

Abstract
Gut microbiome is gaining interest because of its links with several diseases, including colorectal cancer (CRC), as well as the possibility of being used to obtain non-intrusive predictive disease biomarkers. Here we performed a meta-analysis of 1042 fecal metagenomic samples from seven publicly available studies. We used an interpretable machine learning approach based on functional profiles, instead of the conventional taxonomic profiles, to produce a highly accurate predictor of CRC with better precision than those of previous proposals. Moreover, this approach is also able to discriminate samples with adenoma, which makes this approach very promising for CRC prevention by detecting early stages in which intervention is easier and more effective. In addition, interpretable machine learning methods allow extracting features relevant for the classification, which reveals basic molecular mechanisms accounting for the changes undergone by the microbiome functional landscape in the transition from healthy gut to adenoma and CRC conditions. Functional profiles have demonstrated superior accuracy in predicting CRC and adenoma conditions than taxonomic profiles and additionally, in a context of explainable machine learning, provide useful hints on the molecular mechanisms operating in the microbiota behind these conditions.
Subject terms: Cancer, Computational biology and bioinformatics, Genetics, Microbiology, Systems biology, Diseases, Oncology
Introduction
In recent years the study of the microbiome has progressively gained interest, especially in the context of human health1–4. Microbial abundance profiles based on 16S rRNA genes have been used to study microbiomes, although whole genome sequencing (WGS) is becoming increasingly popular nowadays due to the decreasing sequencing costs5,6. Contrary to 16S rRNA data, WGS microbiome data provides the real gene composition in the bacterial pool of each sample, which allows identifying strain-specific genomic traits7,8. During the last years, microbiome WGS has been used to explore microbiome–host interactions within a disease context by means of metagenome-wide association studies, that allow studying gut microbiome alterations characteristic of different pathologic conditions3,9–17. In particular, recent evidence suggests that the human gut microbiome could be a relevant factor in human diseases18,19. In fact, the existence of carcinogenic mechanisms mediated by bacterial organisms has recently been proposed20–22. And, more specifically, it has been suggested that the gut microbiome could play a relevant role in the development of colorectal cancer (CRC)15,16,23–25. Due to this, the gut microbiome has been proposed as a potential diagnostic tool for CRC16,17,26,27. Nevertheless, its reproducibility and the predictive accuracy of the microbial gene signatures across different cohorts have been questioned28,29. The increasing availability of whole metagenome shotgun datasets of CRC cohorts15–17,26,27 facilitates large-scale multi-population exploratory studies of the CRC-associated microbiome at the resolution level of strain30,31. In two recent studies, a combined analysis of heterogeneous CRC cohorts was able to build accurate disease predictive models that open the door to the use of gut microbiota for future clinical prognostic tests28,29. The subsequent meta-analysis of the functional potential in the strains of the signature found gluconeogenesis and putrefaction and fermentation pathways associated with CRC, in coherence with the current knowledge on microbial metabolites implicated in carcinogenesis32.
It is important to note that the current approaches used to obtain biomarkers with predictive power use microbial strain or gene signatures as features to train a predictive model. Since genes or strains do not have a clear interpretability by themselves, the interpretation of the results of the classification produced by the model relies on the analysis of the potential functionalities encoded by these features. In other words, the predictive model is built using features that need to be interpreted a posteriori33. In fact, this is a relatively common problem with many current machine learning techniques, which have evolved in recent years to enable robust association of biological signals with measured phenotypes but, in many cases, such approaches are unable to identify causal relationships34,35. However, the interpretability of models, especially in a clinical context, is becoming an increasingly important issue34–36. The use of features with a direct functional interpretation has been suggested as crucial for the interpretability of the models37. In a recent study, gene profiles derived from WGS of samples of the MetaSub project38 were initially transformed into functional profiles, which account for bacterial metabolism and other cell functionalities, and have subsequently been used as features to build a city classification machine learning algorithm39. Since the features are informative by themselves, their relevance in the classification provides an immediate interpretability to the prediction model built.
Here, we propose an interpretable machine learning approach in which functional profiles of microbiota samples, with a direct interpretation, are first obtained from shotgun sequencing and subsequently used as features for predicting CRC in the patient donor of the sample. Moreover, in the prediction schema proposed, a feature relevance method allows extracting the most important functional features that account for the classification. Thus, any sample is described as a collection of functional modules contributed by the different bacterial species present in it, which account for the potential functional activities that the bacterial population in the sample, as a whole, can perform.
Results
Data-driven analysis of the interpretability
All the projects in Table 1 were preprocessed as described in methods and the corresponding taxonomic and functional profiles were obtained for all the samples. Supplementary Table S1 contains the list of taxonomic features, Supplementary Table S2 the list of KEGG functional features and Supplementary Table S3 the list of eggNOG functional features selected by the model. Also, Krona representations, allowing the exploration of these features at different hierarchical levels, are available for taxonomic (Supplementary Fig. S1) and KEGG (Supplementary Fig. S2) features, respectively.
Table 1.
Datasets used in the study.
| Project ID | Dataset name | References | Samples | Mean aligned reads |
|---|---|---|---|---|
| PRJNA389927 | Hannigan | 40 | 82 | 2.308.712 |
| PRJEB12449 | Vogtmann | 42 | 104 | 3.897.639 |
| PRJEB6070 | Zeller | 16 | 199 | 4.517.730 |
| PRJEB7774 | Feng | 15 | 132 | 9.154.788 |
| PRJEB10878 | Yu | 17 | 128 | 18.372.510 |
| PRJNA447983 | Thomas0 | 29 | 124 | 14.841.290 |
| PRJEB27928 | Thomas1 | 29 | 82 | 6.518.536 |
A stability test was conducted for each project and profile used as described above and the results were congruent with previous observations: all the profiles are quite stable (stability score beyond 0.4 with small CI) except for the Hannigan project40, which happens to be the only project not sequenced at high depth (Fig. 1). In addition, the test for the cross-validation strategy was computed in order to detect the differences in the selection stability when training with more data. As can be seen in (Fig. 1), both analyses follow the same trend and, as expected, the larger the sample size, the greater the stability. The rank stability analysis (Fig. 2) follows the same pattern as the stability analysis which indicates that the features selected and their relevance are stable for each profile and experiment (Hannigan being again the exception) Note that the area under the receiving operating curve (AUROC) follows a similar trend, although not as pronounced, when the mean of the 20 repeated tenfold cross-validation strategy was compared with the average of the test splits over the stability splitting strategy (Fig. 3). Therefore, it can be concluded that under controlled experiments the greater the number of samples, the better the model performs.
Figure 1.
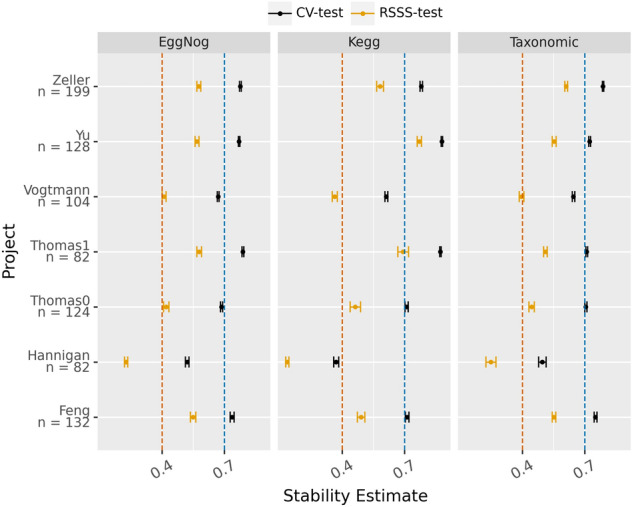
Stability estimate along with the confidence interval (alpha = 0.05) for the Random Stability Sub Sampling (RSSS-test) and 20-times tenfold cross validation (CV-test) splitting schemas, for each metagenomic profile (KEGG, eggNog and taxonomic) and project. The vertical bars indicate the theoretical thresholds on the effect size: below 0.4 represent bad agreement, between o.4 and 0.7 refers to a good enough agreement and scores above 0.7 represent a near perfect agreement.
Figure 2.
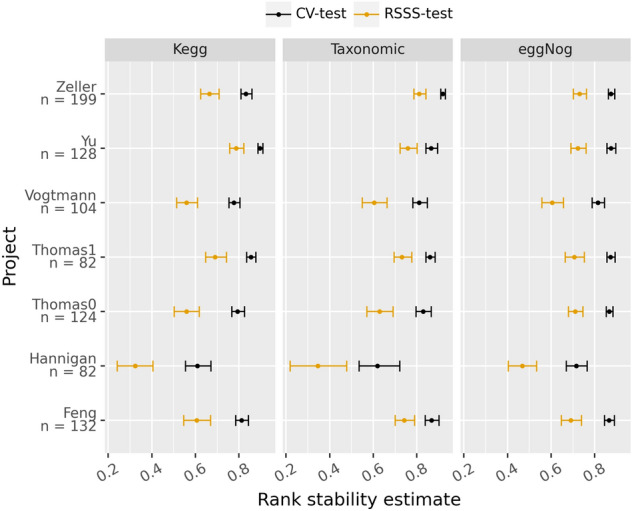
Rank stability estimate (the mean of all the hyperbolic-weighted tau pairwise rank comparisons) along with the 0.25 and 0.75 quantiles for the Random Stability Sub Sampling (RSSS-test) and 20-times tenfold cross validation (CV-test) splitting schemas for each metagenomic profile (KEGG, eggNog and taxonomic) and project.
Figure 3.
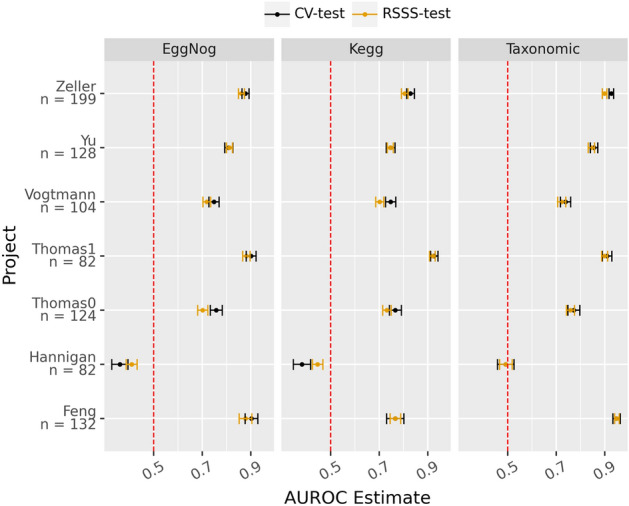
Mean of the area under the receiver operating characteristic curve (AUROC) along with the 0.25 and 0.75 quantiles for the Random Stability Sub Sampling (RSSS-test) and 20-times tenfold cross validation (CV-test) splitting schemas when discriminating between CRC and healthy samples for each metagenomic profile (KEGG, eggNog and taxonomic) and project.
Performance analysis
A comparison of the method proposed here with those already published in the literature has been conducted using a performance validation schema previously proposed29, that consists of measuring the AUROC across the following data-splitting scenarios: (1) a 20-times repeated tenfold cross-validation for each project, (2) a cross-dataset prediction, which consists of training our model over one dataset to predict the rest, and (3) a leave-one-project-out (LOPO) design, where any given study is predicted with a model trained using the remaining projects.
However, the reference methodology conducts an out-of-training feature selection consisting of a two-step process that first preselects those features that are biologically more appropriate for gut-based microbiome analyses and secondly removes those features that are not statistically relevant (FDR correction of discrete correlations) using the whole dataset (the result of joining all studies). In order to have a fair comparison, we have performed two LOPO validation procedures: the first one does not perform any out-of-training feature selection (non-LOPO), thus leading to results more closely aligned to the original intent of checking the cross-dataset variations, while the second procedure uses an out-of-training feature selection (o-LOPO), making the model proposed here directly comparable to the reference methodology29.
The results (Fig. 4) showcases: (1) the difficulties to generalize what the model learned in one dataset to others (non-LOPO off-diagonal scores), (2) a good intra-project performance (non-LOPO diagonal) except for the low-depth project, and (3) a good out-of-project performance that can be achieved by aggregating information from different projects. Overall, these results are congruent with previous observations29. Interestingly, we have found that the performance of the LOPO analysis in the model used here without pre-training is quite similar to the reference model29. However, when all the datasets are used for out-of-training feature selection the model used here behaves significantly better than the reference. These facts imply that not all the dataset-based signatures found by the model are shareable across the projects but rather a signature is learned by joining projects (increase in LOPO scores) as previously observed29. However, the model presented here makes a better use of increasing quantities of information: once noise has been filtered out through feature selection, the more depth in a profile the better the results.
Figure 4.
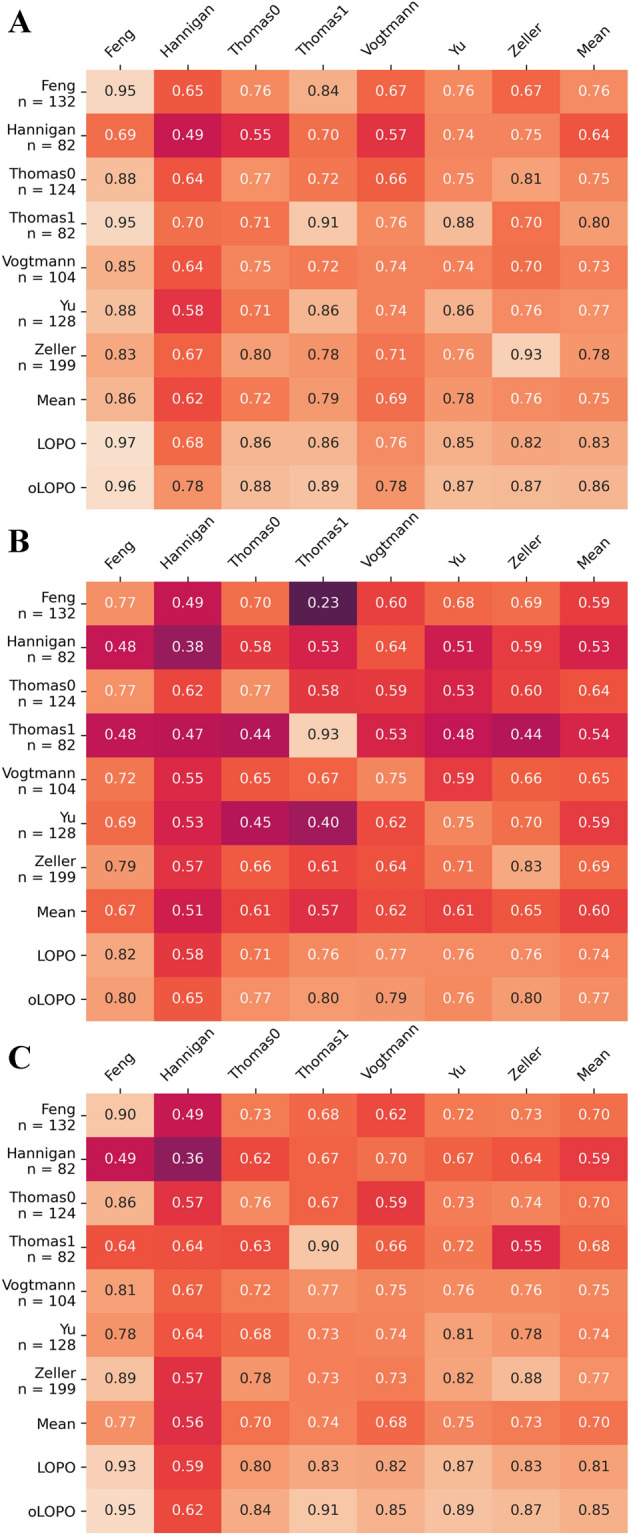
Cross-prediction matrix that measures the performance of the proposed model in terms of the area under the receiver operating characteristic curve (AUROC) for (A) taxonomic, (B) KEGG and (C) eggNog metagenomic profiles. The diagonal represents the intra-project performance by reporting the mean of the AUROC of 20-times tenfold cross validation, whereas the off-diagonal shows the cross-dataset performance, i.e. train with the model indicated in the rows and test over the project in the columns. Finally, the Leave one Project Out (LOPO) row reports the performance of predicting the dataset referred to in the columns while training with the other datasets, whereas the oLOPO row is the same experiment but using the functional signature learned during the LOPO procedure.
Signature computation and validation
For the interpretation analysis, a consensus signature has been built for each profile by combining the learned signature for each LOPO procedure as follows: (1) rescale the feature relevance for each LOPO run to [0, 1], (2) aggregate the score for each feature across all the runs and iii) divide it by (p − nz(i) + 1), where p is the number of projects and nz(i) is the number of projects where feature i is non-zero. Note that a feature that does not pass the FDR-based selection is assigned a score of 0.
Finally, we have evaluated the significance of the o-LOPO scores for the consensus signature by means of the permutations tests technique technique41: for each profile we repeat the o-LOPO validation procedure for our pipeline 100 times while randomly permuting the outcomes. Then a p-value is computed by checking the percentage of runs where the trained model scored greater than the non-permuted score. As can be seen in Fig. 5 we can be confident that our model o-LOPO validation scores are significant (α = 0.05) for all the profiles. Note that the features where the pipeline is trained are fixed by keeping only those with a non-zero relevance score for the consensus signature.
Figure 5.

Significance of the cross-validated score through the use of the target permutation technique for each metagenomic profile (KEGG, eggNog and taxonomic). The p-value approximates the probability that the score for each profile would be the result of chance. The number of permutations is 100 for each profile using the consensus signature previously learnt and a 100-times tenfold cross-validation schema. Note that the worst outcome is 1 and the best is ~ 0.009. The vertical lines for each profile report the true score without permuting the outcome (being CRC or healthy) and the luck threshold (in black), whereas the continuous color lines show the permutation scores distribution (i.e. the null distribution) for each profile.
Adenoma analysis
In order to test how the proposed methodology can model unseen (but related) conditions a statistical test over the predicted probabilities of adenoma samples was proposed. The test procedure consists of the following steps: (1) a sample-wise concatenation of all datasets is carried out to construct three sets. All healthy and CRC (no comorbidities are considered here) samples are collected into the training set, which is further randomly divided into learning and validation sets with 0.7/0.3 the sample size. Finally, the test set is built with the samples not included in the previous sets (other diseases, adenomas and comorbidities including colorectal cancer). (2) The pipeline is first trained with the learning set and further used to compute the probabilities for all the samples of the validation and test sets. Then the distribution of the probabilities of the healthy samples is compared against the distribution of the adenoma samples that are not explicitly labeled as being small, using a Mann–Whitney rank test (healthy < adenoma). Other comparisons are also carried out, which include: the non-small adenoma distribution against the samples with a CRC condition (adenoma < tumor), the small adenoma versus non-small adenoma (small adenoma < adenoma) and healthy (small adenoma < healthy). (3) Steps (1) and (2) are repeated 100 times and the frequencies for the test being passed with a significance of < 0.05 (i.e. a hit ratio) are assessed. Note that the splitting criteria has been specifically chosen to be suitable for comparing such distributions, as the validation and test splits were built with data unseen for the model, thus forcing the independence between both sets and the split where the model has been fitted.
As depicted in Fig. 6 all the profiles perform well (100% hit rate) for the healthy < tumor test comparison. This is the expected behavior since we know from the performance analysis that all the profiles separate healthy from tumor. However, the eggNOG and KEGG functional profiles behave more like a risk score in terms of being CRC, due to the fact that the models based on those profiles assign a probability mass consistently higher to those samples more prone to having it (non-small adenoma) than healthy samples. Furthermore, the eggNOG profile achieves a 100% hit ratio for all the performed tests, except healthy < small adenoma. Thus, although the taxonomic profile and the eggNOG profile perform similarly from a healthy/CRC classification point of view, the former lacks the ability to see any difference between non-small adenoma and healthy samples, as evidenced by the hit ratio of the taxonomic profile in the healthy < adenoma test.
Figure 6.
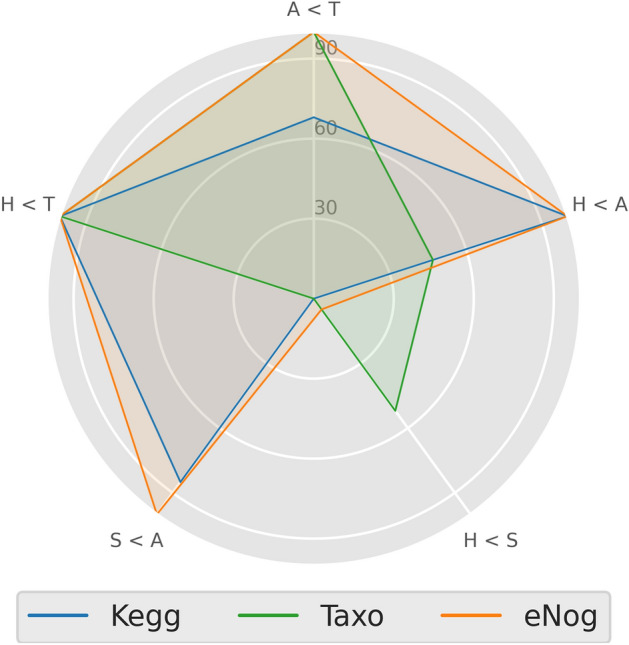
Radar plot with the performances of the different comparisons of the distribution of the probabilities between pairs of categories of samples using a Mann–Whitney rank test. Comparisons are clockwise: Adenoma < Tumor (A < T), healthy < Adenoma (H < A), healthy < small adenoma (H < S), Small adenoma < Adenoma (S < A) and healthy < tumor (H < T). Models were trained with Taxonomic (Taxo) and functional features (KEGG and eggNOG).
Discussion
This study uses a comprehensive collection of the cohorts of CRC (listed in Table 1). Here, three different types of microbiome profiles (taxonomic and two functional ones based on KEGG and on eggNOG annotations) have been analyzed in an interpretable machine learning framework that has demonstrated to outperform other previous class predictors previously reported. As previously reported16,29, predictors render better predictions in the condition in which they were trained than in other conditions, independently of the type of profile used (see Fig. 4). The project PRJEB12449, which resulted with the worst performance, was frozen for more than 25 years before it was sequenced42. This most probably compromised the quality of the results43 and, actually, was described as technically flawed by previous studies28,29.
One of the most interesting properties of this approach is its immediate interpretability. Thus, the features chosen by the model that optimize the discrimination between the conditions compared account for the functionalities that operate differentially among both conditions.
At taxonomic level there are two species that clearly are relevant for the classification: Parvimonas micra and Fusobacterium nucleatum, which are represented by the four more relevant features (see Table 2). These two bacterial species have been related to CRC in numerous publications44 and are known CRC biomarkers16,26,29. In addition, F. nucleatum is known to promote chemoresistance in Colorectal Cancer cells by inhibiting apoptosis45 or by modulating autophagy46. Supplementary Table S1 lists the complete set of taxonomical features with the relevance assigned by the model. Actually, all the bacterial species listed in reviews as associated to CRC were selected by the model (some of them, like Parvimonas, Fusobacterium, Porphyromonas, Gemella, Streptococcus or Clostridium in Table 2 and the rest in preeminent positions in the relevance rank listed in Supplementary Table S1)44.
Table 2.
The 20 most relevant taxons selected by the machine learning method used.
| Relevance score | Name | Taxon ID |
|---|---|---|
| 2.41252 | Parvimonas micra | 33,033 |
| 1.59494 | Fusobacterium nucleatum subsp. animalis 7_1 | 457,405 |
| 1.56152 | Fusobacterium nucleatum subsp. animalis 4_8 | 469,607 |
| 1.28725 | Fusobacterium nucleatum subsp. animalis | 76,859 |
| 0.96797 | Porphyromonas asaccharolytica DSM 20707 | 879,243 |
| 0.81790 | Fusobacterium nucleatum subsp. nucleatum ATCC 23726 | 525,283 |
| 0.70496 | Dialister pneumosintes | 39,950 |
| 0.62700 | Fusobacterium nucleatum subsp. polymorphum | 76,857 |
| 0.61251 | Gemella morbillorum | 29,391 |
| 0.57994 | Fusobacterium necrophorum subsp. funduliforme | 143,387 |
| 0.53820 | Clostridium sporogenes | 1509 |
| 0.53034 | Streptococcus anginosus C238 | 862,971 |
| 0.50665 | Longibaculum sp. KGMB06250 | 2,584,943 |
| 0.49567 | Anaerostipes hadrus | 649,756 |
| 0.47842 | Citrobacter freundii | 546 |
| 0.46420 | Fusobacterium nucleatum subsp. vincentii | 155,615 |
| 0.44508 | Streptococcus pseudoporcinus | 361,101 |
| 0.44365 | Blautia hansenii DSM 20583 | 537,007 |
| 0.43355 | Fusobacterium nucleatum subsp. vincentii 3_1_36A2 | 469,604 |
| 0.41181 | Fusobacterium nucleatum subsp. vincentii 3_1_27 | 469,602 |
The analysis of functional profiles is even more interesting from the point of view of interpretability. Table 3 lists the 20 most relevant KEGG features selected by the model (see all the KEGG functional features in Supplementary Table S2). Interestingly, the most relevant feature is the Methylaspartate mutase sigma subunit (K01846), whose high activity is related to high probability of CRC according to the model. It has been described that cancer cells undergo modifications that include increased glutamine catabolism and over-expression of enzymes involved in glutaminolysis, including glutaminase47, which is liberated to the gut48 and promotes the proliferation of bacteria containing this bacterial module. Another known enzyme related to cancer present in Table 3 is Heptose II phosphotransferase (K02850). This enzyme, located in the Lipopolysaccharide biosynthesis (KO00540) pathway, is associated with CRC in high values. Actually, the presence of lipopolysaccharides produced in the surface of Gram- bacteria has been reported to induce an inflammatory response as well as to stimulate the proliferation of colon carcinoma49,50. Another relevant feature that discriminates healthy from CRC samples is Manganese/zinc/iron transport system permease protein (K11708). This transporter increases its number in excess iron conditions that are known to promote colorectal carcinogenesis51. Methyltransferase (K16168), related to polyketide synthesis, is the next most relevant feature. It has recently been described that a class of molecules, colibactins, are produced from the gene cluster called the polyketide synthase island that occurs in certain strains of Escherichia coli prevalent in the microbiota of CRC patients52.
Table 3.
The 20 most relevant KEGG features selected by the machine learning method used here.
| Relevance score | Name | KEGG ID |
|---|---|---|
| 1.50544 | glmS, mutS, mamA; methylaspartate mutase sigma subunit [EC:5.4.99.1] | K01846 |
| 1.34932 | mal; methylaspartate ammonia-lyase [EC:4.3.1.2] | K04835 |
| 0.77486 | rocR; arginine utilization regulatory protein | K06714 |
| 0.73183 | pldB; lysophospholipase [EC:3.1.1.5] | K01048 |
| 0.73001 | 6GAL; galactan endo-1,6-beta-galactosidase [EC:3.2.1.164] | K18579 |
| 0.72130 | pdaA; peptidoglycan-N-acetylmuramic acid deacetylase [EC:3.5.1.-] | K01567 |
| 0.71882 | MARS, metG; methionyl-tRNA synthetase [EC:6.1.1.10] | K01874 |
| 0.70670 | thiQ; thiamine transport system ATP-binding protein [EC:7.6.2.15] | K02062 |
| 0.70362 | epr; minor extracellular protease Epr [EC:3.4.21.-] | K13277 |
| 0.66044 | kamA; lysine 2,3-aminomutase [EC:5.4.3.2 | K01843 |
| 0.62861 | E2.1.1.77, pcm; protein-L-isoaspartate(D-aspartate) O-methyltransferase [EC:2.1.1.77] | K00573 |
| 0.62662 | troC, mntC, znuB; manganese/zinc/iron transport system permease protein | K11708 |
| 0.62170 | glpX; fructose-1,6-bisphosphatase II [EC:3.1.3.11] | K02446 |
| 0.61670 | bpsB, srsB; methyltransferase | K16168 |
| 0.61284 | spoIIP; stage II sporulation protein P | K06385 |
| 0.60627 | waaY, rfaY; heptose II phosphotransferase [EC:2.7.1.-] | K02850 |
| 0.60596 | FBA, fbaA; fructose-bisphosphate aldolase, class II [EC:4.1.2.13] | K01624 |
| 0.60594 | rgpF; rhamnosyltransferase [EC:2.4.1.-] | K07272 |
| 0.59660 | tex; protein Tex | K06959 |
| 0.58759 | murA; UDP-N-acetylglucosamine 1-carboxyvinyltransferase [EC:2.5.1.7] | K00790 |
Metabolomic measurements in CRC also support the feature selection carried out by the model. A recent review on metabolic alterations in CRC provides a list of metabolites systematically altered in this cancer type53. Table 4 shows the metabolites most frequently reported as differentially deregulated in CRC. All of them are products of the KEGG orthologs selected by the model as most relevant features.
Table 4.
Metabolites described as systematically deregulated in cancer and their relevance in the model using KEGG functional features.
| Metabolite_name | HMDB_ID | KEGG_conpound_ID | Frequency | model_KEGG_KO_score |
|---|---|---|---|---|
| Glycine | HMDB0000123 | C00037 | 24 | 4.823568715 |
| L-Valine | HMDB0000883 | C00183 | 23 | 0.6930878983 |
| L-Alanine | HMDB0000161 | C00041 | 22 | 3.22739351 |
| L-Lactic acid | HMDB0000190 | C00186 | 22 | 0.5279944356 |
| L-Phenylalanine | HMDB0000159 | C00079 | 20 | 2.117857375 |
| L-Proline | HMDB0000162 | C00148 | 20 | 1.434113835 |
| L-Leucine | HMDB0000687 | C00123 | 20 | 0.3006354234 |
| L-Glutamic acid | HMDB0000148 | C00025 | 17 | 13.32534903 |
| Taurine | HMDB0000251 | C00245 | 16 | 0.9103833031 |
| Palmitic acid | HMDB0000220 | C00249 | 15 | 0.3209053449 |
| L-Methionine | HMDB0000696 | C00073 | 15 | 4.503947812 |
| Glycerol | HMDB0000131 | C00116 | 14 | 1.125734858 |
| L-Tyrosine | HMDB0000158 | C00082 | 14 | 1.718910949 |
| L-Threonine | HMDB0000167 | C00188 | 14 | 1.354361221 |
| L-Isoleucine | HMDB0000172 | C00407 | 14 | 0.3873029906 |
| L-Serine | HMDB0000187 | C00065 | 14 | 2.042185229 |
| L-Aspartic acid | HMDB0000191 | C00049 | 14 | 3.752760624 |
| D-Glucose | HMDB0000122 | C00221 | 13 | 0.9079622305 |
| L-Lysine | HMDB0000182 | C00047 | 12 | 1.845182919 |
| L-Arginine | HMDB0000517 | C00062 | 12 | 1.818335229 |
| L-Glutamine | HMDB0000641 | C00064 | 12 | 5.074283424 |
| Choline | HMDB0000097 | C00114 | 11 | 0.3910553328 |
| L-Asparagine | HMDB0000168 | C00152 | 11 | 0.7764985602 |
| myo-Inositol | HMDB0000211 | C00137 | 11 | 0.3847705919 |
| Succinic acid | HMDB0000254 | C00042 | 11 | 1.819066672 |
| L-Tryptophan | HMDB0000929 | C00078 | 11 | 1.028295479 |
| Acetic acid | HMDB0000042 | C00033 | 10 | 4.986949589 |
| Uridine | HMDB0000296 | C00299 | 10 | 1.376520768 |
HMBD is the identifier of the metabolome database (https://hmdb.ca/) and the Frequency column denotes the number of studies in which the metabolite was found as deregulated according to a recent review53. The metabolite scores were calculated by adding the KEGG_KO’s scores, from the machine learning model, for each of the metabolites.
Another functional perspective is provided by the eggNOG features. Table 5 lists the 20 most relevant features (Supplementary Table S3 lists all the relevant eggNOG features). This type of features represents orthologous groups of proteins and constitute an interesting integration of function and taxonomy, given that protein families have a taxonomic-dependent distribution but, at the same time, play different roles in the bacterial biology54. Unfortunately, many bacterial proteins are still poorly annotated and about one third of the eggNOG features in the table are of unknown function. Interestingly, more than third are membrane proteins, which suggests that interaction of bacteria with the intestine cell could be playing a relevant role in CRC. Also, Methylaspartate mutase, E subunit, which correspond to two KEGG features with the best scores (Table 3).
Table 5.
The 20 most relevant eggnog features selected by the model.
| Score | Feature ID (eggNOG 4.5) | Taxonomic Level | Description |
|---|---|---|---|
| 2.47062 | 08XIZ | bactNOG | Integral membrane protein TIGR02185 |
| 2.30583 | 06J4I | bactNOG | N/A |
| 2.22012 | 0NI2F | firmNOG | Integral membrane protein TIGR02185 |
| 1.93081 | 00DN8 | actNOG | One of the primary rRNA binding proteins, it binds directly to 16S rRNA where it nucleates assembly of the head domain of the 30S subunit. Is located at the subunit interface close to the decoding center, probably blocks exit of the E-site tRNA (By similarity) |
| 1.92116 | 0NTFT | firmNOG | N/A |
| 1.76985 | 0Y9D1 | NOG | N/A |
| 1.73496 | 0EX7J | cloNOG | N/A |
| 1.47985 | 05DDE | bactNOG | Outer membrane autotransporter barrel domain-containing protein |
| 1.46286 | 057E2 | bacteNOG | DNA binding protein, excisionase family |
| 1.4445 | 0587E | bacteNOG | Protein of unknown function (DUF1446) |
| 1.37804 | 06F02 | bactNOG | N/A |
| 1.34625 | 05CMH | bactNOG | DEHYDRATASE |
| 1.33923 | 08NTT | bactNOG | N/A |
| 1.33323 | 079XJ | bactNOG | Major outer membrane protein |
| 1.31216 | 08C1U | bactNOG | N/A |
| 1.29914 | 08HM2 | bactNOG | Cell wall binding repeat 2-containing protein |
| 1.26147 | 059H0 | bacteNOG | Methylaspartate mutase, E subunit |
| 1.25394 | 05DCU | bactNOG | 2-Hydroxyglutaryl-CoA dehydratase |
| 1.24681 | 08BIB | bactNOG | Hypothetical bacterial integral membrane protein (Trep_Strep) |
| 1.23689 | 07I7J | bactNOG | s-layer protein |
Although an extensive description of the features selected by the model is beyond the scope of this paper, it is worth noting that the results obtained fully agree with the findings of functional analysis done in previous reports28,29.
Finally, a relevant aspect addressed in the study is the possibility of cancer interception by predicting CRC in early stages55. That would be the case of predicting adenomas. Actually, when the relative performance of the statistical test over the predicted probabilities of adenoma samples based either on functional or on taxonomic features is compared (Fig. 6) all the profiles distinguish tumors from normal samples with a 100% hit rate. However, functional profiles still show an excellent performance in distinguishing between CRC and adenoma samples and even adenoma from small adenoma samples, while taxonomic profiles fail to distinguish between these conditions. These observations suggest that the transition from normal condition to adenoma and CRC is not well defined in terms of strain abundances but there is a clear change at the level of functional activities of the bacteria in the sample that is better captured by functional profiles than by taxonomic profiles, which probably change at later stages, close to the CRC condition. This opens an interesting window of opportunity for clinical applications, as it has previously been suggested28,29, given that sequencing prices are plummeting to levels that obtaining taxonomic profiles result cost-effective in clinics. A trained predictor could systematically be used to detect in early phases individuals in risk of CCR. The results could be prospectively used to re-train the predictor.
Interpretability of the predictive models is becoming a major issue, especially in biomedicine33,34,36. The idea of using features with full biological meaning to gain interpretability in the machine learning methodology used has recently been proposed as a “white box” strategy37 and has successfully been used for the first time in the analysis of urban microbiota39 in the context of the METASub project38.
Conclusions
The interpretable machine learning approach proposed here has demonstrated a more consistent performance in comparison to other approaches previously proposed when dealing with different CRC-based problems, while providing straightforward interpretations. Moreover, it demonstrated a better resolution not only with respect to the separation between healthy and CRC samples, but it is also able to discriminate samples with adenoma, being a promising tool for CRC prevention by detecting early stages in which intervention is easier and more effective. And finally, the model has a biological interpretation that provides important clues to better understand the mechanistic implications of the gut microbiota in CRC as well as in the previous stages of adenoma, which can have an interesting potential in preventive medicine and, specifically, in cancer interception55.
Methods
Data description
A total of 1042 fecal metagenomic whole genome sequencing (WGS) samples were analyzed. The samples were downloaded from the European Nucleotide Archive projects: PRJEB10878, PRJEB12449, PRJEB27928, PRJEB6070, PRJEB7774, PRJNA389927 and PRJNA447983. Sample metadata were obtained from the different supplementary tables of the corresponding publications16,28,56 and complemented in the possible using the R57 package curatedMetagenomicData58 available in Bioconductor59. Table 1 lists the experiments used in this study.
Bacterial whole genome sequence data processing
Whole genome sequencing data was managed using the NGLess-Profiler60 package. Raw sequencing data preprocessing and quality control was carried out using a version of the human-gut.ngl pipeline. The substrim built-in function was used to discard reads that do not meet the basic quality filter of being longer than 45 bases and having all bases with a Phred score over 25. To prevent potential contaminations with human genome sequences the reads were mapped against the human genome hg19. All reads mapping the human genome were discarded. SAMtools61 and BWA62 were used to handle and map reads, respectively.
Functional profiles
Strain functional profiles are generated by assessing the gene coverage for KEGG63 functional orthologs and eggNOG54 ortholog groups. Ortholog genes are the basic feature used here, and each sample is described as a vector of features, or feature profile. The representation of each feature of the profile in any sample is estimated from the number of reads mapping on the corresponding gene. These counts were obtained by mapping the reads that passed the filters mentioned above, using the integrated gene catalog of the human gut64. The NGLess built-in function count was used with the default values, applying the scaled normalization that consists of dividing the raw count by the size of the feature and then scaled up so that the total number of counts is similar to the total raw count.
Taxonomic profiles
Strain taxonomic profiles were obtained using the Centrifuge application65. The centrifuge-download command was used to download the reference genomes of archea, bacteria, virus and vertebrate mammalian (human and mouse) taxons. The reads of each sample were mapped over the reference genomes. Taxons are here the features that describe each sample. The taxonomic profile consists of vectors composed by the relative representation of each genome (taxon) in the sample, which is obtained by normalizing the number of reads mapping on them by the respective genome lengths.
Machine learning approach for tumor status prediction
For tumor status prediction a combination of classical machine learning techniques with a novel algorithm, the explainable boosting machine (EBM)66–68, were used for tumor status prediction with an aim towards interpretability. EBMs are state-of-the-art supervised learning ML whitebox models, also known as glassbox models, specifically designed for being highly explainable without losing predictive power.
The classification pipeline is sequentially constructed by concatenating the following methods: first the profile features are transformed using a logarithmic approach (), then a feature selection based on the ANOVA F-test with FDR correction () is applied, followed by feature-wise discretization (2/20 bins for taxonomic/functional) and finally the EBM classifier is trained with the remaining features.
Explainable boosting machine
Explainable boosting machines are a new type of Generalized Additive Models (GAMs)69, which are constructed by combining different ensemble-based techniques, such as bagging and boosting, with a feature learning algorithm that leads to highly interpretable models.
GAMs can be concisely written as:
where g refers to the link function (e.g. logit in classification problems), alludes to the outputs/labels and represents the (shape) function learned for each feature . Traditionally the shape functions are either splines or polynomials69, which lead to interpretable models that lack predictive power66.
In the EBM algorithm each shape function is basically an ensemble of gradient boosted trees (GBT) constructed by iterating through all the features sequentially. For each feature a shallow tree is fitted, using only the selected feature, while the residuals are updated in a boosting-like fashion. Thus, each trained tree can only use the feature it was trained for, which allows the model to learn its contribution in a very precise way, while maintaining a global approximation by means of the residuals. These steps are repeated thousands of times by iterating through the data using such a small learning rate (GBT training) that the order of how the features are learned is not important. At the end, when all the iterations have been exhausted, the model builds a graph by aggregating all the trained trees for each feature, then the shape functions are combined together in order to assemble the final decision.
Despite the sophisticated of the learning procedure, the resulting model has all the intelligibility advantages of GAMs, since each shape function can be inspected in order to understand its contribution towards the final prediction. Furthermore, the repeated round-robin cycling over the features, along with the small learning rate, mitigates the effects of collinearity in feature-space (a common problem in biology), which leads to a fairer spread of the contributions (- like)67.
Explainability
The intelligibility of our model is driven by the combination of the different layers of the pipeline: the logarithmic preprocessing mitigates the skewness towards large values (frequent in biological multi project analyses), whereas the FDR-based selection drastically reduces the feature space by filtering out spurious relations with the outcome and, finally, the EBM learns a fair feature-attribution score, indeed a proper mathematical function (from now on, the learning graph or to simplify the graph), that fosters the biological interpretation of the problem.
As mentioned above, the EBM learns a graph for each feature and the learned representation can be very useful to complement decision-making in clinical scenarios due to the direct interpretability of the output68.
Moreover, the graph represents a visualization of the individualized feature contributions of each sample in the training dataset. Thus, the model constructs global (graphs) explanations on top of sample-wise explanations (local) without relying on external model-agnostic explainers, such as SHAP (SHapley Additive exPlanations)70 or local interpretable model-agnostic explanations (LIME)71. Note that a global relevance score can be computed by aggregating the absolute local attributions, which can be used to rank the feature importance for CRC prediction.
Data-driven interpretability analysis
In order to check that the model delivers consistent results from an explainability point of view the stability of both, the feature selection and ranking methods of our pipeline were tested. Explanation-based performance tests are needed to account for the stochastic nature of the learning methodologies that drive both predictions and biological interpretations, the presence of technological and experimental noise, data sampling bias, etc.
On the one hand, the performance of the feature selection procedure is tested using the Nogueira stability measure and the associated statistical tests72, which consists of splitting any given dataset into 100 training and test subsets of half the total sample size, fitting the model on the training set while accounting for the features selected, and finally computing the stability measure using estimations of the variances of the selection of each feature. The final results are: (1) a stability score (SS) which ranges from 0 (random guessing) to 1 (perfect agreement), (2) a confidence interval for the score and iii) two theoretical thresholds on the effect size (below 0.4 represent bad agreement, up to 0.7 refers to a good enough agreement and scores higher than 0.7 represent a near perfect agreement).
On the other hand, the performance of the EBM relevance ranking method is tested using the hyperbolic-weighted tau (hwt) statistic73 which measures the correlation between a pair of rankings providing a good tradeoff between lessening the effects of the uninformative parts of a ranking and penalizing the deviations in the informative sections74. The test is a variation of Kendall’s tau, where the correlation between two rankings is corrected using an additive hyperbolic function that penalizes more the discrepancies on the top of the rank than those on the tail. The rank performance is estimated by building a distribution of all the possible hwt pairwise comparisons between the EBM rankings of each data partition (using the same splitting schema as in the stability analysis): a point estimate of 1 represents a perfect agreement, 0 is the score of two random uncorrelated rankings, whereas − 1 represents two opposite rankings.
Software
The Machine Learning and statistical methods have been implemented in Python 3.7 on top of the scikit-learn75 (version 0.23.0), Numpy76 (version 1.18.4) and SciPy77 (version 1.4.1) libraries, whereas the EBMs have been trained and inspected using the InterpretML78 framework (version 0.1.22).
Supplementary Information
Acknowledgements
This work is supported by Grants PID2020-117979RB-I00 from the Spanish Ministry of Science and Innovation and PI20/01305 and IMP/0019 from the ISCIII, both co-funded with European Regional Development Funds (ERDF) as well as H2020 Programme of the European Union Grants Marie Curie Innovative Training Network "Machine Learning Frontiers in Precision Medicine" (MLFPM) (GA 813533) and “ELIXIR-EXCELERATE fast-track ELIXIR implementation and drive early user exploitation across the life sciences” (GA 676559). The postdoctoral contract for CL PAIDI2020- DOC_00350 is funded by Junta de Andalucía and co-funded by the European Social Fund (FSE) 2014-2020.
Author contributions
Conceptualization: C.L., C.S.C.S., J.D. Data curation: C.S.C.S. Formal analysis: C.L. Funding acquisition: J.D., C.L. Investigation: C.L., C.S.C.S., M.P.C., J.D. Project administration: J.D. Supervision: J.D. Writing—original draft: J.D. Writing—review and editing: all. All authors read and approved the final manuscript.
Data availability
The datasets analyzed during the current study are available in the NCBI repository: PRJNA389927, https://www.ncbi.nlm.nih.gov/bioproject/389927. PRJEB12449, https://www.ncbi.nlm.nih.gov/bioproject/310722. PRJEB6070, https://www.ncbi.nlm.nih.gov/bioproject/266076. PRJEB7774, https://www.ncbi.nlm.nih.gov/bioproject/277324. PRJEB10878, https://www.ncbi.nlm.nih.gov/bioproject/297543. PRJNA447983, https://www.ncbi.nlm.nih.gov/bioproject/447983. PRJEB27928, https://www.ncbi.nlm.nih.gov/bioproject/486129. The code used in this work can be found at: https://github.com/babelomics/metagenomic-crc.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Carlos S. Casimiro-Soriguer and Carlos Loucera.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-04182-y.
References
- 1.Cho I, Blaser MJ. The human microbiome: At the interface of health and disease. Nat. Rev. Genet. 2012;13(4):260. doi: 10.1038/nrg3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, Chinwalla AT, et al. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490(7418):55. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 4.Findley K, Williams DR, Grice EA, Bonham VL. Health disparities and the microbiome. Trends Microbiol. 2016;24(11):847–850. doi: 10.1016/j.tim.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snel B, Bork P, Huynen MA. Genome phylogeny based on gene content. Nat. Genet. 1999;21(1):108. doi: 10.1038/5052. [DOI] [PubMed] [Google Scholar]
- 6.Zaneveld JR, Lozupone C, Gordon JI, Knight R. Ribosomal RNA diversity predicts genome diversity in gut bacteria and their relatives. Nucleic Acids Res. 2010;38(12):3869–3879. doi: 10.1093/nar/gkq066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, et al. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428(6978):37. doi: 10.1038/nature02340. [DOI] [PubMed] [Google Scholar]
- 8.Quince C, Walker AW, Simpson JT, Loman NJ, Segata N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017;35(9):833. doi: 10.1038/nbt.3935. [DOI] [PubMed] [Google Scholar]
- 9.Börnigen D, Morgan XC, Franzosa EA, Ren B, Xavier RJ, Garrett WS, et al. Functional profiling of the gut microbiome in disease-associated inflammation. Genome Med. 2013;5(7):65. doi: 10.1186/gm469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scholz M, Ward DV, Pasolli E, Tolio T, Zolfo M, Asnicar F, et al. Strain-level microbial epidemiology and population genomics from shotgun metagenomics. Nat. Methods. 2016;13(5):435. doi: 10.1038/nmeth.3802. [DOI] [PubMed] [Google Scholar]
- 11.Lloyd-Price J, Mahurkar A, Rahnavard G, Crabtree J, Orvis J, Hall AB, et al. Strains, functions and dynamics in the expanded Human Microbiome Project. Nature. 2017;550(7674):61. doi: 10.1038/nature23889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lynch SV, Pedersen O. The human intestinal microbiome in health and disease. N. Engl. J. Med. 2016;375(24):2369–2379. doi: 10.1056/NEJMra1600266. [DOI] [PubMed] [Google Scholar]
- 13.Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, et al. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature. 2013;498(7452):99. doi: 10.1038/nature12198. [DOI] [PubMed] [Google Scholar]
- 14.Bedarf JR, Hildebrand F, Coelho LP, Sunagawa S, Bahram M, Goeser F, et al. Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson’s disease patients. Genome Med. 2017;9(1):39. doi: 10.1186/s13073-017-0428-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng Q, Liang S, Jia H, Stadlmayr A, Tang L, Lan Z, et al. Gut microbiome development along the colorectal adenoma–carcinoma sequence. Nat. Commun. 2015;6:6528. doi: 10.1038/ncomms7528. [DOI] [PubMed] [Google Scholar]
- 16.Zeller G, Tap J, Voigt AY, Sunagawa S, Kultima JR, Costea PI, et al. Potential of fecal microbiota for early-stage detection of colorectal cancer. Mol. Syst. Biol. 2014;10:11. doi: 10.15252/msb.20145645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu J, Feng Q, Wong SH, Zhang D, yi Liang Q, Qin Y, et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut. 2017;66(1):70–78. doi: 10.1136/gutjnl-2015-309800. [DOI] [PubMed] [Google Scholar]
- 18.Jie Z, Xia H, Zhong S-L, Feng Q, Li S, Liang S, et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017;8(1):845. doi: 10.1038/s41467-017-00900-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pasolli E, Truong DT, Malik F, Waldron L, Segata N. Machine learning meta-analysis of large metagenomic datasets: tools and biological insights. PLoS Comput. Biol. 2016;12(7):e1004977. doi: 10.1371/journal.pcbi.1004977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cougnoux A, Dalmasso G, Martinez R, Buc E, Delmas J, Gibold L, et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut. 2014;63(12):1932–1942. doi: 10.1136/gutjnl-2013-305257. [DOI] [PubMed] [Google Scholar]
- 21.Wu S, Rhee K-J, Albesiano E, Rabizadeh S, Wu X, Yen H-R, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009;15(9):1016. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chung L, Orberg ET, Geis AL, Chan JL, Fu K, Shields CED, et al. Bacteroides fragilis toxin coordinates a pro-carcinogenic inflammatory cascade via targeting of colonic epithelial cells. Cell Host Microbe. 2018;23(2):203–214.e55. doi: 10.1016/j.chom.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14(2):207–215. doi: 10.1016/j.chom.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14(2):195–206. doi: 10.1016/j.chom.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Snoek, J., Larochelle, H. & Adams, R. P. Practical bayesian optimization of machine learning algorithms. In: Advances in neural information processing systems. 2951–2959 (2012).
- 26.Baxter NT, Ruffin MT, Rogers MA, Schloss PD. Microbiota-based model improves the sensitivity of fecal immunochemical test for detecting colonic lesions. Genome Med. 2016;8(1):37. doi: 10.1186/s13073-016-0290-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zackular JP, Rogers MA, Ruffin MT, Schloss PD. The human gut microbiome as a screening tool for colorectal cancer. Cancer Prev. Res. 2014;7(11):1112–1121. doi: 10.1158/1940-6207.CAPR-14-0129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wirbel J, Pyl PT, Kartal E, Zych K, Kashani A, Milanese A, et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat. Med. 2019;25(4):679. doi: 10.1038/s41591-019-0406-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas AM, Manghi P, Asnicar F, Pasolli E, Armanini F, Zolfo M, et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med. 2019;25(4):667. doi: 10.1038/s41591-019-0405-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Segata N. On the road to strain-resolved comparative metagenomics. MSystems. 2018;3(2):e00190–e217. doi: 10.1128/mSystems.00190-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Truong DT, Tett A, Pasolli E, Huttenhower C, Segata N. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 2017;27(4):626–638. doi: 10.1101/gr.216242.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerner EW, Meyskens FL., Jr Polyamines and cancer: Old molecules, new understanding. Nat. Rev. Cancer. 2004;4(10):781. doi: 10.1038/nrc1454. [DOI] [PubMed] [Google Scholar]
- 33.Chakraborty, S., Tomsett, R., Raghavendra, R., Harborne, D., Alzantot, M., Cerutti, F., et al. Interpretability of deep learning models: a survey of results. In: 2017 IEEE SmartWorld, Ubiquitous Intelligence & Computing, Advanced & Trusted Computed, Scalable Computing & Communications, Cloud & Big Data Computing, Internet of People and Smart City Innovation (SmartWorld/SCALCOM/UIC/ATC/CBDCom/IOP/SCI). 1–6 (IEEE, 2017).
- 34.Murdoch WJ, Singh C, Kumbier K, Abbasi-Asl R, Yu B. Definitions, methods, and applications in interpretable machine learning. Proc. Natl. Acad. Sci. 2019;116(44):22071–22080. doi: 10.1073/pnas.1900654116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen L, Lu X. Making deep learning models transparent. J. Med. Artif. Intell. 2018;1:5. [Google Scholar]
- 36.Michael KY, Ma J, Fisher J, Kreisberg JF, Raphael BJ, Ideker T. Visible machine learning for biomedicine. Cell. 2018;173(7):1562–1565. doi: 10.1016/j.cell.2018.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang JH, Wright SN, Hamblin M, McCloskey D, Alcantar MA, Schrübbers L, et al. A white-box machine learning approach for revealing antibiotic mechanisms of action. Cell. 2019;177(6):1649–1661.e9. doi: 10.1016/j.cell.2019.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mason C, Afshinnekoo E, Ahsannudin S, Ghedin E, Read T, Fraser C, et al. The metagenomics and metadesign of the subways and urban biomes (MetaSUB) international consortium inaugural meeting report. MICROBIOME. 2016;4(1):24. doi: 10.1186/s40168-016-0168-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casimiro-Soriguer CS, Loucera C, Perez Florido J, López-López D, Dopazo J. Antibiotic resistance and metabolic profiles as functional biomarkers that accurately predict the geographic origin of city metagenomics samples. Biol. Direct. 2019;14(1):15. doi: 10.1186/s13062-019-0246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hannigan GD, Duhaime MB, Ruffin MT, Koumpouras CC, Schloss PD. Diagnostic potential and interactive dynamics of the colorectal cancer virome. MBio. 2018;9:6. doi: 10.1128/mBio.02248-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ojala M, Garriga GC. Permutation tests for studying classifier performance. J. Mach. Learn. Res. 2010;11:6. [Google Scholar]
- 42.Vogtmann E, Hua X, Zeller G, Sunagawa S, Voigt AY, Hercog R, et al. Colorectal cancer and the human gut microbiome: reproducibility with whole-genome shotgun sequencing. PLoS ONE. 2016;11(5):e0155362. doi: 10.1371/journal.pone.0155362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Voigt AY, Costea PI, Kultima JR, Li SS, Zeller G, Sunagawa S, et al. Temporal and technical variability of human gut metagenomes. Genome Biol. 2015;16(1):73. doi: 10.1186/s13059-015-0639-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ternes D, Karta J, Tsenkova M, Wilmes P, Haan S, Letellier E. Microbiome in colorectal cancer: How to get from meta-omics to mechanism? Trends Microbiol. 2020;28(5):401–423. doi: 10.1016/j.tim.2020.01.001. [DOI] [PubMed] [Google Scholar]
- 45.Zhang S, Yang Y, Weng W, Guo B, Cai G, Ma Y, et al. Fusobacterium nucleatum promotes chemoresistance to 5-fluorouracil by upregulation of BIRC3 expression in colorectal cancer. J. Exp. Clin. Cancer Res. 2019;38(1):1–13. doi: 10.1186/s13046-018-0985-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell. 2017;170(3):548–563.e16. doi: 10.1016/j.cell.2017.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fazzari J, Linher-Melville K, Singh G. Tumour-derived glutamate: linking aberrant cancer cell metabolism to peripheral sensory pain pathways. Curr. Neuropharmacol. 2017;15(4):620–636. doi: 10.2174/1570159X14666160509123042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weir TL, Manter DK, Sheflin AM, Barnett BA, Heuberger AL, Ryan EP. Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PLoS ONE. 2013;8(8):e70803. doi: 10.1371/journal.pone.0070803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kojima M, Morisaki T, Izuhara K, Uchiyama A, Matsunari Y, Katano M, et al. Lipopolysaccharide increases cyclo-oxygenase-2 expression in a colon carcinoma cell line through nuclear factor-κB activation. Oncogene. 2000;19(9):1225. doi: 10.1038/sj.onc.1203427. [DOI] [PubMed] [Google Scholar]
- 50.Yoshioka T, Morimoto Y, Iwagaki H, Itoh H, Saito S, Kobayashi N, et al. Bacterial lipopolysaccharide induces transforming growth factor β and hepatocyte growth factor through Tolllike receptor 2 in cultured human colon cancer cells. J. Int. Med. Res. 2001;29(5):409–420. doi: 10.1177/147323000102900505. [DOI] [PubMed] [Google Scholar]
- 51.Ng O. Iron, microbiota and colorectal cancer. Wien. Med. Wochenschr. 2016;166(13–14):431–436. doi: 10.1007/s10354-016-0508-4. [DOI] [PubMed] [Google Scholar]
- 52.Bleich RM, Arthur JC. Revealing a microbial carcinogen. Science. 2019;363(6428):689–690. doi: 10.1126/science.aaw5475. [DOI] [PubMed] [Google Scholar]
- 53.Tian J, Xue W, Yin H, Zhang N, Zhou J, Long Z, et al. Differential metabolic alterations and biomarkers between gastric cancer and colorectal cancer: A systematic review and meta-analysis. Onco Targets Ther. 2020;13:6093–6108. doi: 10.2147/OTT.S247393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huerta-Cepas, J., Szklarczyk, D., Heller, D., Hernández-Plaza, A., Forslund, S. K., Cook, H., et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res.47(D1), D309–D14 (2019). [DOI] [PMC free article] [PubMed]
- 55.Beane J, Campbell JD, Lel J, Vick J, Spira A. Genomic approaches to accelerate cancer interception. Lancet Oncol. 2017;18(8):e494–e502. doi: 10.1016/S1470-2045(17)30373-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Costea PI, Coelho LP, Sunagawa S, Munch R, Huerta-Cepas J, Forslund K, et al. Subspecies in the global human gut microbiome. Mol. Syst. Biol. 2017;13(12):960. doi: 10.15252/msb.20177589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.R Core Team: R: A Language and Environment for Statistical Computing. http://www.R-project.org (2021).
- 58.Pasolli E, Schiffer L, Manghi P, Renson A, Obenchain V, Truong DT, et al. Accessible, curated metagenomic data through ExperimentHub. Nat. Methods. 2017;14(11):1023. doi: 10.1038/nmeth.4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huber W, Carey VJ, Gentleman R, Anders S, Carlson M, Carvalho BS, et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods. 2015;12(2):115. doi: 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coelho LP, Alves R, Monteiro P, Huerta-Cepas J, Freitas AT, Bork P. NG-meta-profiler: Fast processing of metagenomes using NGLess, a domain-specific language. Microbiome. 2019;7(1):84. doi: 10.1186/s40168-019-0684-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42(Database issue):D199–205. doi: 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, et al. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014;32(8):834. doi: 10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- 65.Kim D, Song L, Breitwieser FP, Salzberg SL. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 2016;26(12):1721–1729. doi: 10.1101/gr.210641.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lou, Y., Caruana, R. & Gehrke, J. Intelligible models for classification and regression. In: Proceedings of the 18th ACM SIGKDD International Conference on KNOWLEDGE DISCovery and Data Mining. 150–158 (2012).
- 67.Lou, Y., Caruana, R., Gehrke, J. & Hooker, G. Accurate intelligible models with pairwise interactions. In: Proceedings of the 19th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. 623–631 (2013).
- 68.Caruana, R., Lou, Y., Gehrke, J., Koch, P., Sturm, M. & Elhadad, N. Intelligible models for healthcare: Predicting pneumonia risk and hospital 30-day readmission. In: Proceedings of the 21th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. 1721–1730 (2015).
- 69.Hastie T, Tibshirani R. Generalized additive models: Some applications. J. Am. Stat. Assoc. 1987;82(398):371–386. [Google Scholar]
- 70.Lundberg, S. M. & Lee, S. -I. A unified approach to interpreting model predictions. In: Advances in Neural Information Processing Systems. 4765–4774 (2017).
- 71.Ribeiro, M. T., Singh, S. & Guestrin, C. Why should I trust you? Explaining the predictions of any classifier. In: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. 1135–1144 (ACM, 2016).
- 72.Nogueira S, Sechidis K, Brown G. On the stability of feature selection algorithms. J. Mach. Learn. Res. 2017;18(1):6345–6398. [Google Scholar]
- 73.Shieh GS. A weighted Kendall's tau statistic. Stat. Probab. Lett. 1998;39(1):17–24. [Google Scholar]
- 74.Vigna, S. A weighted correlation index for rankings with ties. In: Proceedings of the 24th International Conference on World Wide Web. 1166–1176 (2015).
- 75.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011;12(Oct):2825–2830. [Google Scholar]
- 76.Harris CR, Millman KJ, van der Walt SJ, Gommers R, Virtanen P, Cournapeau D, et al. Array programming with NumPy. Nature. 2020;585(7825):357–362. doi: 10.1038/s41586-020-2649-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods. 2020;17(3):261–272. doi: 10.1038/s41592-019-0686-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nori, H., Jenkins, S., Koch, P. & Caruana, R. InterpretML: A unified framework for machine learning interpretability. arXiv:190909223 (2019).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets analyzed during the current study are available in the NCBI repository: PRJNA389927, https://www.ncbi.nlm.nih.gov/bioproject/389927. PRJEB12449, https://www.ncbi.nlm.nih.gov/bioproject/310722. PRJEB6070, https://www.ncbi.nlm.nih.gov/bioproject/266076. PRJEB7774, https://www.ncbi.nlm.nih.gov/bioproject/277324. PRJEB10878, https://www.ncbi.nlm.nih.gov/bioproject/297543. PRJNA447983, https://www.ncbi.nlm.nih.gov/bioproject/447983. PRJEB27928, https://www.ncbi.nlm.nih.gov/bioproject/486129. The code used in this work can be found at: https://github.com/babelomics/metagenomic-crc.