interest [139]. The safety of this approach has been established in numerous clinical trials for neurodegenerative diseases [140–143] and AAVs delivering specific genes are now FDA-approved drugs [144, 145]. Nonetheless, caution is still needed [146, 147].
Additional approaches for gene delivery might also be considered, including lipid-containing nanoparticle-mediated delivery of chemically-modified mRNAs to afflicted neurons akin to the technology that has produced highly effective mRNA vaccines (Figure 2C) [15, 148, 149]. Antisense oligonucleotides (ASOs) have also been used to increase the expression of certain proteins by facilitating specific splicing events for productive gene expression (Figure 2C) [150, 151]. Alternatively, small-activating RNAs (saRNAs) can be used to activate the expression of target genes with the RNA-induced transcriptional activation complex (Figure 2C) [152, 153]. These approaches could be used to supplement production of NIRs, which undergo age- and disease-related changes in expression levels [52, 56, 154].
Perspectives
It is now clear that NIRs deliver a range of beneficial chaperone and dissolvase activities that could, if channeled appropriately, provide profound therapeutic effects for several presently fatal neurodegenerative disorders, including ALS, FTD, MSP, LATE, and tauopathies [16, 112].
However, several challenges lie ahead. These include ensuring that elevating or enhancing NIR activity does not result in unanticipated off-target effects or undesirable effects such as disturbing the nuclear:cytoplasmic ratio of various cargo in a manner that is detrimental to cell viability. Likewise, some cytoplasmic condensates formed by RBPs with PrLDs, such as TDP-43 myogranules [155] or RNA-transport granules [34, 135, 156, 157] serve beneficial functions, which ideally would not be perturbed by elevated NIR activity. Indeed, it will be important to disperse pathological condensates and simultaneously preserve beneficial condensates. Despite these challenges, the ability of NIRs to reverse the cytoplasmic mislocalization and aggregation of specific cargo and restore their nuclear localization and function could enable the development of powerful therapeutics, which warrants intense investigation.
Research Highlights.
Nuclear-import receptors (NIRs) are potent chaperones for their NLS-bearing cargo
NIRs prevent and reverse deleterious phase transitions of disease-linked cargo
NIRs prevent toxicity of arginine-rich, dipeptide-repeat proteins
Multiple NIR family members can prevent and reverse FUS phase separation
Enhancing NIR activity could be therapeutic for neurodegenerative disorders
Acknowledgements
We thank Katie Copley and Zarin Tabassum for feedback on the manuscript. H.M.O. is supported by an AstraZeneca post-doctoral fellowship. C.M.F. is supported by National Institutes of Health (NIH) grants T32GM008275 and F31NS111870. J.S. is supported by grants from Target ALS, The Association for Frontotemporal Degeneration, the Amyotrophic Lateral Sclerosis Association, the Office of the Assistant Secretary of Defense for Health Affairs through the Amyotrophic Lateral Sclerosis Research Program (W81XWH-20-1-0242 and W81XWH-17-1-0237), the G. Harold and Leila Y. Mathers Foundation, Sanofi, and NIH grants R01GM099836, R21AG061784, R21AG065854, and R21NS090205.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
H.M.O. and C.M.F. have nothing to declare. J.S. is a consultant for Dewpoint Therapeutics, Maze Therapeutics, Vivid Sciences, Korro Bio, ADRx, and RBNC Therapeutics.
References
- [1].Portz B, Lee BL, Shorter J. FUS and TDP-43 Phases in Health and Disease. Trends Biochem Sci. 2021;46:550–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cushman M, Johnson BS, King OD, Gitler AD, Shorter J. Prion-like disorders: blurring the divide between transmissibility and infectivity. J Cell Sci. 2010;123:1191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].King OD, Gitler AD, Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012;1462:61–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. J Cell Biol. 2013;201:361–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].March ZM, King OD, Shorter J. Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res. 2016;1647:9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Guo L, Shorter J. Biology and Pathobiology of TDP-43 and Emergent Therapeutic Strategies. Cold Spring Harb Perspect Med. 2017;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Harrison AF, Shorter J. RNA-binding proteins with prion-like domains in health and disease. Biochem J. 2017;474:1417–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lee EB, Lee VM, Trojanowski JQ. Gains or losses: molecular mechanisms of TDP43-mediated neurodegeneration. Nat Rev Neurosci. 2011;13:38–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kim G, Gautier O, Tassoni-Tsuchida E, Ma XR, Gitler AD. ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron. 2020;108:822–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jackrel ME, Shorter J. Engineering enhanced protein disaggregases for neurodegenerative disease. Prion. 2015;9:90–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Jackrel ME, Shorter J. Protein-Remodeling Factors As Potential Therapeutics for Neurodegenerative Disease. Front Neurosci. 2017;11:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Shorter J. Hsp104: a weapon to combat diverse neurodegenerative disorders. Neurosignals. 2008;16:63–74. [DOI] [PubMed] [Google Scholar]
- [14].Shorter J. Designer protein disaggregases to counter neurodegenerative disease. Curr Opin Genet Dev. 2017;44:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shorter J. Engineering therapeutic protein disaggregases. Mol Biol Cell. 2016;27:1556–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Guo L, Fare CM, Shorter J. Therapeutic Dissolution of Aberrant Phases by Nuclear-Import Receptors. Trends Cell Biol. 2019;29:308–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Darling AL, Shorter J. Combating deleterious phase transitions in neurodegenerative disease. Biochim Biophys Acta Mol Cell Res. 2021;1868:118984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fare CM, Shorter J. (Dis)Solving the problem of aberrant protein states. Dis Model Mech. 2021;14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Guo L, Kim HJ, Wang H, Monaghan J, Freyermuth F, Sung JC, et al. Nuclear-Import Receptors Reverse Aberrant Phase Transitions of RNA-Binding Proteins with Prion-like Domains. Cell. 2018;173:677–92 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Springhower CE, Rosen MK, Chook YM. Karyopherins and condensates. Curr Opin Cell Biol. 2020;64:112–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chook YM, Suel KE. Nuclear import by karyopherin-betas: recognition and inhibition. Biochim Biophys Acta. 2011;1813:1593–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Soniat M, Chook YM. Nuclear localization signals for four distinct karyopherin-beta nuclear import systems. Biochem J. 2015;468:353–62. [DOI] [PubMed] [Google Scholar]
- [23].Allegretti M, Zimmerli CE, Rantos V, Wilfling F, Ronchi P, Fung HKH, et al. In-cell architecture of the nuclear pore and snapshots of its turnover. Nature. 2020;586:796–800. [DOI] [PubMed] [Google Scholar]
- [24].Mohr D, Frey S, Fischer T, Guttler T, Gorlich D. Characterisation of the passive permeability barrier of nuclear pore complexes. EMBO J. 2009;28:2541–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Frey S, Gorlich D. A saturated FG-repeat hydrogel can reproduce the permeability properties of nuclear pore complexes. Cell. 2007;130:512–23. [DOI] [PubMed] [Google Scholar]
- [26].Frey S, Richter RP, Gorlich D. FG-rich repeats of nuclear pore proteins form a three-dimensional meshwork with hydrogel-like properties. Science. 2006;314:815–7. [DOI] [PubMed] [Google Scholar]
- [27].Labokha AA, Gradmann S, Frey S, Hulsmann BB, Urlaub H, Baldus M, et al. Systematic analysis of barrier-forming FG hydrogels from Xenopus nuclear pore complexes. EMBO J. 2013;32:204–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Celetti G, Paci G, Caria J, VanDelinder V, Bachand G, Lemke EA. The liquid state of FG-nucleoporins mimics permeability barrier properties of nuclear pore complexes. J Cell Biol. 2020;219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Frey S, Rees R, Schunemann J, Ng SC, Funfgeld K, Huyton T, et al. Surface Properties Determining Passage Rates of Proteins through Nuclear Pores. Cell. 2018;174:202–17 e9. [DOI] [PubMed] [Google Scholar]
- [30].Maharana S, Wang J, Papadopoulos DK, Richter D, Pozniakovsky A, Poser I, et al. RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science. 2018;360:918–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mann JR, Gleixner AM, Mauna JC, Gomes E, DeChellis-Marks MR, Needham PG, et al. RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron. 2019;102:321–38 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yu H, Lu S, Gasior K, Singh D, Vazquez-Sanchez S, Tapia O, et al. HSP70 chaperones RNA-free TDP-43 into anisotropic intranuclear liquid spherical shells. Science. 2021;371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Yoshizawa T, Ali R, Jiou J, Fung HYJ, Burke KA, Kim SJ, et al. Nuclear Import Receptor Inhibits Phase Separation of FUS through Binding to Multiple Sites. Cell. 2018;173:693–705 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Qamar S, Wang G, Randle SJ, Ruggeri FS, Varela JA, Lin JQ, et al. FUS Phase Separation Is Modulated by a Molecular Chaperone and Methylation of Arginine Cation-pi Interactions. Cell. 2018;173:720–34 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Baade I, Hutten S, Sternburg EL, Porschke M, Hofweber M, Dormann D, et al. The RNA-binding protein FUS is chaperoned and imported into the nucleus by a network of import receptors. J Biol Chem. 2021;296:100659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hofweber M, Hutten S, Bourgeois B, Spreitzer E, Niedner-Boblenz A, Schifferer M, et al. Phase Separation of FUS Is Suppressed by Its Nuclear Import Receptor and Arginine Methylation. Cell. 2018;173:706–19 e13. [DOI] [PubMed] [Google Scholar]
- [37].Hutten S, Usluer S, Bourgeois B, Simonetti F, Odeh HM, Fare CM, et al. Nuclear Import Receptors Directly Bind to Arginine-Rich Dipeptide Repeat Proteins and Suppress Their Pathological Interactions. Cell Rep. 2020;33:108538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Niaki AG, Sarkar J, Cai X, Rhine K, Vidaurre V, Guy B, et al. Loss of Dynamic RNA Interaction and Aberrant Phase Separation Induced by Two Distinct Types of ALS/FTD-Linked FUS Mutations. Mol Cell. 2020;77:82–94 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rhine K, Makurath MA, Liu J, Skanchy S, Lopez C, Catalan KF, et al. ALS/FTLD-Linked Mutations in FUS Glycine Residues Cause Accelerated Gelation and Reduced Interactions with Wild-Type FUS. Mol Cell. 2020;80:666–81 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gonzalez A, Mannen T, Cagatay T, Fujiwara A, Matsumura H, Niesman AB, et al. Mechanism of karyopherin-beta2 binding and nuclear import of ALS variants FUS(P525L) and FUS(R495X). Sci Rep. 2021;11:3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jakel S, Mingot JM, Schwarzmaier P, Hartmann E, Gorlich D. Importins fulfil a dual function as nuclear import receptors and cytoplasmic chaperones for exposed basic domains. EMBO J. 2002;21:377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chou CC, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F, et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci. 2018;21:228–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Miyake Y, Keusch JJ, Decamps L, Ho-Xuan H, Iketani S, Gut H, et al. Influenza virus uses transportin 1 for vRNP debundling during cell entry. Nat Microbiol. 2019;4:578–86. [DOI] [PubMed] [Google Scholar]
- [44].King MR, Petry S. Phase separation of TPX2 enhances and spatially coordinates microtubule nucleation. Nat Commun. 2020;11:270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jackel S, Summerer AK, Thommes CM, Pan X, Voigt A, Schulz JB, et al. Nuclear import factor transportin and arginine methyltransferase 1 modify FUS neurotoxicity in Drosophila. Neurobiol Dis. 2015;74:76–88. [DOI] [PubMed] [Google Scholar]
- [46].Jaru-Ampornpan P, Shen K, Lam VQ, Ali M, Doniach S, Jia TZ, et al. ATP-independent reversal of a membrane protein aggregate by a chloroplast SRP subunit. Nat Struct Mol Biol. 2010;17:696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Huang L, Agrawal T, Zhu G, Yu S, Tao L, Lin J, et al. DAXX represents a new type of protein-folding enabler. Nature. 2021;doi: 10.1038/s41586-021-03824-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhu G, Harischandra DS, Ghaisas S, Zhang P, Prall W, Huang L, et al. TRIM11 Prevents and Reverses Protein Aggregation and Rescues a Mouse Model of Parkinson’s Disease. Cell Rep. 2020;33:108418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Baker JD, Shelton LB, Zheng D, Favretto F, Nordhues BA, Darling A, et al. Human cyclophilin 40 unravels neurotoxic amyloids. PLoS Biol. 2017;15:e2001336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Yoshizawa T, Guo L. Karyopherin-betas play a key role as a phase separation regulator. J Biochem. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Mikhaleva S, Lemke EA. Beyond the Transport Function of Import Receptors: What’s All the FUS about? Cell. 2018;173:549–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Pasha T, Zatorska A, Sharipov D, Rogelj B, Hortobagyi T, Hirth F. Karyopherin abnormalities in neurodegenerative proteinopathies. Brain. 2021;doi: 10.1093/brain/awab201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci USA. 2011;108:260–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci. 2015;18:1226–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Vihola A, Palmio J, Danielsson O, Penttila S, Louiselle D, Pittman S, et al. Novel mutation in TNPO3 causes congenital limb-girdle myopathy with slow progression. Neurol Genet. 2019;5:e337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Nishimura AL, Zupunski V, Troakes C, Kathe C, Fratta P, Howell M, et al. Nuclear import impairment causes cytoplasmic trans-activation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain. 2010;133:1763–71. [DOI] [PubMed] [Google Scholar]
- [57].Zhang K, Daigle JG, Cunningham KM, Coyne AN, Ruan K, Grima JC, et al. Stress Granule Assembly Disrupts Nucleocytoplasmic Transport. Cell. 2018;173:958–71 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Solomon DA, Stepto A, Au WH, Adachi Y, Diaper DC, Hall R, et al. A feedback loop between dipeptide-repeat protein, TDP-43 and karyopherin-alpha mediates C9orf72-related neurodegeneration. Brain. 2018;141:2908–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Troakes C, Hortobagyi T, Vance C, Al-Sarraj S, Rogelj B, Shaw CE. Transportin 1 colocalization with Fused in Sarcoma (FUS) inclusions is not characteristic for amyotrophic lateral sclerosis-FUS confirming disrupted nuclear import of mutant FUS and distinguishing it from frontotemporal lobar degeneration with FUS inclusions. Neuropathol Appl Neurobiol. 2013;39:553–61. [DOI] [PubMed] [Google Scholar]
- [60].Neumann M, Valori CF, Ansorge O, Kretzschmar HA, Munoz DG, Kusaka H, et al. Transportin 1 accumulates specifically with FET proteins but no other transportin cargos in FTLD-FUS and is absent in FUS inclusions in ALS with FUS mutations. Acta Neuropathol. 2012;124:705–16. [DOI] [PubMed] [Google Scholar]
- [61].Dormann D, Madl T, Valori CF, Bentmann E, Tahirovic S, Abou-Ajram C, et al. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J. 2012;31:4258–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Zhang ZC, Chook YM. Structural and energetic basis of ALS-causing mutations in the atypical proline-tyrosine nuclear localization signal of the Fused in Sarcoma protein (FUS). Proc Natl Acad Sci U S A. 2012;109:12017–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A, et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010;29:2841–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Naruse H, Ishiura H, Mitsui J, Date H, Takahashi Y, Matsukawa T, et al. Molecular epidemiological study of familial amyotrophic lateral sclerosis in Japanese population by whole-exome sequencing and identification of novel HNRNPA1 mutation. Neurobiol Aging. 2018;61:255.e9–.e16. [DOI] [PubMed] [Google Scholar]
- [65].Liu Q, Shu S, Wang RR, Liu F, Cui B, Guo XN, et al. Whole-exome sequencing identifies a missense mutation in hnRNPA1 in a family with flail arm ALS. Neurology. 2016;87:1763–9. [DOI] [PubMed] [Google Scholar]
- [66].Bosco DA, Lemay N, Ko HK, Zhou H, Burke C, Kwiatkowski TJ Jr., et al. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet. 2010;19:4160–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Beijer D, Kim H, Guo L, O’Donovan K, Mademan I, Deconinck T, et al. DEFINING THE DIVERSITY OF HNRNPA1 MUTATIONS IN CLINICAL PHENOTYPE AND PATHOMECHANISM. medRxiv. 2021:2021.02.02.21250330. [Google Scholar]
- [68].Kim HJ, Mohassel P, Donkervoort S, Guo L, O’Donovan K, Coughlin M, et al. Specific heterozygous frameshift variants in hnRNPA2B1 cause early-onset oculopharyngeal muscular dystrophy. medRxiv. 2021:2021.04.08.21254942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Goodman LD, Cope H, Nil Z, Ravenscroft TA, Charng WL, Lu S, et al. TNPO2 variants associate with human developmental delays, neurologic deficits, and dysmorphic features and alter TNPO2 activity in Drosophila. Am J Hum Genet. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Odeh HM, Shorter J. Arginine-rich dipeptide-repeat proteins as phase disruptors in C9-ALS/FTD. Emerg Top Life Sci. 2020;4:293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALSFTD. Neuron. 2011;72:257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Gitler AD, Tsuiji H. There has been an awakening: Emerging mechanisms of C9orf72 mutations in FTD/ALS. Brain research. 2016;1647:19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Iacoangeli A, Al Khleifat A, Jones AR, Sproviero W, Shatunov A, Opie-Martin S, et al. C9orf72 intermediate expansions of 24–30 repeats are associated with ALS. Acta Neuropathol Commun. 2019;7:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Mori K, Arzberger T, Grässer FA, Gijselinck I, May S, Rentzsch K, et al. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol. 2013;126:881–93. [DOI] [PubMed] [Google Scholar]
- [76].Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–8. [DOI] [PubMed] [Google Scholar]
- [77].Zu T, Liu Y, Bañez-Coronel M, Reid T, Pletnikova O, Lewis J, et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc Natl Acad Sci USA. 2013;110:E4968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Mizielinska S, Grönke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science (New York, NY). 2014;345:1192–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron. 2014;84:1213–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Lee K-H, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell. 2016;167:774–88.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Zhang YJ, Guo L, Gonzales PK, Gendron TF, Wu Y, Jansen-West K, et al. Heterochromatin anomalies and double-stranded RNA accumulation underlie C9orf72 poly(PR) toxicity. Science. 2019;363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Cook CN, Wu Y, Odeh HM, Gendron TF, Jansen-West K, Del Rosso G, et al. C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Sci Transl Med. 2020;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovicic A, et al. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep. 2016;6:20877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee K-H, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525:129–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Hayes LR, Duan L, Bowen K, Kalab P, Rothstein JD. C9orf72 arginine-rich dipeptide repeat proteins disrupt karyopherin-mediated nuclear import. eLife. 2020;9:e51685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Boeynaems S, Holehouse AS, Weinhardt V, Kovacs D, Van Lindt J, Larabell C, et al. Spontaneous driving forces give rise to protein-RNA condensates with coexisting phases and complex material properties. Proc Natl Acad Sci U S A. 2019;116:7889–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Khosravi B, Hartmann H, May S, Möhl C, Ederle H, Michaelsen M, et al. Cytoplasmic poly-GA aggregates impair nuclear import of TDP-43 in C9orf72 ALS/FTLD. Hum Mol Genet. 2017;26:790–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].McEachin ZT, Gendron TF, Raj N, García-Murias M, Banerjee A, Purcell RH, et al. Chimeric Peptide Species Contribute to Divergent Dipeptide Repeat Pathology in c9ALS/FTD and SCA36. Neuron. 2020;107:292–305.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Park JH, Chung CG, Park SS, Lee D, Kim KM, Jeong Y, et al. Cytosolic calcium regulates cytoplasmic accumulation of TDP-43 through Calpain-A and Importin α3. eLife. 2020;9:e60132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Todd TW, McEachin ZT, Chew J, Burch AR, Jansen-West K, Tong J, et al. Hexanucleotide Repeat Expansions in c9FTD/ALS and SCA36 Confer Selective Patterns of Neurodegeneration In Vivo. Cell Rep. 2020;31:107616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].O’Reilly AJ, Dacks JB, Field MC. Evolution of the Karyopherin-β Family of Nucleocytoplasmic Transport Factors; Ancient Origins and Continued Specialization. PLOS ONE. 2011;6:e19308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Arnold M, Nath A, Hauber J, Kehlenbach RH. Multiple importins function as nuclear transport receptors for the Rev protein of human immunodeficiency virus type 1. J Biol Chem. 2006;281:20883–90. [DOI] [PubMed] [Google Scholar]
- [94].Baake M, Bauerle M, Doenecke D, Albig W. Core histones and linker histones are imported into the nucleus by different pathways. Eur J Cell Biol. 2001;80:669–77. [DOI] [PubMed] [Google Scholar]
- [95].Ivic N, Potocnjak M, Solis-Mezarino V, Herzog F, Bilokapic S, Halic M. Fuzzy Interactions Form and Shape the Histone Transport Complex. Mol Cell. 2019;73:1191–203 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Jakel S, Albig W, Kutay U, Bischoff FR, Schwamborn K, Doenecke D, et al. The importin beta/importin 7 heterodimer is a functional nuclear import receptor for histone H1. EMBO J. 1999;18:2411–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Muhlhausser P, Muller EC, Otto A, Kutay U. Multiple pathways contribute to nuclear import of core histones. EMBO Rep. 2001;2:690–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Waldmann I, Walde S, Kehlenbach RH. Nuclear import of c-Jun is mediated by multiple transport receptors. J Biol Chem. 2007;282:27685–92. [DOI] [PubMed] [Google Scholar]
- [99].Freedman ND, Yamamoto KR. Importin 7 and importin alpha/importin beta are nuclear import receptors for the glucocorticoid receptor. Mol Biol Cell. 2004;15:2276–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Bourgeois B, Hutten S, Gottschalk B, Hofweber M, Richter G, Sternat J, et al. Nonclassical nuclear localization signals mediate nuclear import of CIRBP. Proc Natl Acad Sci U S A. 2020;117:8503–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell. 2015;162:1066–77. [DOI] [PubMed] [Google Scholar]
- [102].Angelini C, Marozzo R, Pinzan E, Pegoraro V, Molnar MJ, Torella A, et al. A new family with transportinopathy: increased clinical heterogeneity. Ther Adv Neurol Disord. 2019;12:1756286419850433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Pál E, Zima J, Hadzsiev K, Ito YA, Hartley T, Boycott KM, et al. A novel pathogenic variant in TNPO3 in a Hungarian family with limb-girdle muscular dystrophy 1F. Eur J Med Genet. 2019;62:103662. [DOI] [PubMed] [Google Scholar]
- [104].Rodríguez-Mora S, De Wit F, García-Perez J, Bermejo M, López-Huertas MR, Mateos E, et al. The mutation of Transportin 3 gene that causes limb girdle muscular dystrophy 1F induces protection against HIV-1 infection. PLoS Pathog. 2019;15:e1007958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Melià MJ, Kubota A, Ortolano S, Vílchez JJ, Gámez J, Tanji K, et al. Limb-girdle muscular dystrophy 1F is caused by a microdeletion in the transportin 3 gene. Brain. 2013;136:1508–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Torella A, Fanin M, Mutarelli M, Peterle E, Del Vecchio Blanco F, Rispoli R, et al. Next-generation sequencing identifies transportin 3 as the causative gene for LGMD1F. PLoS One. 2013;8:e63536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Costa R, Rodia MT, Vianello S, Santi S, Lattanzi G, Angelini C, et al. Transportin 3 (TNPO3) and related proteins in limb girdle muscular dystrophy D2 muscle biopsies: A morphological study and pathogenetic hypothesis. Neuromuscul Disord. 2020;30:685–92. [DOI] [PubMed] [Google Scholar]
- [108].Kimura M, Morinaka Y, Imai K, Kose S, Horton P, Imamoto N. Extensive cargo identification reveals distinct biological roles of the 12 importin pathways. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Kataoka N, Bachorik JL, Dreyfuss G. Transportin-SR, a nuclear import receptor for SR proteins. J Cell Biol. 1999;145:1145–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Lester E, Ooi FK, Bakkar N, Ayers J, Woerman AL, Wheeler J, et al. Tau aggregates are RNA-protein assemblies that mislocalize multiple nuclear speckle components. Neuron. 2021;109:1675–91 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].McMillan PJ, Strovas TJ, Baum M, Mitchell BK, Eck RJ, Hendricks N, et al. Pathological tau drives ectopic nuclear speckle scaffold protein SRRM2 accumulation in neuron cytoplasm in Alzheimer’s disease. Acta Neuropathol Commun. 2021;9:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Gorenberg EL, Shorter J. Tau heckles speckles: A pathogenic mechanism in tauopathy? Neuron. 2021;109:1585–7. [DOI] [PubMed] [Google Scholar]
- [113].Safari MS, King MR, Brangwynne CP, Petry S. Interaction of spindle assembly factor TPX2 with importins-alpha/beta inhibit protein phase separation. J Biol Chem. 2021:100998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Kimura M, Imai K, Morinaka Y, Hosono-Sakuma Y, Horton P, Imamoto N. Distinct mutations in importin-β family nucleocytoplasmic transport receptors transportin-SR and importin-13 affect specific cargo binding. Scientific Reports. 2021;11:15649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Kosyna FK, Depping R. Controlling the Gatekeeper: Therapeutic Targeting of Nuclear Transport. Cells. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Floer M, Blobel G, Rexach M. Disassembly of RanGTP-karyopherin beta complex, an intermediate in nuclear protein import. J Biol Chem. 1997;272:19538–46. [DOI] [PubMed] [Google Scholar]
- [117].Lee BJ, Cansizoglu AE, Suel KE, Louis TH, Zhang Z, Chook YM. Rules for nuclear localization sequence recognition by karyopherin beta 2. Cell. 2006;126:543–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Smith KM, Tsimbalyuk S, Edwards MR, Cross EM, Batra J, Soares da Costa TP, et al. Structural basis for importin alpha 3 specificity of W proteins in Hendra and Nipah viruses. Nat Commun. 2018;9:3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Koyama M, Matsuura Y. Crystal structure of importin-α3 bound to the nuclear localization signal of Ran-binding protein 3. Biochemical and biophysical research communications. 2017;491:609–13. [DOI] [PubMed] [Google Scholar]
- [120].Jung H, Takeshima T, Nakagawa T, MacMillan KS, Wynn RM, Wang H, et al. The structure of importin α and the nuclear localization peptide of ChREBP, and small compound inhibitors of ChREBP-importin α interactions. Biochem J. 2020;477:3253–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Kirby TW, Pedersen LC, Gabel SA, Gassman NR, London RE. Variations in nuclear localization strategies among pol X family enzymes. Traffic. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Kirby TW, Gassman NR, Smith CE, Pedersen LC, Gabel SA, Sobhany M, et al. Nuclear Localization of the DNA Repair Scaffold XRCC1: Uncovering the Functional Role of a Bipartite NLS. Sci Rep. 2015;5:13405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Jeong SA, Kim K, Lee JH, Cha JS, Khadka P, Cho HS, et al. Akt-mediated phosphorylation increases the binding affinity of hTERT for importin α to promote nuclear translocation. J Cell Sci. 2015;128:2287–301. [DOI] [PubMed] [Google Scholar]
- [124].Neef DW, Jaeger AM, Gomez-Pastor R, Willmund F, Frydman J, Thiele DJ. A direct regulatory interaction between chaperonin TRiC and stress-responsive transcription factor HSF1. Cell Rep. 2014;9:955–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Vigh L, Literati PN, Horvath I, Torok Z, Balogh G, Glatz A, et al. Bimoclomol: a nontoxic, hydroxylamine derivative with stress protein-inducing activity and cytoprotective effects. Nat Med. 1997;3:1150–4. [DOI] [PubMed] [Google Scholar]
- [126].Hargitai J, Lewis H, Boros I, Rácz T, Fiser A, Kurucz I, et al. Bimoclomol, a heat shock protein co-inducer, acts by the prolonged activation of heat shock factor-1. Biochemical and biophysical research communications. 2003;307:689–95. [DOI] [PubMed] [Google Scholar]
- [127].Kieran D, Kalmar B, Dick JR, Riddoch-Contreras J, Burnstock G, Greensmith L. Treatment with arimoclomol, a coinducer of heat shock proteins, delays disease progression in ALS mice. Nat Med. 2004;10:402–5. [DOI] [PubMed] [Google Scholar]
- [128].Ahmed M, Machado PM, Miller A, Spicer C, Herbelin L, He J, et al. Targeting protein homeostasis in sporadic inclusion body myositis. Sci Transl Med. 2016;8:331ra41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Jackrel ME, Shorter J. Shock and awe: unleashing the heat shock response to treat Huntington disease. J Clin Invest. 2011;121:2972–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Auluck PK, Bonini NM. Pharmacological prevention of Parkinson disease in Drosophila. Nat Med. 2002;8:1185–6. [DOI] [PubMed] [Google Scholar]
- [131].Burslem GM, Crews CM. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell. 2020;181:102–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Levone BR, Lenzken SC, Antonaci M, Maiser A, Rapp A, Conte F, et al. FUS-dependent liquid-liquid phase separation is important for DNA repair initiation. J Cell Biol. 2021;220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Mitra J, Guerrero EN, Hegde PM, Liachko NF, Wang H, Vasquez V, et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc Natl Acad Sci U S A. 2019;116:4696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW, et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron. 2014;81:536–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Gopal PP, Nirschl JJ, Klinman E, Holzbaur EL. Amyotrophic lateral sclerosis-linked mutations increase the viscosity of liquid-like TDP-43 RNP granules in neurons. Proc Natl Acad Sci U S A. 2017;114:E2466–e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Hallegger M, Chakrabarti AM, Lee FCY, Lee BL, Amalietti AG, Odeh HM, et al. TDP-43 condensation properties specify its RNA-binding and regulatory repertoire. Cell. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Deverman BE, Ravina BM, Bankiewicz KS, Paul SM, Sah DWY. Gene therapy for neurological disorders: progress and prospects. Nature reviews Drug discovery. 2018;17:641–59. [DOI] [PubMed] [Google Scholar]
- [138].Bravo-Hernandez M, Tadokoro T, Navarro MR, Platoshyn O, Kobayashi Y, Marsala S, et al. Spinal subpial delivery of AAV9 enables widespread gene silencing and blocks motoneuron degeneration in ALS. Nat Med. 2020;26:118–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Naldini L Gene therapy returns to centre stage. Nature. 2015;526:351–60. [DOI] [PubMed] [Google Scholar]
- [140].LeWitt PA, Rezai AR, Leehey MA, Ojemann SG, Flaherty AW, Eskandar EN, et al. AAV2-GAD gene therapy for advanced Parkinson’s disease: a double-blind, sham-surgery controlled, randomised trial. The Lancet Neurology. 2011;10:309–19. [DOI] [PubMed] [Google Scholar]
- [141].Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, et al. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. The New England journal of medicine. 2017;377:1713–22. [DOI] [PubMed] [Google Scholar]
- [142].Warren Olanow C, Bartus RT, Baumann TL, Factor S, Boulis N, Stacy M, et al. Gene delivery of neurturin to putamen and substantia nigra in Parkinson disease: A double-blind, randomized, controlled trial. Annals of neurology. 2015;78:248–57. [DOI] [PubMed] [Google Scholar]
- [143].Kuzmin DA, Shutova MV, Johnston NR, Smith OP, Fedorin VV, Kukushkin YS, et al. The clinical landscape for AAV gene therapies. Nature reviews Drug discovery. 2021;20:173–4. [DOI] [PubMed] [Google Scholar]
- [144].Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nature reviews Drug discovery. 2019;18:358–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Mendell JR, Al-Zaidy SA, Rodino-Klapac LR, Goodspeed K, Gray SJ, Kay CN, et al. Current Clinical Applications of In Vivo Gene Therapy with AAVs. Mol Ther. 2021;29:464–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Finkel RS, Fischbeck KH. Maybe too much of a good thing in gene therapy. Nat Neurosci. 2021;24:901–2. [DOI] [PubMed] [Google Scholar]
- [147].Van Alstyne M, Tattoli I, Delestree N, Recinos Y, Workman E, Shihabuddin LS, et al. Gain of toxic function by long-term AAV9-mediated SMN overexpression in the sensorimotor circuit. Nat Neurosci. 2021;24:930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Kormann MS, Hasenpusch G, Aneja MK, Nica G, Flemmer AW, Herber-Jonat S, et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat Biotechnol. 2011;29:154–7. [DOI] [PubMed] [Google Scholar]
- [149].Pardi N, Weissman D. Development of vaccines and antivirals for combating viral pandemics. Nat Biomed Eng. 2020;4:1128–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Han Z, Chen C, Christiansen A, Ji S, Lin Q, Anumonwo C, et al. Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome. Sci Transl Med. 2020;12. [DOI] [PubMed] [Google Scholar]
- [151].Lim KH, Han Z, Jeon HY, Kach J, Jing E, Weyn-Vanhentenryck S, et al. Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression. Nat Commun. 2020;11:3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Roberts TC, Langer R, Wood MJA. Advances in oligonucleotide drug delivery. Nature reviews Drug discovery. 2020;19:673–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Zhao X, Reebye V, Hitchen P, Fan J, Jiang H, Saetrom P, et al. Mechanisms involved in the activation of C/EBPalpha by small activating RNA in hepatocellular carcinoma. Oncogene. 2019;38:3446–57. [DOI] [PubMed] [Google Scholar]
- [154].Berchtold NC, Cribbs DH, Coleman PD, Rogers J, Head E, Kim R, et al. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc Natl Acad Sci U S A. 2008;105:15605–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Vogler TO, Wheeler JR, Nguyen ED, Hughes MP, Britson KA, Lester E, et al. TDP-43 and RNA form amyloid-like myo-granules in regenerating muscle. Nature. 2018;563:508–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Yasuda K, Clatterbuck-Soper SF, Jackrel ME, Shorter J, Mili S. FUS inclusions disrupt RNA localization by sequestering kinesin-1 and inhibiting microtubule detyrosination. J Cell Biol. 2017;216:1015–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Yasuda K, Zhang H, Loiselle D, Haystead T, Macara IG, Mili S. The RNA-binding protein Fus directs translation of localized mRNAs in APC-RNP granules. J Cell Biol. 2013;203:737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]