

Abstract
Background & Aims
Intrahepatic vascular resistance is increased in early non-alcoholic fatty liver disease (NAFLD), potentially leading to tissue hypoxia and triggering disease progression. Hepatic vascular hyperreactivity to vasoconstrictors has been identified as an underlying mechanism. This study investigates vasoconstrictive agonism and antagonism in 2 models of early NAFLD and in non-alcoholic steatohepatitis (NASH).
Methods
The effects of endothelin-1 (ET-1), angiotensin II (ATII) and thromboxane A2 (TxA2) agonism and antagonism were studied by in situ ex vivo liver perfusion and preventive/therapeutic treatment experiments in a methionine-choline-deficient diet model of steatosis. Furthermore, important results were validated in Zucker fatty rats after 4 or 8 weeks of high-fat high-fructose diet feeding. In vivo systemic and portal pressures, ex vivo transhepatic pressure gradients (THPG) and transaminase levels were measured. Liver tissue was harvested for structural and mRNA analysis.
Results
The THPG and consequent portal pressure were significantly increased in both models of steatosis and in NASH. ET-1, ATII and TxA2 increased the THPG even further. Bosentan (ET-1 receptor antagonist), valsartan (ATII receptor blocker) and celecoxib (COX-2 inhibitor) attenuated or even normalised the increased THPG in steatosis. Simultaneously, bosentan and valsartan treatment improved transaminase levels. Moreover, bosentan was able to mitigate the degree of steatosis and restored the disrupted microvascular structure. Finally, beneficial vascular effects of bosentan endured in NASH.
Conclusions
Antagonism of vasoconstrictive mediators improves intrahepatic vascular function. Both ET-1 and ATII antagonists showed additional benefit and bosentan even mitigated steatosis and structural liver damage. In conclusion, vasoconstrictive antagonism is a potentially promising therapeutic option for the treatment of early NAFLD.
Lay summary
In non-alcoholic fatty liver disease (NAFLD), hepatic blood flow is impaired and the blood pressure in the liver blood vessels is increased as a result of an increased response of the liver vasculature to vasoconstrictors. Using drugs to block the constriction of the intrahepatic vasculature, the resistance of the liver blood vessels decreases and the increased portal pressure is reduced. Moreover, blocking the vasoconstrictive endothelin-1 pathway restored parenchymal architecture and reduced disease severity.
Keywords: non-alcoholic fatty liver disease, transhepatic pressure gradient, portal hypertension, endothelin-1, thromboxane A2, angiotensin II
Abbreviations: ALT, alanine aminotransferase; ARB, angiotensin receptor blocker; AST, aspartate aminotransferase; ATII, angiotensin II; COX, cyclooxygenase; ET, endothelin; HFHFD, high-fat high-fructose diet; IHVR, intrahepatic vascular resistance; Jak2, Janus-kinase-2; MCD, methionine-choline deficient diet; Mx, methoxamine; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; NO, nitric oxide; PP, portal pressure; PR, pulse rate; SEM, scanning electron microscopy; TEM, transmission electron microscopy; TBW, total body weight; TxA2, thromboxane A2; TXAS, thromboxane synthase; ZFR, Zucker fatty rats
Graphical abstract

Highlights
-
•
The transhepatic pressure gradient and thus portal pressure are increased in severe hepatic steatosis.
-
•
Vasoconstrictor antagonists attenuate the transhepatic gradient to near normal in steatosis.
-
•
Vasoconstrictor antagonists attenuate the transhepatic gradient in steatosis.
-
•
Bosentan and valsartan attenuate increased transaminase levels in severe steatosis.
-
•
Bosentan treatment decreases steatosis and restores the microvascular architecture.
Introduction
Non-alcoholic fatty liver disease (NAFLD) represents a spectrum of liver disease primarily characterised by macrovesicular fat accumulation in hepatocytes. Isolated steatosis can progress to non-alcoholic steatohepatitis (NASH), indicating the presence of inflammation and hepatocellular damage, with or without concurrent fibrosis and eventually leading to hepatocellular carcinoma and/or cirrhosis and its complications. Despite the increasing prevalence and associated morbidity and mortality, no pharmacological therapy is currently approved for the treatment of NAFLD.1 A better understanding of the pathophysiology of NAFLD is pivotal in the search for therapeutic targets.
The hepatic vasculature might play an important role in the progression of NAFLD.2,3 In both humans and animal models, increased intrahepatic vascular resistance (IHVR) has been observed in early NAFLD.[4], [5], [6], [7], [8] Via impaired intrahepatic blood flow and thereby inadequate hepatic blood perfusion, subsequent relative hypoxia of the liver might trigger disease progression.2
Structurally, architectural disruption of the normal hepatic vasculature and swelling of hepatocytes due to intracellular fat accumulation and ballooning (degeneration of the cell-shaping cytoskeleton) leads to compression of the sinusoids and increases IHVR.6,9,10 Functionally, disturbances in concentrations of and reactivity to vaso-active mediators result in intrahepatic vascular dysfunction. Hepatic vascular dysfunction means the inability of the hepatic vasculature to adapt its tone adequately, in favour of vasoconstriction rather than vasodilation. This might be a result of disturbances in either the concentrations of vasoactive substances or the reactivity of the hepatic vascular tone to these stimuli.[6], [7], [8]
So far, and in line with findings in other causes of portal hypertension like cirrhosis, research has mainly focused on impaired vasodilation as the main mechanism underlying vascular dysfunction. However, in a previous study, the effects of nitric oxide (NO) blockage on the transhepatic pressure gradient (THPG) during perfusion experiments were little and constrained to higher flows. Stimulation of NO production via acetylcholine or direct administration of the NO donor sodium nitroprusside did not affect the elevated THPG.7 Meanwhile, the possible role of vasoconstrictors and hyperreactivity thereto has only recently gained more attention.7 Therefore, the aim of this study was to elucidate the role of various endogenous vasoconstrictors in the increased IHVR in early NAFLD.
Materials and methods
Animal models
Male Wistar Han rats (Charles River, France; 200-250 g) were fed a control diet (ICN Biomedicals SA, Asse, Belgium) or a methionine-choline-deficient diet (MCDD, Envigo RMS B.V., Indianapolis, Indiana USA) for 4 weeks, which is known to induce severe steatosis in the absence of NASH in this rat strain.6,7
To study the effects of in vivo treatment with vasoconstrictor antagonists, rats (n = 6-8/group) were gavaged daily during the complete 4 weeks of diet as a preventive treatment or during the last 2 out of 4 weeks of diet as a therapeutic treatment (Fig. S1). The following treatments were studied: 100 mg/kg/d bosentan (dual endothelin receptor antagonist), 30 mg/kg/d valsartan (angiotensin receptor blocker [ARB]) or 30 mg/kg/d celecoxib (cyclooxygenase [COX]-2-inhibitor). Doses were based on literature.[11], [12], [13] All details on drugs and doses are described in the supplementary materials and methods.
To exclude model specificity, the most important results were tested in a second model. Male Zucker fatty rats (ZFR, fa/fa) (Charles River, Italy; 7 weeks old, n = 5-7/group) were fed a high-fat high-fructose diet (HFHFD, D16042610, Research Diets, New Brunswick, NJ, USA) and compared to lean control rats (fa/+) fed a control diet during 4 weeks. Additionally, ZFR were treated orally with bosentan (100 mg/kg/d) or placebo preventively during the complete 4 weeks of HFHFD (Fig. S1).
Additionally, to investigate whether the alterations were also relevant at the stage of steatohepatitis, ZFR fed 8 weeks of HFHFD (to establish NASH) were treated orally with bosentan (100 mg/kg/d) or placebo during the complete 8 weeks of diet (preventive setting) and compared to lean controls fed a control diet for 8 weeks.
All animals were kept in a 12 h:12 h light/dark cycle with controlled temperature and humidity in enriched cages of 2 animals with free access to food and water. All animals were treated according to the ARRIVE guidelines.14 The protocols were approved by the Antwerp University ethical committee on animal experiments (ECD 2016-66 and 2020-14).
Ex vivo liver perfusion flow-pressure measurements
The IHVR was studied by measuring the THPG directly by in situ ex vivo liver perfusion experiments.7 Briefly, after anaesthesia (see supplementary materials and methods), the abdomen was opened via median laparotomy, vascular structures were identified and heparin (1,400 U/kg) was injected intravenously in the caudal caval vein. Subsequently, the portal vein was cannulated with a 14 G catheter, the thoracic cavity was opened and the suprahepatic caval vein was cannulated through the right atrium with a 16 G catheter. Next, the liver was perfused in a single-pass way by oxygenated Krebs-Ringer (Krebs) solution (37ᵒC) and the catheters were connected to pressure and flow monitoring equipment (Powerlab 8/30 and LabChart 7, AD Instruments, Oxford, UK). In all experiments, the portal (inflow) and caval (outflow) pressure, that was kept at a constant level of -1 mmHg, were measured continuously and the THPG was calculated by subtracting the outflow from the inflow pressure.7
Dose-response experiments
In dose-response experiments, the following vasoconstrictors and antagonists were tested at a constant flow rate of 30 ml/min: methoxamine (Mx, α1-adrenergic agonist 1-300 μM); BQ-123 (endothelin [ET]A-receptor antagonist, 1.3-400 nM) and BQ-788 (ETB-receptor antagonist, 0.12-36 nM) in the presence of endothelin-1 (ET-1, 0.1 nM); angiotensin II (ATII, 0.003-1 μM); valsartan (ARB, 0.2-60 μM) in the presence of ATII (10 nM); U46619 (thromboxane A2 [TxA2] agonist, 0.35-105 nM); SC-560 (COX-1 inhibitor, 0.3-1 μM) and SC-236 (COX-2-inhibitor, 0.003-1 μM) in the presence of Mx (10 μM). Antagonists were always studied in the presence of their vasoconstrictive agonist because the basal THPG in the absence of a vasoconstrictor is too low to detect an effect on the THPG. Mx was used with COX-inhibition instead of the TxA2 agonist, because TxA2 is only synthesized downstream of COX.
Flow-pressure experiments after in vivo treatment
After the evaluation of vasoconstrictors and their antagonists ex vivo, the effects of in vivo oral preventive/therapeutic bosentan, valsartan, celecoxib and placebo treatment were studied in flow-pressure measurements of the hepatic vasculature, in which the flow was gradually increased from 10 to 50 ml/min, at the doses described above (n = 6-8/group).
In vivo pressure measurements
Under anaesthesia, a tracheal tube (PE 240 ID 1.67 mm OD 2.42 mm Intramedic Clay Adams brand non-radiopaque polyethylene tubing) was inserted by tracheostomy. A 24 G catheter was inserted into the carotid artery. The abdomen was subsequently opened by median incision. The portal vein was exposed and canulated with a 24 G catheter under microscopy. The inferior caval vein was canulated with a 22 G catheter, which was advanced with the tip into the retrohepatic part of the inferior caval vein. The different catheters were connected to an in-house pressure monitoring equipment. Carotid artery pressure (mean arterial blood pressure), pulse rate (PR), portal pressure (PP) and inferior caval vein pressure were measured.
Blood analysis
Before liver perfusion, venous blood was drawn from the caval vein and samples were centrifuged for 15 min at 3,500 revolution/min. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) levels were measured via photometry (Atellica, Siemens Healthcare Diagnostics, Deerfield, CT, USA).
Histology
Rats were weighed, anaesthetised and sacrificed. Livers were weighed, random samples were fixed in 4% formaldehyde and embedded in paraffin. Subsequently, 5 μm sections were stained with haematoxylin-eosin, Masson’s trichrome, picrosirius red and reticulin according to standard laboratory protocols. The degree of steatosis was morphometrically measured as the percentage of fat over total liver surface at 10x magnification images of haematoxylin-eosin-stained liver samples, using ImageJ software (Bethesda, MD, USA).
Vascular corrosion casting
Rats were sacrificed and vascular corrosion casts were prepared as previously described6 (Fig. S2) and examined systematically by scanning electron microscopy (SEM) (Jeol JSM 5600 LV, Jeol).
Transmission electron microscopy
Rats were sacrificed and underwent liver perfusion fixation with glutaraldehyde. Tissues were fixed in 0.1 M sodium cacodylate-buffered (pH 7.4) 2.5% glutaraldehyde solution at room temperature, and washed 3x in 0.1 M sodium cacodylate-buffered (pH 7.4) 7.5% saccharose solution. Post-fixation was performed by incubating cells for 2 h with 1% OsO4 solution. After dehydration in an ethanol gradient, samples were embedded in EM-bed812. Ultrathin sections were stained with uranyl acetate and lead citrate, and examined in a Tecnai G2 Spirit Bio TWIN microscope (Fei, Europe BV, Zaventem, Belgium) at 120 kV.
mRNA analysis
Random liver samples of rats sacrificed for histology were processed as described before.15 Results were normalised in the nSolver Analysis Software (NanoString Technologies) by the geometric mean of 5 housekeeping genes (Table S1).
Statistics
All data are presented as the mean ± standard error of the mean. p values <0.05 were considered significantly different.
The THPG data were analysed using a generalised estimating equation model followed by least significant difference post hoc testing when appropriate, using SPPS v24.0 (IBM, Armonk, NY, USA).
Concerning mRNA analysis, heat maps were generated using the nSolver Analysis Software (NanoString). Normalised gene counts and all other results were analysed with a two-way ANOVA (with the diet as the first factor [between], the treatment used as the second factor [within]) and Scheffé post hoc testing when appropriate, using SPSS.
Results
Early NAFLD
After 4 weeks of MCDD, animals developed marked hepatomegaly (liver/total body weight (TBW)-ratio: steatosis 5.4±0.2% vs. controls 3.2±0.1%, p <0.01 [Table S2]). In ZFR after 4 weeks of HFHFD, body and liver weight increased significantly compared to controls (ZFR 468.1±10.3 g vs. controls 294.4±8.9 g, p <0.001; liver/TBW-ratio: ZFR 4.6±0.1% vs. controls 3.9±0.2%, p <0.05 [Table S3]).
The increased liver volume corresponded with histology, confirming moderate to severe steatosis in all MCDD-fed rats without microscopic features of inflammation, ballooning (Fig. 1) or fibrosis (Fig. S3). These results confirm the presence of severe steatosis without any histological features of steatohepatitis after 4 weeks of MCDD. Likewise, histology in ZFR demonstrated moderate hepatic steatosis without features of NASH or fibrosis after 4 weeks of HFHFD (Fig. S4).
Fig. 1.

Histological liver sections (H&E stain, original magnification 10x) of rats fed control or methionine-choline-deficient diet (steatosis) after preventive placebo, bosentan, valsartan or celecoxib treatment.
Structural assessment of early NAFLD
In controls, SEM of liver vascular corrosion casts demonstrated a regular pattern of the sinusoids organised in lobules, with small sinusoids that have even diameters. In the MCDD-fed group, the regular sinusoidal pattern was disrupted, resulting in a disorganised tangle of vessels. The sinusoids in this irregular organised pattern have uneven and larger diameters. Furthermore, many vessels appear to branch into dead-ending dilated vessel stumps, known as blebs (Fig. 2).
Fig. 2.
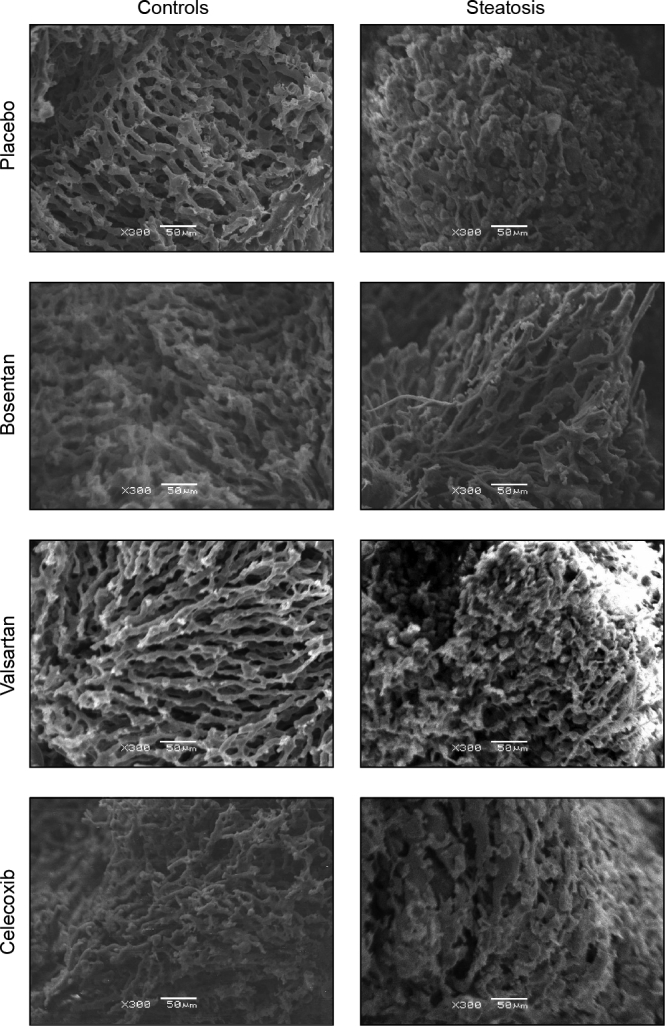
Representative scanning electron microscopy images of hepatic vascular corrosion casts of rats fed control or methionine-choline-deficient diet (steatosis) after preventive placebo, bosentan, valsartan or celecoxib treatment (original magnification 300x).
In controls, sinusoids are regular with small and even diameters. In steatosis, the number of sinusoids is reduced, the vascular pattern is irregular and the remaining sinusoids appear to have uneven and larger diameters with numerous blebs (dead-ending vessel stumps). After bosentan treatment, the number of sinusoids remains reduced, but the diameter is more comparable to control livers and the pattern appears more regular with less blebs.
Besides our SEM images, additional transmission electron microscopy (TEM) of liver tissue was performed to evaluate the morphology of the liver sinusoids and sinusoidal endothelial cells. In 4-week HFHFD-fed ZFR, it can be observed that the sinusoids are compressed, the endothelium is thicker and fenestrae appear to be more constricted compared to controls (Fig. S5). These findings are in in line with observations on histology and SEM.3,4
Liver perfusion experiments
In all experiments, the baseline THPG in rats with steatosis was significantly increased compared to controls independent of flow, both in MCDD-fed rats (steatosis 8.4±0.3 mmHg vs. controls 6.6 ± 0.5 mmHg at 30 ml/min, p <0.01 [Fig. S6]) and ZFR (ZFR 7.4±0.5 mmHg vs. controls 5.1±0.6 mmHg at 30 ml/min, p <0.01 [Fig. S7A]).
There was non-significant hyperreactivity to Mx in ZFR compared to controls (ΔEmax: ZFR +8.8±1.0 mmHg vs. controls +6.3±0.9 mmHg, p = 0.092 [Fig. S7B+C]).
Haemodynamic measurements
Although statistical significance was not reached (which could be related to the small numbers), the in vivo THPG was numerically higher in steatotic compared to control livers in vivo (steatosis 5.0±0.6 mmHg vs. controls 3.7±0.3 mmHg, p = 0.062) and again lower in bosentan-treated livers, with values close to those in controls (4.1±0.2 mmHg, p = 0.610 compared to controls)(Table S4).
Endothelin-1-mediated mechanisms
Liver perfusion experiments: dose-response curves
We previously demonstrated hyperreactivity of the hepatic vasculature to ET-1 in MCDD-fed rats.7
Blocking the ETA-receptor by the addition of BQ-123 in increasing doses to the perfusion fluid (which contains 0.1 nM ET-1) at a constant flow of 30 ml/min, induced a significant decline in THPG compared to perfusion by Krebs (containing 0.1 nM ET-1 alone) without BQ-123. The decrease of the THPG by BQ-123 was more pronounced in MCDD-fed rat livers compared to controls and was most obvious at a dose of 400 nM BQ-123 (steatosis + placebo 24.9±4.5 mmHg vs. steatosis + BQ-12316.1±1.1 mmHg, p <0.05). BQ-123 even restored the THPG of MCDD-fed rats to the level of controls from a dose of 13 nM BQ-123 onwards (steatosis with 13 nM BQ-132 and 0.1 nM ET-1 14.2±0.7 mmHg vs. controls with 0.1 nM ET-1 13.3±1.2 mmHg, p = 0.307 [Fig. 3A]). Blocking the ETB-receptor by BQ-788 did not change the THPG (Fig. 3B). Because the effects of ET-1 have some delay, an upward slope of the placebo-treated groups is observed in the dose-response curves of these figures.
Fig. 3.

Transhepatic pressure gradient with modulation of endothelin-1 pathways.
(A) Dose-response curves of the transhepatic pressure gradient to ETA-receptor blocker BQ-123 with endothelin-1. (B) Dose-response curves of the transhepatic pressure gradient to ETB-receptor blocker BQ-788 with endothelin-1. (C) Flow-pressure curves of control and steatotic rat hepatic vasculature after preventive bosentan. (D) Flow-pressure curves of control and steatotic rat hepatic vasculature after therapeutic bosentan. All data were analysed using the generalised estimating equation model. Significances between controls with or without compound/treatment are indicated by black signs. Significances between steatosis with or without compound/treatment are indicated by red signs. ∗p <0.05; ∗∗p <0.01; n.s., not significant.
Liver perfusion experiments: flow-pressure curves
In line with the decreased THPG after ETA antagonism, preventive oral treatment of MCDD-fed rats with bosentan significantly decreased and normalised the THPG to the level of control livers (controls + bosentan 12.6±0.3 mmHg vs. controls + placebo 12.6±0.5 mmHg, p = 0.547; steatosis + bosentan 13.4±0.4 mmHg vs. steatosis + placebo 15.4±0.5 mmHg at 50 ml/min, p <0.01 [Fig. 3C]). In therapeutically bosentan-treated rats, we observed a similar decline of the THPG. The difference between control and MCDD-fed rat livers became non-significant, meaning that bosentan normalised the THPG in MCDD-fed rat livers (controls + placebo 13.6±0.6 mmHg vs. steatosis + bosentan 14.6±0.3 mmHg at 50 ml/min, p = 0.085 [Fig. 3D]).
After preventive bosentan treatment, the THPG in 4-week HFHFD-fed ZFR showed lower values at higher flows compared to placebo-treated ZFR, most prominent at 45 ml/min, but without reaching statistical significance (bosentan-treated 9.9±0.6 mmHg vs. placebo-treated 11.4±1.0 mmHg at 45 ml/min, p = 0.162 [Fig. S7A]).
Finally, preventive bosentan treatment could also attenuate the increased PP in vivo in rats with MCDD-induced steatosis (bosentan-treated 4.8±0.2 mmHg vs. placebo-treated 5.7±0.7 mmHg, p = 0.209 [Table S4]).
Weight
Liver/TBW-ratio were lower in bosentan-treated (4.6±0.1%) compared to placebo-treated MCDD-fed rats (5.4±0.2% p <0.05 [Table S2]). Bosentan treatment did not decrease liver weight in ZFR (Table S3).
Blood analysis
ALT levels were significantly increased in MCDD-fed rats (94.9±14.0 U/L) compared to controls (43.6±3.1 U/L), whereas preventive bosentan-treated rats with steatosis showed ALT levels comparable to control rats (48.4±3.0 U/L, p <0.05 compared to placebo-treated steatosis and p = 0.292 compared to controls [Fig. 4A]). A non-significant reduction in AST was observed after preventive bosentan treatment in steatosis (Fig. 4B). In therapeutically bosentan-treated rats with steatosis, we observed a trend towards decreased transaminase levels (Fig. 4C,D).
Fig. 4.

Serum ALT and AST in rats fed control or methionine-choline-deficient diet (steatosis) after preventive/therapeutic placebo, bosentan, valsartan or celecoxib treatment.
Statistic comparison by two-way ANOVA and post hoc Scheffé. ∗p <0.05; ∗∗p <0.01; ∗∗∗p <0.001. ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Likewise, ALT and AST levels were significantly increased in ZFR compared to controls (ALT: ZFR 119.2±12.1 vs. controls 31.0±7.0, p <0.05; AST: ZFR 257.0±40.1 vs. controls 70.5±5.1, p <0.001 [Fig. S8]). Preventive bosentan treatment significantly decreased AST levels to the level of controls (bosentan-treated ZFR 69.0±9.0 U/L, p <0.001 compared to placebo-treated ZFR and p = 1.00 compared to controls) and induced non-significantly decreased ALT levels (Fig. S8). It should be noted that blood samples of the ZFR were more difficult to analyse due to a high lipid content in the blood, resulting in a small sample size.
Microscopy and SEM
The degree of steatosis (% fat of total liver surface) was significantly lower in preventively bosentan-treated rats compared to placebo after 4 weeks of MCDD (bosentan-treated 55.2±1.8% vs. placebo-treated 61.1±1.2%, p <0.05). A trend towards a lower degree of steatosis was also observed with therapeutic bosentan treatment after 4 weeks of MCDD (bosentan-treated 60.0±1.9% vs. placebo-treated 64.0±2.7%, p = 0.25). Results were repeated in ZFR after 4 weeks of HFHFD (Fig. S4).
SEM of vascular corrosion casts of MCDD-fed rat livers after bosentan treatment demonstrated a remarkable improvement of the sinusoidal organisation compared to placebo-treated MCDD-fed rat livers, with a more regular and untangled pattern, reminiscent to that of controls, and a decrease in the number of blebs (Fig. 2).
Transcriptomics
Hepatic mRNA expression of Edn1 (encoding preproendothelin-1) was upregulated in MCDD-fed rats, while Ece1 gene expression was downregulated. Preventive bosentan treatment normalised Edn1 expression (Fig. 5).
Fig. 5.

Effect of bosentan, celecoxib and valsartan treatment on mRNA expression of genes related to endothelin-1-, angiotensin II- and cyclooxygenase-related pathways. Heat maps were generated using the nSolver Analysis Software (NanoString). Normalised gene counts and all other results were analysed with a two-way ANOVA (with the diet as the first factor [between], the treatment used as the second factor [within]) and Scheffé post hoc testing when appropriate.
BOS, bosentan; CD, control diet; CEL, celecoxib; MCDD, methionine-choline-deficient diet; VAL, valsartan.
Angiotensin II-mediated mechanisms
Liver perfusion experiments: dose-response curves
ATII induced an initial increase of the THPG with a maximum effect at 0.01 μM, and subsequently a gradual decrease of the THPG at increasing doses both in control and in MCDD-fed rats (controls: 12.6±1.7 with 0.01 μM ATII, 187.4% maximal increase from baseline; steatosis: 16.6±2.0 mmHg with 0.01 μM ATII, 199.6% maximal increase from baseline [Fig. 6A]).
Fig. 6.

Transhepatic pressure gradient with modulation of angiotensin II pathways.
(A) Dose-response curves of the transhepatic pressure gradient to angiotensin II or Krebs in livers of control and methionine-choline-deficient diet-fed rats (steatosis). (B) Dose-response curves of the transhepatic pressure gradient to the angiotensin receptor blocker valsartan or Krebs with angiotensin II. (C) Flow-pressure curves of control and steatotic hepatic vasculature after preventive valsartan. (D) Flow-pressure curves after therapeutic valsartan. All data were analysed using the generalised estimating equation model. Significances between controls with or without compound/treatment are indicated by black signs. Significances between steatosis with or without compound/treatment are indicated by purple signs. ∗p <0.05; ∗∗p <0.01; ∗∗∗p <0.001; n.s., not significant.
The response of the THPG to a constant dose of ATII in MCDD-fed rat livers was shifted downwards and the downward slope was increased by increasing doses of valsartan (controls: 15.0±0.4 mmHg with 10 nM ATII to 12.8±0.6 mmHg with 10 nM ATII and 0.2 μM valsartan, p <0.001; steatosis: 18.6±0.6 mmHg with 10 nM ATII to 16.9±0.8 mmHg with 10 nM ATII and 0.2 μM valsartan, p = 0.064). The THPG curve of ATII and valsartan-perfused MCD-fed rat livers diverged from the THPG curve in MCD-fed rat livers that were perfused with ATII alone and decreased to the level of (and below) the THPG of control livers perfused with ATII alone (controls with 10 nM ATII 10.1±0.7 mmHg vs. steatosis with 10 nM ATII and 6 μM valsartan 9.6±0.4 mmHg, p = 0.536 [Fig. 6B]).
Liver perfusion experiments: flow-pressure curves
In all rats, preventive valsartan treatment resulted in a significant decrease of the THPG compared to placebo, most pronounced at 50 ml/min (valsartan-treated steatosis 13.5±0.6 mmHg vs. placebo-treated steatosis 15.6±0.6 mmHg, p <0.01; valsartan-treated controls 11.4±0.6 mmHg vs. placebo-treated controls 13.4±0.4 mmHg, p <0.001). Moreover, the THPG in MCD-fed rats treated with valsartan was normalised to the level of control livers treated with placebo (Fig. 6C). Likewise, therapeutic valsartan treatment induced a significant decrease of the THPG in control livers, but not in the livers of MCD-fed rats (Fig. 6D).
Weight
Liver weight and liver/TBW-ratio were lower in valsartan-treated compared to placebo-treated MCD-fed rats (liver/TBW: valsartan-treated 4.6±0.2% vs. placebo-treated 5.4±0.2%, p <0.05 [Table S2]).
Blood analysis
In valsartan-treated MCD-fed rats, ALT and AST levels were significantly reduced and normalised (preventively valsartan-treated 80.8±30.4 U/L ALT and 63.0±15.0 U/L AST vs. placebo-treated 142.3±16.4 U/L ALT [p <0.05] and 144.2±22.2 U/L AST [p <0.01][Fig. 4E-H]).
Microscopy and SEM
Despite lower liver weight and transaminase levels, valsartan treatment did not affect liver histology or the hepatic microvasculature as observed by SEM in MCD-fed rats (Fig. 1, Fig. 2 and Fig. S3).
Transcriptomics
Hepatic mRNA expression of Agt and Agtr1a were downregulated while Ace expression was upregulated in MCD-fed rats (Fig. 5). Preventive valsartan treatment increased the transcription of Agtr1a.
Cyclooxygenase-related mechanisms
Liver perfusion experiments: dose-response curves
The TXA2 agonist U46619 significantly increased the THPG dose-dependently compared to Krebs (controls 6.6±0.8 with Krebs to 14.8±1.5 mmHg with 10.5 nM U46619, p <0.001; steatosis 8.6±0.7 with Krebs to 17.2±1.9 mmHg with 10.5 nM U46619, p <0.001 [Fig. 7A]).
Fig. 7.

Transhepatic pressure gradient with modulation of thromboxane pathways.
(A) Dose-response curves of the transhepatic pressure gradient to thromboxane agonist U-46619 or Krebs in control and methionine-choline-deficient diet-fed rats (steatosis). (B) Dose-response curves of the transhepatic pressure gradient to COX-1 antagonist SC-560 or Krebs with methoxamine. (C) Dose-response curves of the transhepatic pressure gradient to COX-2 antagonist SC-236 or Krebs with methoxamine. (D) Flow-pressure curves of control and steatotic hepatic vasculature with preventive celecoxib. (E) Flow-pressure curves with therapeutic celecoxib. All data were analysed using the generalised estimating equation model. Significances between controls with or without compound/treatment are indicated by black signs. Significances between steatosis with or without compound/treatment are indicated by purple signs. ∗p <0.05; ∗∗p <0.01; ∗∗∗p <0.001; n.s., not significant.
The THPG in the presence of Mx was not significantly affected by SC-560 (Fig. 7B). However, SC-236 significantly attenuated the raised THPG in MCD-fed rats while it had no significant effect in controls (steatosis: 15.9±1.3 with Krebs+Mx to 12.4±0.5 mmHg with 0.3 μM SC-236+Mx, p <0.01[Fig. 7C]). It should be noted that the slight decrease of the THPG over time is caused by the time-dependent attenuation of vasoconstriction by Mx.
Liver perfusion experiments: flow-pressure curves
Preventive celecoxib treatment attenuated the THPG in MCD-fed rats (celecoxib-treated 13.7±0.6 vs. placebo-treated 15.0±0.5 mmHg at 50 ml/min, p <0.01). On the contrary, in controls, an increased THPG was observed with celecoxib (celecoxib-treated 14.4±1.1 vs. placebo-treated 11.9±0.4 mmHg at 50 ml/min, p <0.001 [Fig. 7D]). In therapeutically celecoxib-treated rats, the THPG in MCD-fed rats was slightly but not significantly decreased compared to placebo-treated animals. After therapeutic treatment, the THPG in celecoxib-treated control rats reduced compared to placebo-treated control rats (Fig. 7E).
Weight
Liver weight and liver/TBW-ratio did not change with celecoxib treatment (Table S2).
Blood analysis
ALT and AST levels were not changed by celecoxib treatment (Fig. 4I-L).
Microscopy and SEM
Microscopically, celecoxib treatment did not affect the histology or microstructure on SEM in MCD-fed rat livers (Fig. 1, Fig. 2 and Fig. S3).
Transcriptomics
Hepatic mRNA expression of genes related to COX-pathways were not significantly altered in MCD-fed rats, besides the downregulation of Ptgir (encoding the vasodilatory prostacyclin receptor) which was not improved by celecoxib treatment (Fig. 5).
NASH
Establishing NASH
Both TBW and liver weight were significantly increased in 8-week HFHFD-fed ZFR compared to lean controls, with the latter even more pronounced (liver/TBW-ratio: ZFR 4.4±0.3% vs. controls 3.0±0.2%, p <0.05 [Table S3]).
Blood analysis demonstrated non-significantly increased transaminase levels in HFHFD-fed rats compared to controls (ALT: NASH 201.7±77.7 vs. controls 88.3±5.4 U/L, p = 0.713; AST: NASH 174.5±100.5 vs. controls 94.0±7.3 U/L, p = 0.822), although we must remark that blood samples of the former were more difficult to analyse due to a high lipid content in the blood, which makes the group smaller and probably leaves out the samples with the highest transaminase levels (Fig. 8A).
Fig. 8.

Lean Zucker rats on control diet (controls) compared to 8-week high-fat high-fructose diet-fed Zucker fatty rats (NASH) receiving bosentan or placebo treatment.
(A) Serum ALT and AST levels. Statistical comparison by two-way ANOVA and post hoc Scheffé. n.s., not significant. (B) Liver histological liver sections (H&E and Masson’s trichrome stains [TM], original magnification 10x). (C) Flow-pressure curves of the transhepatic pressure gradient, analysed using the generalised estimating equation model. Significances between controls and NASH are indicated by black signs. Significances between NASH with placebo or bosentan are indicated by purple signs. ∗∗∗p <0.001. ALT, alanine aminotransferase; AST, aspartate aminotransferase; NASH, non-alcoholic steatohepatitis.
In HFHFD-fed ZFR, histology demonstrated the presence of NASH with significant microvesicular steatosis and prominent ballooning, without the development of fibrosis. Lean Zucker rats on a control diet had normal liver histology without any steatosis, inflammation or fibrosis (Fig. 8B).
Liver perfusion in NASH
In line with our findings in different models of steatosis, in ZFR after 8 weeks of HFHFD, the THPG was significantly increased compared to controls at all flows (NASH 7.6±0.3 mmHg vs. controls 5.7±0.2 mmHg at 30 ml/min, p <0.001 [Fig. 8C]).
Effects of bosentan in NASH
ZFR treated with bosentan during 8 weeks of HFHFD demonstrated attenuation of the increased THPG, which was significantly different from placebo-treated ZFR from a flow of 30 ml/min onwards (bosentan-treated 6.3±0.4 mmHg vs. placebo-treated 7.6±0.3 mmHg at 30 ml/min, p <0.001 [Fig. 8C]). Bosentan treatment did not reduce transaminase levels. Histologically, treatment with bosentan reduced the degree of steatosis and resulted in less ballooning (Fig. 8B).
Discussion
In this study we investigated the role of vasoconstrictors on the increased IHVR in severe steatosis. Our current data repetitively demonstrate a significant increase of the THPG in early NAFLD, when steatohepatitis or fibrosis have not yet developed. We were also able to demonstrate a consequent increase in PP. This confirms previous data demonstrating the presence of some degree of portal hypertension in early NAFLD, both in animal models and in patients,[4], [5], [6], [7], [8] and implies that the presence of an increased IHVR is a relevant early event in NAFLD pathogenesis.
Although no animal model recapitulates human NAFLD completely and every model has its advantages and limitations, the MCD in male Wistar rats is an adequate model to study the intrahepatic features and mechanisms of NAFLD,16 but without any other traits of the metabolic syndrome as can be observed in human NAFLD. Therefore, some experiments were repeated in ZFR after 4 weeks of HFHFD, a model that resembles the human phenotype of NAFLD more completely with the presence of obesity and systemic insulin resistance. These rats had a significantly increased THPG compared to controls and manifested vasoconstrictor hyperreactivity as repeatedly observed in MCD-fed rats,6,7,17 supporting the concept that these are disease-related phenomena rather than model-specific. The confirmation of an increased PP in steatosis in vivo showed that the increased IHVR indeed results in an increased PP, hence the results of the ex vivo liver perfusion experiments are clinically relevant and not limited to this experimental setting. Having confirmed the increased IHVR in 2 different models of early NAFLD, we subsequently studied the role of different endogenous vasoconstrictive mechanisms.
ET-1 is produced by the endothelium and induces vasoconstriction by acting on the ETA and ETB2 receptors while promoting vasodilation by acting on the ETB1 receptor. As demonstrated previously by our research group, the reactivity to ET-1 is significantly increased in MCD-fed rat livers compared to controls.7 In this study, we demonstrated that this vascular hyperreactivity appears to be ETA-receptor mediated. Moreover, hepatic Edn1 mRNA expression, the rate-limiting step in ET-1 production, was upregulated in MCD-fed rats, suggesting increased ET-1 production. Likewise, serum levels of ET-1 were previously found to be increased in steatosis.6 Preventive bosentan treatment not only normalised intrahepatic vascular function, it also normalised Edn1 mRNA expression. All these data point towards a pivotal role of ET-1 via its ETA-receptor in the increased IHVR in early NAFLD.
Two other vasoconstrictors, ATII and thromboxane agonist U46619, increased the THPG in control and MCD-fed rat livers but, unlike ET-1, there was no hepatic vascular hyperreactivity to these mediators in steatosis. Of note, upon continuous but increasing administration of ATII, a gradual decrease of the THPG was observed in both groups, likely reflecting the known desensitisation of ATII receptors.18
Even without vascular hyperreactivity, blocking the ATII receptor by valsartan administration during ex vivo liver perfusion normalised the increased THPG in severe steatosis. COX-2-, but not COX-1-inhibition was able to restore the increased IHVR in steatosis to normal values in ex vivo experiments, in line with our previous findings that COX-2 is more important in the systemic haemodynamics in early NAFLD.7
In our study, mRNA expression of Ace (encoding angiotensin I converting enzyme) was upregulated, implying increased formation of active ATII, contributing to an increased IHVR. Increased serum ATII levels have been reported in patients with NAFLD.19 Expression of Agt (encoding angiotensinogen) and Atgr14a (encoding angiotensin II receptor) were downregulated, suggesting the presence of a compensatory mechanism to decrease the production of endogenous ATII in the case of increased THPG in steatosis. COX converts arachidonic acid to prostanoids and, with the help of thromboxane synthase (TXAS), to the vasoconstrictor TxA2. Since COX also produces some vasodilatory prostanoids, an increased production of TxA2 as a cause of the increased IHVR in early NAFLD was not excluded in this study, although Tbxas1 expression (encoding TXAS) was not altered. Nevertheless, previous results demonstrated increased levels of circulating TxA2, elevated TxA2 receptor expression and significantly higher TXAS expression in steatosis.6 Therefore, TxA2 might still be a mediator of the increased IHVR.
Taken together, all 3 vasoconstrictive pathways investigated could be involved in the increased IHVR in early NAFLD since antagonism decreased THPG. The effects of ET-1, due to concurrent intrahepatic vascular hyperreactivity in steatosis, seem to be most important.
In 2 animal models of severe steatosis, we demonstrated that preventive and therapeutic administration of ET-1 antagonist bosentan normalised THPG and PP in vivo, implying normalisation of the IHVR. Likewise, even without the presence of vascular hyperreactivity to ATII, blocking the ATII receptor by preventive, but not therapeutic oral valsartan treatment in vivo decreased the elevated THPG in steatosis significantly. Apparently, valsartan is not potent enough to have an effect on the IHVR when steatosis has already been established by a strong driver like the MCD. Finally, oral celecoxib treatment could also decrease the THPG in MCD-fed rats. In controls, therapeutic celecoxib treatment decreased the THPG as well. Paradoxically, preventive celecoxib treatment increased the THPG in controls. To explain these results, we hypothesise that there might be a predominant COX-mediated vasodilatory mechanism in healthy livers to maintain the physiological perfusion of the liver. COX is not only involved in TXA2 production, but is responsible for the production of other vasoactive prostanoids like vasodilatory prostacyclin. Therefore, COX-2-inhibition illustrates that timing of a certain intervention in the disease course is of importance since it can affect the outcome substantially. Comparable to our data, indomethacin (a non-selective COX-inhibitor) improved vascular dysfunction, but TXA2 antagonism had no effect in rats with HFD-induced steatosis.17
Besides the beneficial effects on the increased THPG in steatosis, additional observations underline the importance of intrahepatic vascular function in the pathophysiology of NAFLD. In parallel with the haemodynamic improvements, both bosentan and valsartan treatment caused a normalisation of serum transaminases in MCD-induced steatosis. Moreover, improvement of transaminase levels with bosentan treatment was confirmed in the ZFR model. By contrast, COX-2 inhibition could not improve transaminase levels, suggesting that the beneficial effect of celecoxib on the IHVR is overruled by other mechanisms in vivo.
Bosentan treatment in steatosis caused lower liver weight, as well as a significant decline in the histological degree of steatosis. Above all, bosentan was able to reverse the substantial alterations that occur in the hepatic microvasculature in early NAFLD, normalising the sinusoidal diameter and making the microvascular pattern more regular with a reduced number of blebs. A decrease of the histological degree of steatosis after bosentan treatment was also demonstrated in ZFR, proving again that these findings are not model-specific.
Valsartan treatment resulted in an improvement of liver weight, which did, however, not translate into an improvement of the histological degree of steatosis. In previous research, ARBs improved the degree of steatosis in different mouse models of NAFLD,20 but this finding was not supported by all studies.21 ARBs are believed to exert their beneficial effects mainly through inflammation- and fibrosis-related pathways,22 which might in part explain why we did not observe any histological improvement in early NAFLD. Likewise, no effect on liver histology could be observed after treatment with celecoxib. In previous research, nimesulide (COX-2-inhibitor) attenuated the increased hepatic triglyceride content23 and celecoxib attenuated the degree of steatosis and inflammation in different preclinical NAFLD models.24 While we did not demonstrate histological improvement in our model, it should be noted that our treatments are shorter than those in the aforementioned studies.23,24 Furthermore, MCD is a powerful driver of steatosis and steatohepatitis, so it can also be hypothesised that the effects of celecoxib are too weak to counteract its effects.
As in steatosis, an increased transhepatic pressure gradient could also be observed in a model of NASH. As the increase in THPG was similar to early NAFLD, it is implied that the described phenomena are indeed early events. Moreover, the beneficial effects of bosentan were not limited to both models of severe steatosis, but could be repeated in a model of NASH, both in reducing the histological severity of disease and attenuating the THPG. This demonstrates the clinical relevance of the findings as they are not limited to isolated steatosis but remain present further along the spectrum of NAFLD. In previous research, bosentan treatment improved the severity of liver disease in patients with cirrhosis,25 implying it might even be safe and useful in later stages of NAFLD.
In conclusion, vasoconstrictive mechanisms appear to dominate intrahepatic vascular dysfunction in early NAFLD and their modulation can reduce disease severity, with therapeutic impacts beyond reducing IHVR and portal hypertension. Importantly, whilst MCD feeding, a strong driver of NAFLD, persisted during treatment, bosentan still had significant effects both functionally as well as structurally. This implies that it is a potent pathway that deserves to be further explored clinically for its therapeutic potential. To the best of our knowledge, bosentan has so far not been clinically evaluated for the treatment of NAFLD/NASH. Preclinical data suggest that bosentan can also improve several components of the metabolic syndrome, e.g. insulin resistance, dyslipidaemia and adipose tissue inflammation.26 It has already been successfully introduced into clinics for pulmonary hypertension, making it an attractive molecule to be studied and potentially repurposed in NAFLD, although several case studies have reported potential hepatotoxicity.27 Fortunately, severe hepatotoxicity is rare. Adverse events related to bosentan are usually mild and self-limiting and can be detected early by regular serum transaminase testing.28 In this study, histological liver cell damage or increased serum transaminase levels, as expected with bosentan hepatotoxicity, were not observed in any rat after 4 or 8 weeks of bosentan treatment. Furthermore, macitentan, another endothelin receptor antagonist, has been used safely in advanced cirrhosis.29
The role of the renin-angiotensin system on hepatic fibrogenesis, which subsequently increases portal hypertension, is already known. Furthermore, components of the renin-angiotensin system also directly influence both liver as well as splanchnic vasculature, contributing to portal hypertension.30 Moreover, in patients with cirrhosis, angiotensin 1-7 and ACE2 activity were shown to be increased and correlated with disease severity.31 This observation could be the result of the angiotensin-induced alterations of the hepatic transcription of the Janus-kinase-2 (Jak2) pathway, which were demonstrated both in a mouse model of cirrhosis and in human cirrhosis.32 Deletion of Jak2 in a mouse model of cirrhosis could attenuate fibrosis and decrease portal pressure.33 These observations might in part explain the functional component of the increased THPG and demonstrate the clinical relevance of angiotensin-mediated mechanisms.
In line with what we observed with valsartan in our rat steatosis model, olmesartan (an ARB) could significantly decrease the hepatic venous pressure gradient in patients with cirrhotic portal hypertension.34 Besides, candesartan (another ARB) decreased levels of hyaluronic acid in patients with cirrhotic portal hypertension, suggesting reduced fibrogenesis.35 Despite their reported anti-fibrotic effects, ARBs are not routinely used in clinical practice in patients without coexisting systemic arterial hypertension because of conflicting data concerning histological improvement of NAFLD in humans.36 Adding to the described anti-inflammatory and anti-fibrotic effects, our data show that its effects on the increased THPG in very early NAFLD might play an additional role in the beneficial effects of ARBs. Because of the absence of effects on liver damage in our model, time-dependent divergent effects in controls and well-known cardiovascular side-effects, celecoxib might be a less suitable therapeutic agent.
Conclusions
Our study explored the role of vasoconstriction in the increased THPG in NAFLD. We confirmed the presence of an increased IHVR and vasoconstrictor hyperreactivity in 2 rat models of early NAFLD with severe steatosis and in a model of NASH. This is accompanied by a marked disruption of normal sinusoidal architecture. While celecoxib treatment decreased the THPG in steatosis slightly, it failed to demonstrate any other beneficial effects on the liver. Bosentan and valsartan treatment caused a reduction of the THPG, attenuated transaminase levels and improved liver weight. Moreover, bosentan treatment improved hepatic steatosis, largely restored the disrupted microvasculature and demonstrated beneficial vascular and histological effects in a model of NASH. Therefore, inhibiting vasoconstrictive pathways in early NAFLD, particularly with bosentan, seems a promising therapeutic approach.
Financial support
Sven M. Francque received a senior clinical research fellowship from the Research Foundation Flanders (1802154N). The project was also partially funded by the Antwerp University (GOA 2018, ID36572). The funders had no role in the study design; in the collection, analysis and interpretation of data; in the writing of the report; and in the decision to submit the article for publication.
Authors’ contributions
CRediT authorship contributions Denise van der Graaff (Conceptualization: Equal; Data curation: Lead; Formal analysis: Lead; Investigation: Lead; Methodology: Equal; Visualization: Lead; Writing – original draft: Lead); Shivani Chotkoe (Data curation: Equal); Benedicte De Winter (Conceptualization: Equal; Methodology: Equal; Supervision: Equal; Writing – review & editing: Equal); Joris De Man (Conceptualization: Equal; Methodology: Equal; Supervision: Equal; Writing – review & editing: Equal); Christophe Casteleyn (Methodology: Equal; Writing – review & editing: Equal); Jean-Pierre Timmermans (Methodology: Equal); Isabel Pintelon (Data curation: Equal; Methodology: Equal); Luisa Vonghia, MD, PhD (Methodology: Equal; Supervision: Equal; Writing – review & editing: Equal); Wilhelmus J Kwanten, MD, PhD (Conceptualization: Equal; Methodology: Equal; Supervision: Equal; Writing – review & editing: Equal); Sven Francque (Conceptualization: Equal; Funding acquisition: Lead; Methodology: Equal; Supervision: Lead; Writing – review & editing: Equal). The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Data availability statement
Due to the confidentiality of data, the data which support the findings of this study (other than those that can be found within the article) are only available in a redacted form upon request.
Conflict of interest
Denise van der Graaff declares no conflict of interest. Shivani Chotkoe declares no conflict of interest. Benedicte De Winter declares no conflict of interest. Joris De Man declares no conflict of interest. Christophe Casteleyn declares no conflict of interest. Jean-Pierre Timmermans declares no conflict of interest. Isabel Pintelon declares no conflict of interest. Luisa Vonghia declares no conflict of interest. Wilhelmus J. Kwanten is co-inventor of a patent on the use lipopigment imaging for disease filed by MIT/MGH. Sven Francque has acted as advisor and/or lecturer for Roche, Gilead, Abbvie, Bayer, BMS, MSD, Janssen, Actelion, Astellas, Genfit, Inventiva, Intercept, Genentech, Galmed, Promethera, Coherus, NGM Bio and Julius Clinical.
Please refer to the accompanying ICMJE disclosure forms for further details.
Acknowledgements
The authors thank Petra Aerts, Hannah Ceuleers, Paul Fransen Ilse Goolaerts, Angelika Jürgens, Philip Plaeke, Rita Van Den Bossche, Mandy Vermont, Marleen Vinckx, and Lieve Vits for their technical support.
All authors have access to the study data and have reviewed and approved the final manuscript.
Footnotes
The work was performed in all mentioned insitutions.
Author names in bold designate shared co-first authorship
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jhepr.2021.100412.
Supplementary data
The following are the supplementary data to this article:
References
- 1.Francque S., Vonghia L. Pharmacological treatment for non-alcoholic fatty liver disease. Adv Ther. 2019;36:1052–1074. doi: 10.1007/s12325-019-00898-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Van der Graaff D., Kwanten W.J., Francque S.M. The potential role of vascular alterations and subsequent impaired liver blood flow and hepatic hypoxia in the pathophysiology of non-alcoholic steatohepatitis. Med Hypoth. 2018;122:188–197. doi: 10.1016/j.mehy.2018.11.014. [DOI] [PubMed] [Google Scholar]
- 3.Baffy G. Origins of portal hypertension in nonalcoholic fatty liver disease. Dig Dis Sci. 2018;63:563–576. doi: 10.1007/s10620-017-4903-5. [DOI] [PubMed] [Google Scholar]
- 4.Francque S., Wamutu S., Chatterjee S., Van Marck E., Herman A., Ramon A., et al. Non-alcoholic steatohepatitis induces non-fibrosis-related portal hypertension associated with splanchnic vasodilation and signs of a hyperdynamic circulation in vitro and in vivo in a rat model. Liver Int. 2010;30:365–375. doi: 10.1111/j.1478-3231.2009.02136.x. [DOI] [PubMed] [Google Scholar]
- 5.Francque S., Verrijken A., Mertens I., Hubens G., Van Marck E., Pelckmans P., et al. Noncirrhotic human nonalcoholic fatty liver disease induces portal hypertension in relation to the histological degree of steatosis. Eur J Gastroenterol Hepatol. 2010;22:1449–1457. doi: 10.1097/MEG.0b013e32833f14a1. [DOI] [PubMed] [Google Scholar]
- 6.Francque S., Laleman W., Verbeke L., Van Steenkiste C., Casteleyn C., Kwanten W., et al. Increased intrahepatic resistance in severe steatosis: endothelial dysfunction, vasoconstrictor overproduction and altered microvascular architecture. Lab Invest. 2012;92:1428–1439. doi: 10.1038/labinvest.2012.103. [DOI] [PubMed] [Google Scholar]
- 7.Van der Graaff D., Kwanten W.J., Couturier F.J., Govaerts J.S., Verlinden W., Brosius I., et al. Severe steatosis induces portal hypertension by systemic arterial hyporeactivity and hepatic vasoconstrictor hyperreactivity in rats. Lab Investig. 2018;98:1263–1275. doi: 10.1038/s41374-017-0018-z. [DOI] [PubMed] [Google Scholar]
- 8.Pasarín M., La Mura V., Gracia-Sancho J., García-Calderó H., Rodríguez-Vilarrupla A., García-Pagán J.C., et al. Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PloS One. 2012;7 doi: 10.1371/journal.pone.0032785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCuskey R.S., Ito Y., Robertson G.R., McCuskey M.K., Perry M., Farrell G.C. Hepatic microvascular dysfunction during evolution of dietary steatohepatitis in mice. Hepatology. 2004;40:386–393. doi: 10.1002/hep.20302. [DOI] [PubMed] [Google Scholar]
- 10.Lefere S., Van de Velde F., Hoorens A., Raevens S., Van Campenhout S., Vandierendonck A., et al. Angiopoietin-2 promotes pathological angiogenesis and is a therapeutic target in murine nonalcoholic fatty liver disease. Hepatology. 2019;69:1087–1104. doi: 10.1002/hep.30294. [DOI] [PubMed] [Google Scholar]
- 11.Olapeju B.I., Inanemo O.E.K., Enitome B.E. Effects of amlodipine and valsartan on glibenclamide-treated streptozotocin-induced diabetic rats. Biomed Pharmacother. 2018;106:566–574. doi: 10.1016/j.biopha.2018.06.152. [DOI] [PubMed] [Google Scholar]
- 12.Kianian F., Seifi B., Kadkhodaee M., Sajedizadeh A., Ahghari P. Protective effects of celecoxib on ischemia reperfusion-induced acute kidney injury: comparing between male and female rats. Iran J Basic Med Sci. 2019;22:43–48. doi: 10.22038/ijbms.2018.29644.7156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu S., Lin T., Wang S., Chuang C., Lee F., Huang H. Endothelin receptor blockers reduce shunting and angiogenesis in cirrhotic rats. Eur J Clin Investig. 2016;46:572–580. doi: 10.1111/eci.12636. [DOI] [PubMed] [Google Scholar]
- 14.Percie du Sert N., Ahluwalia A., Alam S., Avey M.T., Baker M., Browne W.J., et al. Reporting animal research: explanation and elaboration for the ARRIVE guidelines 2.0. Plos Biol. 2020;18 doi: 10.1371/journal.pbio.3000411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Herck M.A., Vonghia L., Kwanten W.J., Julé Y., Vanwolleghem T., Ebo D.G., et al. Diet reversal and immune modulation show key role for liver and adipose tissue T cells in murine nonalcoholic steatohepatitis. Cell Mol Gastroenterol Hepatol. 2020;10:467–490. doi: 10.1016/j.jcmgh.2020.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hebbard L., George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2011;8:35–44. doi: 10.1038/nrgastro.2010.191. [DOI] [PubMed] [Google Scholar]
- 17.Gonzalez-Paredes F.J., Hernández Mesa G., Morales Arraez D., Marcelino Reyes R., Abrante B., Diaz-Flores F., et al. Contribution of cyclooxygenase end products and oxidative stress to intrahepatic endothelial dysfunction in early non-alcoholic fatty liver disease. PloS One. 2016;11:1–15. doi: 10.1371/journal.pone.0156650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hadoke P.W.F. Cirrhosis of the liver and receptor-mediated function in vascular smooth muscle. Pharmacol Ther. 2001;89:233–254. doi: 10.1016/s0163-7258(01)00126-7. [DOI] [PubMed] [Google Scholar]
- 19.Li Y., Xiong F., Xu W., Liu S. Increased serum angiotensin II is a risk factor of nonalcoholic fatty liver disease: a prospective pilot study. Gastroenterol Res Pract. 2019;2019:5647161. doi: 10.1155/2019/5647161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kato J.U.N., Koda M., Kishina M., Tokunaga S., Matono T. Therapeutic effects of angiotensin II type 1 receptor blocker, irbesartan, on non-alcoholic steatohepatitis using FLS-ob/ob male mice. Int J Mol Med. 2012;30:107–113. doi: 10.3892/ijmm.2012.958. [DOI] [PubMed] [Google Scholar]
- 21.Verbeek J., Spincemaille P., Vanhorebeek I., Van den Berghe G., Vander Elst I., Windmolders P., et al. Dietary intervention, but not losartan, completely reverses non-alcoholic steatohepatitis in obese and insulin resistant mice. Lipids Health Dis. 2017;16:46. doi: 10.1186/s12944-017-0432-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park J.G., Mok J.S., Han Y.I., Park T.S., Kang K.W., Choi C.S., et al. Connectivity mapping of angiotensin-PPAR interactions involved in the amelioration of non-alcoholic steatohepatitis by Telmisartan. Sci Rep. 2019;9:4003. doi: 10.1038/s41598-019-40322-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsujimoto S., Kishina M., Koda M., Yamamoto Y. Nimesulide, a cyclooxygenase-2 selective inhibitor, suppresses obesity-related non-alcoholic fatty liver disease and hepatic insulin resistance through the regulation of peroxisome proliferator-activated receptor γ. Int J Mol Med. 2016;38:721–728. doi: 10.3892/ijmm.2016.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J., Liu D., Bai Q., Song J. Celecoxib attenuates liver steatosis and inflammation in non-alcoholic steatohepatitis induced by high-fat diet in rats. Mol Med Rep. 2011;4:811–816. doi: 10.3892/mmr.2011.501. [DOI] [PubMed] [Google Scholar]
- 25.Barth F., Gerber P.J., Reichen J., Dufour J.F., Nicod L.P. Efficiency and safety of bosentan in child C cirrhosis with portopulmonary hypertension and renal insufficiency. Eur J Gastroenterol Hepatol. 2006;18:1117–1119. doi: 10.1097/01.meg.0000231749.60889.f7. [DOI] [PubMed] [Google Scholar]
- 26.Rivera-Gonzalez O., Wilson N.A., Coats L.E., Taylor E.B., Speed J.S. Endothelin receptor antagonism improves glucose handling, dyslipidemia, and adipose tissue inflammation in obese mice. Clin Sci. 2021;135:1773–1789. doi: 10.1042/CS20210549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eriksson C., Gustavsson A., Kronvall T., Tysk C. Hepatotoxicity by bosentan in a patient with portopulmonary hypertension : a case-report and review of the literature. J Gastrointest Liver Dis. 2010;20:77–80. [PubMed] [Google Scholar]
- 28.Aversa M., Porter S., Granton J. Comparative safety and tolerability of endothelin receptor antagonists in pulmonary arterial hypertension. Drug Saf. 2015;38:419–435. doi: 10.1007/s40264-015-0275-y. [DOI] [PubMed] [Google Scholar]
- 29.Sitbon O., Bosch J., Cottreel E., Csonka D., de Groote P., Hoeper M.M., et al. Macitentan for the treatment of portopulmonary hypertension (PORTICO): a multicentre, randomised, double-blind, placebo-controlled, phase 4 trial. Lancet Respir Med. 2019;7:594–604. doi: 10.1016/S2213-2600(19)30091-8. [DOI] [PubMed] [Google Scholar]
- 30.Grace J.A., Herath C.B., Mak K.Y., Burrell L.M., Angus P.W. Update on new aspects of the renin-angiotensin system in liver disease: clinical implications and new therapeutic options. Clin Sci (Lond) 2012;123:225–239. doi: 10.1042/CS20120030. [DOI] [PubMed] [Google Scholar]
- 31.Casey S., Schierwagen R., Mak K.Y., Klein S., Uschner F., Jansen C., et al. Activation of the alternate renin-angiotensin system correlates with the clinical status in human cirrhosis and corrects post liver transplantation. J Clin Med. 2019;8:203–222. doi: 10.3390/jcm8040419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Granzow M., Schierwagen R., Klein S., Kowallick B., Huss S., Linhart M., et al. Angiotensin-II type 1 receptor-mediated Janus kinase 2 activation induces liver fibrosis. Hepatology. 2014;60:334–348. doi: 10.1002/hep.27117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klein S., Rick J., Lehmann J., Schierwagen R., Schierwagen I.G., Verbeke L., et al. Janus-kinase-2 relates directly to portal hypertension and to complications in rodent and human cirrhosis. Gut. 2017;66:145–155. doi: 10.1136/gutjnl-2015-309600. [DOI] [PubMed] [Google Scholar]
- 34.Hidaka H., Kokubu S., Nakazawa T., Okuwaki Y., Ono K., Watanabe M., et al. New angiotensin II type 1 receptor blocker olmesartan improves portal hypertension in patients with cirrhosis. Hepatol Res. 2007;37:1011–1017. doi: 10.1111/j.1872-034X.2007.00165.x. [DOI] [PubMed] [Google Scholar]
- 35.Wang J., Lu W., Li J., Zhang R., Zhou Y., Yin Q., et al. Hemodynamic effects of renin-angiotensin-aldosterone inhibitor and β-blocker combination therapy vs. β-blocker monotherapy for portal hypertension in cirrhosis: a meta-analysis. Exp Ther Med. 2017;13:1977–1985. doi: 10.3892/etm.2017.4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Y., Xu H., Wu W., Ye J., Fang D., Shi D., et al. Clinical application of angiotensin receptor blockers in patients with non-alcoholic fatty liver disease: a systematic review and. Oncotarget. 2018;9:24155–24167. doi: 10.18632/oncotarget.23816. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Due to the confidentiality of data, the data which support the findings of this study (other than those that can be found within the article) are only available in a redacted form upon request.