

Summary
Abnormal activation of SETBP1 due to overexpression or missense mutations occurs frequently in various myeloid neoplasms and associates with poor prognosis. Direct activation of Hoxa9/Hoxa10/Myb transcription by SETBP1 and its missense mutants is essential for their transforming capability; however, the underlying epigenetic mechanisms remain elusive. We found that both SETBP1 and its missense mutant SETBP1(D/N) directly interact with histone methyltransferase MLL1. Using a combination of ChIP-seq and RNA-seq analysis in primary hematopoietic stem and progenitor cells, we uncovered extensive overlap in their genomic occupancy and their cooperation in activating many oncogenic transcription factor genes including Hoxa9/Hoxa10/Myb and a large group of ribosomal protein genes. Genetic ablation of Mll1 as well as treatment with an inhibitor of the MLL1 complex OICR-9429 abrogated Setbp1/Setbp1(D/N)-induced transcriptional activation and transformation. Thus, the MLL1 complex plays a critical role in Setbp1-induced transcriptional activation and transformation and represents a promising target for treating myeloid neoplasms with SETBP1 activation.
Subject areas: Biological sciences, Molecular biology, Cell biology, Cancer
Graphical abstract
Highlights
-
•
SETBP1 and mutant SETBP1(D/N) interact directly with multiple regions of MLL1
-
•
SETBP1/SETBP1(D/N) co-localizes with MLL1 at oncogenes and ribosomal protein genes
-
•
MLL1 is required for SETBP1/SETBP1(D/N)-induced gene activation and AML development
-
•
WDR5 inhibition is a promising strategy for treating AMLs with SETBP1 activation
Biological sciences; Molecular biology; Cell biology; Cancer
Introduction
SETBP1 is a nuclear protein first identified through its interaction with oncoprotein SET (also known as TAF-I) (Minakuchi et al., 2001). Growing evidence suggests its abnormal activation may play an important role in the development of a number of myeloid malignancies. Overexpression of SETBP1 has been found in up to 30% of patients with primary acute myeloid leukemia (AML) (Cristobal et al., 2010; Lucas et al., 2018) and a subset of patients with chronic myeloid leukemia (CML) blast crisis (Oakley et al., 2012). SETBP1 overexpression also has been identified as a signature for leukemia-initiating cells (LICs) in human AMLs (Gentles et al., 2010). In addition, missense mutations in SETBP1 stabilizing its protein have been found to occur frequently in atypical chronic myeloid leukemia (Piazza et al., 2013), chronic myelomonocytic leukemia (Makishima et al., 2013), secondary AML (sAML) (Makishima et al., 2013), and juvenile myelomonocytic leukemia (Sakaguchi et al., 2013) and myelodysplastic syndrome (Damm et al., 2013; Fernandez-Mercado et al., 2013). SETBP1 activation likely plays an important role in driving transformation in these diseases as both wild-type Setbp1 and its missense mutants have been shown to be potent oncogenes in mice (Nguyen et al., 2016; Vishwakarma et al., 2016). SETBP1 activation by overexpression or missense mutations also has been associated with a poor prognosis in many of these diseases, underscoring the need to develop effective targeted therapies. Multiple mechanisms may contribute to the oncogenic function of SETBP1. Early studies suggested that SETBP1 may promote inhibition of PP2A through physical interaction with SET (Cristobal et al., 2010). We have found that SETBP1 can function as an AT-hook transcription factor to activate transcription of the oncogenes Hoxa9/Hoxa10/Myb and also repress the transcription of Runx1 (Nguyen et al., 2016, 2019; Oakley et al., 2012; Vishwakarma et al., 2016). In addition, overexpression of Setbp1 can help confer unlimited self-renewal capability to granulocyte macrophage progenitors, indicating that it may play an important role in LIC self-renewal regulation in myeloid leukemias (Oakley et al., 2012; Ott et al., 2006). Since activation of Hoxa9/Hoxa10/Myb has been shown to be essential for SETBP1 and its missense mutants to induce transformation, blocking their activation represents an attractive strategy for treating myeloid neoplasms with SETBP1 activation. However, the molecular mechanisms responsible for such activation remains largely unknown, which significantly hampers the efforts in designing and testing this strategy.
The MLL1 (also known as KMT2A, MLL, HRX, ALL1) gene encodes a histone methyltransferase homologous to the Drosophila melanogaster Trithorax (Trx) and is widely expressed in many tissues. The full-length MLL1 protein is known to be cleaved by Taspase-1 into two fragments, MLL1-N and MLL1-C (Hsieh et al., 2003; Yokoyama et al., 2002). MLL1-N and MLL1-C normally associate with one another as components of a multiprotein complex (MLL1 complex) with additional core components including MENIN (Yokoyama et al., 2004), PSIP1 (also known as LEDGF) (Yokoyama and Cleary, 2008), ASH2L (Steward et al., 2006), WDR5 (Dou et al., 2006; Ruthenburg et al., 2006), RBBP5 (Cao et al., 2010; Nayak et al., 2014), and DPY30 (Jiang et al., 2011; Patel et al., 2009). The SET domain of MLL1 catalyzes mono-, di-, and tri-methylation of lysine 4 on histone 3 (H3K4); however, recent studies have suggested that this enzymatic activity of MLL1 is not essential for its activation of gene transcription, whereas H4K16 acetylation induced through its recruitment of KAT8 is critical (Mishra et al., 2014). Gene targeting studies in mice have shown that Mll1 is essential for normal fetal and adult hematopoiesis (Jude et al., 2007; McMahon et al., 2007). MLL1 also is involved in chromosomal translocations in AML and acute lymphoblastic leukemia, leading to the production of fusion proteins with the N-terminal part of MLL1 fused with the C terminus of over 90 different fusion partners (Meyer et al., 2018). Transcriptional activation of a number of oncogenes by the MLL1 fusion proteins, including HOXA genes, MEIS1, MYC, and EYA1, have been found to be critical for leukemia transformation (Armstrong et al., 2002; Krivtsov et al., 2006; Wang et al., 2011; Wong et al., 2007; Zuber et al., 2011). A recent study also has suggested that direct activation of ribosome protein genes by MLL1 fusion proteins likely plays an important role in their induction of transformation (Garcia-Cuellar et al., 2016). Besides chromosomal rearrangements, wild-type MLL1 has also been shown recently to be critical for leukemia development induced by NUP98 fusions, NPM1 mutations, and MN1 overexpression (Kuhn et al., 2016; Libbrecht et al., 2021; Xu et al., 2016).
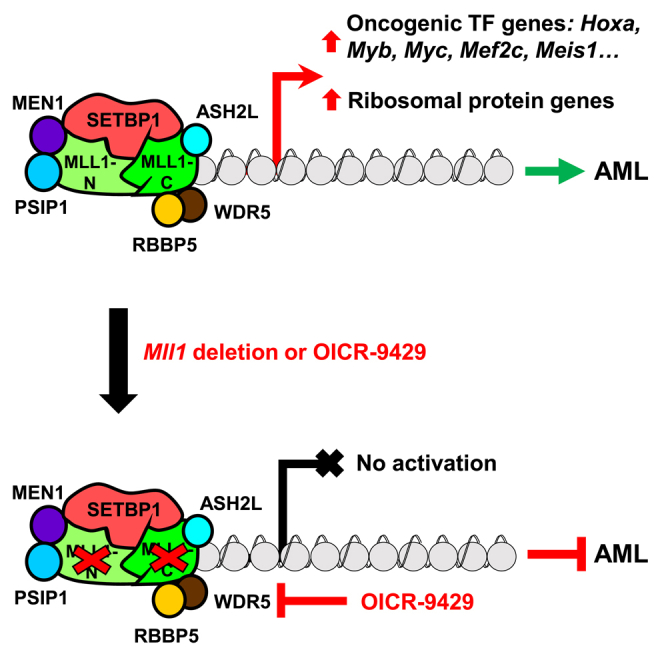
Here we uncovered direct physical interactions of MLL1 with SETBP1 and its missense mutant SETBP1(D/N) and established MLL1 complex as an essential cofactor complex for both to activate target gene transcription and to induce transformation of hematopoietic progenitor cells. By integrating results from RNA-seq and ChIP-seq analysis, we also identify a comprehensive list of SETBP1 targets that are co-regulated by MLL1 and identify ribosome biogenesis as a downstream pathway of SETBP1 activation. Moreover, our study also suggests that blocking the interaction between WDR5 and MLL1 may be effective for treating myeloid neoplasms with SETBP1 activation.
Results
Mll1 is required for SETBP1-induced transcriptional activation of Hoxa9/Hoxa10/Myb
Epigenetic regulators have been implicated in the regulation of self-renewal of LICs (Cozzio et al., 2003; Huntly et al., 2004; Kouzarides, 2007; Lessard and Sauvageau, 2003). In an effort to identify potential epigenetic mechanisms critical for Setbp1-induced self-renewal, we found that the levels of H3K4 tri-methylation at Hoxa9 in Setbp1-immortalized myeloid progenitors decreased significantly after Setbp1 knockdown (Figure 1A), suggesting that a histone methyltransferase is recruited by SETBP1 to the Hoxa9 locus. Histone methyltransferase gene Mll1 is expressed in hematopoietic stem and progenitor cells (Butler et al., 1997; Gu et al., 1992; Jude et al., 2007; Kawagoe et al., 1999; Phillips et al., 2000; Tkachuk et al., 1992) and is known to regulate Hoxa9 transcription in myeloid cells (Milne et al., 2002; Nakamura et al., 2002), suggesting it could cooperate with Setbp1 in activating Hoxa9 transcription. Consistent with this notion, Setbp1 knockdown also significantly reduced the levels of H4K16 acetylation at Hoxa9 (Figure 1B), which is maintained by the MLL1 complex (Mishra et al., 2014). Moreover, significant binding by MLL1 was readily detected at all tested regions of Hoxa9 locus in Setbp1-immortalized cells and displayed a similar pattern as SETBP1 (Figure 1C). Furthermore, resembling Setbp1 knockdown, knockdown of Mll1 in Setbp1-immortalized cells caused significant reduction in the mRNA levels of Hoxa9/Hoxa10/Myb (Figures 1D and S1A), suggesting that Mll1 is critical for SETBP1 to activate their transcription. Similar occupancy by MLL1 at Hoxa9 was also detected in myeloid progenitors immortalized by Setbp1(D/N) (Figure 1E), which contains the missense mutation D868N frequently identified in various myeloid neoplasms (Makishima et al., 2013). Knockdown of either Mll1 or Setbp1(D/N) in these cells also induced a significant reduction in Hoxa9/Hoxa10/Myb mRNA levels (Figures 1F and S1B), suggesting that the Mll1 is also critical for SETBP1 missense mutants to activate transcription. In addition, knockdown of MLL1 in primary leukemia cells from patients with sAML with SETBP1 mutations and patients with primary AML (pAML) with overexpression of wild-type SETBP1 also significantly reduced their HOXA9/HOXA10/MYB mRNA levels (Figures 1G and S1C–S1F), suggesting that the dependency on MLL1 is also conserved in human AML cells with SETBP1 activation. Collectively, these results suggest that MLL1 is a critical cooperating partner for SETBP1 and its missense mutants in activating gene transcription.
Figure 1.

MLL1 is a critical partner for SETBP1/SETBP1(D/N) to activate Hoxa9/Hoxa10/Myb transcription
(A) Chromatin immunoprecipitation (ChIP) analysis of indicated regions of Hoxa9 promoter relative to transcriptional start site using an antibody specific for H3K4me3 in mouse myeloid progenitor cells immortalized by transduction with pMYs-3xFLAG-Setbp1-IRES-GFP virus at 72 h after transduction with a lentiviral shRNA targeting GFP (GFP-sh) and a non-targeting shRNA (NC-sh). Data are represented as mean ± SD (n = 3). ∗, p <0.05; ∗∗, p <0.01; ∗∗∗, p <0.001 (two-tailed Student's t test).
(B) ChIP analysis of indicated regions of Hoxa9 promoter in same cells as in (A) using an H4K16ac-specific antibody. Data are represented as mean ± SD (n = 3). ∗, p <0.05; ∗∗∗, p <0.001 (two-tailed Student's t test).
(C) ChIP analysis of Hoxa9 promoter regions in same cells as in (A) using antibodies specific for MLL1-N and MLL1-C fragments (left panel) and anti-FLAG M2 (right panel). Data are represented as mean ± SD (n = 3).
(D) Left panel, real-time RT-PCR analysis of relative Hoxa9/Hoxa10/Myb mRNA levels in Setbp1-immortalized cells at 72 h after transduction with Mll1-specific shRNAs (Mll1-sh1 and -sh2) or a non-targeting control shRNA (NC-sh). Data are represented as mean ± SD (n = 3). ∗, p <0.05; ∗∗, p <0.01 (two-tailed Student's t test). Right panel, western blotting analysis of the same transduced cells using indicated antibodies.
(E) ChIP analysis of Hoxa9 promoter regions in mouse myeloid progenitor cells immortalized by transduction with pMYs-3xFLAG-Setbp1(D/N)-IRES-GFP virus using antibodies specific for MLL1-N and MLL1-C fragments (left panel) and anti-FLAG M2 (right panel). Data are represented as mean ± SD (n = 3).
(F) Left panel, real-time RT-PCR analysis of relative Hoxa9/Hoxa10/Myb mRNA levels in Setbp1(D/N)-immortalized cells at 72 h after transduction with the same lentiviral shRNAs as in (D). Data are represented as mean ± SD (n = 3). ∗, p <0.05; ∗∗, p <0.01 (two-tailed Student's t test). Right panel, western blotting analysis of the same transduced cells using indicated antibodies.
(G) Left panel, real-time RT-PCR analysis of relative HOXA9, HOXA10, and MYB mRNA levels in primary human sAML cells with a SETBP1 missense mutation (p.D868Y) at 72 h after transduction with indicated negative control or MLL1-specific shRNAs. Data are represented as mean ± SD (n = 3). ∗∗, p <0.01; ∗∗∗, p <0.001 (two-tailed Student's t test). Right panel, Western blotting analysis of same transduced cells using indicated antibodies.
Direct physical interaction between SETBP1/SETBP1(D/N) and MLL1
Similar binding patterns of MLL1 and SETBP1 at Hoxa9 promoter suggest that they may form a complex to activate gene transcription. To test whether SETBP1 and its missense mutants could physically associate with MLL1, we transiently transfected HEK293T cells, which normally express MLL1, with a construct expressing an N-terminally 3xFLAG-tagged SETBP1 or SETBP1(D/N) carrying the recurrent D868N mutation (Makishima et al., 2013) and performed immunoprecipitation studies using the anti-FLAG M2 antibody. The full-length MLL1 protein is normally processed into two proteolytic fragments MLL1-N and MLL1-C (Hsieh et al., 2003; Yokoyama et al., 2002). Both MLL1 fragments were readily detected in immunoprecipitates prepared from Setbp1 and Setbp1(D/N) transfected cells but not in precipitates obtained from empty vector transfected cells (Figure 2A), suggesting that they can physically associate with SETBP1 and its missense mutants. The complex formation of SETBP1/SETBP1(D/N) with MLL1 was confirmed by reverse immunoprecipitation using a MLL1-specific antibody (Figure 2B). Other components of the MLL1 complex including MENIN, PSIP1, ASH2L, and WDR5 were also pulled down together with SETBP1/SETBP1(D/N) (Figure 2C), suggesting that the entire MLL1 complex is associated with SETBP1/SETBP1(D/N). To test whether such association occurs in myeloid cells, we performed co-immunoprecipitation in myeloid progenitors immortalized by 3xFLAG-tagged SETBP1 or SETBP1(D/N). MLL1 was efficiently pulled down in these cells by the anti-FLAG M2 antibody (Figure 2D). In addition, this association was confirmed by the extensive co-localization of MLL1 with SETBP1/SETBP1(D/N) in these cells detected by co-immunofluorescence studies (Figures 2E and 2F). In supporting this physical association in normal hematopoietic stem and progenitor cells, significant co-localization of MLL1 with SETBP1 was also detected in purified mouse Lin−Sca-1+c-Kit+ (LSK) cells (Figure S2). Taken together, these data suggest that SETBP1/SETBP1(D/N) physically associate with the MLL1 complex to activate transcription in myeloid progenitor cells.
Figure 2.

MLL1 physically associates with SETBP1 and SETBP1(D/N)
(A) Western blotting analysis of M2 immunoprecipitates prepared from nuclear extract of HEK293T cells transiently transfected with empty pMYs vector or pMYs construct expressing 3xFLAG-tagged Setbp1 or Setbp1(D/N) using indicated specific antibodies.
(B) Western blotting analysis of immunoprecipitates prepared with an MLL1-N-specific antibody from the same nuclear extracts as in (A) using the indicated specific antibodies.
(C) Western blotting analysis of M2 immunoprecipitates similarly prepared as in (A) using additional specific antibodies.
(D) Western blotting analysis of M2 and IgG immunoprecipitates prepared from myeloid progenitors immortalized by 3xFLAG-tagged Setbp1 or Setbp1(D/N) using indicated specific antibodies.
(E) Immunofluorescence staining of Setbp1-immortalized myeloid progenitors using a SETBP1-specific antibody together with either an MLL1-N-specific or MLL1-C-specific antibody. Nuclei were counterstained with DAPI. Scale bar, 10 μm.
(F) Same immunofluorescence staining as in (D) for Setbp1(D/N)-immortalized myeloid progenitors. Scale bar, 10 μm.
We further tested the possibility that MLL1 may interact directly with SETBP1/SETBP1(D/N). We synthesized five 3xHA-tagged MLL1 fragments derived from both the MLL1-N region (N1-N3) and the MLL1-C region (C1 and C2) and 3x-FLAG-tagged full-length SETBP1 protein by in vitro transcription and translation and tested their interaction in binding assays (Figure 3A). We found that all MLL1 fragments except C1 can interact with SETBP1 and SETBP1(D/N) by immunoprecipitation studies using either the anti-FLAG M2 or an anti-HA antibody (Figures 3B and 3C). These results suggest that multiple regions of MLL1 are in direct contact with SETBP1/SETBP1(D/N) in their interaction.
Figure 3.

Both MLL1-N1 and MLL1-C directly interact with SETBP1 and SETBP1(D/N)
(A) Schematic representation of domain structure of full-length MLL1 protein and the different 3xHA-tagged mouse MLL1 fragments used for co-immunoprecipitation studies to test possible direct interactions with 3xFLAG-tagged SETBP1 and SETBP1(D/N). Results of the co-immunoprecipitation studies shown in (B) and (C) are also summarized, with “+” indicating interaction detected with both SETBP1 and SETBP1(D/N) and “-” indicating no interaction detected.
(B) Co-immunoprecipitation experiments were performed using either anti-FLAG M2 antibody (left panels) or anti-HA antibody (right panels) on mixtures of SETBP1 with different MLL1 fragments. Inputs and immunoprecipitates were subsequently analyzed by western blotting analysis using anti-SETBP1, M2, or anti-HA antibody. SETBP1 protein and all MLL1 fragments were synthesized by in vitro transcription and translation in wheat germ extract.
(C) Same co-immunoprecipitation experiments as in (B) to detect possible direct interaction between SETBP1(D/N) and MLL1 fragments.
Global gene transcription regulated by SETBP1/SETBP1(D/N)
To better understand the significance of MLL1-SETBP1/SETBP1(D/N) interaction in regulating gene transcription, we first mapped the genomic binding profiles of SETBP1 and SETBP1(D/N) in hematopoietic stem and progenitor cells by ChIP sequencing. We transduced mouse LSK cells with pMYs retrovirus expressing 3xFLAG-tagged SETBP1 or SETBP1(D/N) and subsequently sorted the transduced cells based on GFP positivity. After a brief expansion of the cells in methylcellulose medium, the genome-wide binding targets of SETBP1 and SETBP1(D/N) were subsequently identified by ChIP sequencing using the anti-FLAG M2 antibody.
A total of 110,847 and 180,732 peaks were identified (with q-value ≤ 0.05) for SETBP1 and SETBP1(D/N) occupancy, respectively. To focus our analysis on the most significant peaks, the top 5,000 peaks for each occupancy based on q-value were further analyzed. About 18.9% (947 peaks) of the SETBP1 peaks and 18.7% (935 peaks) of the SETBP1(D/N) peaks were mapped at promoter regions. In addition, 50.9% (2545 peaks) of the SETBP1 peaks and 48.1% (2406 peaks) of the SETBP1(D/N) peaks were located at intragenic regions, whereas 30.2% (1508 peaks) of the SETBP1 peaks and 33.2% (1659 peaks) of the SETBP1(D/N) peaks were present in intergenic regions (Figure 4A). A majority of the peaks mapped to intragenic and intergenic regions, suggesting that SETBP1 and SETBP1(D/N) may regulate transcription mostly through binding to enhancers and repressors. Consistent with the presence of three AT-hook DNA-binding motifs in SETBP1 protein, a similar AT-enriched consensus motif was found to associate with over 19% of SETBP1 and SETBP1(D/N) peaks (Figure S3A). A direct comparison of the SETBP1 peaks with the SETBP1(D/N) peaks showed that 43.7% (414 peaks), 18.5% (472 peaks), and 21% (317 peaks) of the SETBP1 peaks overlap with the SETBP1(D/N) peaks at promoter, intragenic, and intergenic regions, respectively (Figure 4A). This significant overlap in binding suggests that both proteins may regulate similar target genes in driving leukemia transformation.
Figure 4.

Genome-wide transcriptional regulation by SETBP1 and SETBP1(D/N) in mouse LSK cells
(A) Upper panels, mapping of top 5,000 peaks (by q-value) from SETBP1 and SETBP1(D/N) ChIP-seq data. Lower panels, Venn diagrams showing overlaps of SETBP1-bound or SETBP1(D/N)-bound peaks in indicated genomic regions. p Values for overlaps were calculated by hypergeometric test.
(B) Pie charts showing the numbers of differentially expressed genes induced by Setbp1 and Setbp1(D/N) expression in mouse LSK cells.
(C) Top GO gene sets with positive enrichment from GSEA analysis of differentially expressed genes in mouse LSK cells induced by ectopic Setbp1 and Setbp1(D/N) expression.
(D) GSEA analysis of differentially expressed genes in mouse LSK cells induced by Setbp1 expression using indicated gene sets.
(E) Genome browser tracks representing SETBP1 and SETBP1(D/N) shared occupancy at indicated loci of oncogenic transcription factor genes and ribosomal protein genes.
In order to determine how genomic occupancy by SETBP1 and SETBP1(D/N) may affect gene expression, we also examined and compared the gene expression profiles of Setbp1- and Setbp1(D/N)-expressing cells with that of control cells infected with empty pMYs virus by RNA-seq analysis. A total of 2,599 and 3,512 differentially expressed genes (DEGs) with expression changes greater than 2-fold were identified for Setbp1- and Setbp1(D/N)-expressing cells, respectively (Figure 4B and Tables S1 and S2). A total of 1,284 and 1,719 genes were upregulated by 2-fold by SETBP1 and SETBP1(D/N) respectively, whereas 1,315 and 1,793 genes were downregulated (Figures 4B and Tables S1 and S2). When the gene expression profiles of Setbp1- and Setbp1(D/N)-expressing cells were directly compared with each other, only 135 genes showed expression changes by 2-fold (Table S3), suggesting that SETBP1 and SETBP1(D/N) regulate similar pathways in driving leukemia transformation. To identify these downstream pathways, we performed gene set enrichment analysis (GSEA) of differentially expressed genes identified for Setbp1- and Setbp1(D/N)-expressing cells in comparison with control cells. Analysis using ontology gene sets showed that both Setbp1 and Setbp1(D/N) significantly upregulated a large number of genes involved in ribosome biogenesis and RNA processing (Figure 4C), indicating that both may considerably promote global protein synthesis. Genes downregulated by Setbp1 and Setbp1(D/N) were found to associate with functions of mature cell types, suggesting that both may also block differentiation (Figure S3B). In supporting the hypothesis that SETBP1 activation confers self-renewal capability to leukemia stem cells, we also found that genes upregulated in hematopoietic stem cells are significantly enriched in Setbp1 or Setbp1(D/N)-expressing cells (Figures 4D and S3C). In line with the Hoxa9 as a critical target for both Setbp1 and Setbp1(D/N) in inducing transformation, genes upregulated by Hoxa9 and Meis1 are also activated by Setbp1 and Setbp1(D/N) (Figures 4D and S3C). Genes positively regulated by MYC are also upregulated in Setbp1 or Setbp1(D/N)-expressing cells (Figures 4D and S3C), suggesting activation of the MYC pathway in these cells. Interestingly, consistent with the notion that MLL1 is a critical partner for SETBP1 and SETBP1(D/N) to activate transcription, we also observed significant positive correlation with genes upregulated in AMLs involving MLL1, including leukemias induced by MLL fusions, NPM1 mutated AMLs, and the NUP98-HOXA9 fusion protein (Figures 4D and S3C).
Next, we tried to correlate gene expression changes with genomic occupancy by SETBP1 and SETBP1(D/N). Since our ChIP-seq results suggest that SETBP1 and SETBP1(D/N) likely regulate transcription primarily through binding to enhancers and repressors, we compared all genes located within +/− 20 kb of the SETBP1 and SETBP1(D/N) peaks to the list of DEGs from our RNA-seq analysis. Over 96% and 98% of DEGs were found to overlap with SETBP1 and SETBP1(D/N) peak genes, respectively (Tables S4 and S5), suggesting that the vast majority of the DEGs are directly regulated at the transcriptional level by the two proteins. Since the DEGs include similar numbers of up- and down-regulated genes, these data also agree with our previous studies in supporting the idea that SETBP1 and SETBP1(D/N) can directly activate and repress gene transcription (Oakley et al., 2012; Vishwakarma et al., 2016). As expected from our previous studies, Hoxa9, Hoxa10, and Myb were found to be directly activated by Setbp1 and Setbp1(D/N) (Figure 4E). In addition, this analysis allowed us to identify a number of oncogenic transcription factor genes as direct targets of Setbp1 and Setbp1(D/N), which have also been found to be activated by MLL1 fusion proteins, including additional HoxA genes (Hoxa1, Hoxa3, Hoxa5, Hoxa6, and Hoxa7), Myc, Eya1, Sox4, Mecom, Meis1, Mef2c, and Lmo2 (Bach et al., 2010; Bindels et al., 2012; Krivtsov et al., 2006; Patel et al., 2014; Wang et al., 2011; Zuber et al., 2011) (Figures 4E and S4). Moreover, we also found significant binding by SETBP1 and SETBP1(D/N) at or near over 48% and 78% of ribosomal protein genes (RPGs), respectively (Figures S4 and S5). These RPGs were also found to be upregulated by RNA-seq analysis (Figure S5), suggesting that SETBP1 and SETBP1(D/N) may increase ribosomal biogenesis in part through direct transcriptional upregulation of RPGs. In supporting the relevance of these targets in human AML development, we found a significant positive correlation between SETBP1 mRNA levels and the mRNA levels of MEF2C and EYA1 in the TCGA human AML samples (Figure S6). Although a significant correlation with SETBP1 expression levels was not detected for other identified target genes, these genes are expressed at relatively high levels in most of the TCGA human AML samples with high SETBP1 expression (Figure S6), suggesting that they could be upregulated by SETBP1 in these samples, even though other mechanisms are also likely involved in regulating their expression.
MLL1 and SETBP1/SETBP1(D/N) co-occupy targets genome-wide
To examine the extent of MLL1-SETBP1/SETBP1(D/N) interaction in regulating gene transcription, ChIP-seq using a previously validated MLL1-N-specific antibody was also performed to determine the genome-wide binding of MLL1 in the mouse LSK cells transduced with pMYs virus expressing SETBP1 and SETBP1(D/N). A similar genomic distribution of MLL1 peaks was observed in both cell populations when the top 5,000 MLL1 peaks were analyzed, with about 27%, 30%, and 42% of the peaks present at promoter, intergenic, and gene body regions, respectively (Figure 5A). When the top 5,000 SETBP1/SETBP1(D/N) and MLL1 peaks were directly compared, over 20% of SETBP1-bound peaks (1,027 peaks) overlap with over 21% of MLL1 peaks (1,063 peaks) in Setbp1-transduced cells (Figure 5B). Similarly, over 23% of SETBP1(D/N)-bound peaks (1,160 peaks) and MLL1-bound peaks (1,186 peaks) overlap in Setbp1(D/N)-transduced cells (Figure 5B). These significant overlaps show that SETBP1/SETBP1(D/N) co-localize with MLL1 on chromatin in agreement with our co-immunoprecipitation data showing their direct physical interactions. Since the MLL1 complex has been shown to induce H3K4me3 and H4K16ac at promoter regions, we also carried out ChIP-seq studies to map their genomic profiles in the same cells. Higher H3K4me3 and H4K16ac occupancies were observed at the promoter region of genes co-bound by SETBP1/SETBP1(D/N) and MLL1 than genes bound by only one of the factors (Figures 5C and S7), suggesting that co-localization of SETBP1/SETBP1(D/N) and MLL1 leads to an increase in these histone modifications of active transcription. More importantly, examination of ChIP-seq peaks at SETBP1/SETBP1(D/N) activation target genes including both oncogenic transcription factor genes (HoxA genes, Myb, Myc, Meis1, Eya1, Mef2c, Sox4, and Mecom) and many RPGs show nearly complete overlap in regional occupancy for MLL1 and SETBP1/SETBP1(D/N) (Figures 5D, 5E, and S4). These results strongly suggest that these activation targets of SETBP1/SETBP1(D/N) are co-regulated by MLL1.
Figure 5.

Co-localization of SETBP1 and SETBP1(D/N) with MLL1 at oncogenic transcription factor genes and ribosome protein genes
(A) Mapping of MLL1-N ChIP-seq peaks in mouse LSK cells overexpressing Setbp1 or Setbp1(D/N).
(B) Venn diagrams showing overlaps of top 5,000 SETBP1-bound or SETBP1(D/N)-bound peaks with top 5,000 MLL1-bound peaks in LSK cells overexpressing Setbp1 or Setbp1(D/N). p Values were calculated using hypergeometric test.
(C) Average binding profiles of H3K4me3 at promoter regions of SETBP1-bound, MLL1-bound, and SETBP1/MLL1 co-bound targets.
(D) Genome browser tracks showing co-localization of SETBP1, MLL1, H3K4me3, and H4K16ac at indicated loci of oncogenic transcription factor gene targets and ribosomal protein gene targets.
(E) Genome browser tracks showing co-localization of SETBP1(D/N), MLL1, H3K4me3, and H4K16ac at the same target loci as in (D).
MLL1 deletion inhibits transformation induced by SETBP1 and SETBP1(D/N) in vitro and in vivo
To further test the requirement for MLL1 using a genetic model, we established Setbp1- and Setbp1(D/N)-immortalized myeloid progenitor cells using a well-established Mll1 conditional knockout (Mll1F) mouse model in which Mll1 could be deleted upon activation of Cre/ERT2 (Cre+) (Jude et al., 2007) (Figure 6A). Purified Lin-Sca1+c-Kit+ (LSK) cells from the bone marrow of Mll1F/F;Cre+ and Mll1+/+;Cre+ mice were transduced with pMYs retrovirus expressing Setbp1 or Setbp1 (D/N) and subsequently cultured in the presence of SCF and IL-3 to establish immortalized myeloid progenitors (Figure 6A). Upon treatment with 4-hydroxytamoxifen (4-OHT), efficient Mll1 deletion was induced in both Setbp1- and Setbp1(D/N)-immortalized Mll1F/F;Cre+ cells (Figure S8A). Consistent with our Mll1 knockdown studies, the colony-forming potential of these cells was significantly reduced, whereas similarly immortalized Mll1+/+;Cre+ cells remained competent in forming colonies upon 4-OHT treatment (Figure 6B), confirming that Mll1 is essential for in vitro transformation induced by Setbp1/Setbp1(D/N). Cytospin analysis further showed that Mll1 deletion induced differentiation of the immortalized Mll1F/F;Cre+ cells (Figure 6C). Also similar to the Mll1 knockdown studies, the mRNA levels of Hoxa9, Hoxa10, and Myb significantly decreased in these cells after Mll1 loss (Figures 6D and 6E). In addition, significant expression reductions upon Mll1 deletion were observed for new oncogenic targets found to be co-regulated by SETBP1/SETBP1(D/N) and MLL1 in our ChIP-seq studies, including Myc, Mef2c, Eya1, and RPGs (Figures 6D and 6E). In contrast, treatment with 4-OHT did not induce any significant decrease in the mRNA levels of these Setbp1 and Setbp1(D/N) targets in immortalized Mll1+/+;Cre+ cells. These data confirm that Mll1 is essential for the transcriptional activation of oncogenic targets by Setbp1 and Setbp1(D/N).
Figure 6.

Mll1 is essential for transformation induced by Setbp1 and Setbp1(D/N)
(A) Schematic diagram showing the experimental design for the generation of Mll1 conditional immortalized and AML cells.
(B) Relative colony formation by Setbp1- and Setbp1(D/N)-immortalized Mll1F/F;Cre+ and Mll1+/+;Cre+ myeloid progenitors at 48 h after treatment with 4-OHT or control ethanol (EtOH). Data are represented as mean ± SD (n = 3). ∗∗∗, p <0.001 (two-tailed Student's t test).
(C) Cytospin analysis of Setbp1- and Setbp1(D/N)-immortalized Mll1F/F;Cre+ and Mll1+/+;Cre+ myeloid progenitors at 72 h after treatment with 4-OHT. Scale bar, 10 μm.
(D) Real-time RT-PCR analysis of indicated SETBP1/MLL1 co-bound targets in Setbp1-immortalized Mll1F/F;Cre+ and Mll1+/+;Cre+ cells at 48 h after treatment with 4-OHT or EtOH. Data are represented as mean ± SD (n = 3).
(E) Real-time RT-PCR analysis of indicated SETBP1(D/N)/MLL1 co-bound targets in Setbp1-immortalized Mll1F/F;Cre+ and Mll1+/+;Cre+ cells at 48 h after treatment with 4-OHT or EtOH. Data are represented as mean ± SD (n = 3).
(F) Survival curves of mice receiving 5 × 105Setbp1-induced (left panel) or Setbp1(D/N)-induced (right panel) Mll1F/F;Cre+ AML cells treated with Tamoxifen or control corn oil. Tamoxifen or corn oil treatments are indicated by dotted lines. p Values were calculated using the log rank test. ∗∗∗, p <0.001.
To test whether Mll1 is required for the maintenance of AML cells induced by Setbp1/Setbp1(D/N) in vivo, we also generated mouse AMLs by transplanting the transduced LSK cells from Mll1F/F;Cre+ and Mll1+/+;Cre+ mice into lethally irradiated recipient mice with supporting bone marrow cells (Figure 6A). The latency and penetrance of leukemia development in the recipient mice are in line with our previous studies using these viruses (Figure S8B) (Nguyen et al., 2016; Vishwakarma et al., 2016). A control transplantation study using LSK cells transduced by empty pMYs virus was not performed because our previous studies had shown that infection of mouse hematopoietic stem and progenitor cells with empty pMYs virus alone is not able to induce leukemia development in our bone marrow transplantation system (Nguyen et al., 2016; Vishwakarma et al., 2016). After AML development, leukemic spleen cells from these primary recipients were transplanted into secondary recipient mice. Starting from day 10 of transplantation, half of the secondary recipients were treated with two injections of Tamoxifen to induce Mll1 deletion in Mll1F/F;Cre+ leukemia cells, whereas the other half were injected with vehicle. Treatment with Tamoxifen did not cause any significant change in the survival of mice receiving Setbp1- or Setbp1(D/N)-induced Mll1+/+;Cre+ leukemia cells compared with treatment with vehicle (Figure S8C), suggesting Tamoxifen has minimal effects on these leukemia cells. In contrast, for mice receiving Mll1F/F;Cre+ leukemia cells, Tamoxifen treatment significantly extended their survival (Figure 6F). Although efficient Mll1 deletion was induced in these mice as expected, genotyping results of the leukemia cells from moribund mice show that most of them contained the floxed allele (Figures S8D and S8E), suggesting that they were derived from leukemia cells that had escaped Tamoxifen-induced deletion. These results strongly suggest that MLL1 is required for the maintenance of AMLs induced by SETBP1 activation.
Inhibition of WDR5 is effective for blocking SETBP1-induced transcriptional activation and transformation
To test the possibility that pharmacological inhibition of the MLL1 complex may be effective for treating AMLs with SETBP1 activation, we treated mouse AML cells induced by overexpression of Setbp1 or Setbp1(D/N) with WDR5 and MENIN inhibitors previously shown to disrupt the MLL1 complex. As expected, treatments with new MENIN inhibitors including MI-3454 and SNDX-5613 reduced the HOXA9/HOXA10/MYB mRNA levels in MLL/AF9-induced mouse AML cells at previously established effective concentrations (Figure S9) (Klossowski et al., 2020). However, these inhibitors did not cause any reduction in the mRNA levels of Hoxa9/Hoxa10/Myb in Setbp1- or Setbp1(D/N)-induced mouse AML cells at the same concentrations after 6 days of treatment (Figure S9). Consistent with the requirement for MEN1 in transcriptional activation of Hoxa9/Hoxa10/Myb, Men1 knockdowns significantly reduced their mRNA levels in Setbp1(D/N)-induced AML cells (Figure S9). These results suggest that transcriptional activation co-regulated by SETBP1/SETBP1(D/N) and the MLL1 complex may be less sensitive to these MENIN inhibitors. In contrast, we found that treatment with the WDR5 inhibitor OICR-9429 efficiently reduced the transcription of oncogenic and ribosomal targets of Setbp1 and Setbp1(D/N) in these mouse AML cells after 3 days of treatment (Figures 7A and 7B). Consistent with these results, OICR-9429 is also capable of significantly reducing the colony-forming potential of these cells (Figures 7C and 7D). A significant reduction in the mRNA levels of Setbp1 and Setbp1(D/N) targets was observed after Wdr5 knockdowns in the same cells (Figures 7E and 7F), suggesting that the effects of the inhibitor are not due to possible off-target activity. These data suggest that WDR5 inhibitors are likely effective for the treatment of myeloid neoplasms with SETBP1 activation.
Figure 7.

Setbp1- and Setbp1(D/N)-induced AML cells are sensitive to WDR5 inhibition
(A) Real-time RT-PCR analysis of indicated SETBP1/MLL1 co-bound targets in Setbp1-induced AML cells at 72 h after treatment with OICR-9429 at indicated concentration or control DMSO. Data are represented as mean ± SD (n = 3).
(B) Real-time RT-PCR analysis of same genes in Setbp1(D/N)-induced AML cells at 72 h after treatment with OICR-9429 or control DMSO. Data are represented as mean ± SD (n = 3).
(C) Relative colony formation by Setbp1-induced AML cells upon treatment with WDR5 inhibitor OICR-9429 at indicated concentration or control DMSO. Data are represented as mean ± SD (n = 3).
(D) Relative colony formation by Setbp1(D/N)-induced AML cells upon treatment with WDR5 inhibitor OICR-9429 at indicated concentration or control DMSO. Data are represented as mean ± SD (n = 3).
(E) Left panel, real-time RT-PCR analysis of relative indicated SETBP1/MLL1 co-bound targets in Setbp1-induced AML cells after transduction with Wdr5-specific shRNAs (Wdr5-sh1 and -sh2) or a non-targeting control shRNA (NC-sh). Data are represented as mean ± SD (n = 3). Right panel, western blotting analysis of the same transduced cells using indicated antibodies.
(F) Same analyses as in (E) for Setbp1(D/N)-induced AML cells.
Discussion
More effective therapies are critically needed for myeloid neoplasms with SETBP1 activation owing to the link with a poor disease prognosis. Transcriptional activation of Hoxa9/Hoxa10/Myb has been found to be essential for SETBP1 and its missense mutants to induce transformation of myeloid progenitors, suggesting that blocking their ability to activate transcription may represent a promising treatment strategy. In our efforts to identify potential epigenetic mechanisms involved in Setbp1-induced transcriptional activation, we identified MLL1 as a key cooperating partner for SETBP1 and its missense mutant SETBP1(D/N) in activating target gene transcription in hematopoietic stem and progenitor cells., This cooperation also causes activation of a number of oncogenic transcription factors and a large group of ribosomal protein genes, in addition to Hoxa9/Hoxa10/Myb. This cooperation involves their direct physical interaction with both MLL1-N and MLL1-C fragments of MLL1 protein and also the recruitment of other major components of the MLL1 complex. This dependence on the MLL1 complex to activate transcription of key targets for transformation also led to our finding that the WDR5 inhibitor, OICR-9429, is effective for treating myeloid neoplasms with SETBP1 activation.
Association of a mutant SETBP1 protein with the MLL1 complex was recently suggested by a study using HEK293T cells; however, the nature of this association was not clear and its functional significance in hematopoietic cells remained completely unknown (Piazza et al., 2018). Here we show that three different regions of MLL1-N and one region of MLL1-C can be efficiently pulled down together with both wild-type SETBP1 and its missense mutant SETBP1(D/N) in immunoprecipitation assay using proteins synthesized through in vitro transcription and translation, suggesting that MLL1-N and MLL1-C can form multiple direct interactions with SETBP1/SETBP1(D/N). This extensive interaction is also supported by the broad overlap between their genomic occupancy observed in our ChIP-seq studies. This finding also suggests that the activity of the MLL1 complex could be potentially modulated by the interaction with SETBP1/SETBP1(D/N) and that SETBP1 may be a regulator of the MLL1 complex in normal cells with high SETBP1 expression such as hematopoietic stem cells (Oakley et al., 2012). Since higher H3K4me3 and H4K16ac occupancies were observed at the promoter region of genes co-bound by SETBP1/SETBP1(D/N) and MLL1 than at that of genes bound by MLL1 only in our ChIP-seq studies, it is possible that the binding of SETBP1/SETBP1(D/N) to MLL1 may help stabilize the interaction of the MLL1 complex to chromatin.
A global view of pathways regulated by SETBP1 has been lacking. Using a combination of RNA-seq and ChIP-seq analysis in mouse LSK cells, our study uncovered many transcriptional targets for both wild-type and missense mutant SETBP1, generating a comprehensive picture of the transformation process induced by SETBP1 activation. Among these, many are known oncogenes in the hematopoietic system, including Myc, Mef2c, Meis1, Eya1, Sox4, Lmo2, and Mecom, suggesting that they also likely contribute to transformation induced by SETBP1 activation, and inhibiting their downstream pathways may prove useful for treating myeloid neoplasms with SETBP1 activation. Interestingly, our studies also identified a large number of ribosome protein genes as direct targets of SETBP1 and SETBP1(D/N), suggesting that ribosomal biogenesis is another significant pathway induced by SETBP1 activation. Ribosome biogenesis is a key process essential for the translation of mRNAs into proteins. Hyperactivation of ribosome biogenesis has been thought to play a critical role in cancer development (Pelletier et al., 2018; Truitt and Ruggero, 2016). Highlighting the importance of RPGs to cancer development, a 50% reduction in only one ribosomal protein has been shown to suppress Myc-induced tumorigenesis (Barna et al., 2008). Thus, activation of ribosome biogenesis likely plays an essential role in transformation induced by SETBP1 activation and such transformed cells may be particularly sensitive to inhibitors of translation. Our results also identify MLL1, in addition to SETBP1 and its missense mutants, as a regulator of these genes as significant binding by MLL1 was also detected at most of these RPGs and their expression decreased upon Mll1 deletion. WDR5 and MYC have been identified recently as critical transcriptional regulators of many RPGs (Aho et al., 2019; Thomas et al., 2019). It was shown that MYC is recruited to RPGs through direct interaction with WDR5, but the mechanism responsible for WDR5 recruitment remains unclear as WDR5 is known to interact with several factors. As recruitment of WDR5 to RPGs can be blocked by a WIN site inhibitor, our findings further suggest that MLL1 may be responsible for the recruitment of WDR5. As Myc is also directly activated by SETBP1/SETBP1(D/N) and MLL1, it is likely that recruitment of MYC also contributes to the activation of RPGs in cells with SETBP1 activation.
The dependence of SETBP1/SETBP1(D/N) on MLL1 to activate oncogenic targets suggested that inhibition of the MLL1 complex could be potentially effective for blocking their induction of transformation. Recent studies have shown that the MLL1 complex can be efficiently inhibited by MENIN and WDR5 inhibitors (Grebien et al., 2015; Klossowski et al., 2020; Krivtsov et al., 2019). However, we found that only the WDR5 inhibitor OICR-9429 but not two different MENIN inhibitors is efficient for inhibiting the transcription induced by SETBP1/SETBP1(D/N) and MLL1. Although the reason for this reduced sensitivity to MENIN inhibitors remains unclear, it is possible that the direct interaction between SETBP1/SETBP1(D/N) and MLL1 may restrict the access of MENIN inhibitors. Future development of WDR5 inhibitors with better pharmacokinetic profiles may significantly improve our treatment for myeloid neoplasms with SETBP1 activation.
Our finding of direct physical interaction between SETBP1 and MLL1 also suggests a potentially underappreciated role of SETBP1 in cancer development. The direct interaction between SETBP1 and the N1 fragment of MLL1-N, which is retained in MLL1 fusion proteins, suggests that SETBP1 may also physically interact with MLL1 fusion proteins. Genes upregulated in AMLs with the NPM1 mutation and NUP98 translocations are also activated by SETBP1 and SETBP1(D/N) and both types of AMLs have been shown to be dependent on the recruitment of the MLL1 complex to activate key transforming genes. The direct physical interaction between SETBP1 and MLL1 further suggests the possibility that SETBP1 may also be involved in the pathogenesis of these AMLs. An increase in MLL1 expression has also been found to contribute to the development of many solid tumors (Bryan et al., 2020; Zhu et al., 2015), further suggesting that the role of SETBP1 in cancer development may not be limited to blood cancers. Given the cooperation between SETBP1 and MLL1 in activating RPGs, one potential function of SETBP1 in solid tumors may be to induce hyperactivation of ribosome biogenesis. These possibilities warrant further investigation.
Limitations of the study
RNA-seq and ChIP-seq studies performed in LSK cells provide valuable insights into the genes and pathways regulated by SETBP1 and its missense mutants in early hematopoietic progenitors. However, these cells are a mixture of long-term and short-term hematopoietic stem cells and multi-potent progenitors, and the SETBP1-induced effects may differ among the different populations. In addition, these downstream effects may also be influenced by the culture condition, which does not fully recapitulate the bone marrow microenvironment in vivo. Performing analysis on freshly purified individual populations will address these issues.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Purified anti-mouse Ly-6G/Ly-6C (Gr1) | Biolegend | Cat# 108402, Clone RB6-8C5; RRID:AB_313367 |
| Purified anti-mouse CD4 | Biolegend | Cat# 100506, Clone RM4-5; RRID:AB_312709 |
| Purified anti-mouse CD8a | Biolegend | Cat# 100702, Clone 53-6.7; RRID:AB_312741 |
| Purified anti-mouse/human CD45R/B220 | Biolegend | Cat# 103202, Clone RA3-6B2; RRID:AB_312987 |
| Purified anti-mouse Ter119 | Biolegend | Cat# 116202, Clone Ter119;RRID:AB_313703 |
| Purified anti-mouse CD11b | Biolegend | Cat# 101202, Clone M1/70; RRID:AB_312785 |
| Purfied anti-mouse CD127 | Biolegend | Cat# 135002, Clone A7R34; RRID:AB_1937287 |
| PE anti-mouse CD117 (c-Kit) | Biolegend | Cat# 105808, Clone 2B8; RRID:AB_313217 |
| APC anti-mouse Ly-6A (Sca-1) | Biolegend | Cat# 108112, Clone D7; RRID:AB_313349 |
| Anti-SETBP1 | ProteinTech | Cat# 16841-1-AP; RRID:AB_2185750 |
| Anti-MLL1-N | Millipore | Cat# 05-764, Clone N4.4; RRID:AB_309976 |
| Anti-MLL1-C | Millipore | Cat# 05-765, Clone 9-12; RRID:AB_11212995 |
| Anti-MLL1 | Bethyl Laboratory | Cat# A300-086A; RRID:AB_242510 |
| Anti-Men1 | Bethyl Laboratory | Cat# A300-105A; RRID:AB_2143306 |
| Anti-H3K4me3 | Abcam | Cat# ab8580; RRID:AB_306649 |
| Anti-H4K16ac | Millipore | Cat# 07-329; RRID:AB_310525 |
| Anti-Beta Actin | Abcam | Cat# ab8224; RRID:AB_449644 |
| Anti-WDR5 | Bethyl Laboratory | Cat# A302-430A; RRID:AB_1944300 |
| Anti-ASH2 | Bethyl Laboratory | Cat# 300-489A; RRID:AB_451024 |
| Anti-HA Tag | Cell Signaling | Cat# 3724S, Clone C29F4; RRID:AB_1549585 |
| Anti-FLAG | Sigma | Cat# F1804, Clone M2; RRID:AB_262044 |
| Anti-HA Tag | ProteinTech | Cat# 66006-1-Ig; RRID:AB_2857911 |
| Anti-PSIP1 | Cell Signaling | Cat# 2088S, Clone C57G11; RRID:AB_2171216 |
| Chemicals, peptides, and recombinant proteins | ||
| (Z)-4-Hydroxytamoxifen (4-OHT) | Sigma | Cat# H7904-5MG |
| Tamoxifen, free base (TAM) | MP Biomedicals | Cat# 156738-1GM |
| MI-3454 | MedChemExpress | Cat# HY-136360 |
| SNDX-5613 | MedChemExpress | Cat# HY-136175 |
| OICR-9429 | Selleck Chemicals | Cat# S7833 |
| Critical commercial assays | ||
| RNeasy Mini Kit | Qiagen | Cat# 74104 |
| Ipure Kit V2 | Diagenode | Cat# C03010015 |
| iDeal ChIP-seq Kit (TF) | Diagenode | Cat# C01010054 |
| iDeal ChIP-seq Kit (Histones) | Diagenode | Cat# C01010051 |
| iDeal ChIP-qPCR | Diagenode | Cat# C01010180 |
| Nuclear Complex Co-IP Kit | Active Motif | Cat# 54001 |
| DNA SMART ChIP-Seq | Takara Bio | Cat# 634866 |
| TruSeq Stranded mRNA Library Prep Kit | Illumina | Cat# 20020594 |
| TnT® Coupled Wheat Germ Extract System | Promega | Cat# L4140 |
| Invitrogen™ SuperScript™ IV First-Strand Synthesis System | Invitrogen | Cat# 18091050 |
| Deposited data | ||
| RNA-seq and ChIP-seq data | NCBI Gene Expression Ominibus | GSE167301 |
| Experimental models: Cell lines | ||
| Hs27 | ATCC | CRL-1634 |
| HEK293T | ATCC | CRL-11268 |
| Plat-E | Cell Biolabs | RV-101 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 | Charles River | Cat# 027 |
| Mouse: Mll1F/F | Dr. Patricia Ernst | Jude et al. (2007) |
| Recombinant DNA | ||
| pCMVTNT | Promega | Cat# L5620 |
| pMYs-IRES-GFP | Cell Biolabs | RTV-021 |
| Software and algorithms | ||
| GSEA v4.0.3 | Broad Institute | |
| Prism 9 | GraphPad | |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact, Dr. Yang Du (yang.du@usuhs.edu).
Materials availability
Reagents generated in this study are available from the lead contact under Materials Transfer Agreements.
Experimental model and subject details
Mice
C57BL/6 female mice (8–12 weeks old) were purchased from Charles River Laboratories (Wilmington, MA). Mll1 conditional mice were described previously (Jude et al., 2007). Mice were maintained in the animal facility of Department of Laboratory Animal Resources at Uniformed Services University of the Health Sciences (USUHS, Bethesda, MD). All mouse experiments were carried out according to protocols approved by the USUHS Institutional Animal Care and Use Committee.
Transformation of mouse hematopoietic stem and progenitor cells
Freshly purified Lin−Sca-1+c-kit+ (LSK) cells from female C57BL/6 or Mll1 conditional mice were initially cultured in DMEM media with 15% heat inactivated fetal bovine serum, and mouse cytokines (100 ng/mL SCF, 6 ng/mL IL-3, and 10 ng/mL IL-6) for 24 h to stimulate cell proliferation, and subsequently infected twice with high-titer pMYs retroviruses (pMYs-Setbp1-IRES-GFP, pMYs-Setbp1[D/N]-IRES-GFP, pMYs-3xFLAG-Setbp1-IRES-GFP, or pMYs-3xFLAG-Setbp1(D/N)-IRES-GFP) on retronectin-coated plates (Takara Bio, Mountain View, CA) for 48 h. For in vitro transformation, transduced cells were sorted based on GFP positivity and plated at 5 × 104 cells in a 35 mm dish containing MethoCult M3134 (StemCell Technologies, Cambridge, MA) supplemented with IMDM, heat inactivated 15% fetal bovine serum, 1X penicillin/streptomycin, and mouse cytokines (100 ng/mL SCF, 6 ng/mL IL-3, and 10 ng/mL IL-6). After 5–7 days, individual colonies were picked and passaged in IMDM supplemented with heat inactivated 20% horse serum, 1X penicillin/streptomycin, and mouse cytokines (50 ng/mL SCF and 10 ng/mL IL-3) for 3 weeks to establish in vitro transformed mouse LSK cells. For generation of primary leukemia, freshly transduced LSK cells were injected into the tail vein of each lethally irradiated (2 × 500 rads from 137Cs source) 8–12 week old C57BL/6 female recipient mice along with 7.5 × 105 supporting bone marrow cells from un-irradiated C57BL/6 mice. After disease onset, frozen viable stocks of the bone marrow and spleen cells were preserved for secondary transplantation experiments.
Patient samples
Primary human AML cells were collected after signing informed consent, according to the protocols approved by the Institutional Review Board of Cleveland Clinic and Ohio State University in accordance with the Declaration of Helsinki. Viably frozen cells after thawing were expanded on irradiated (4,000 rad from 137Cs source) monolayer of Hs27 cells (ATCC, Manassas, VA) in DMEM supplemented with 15% FBS, 50 μM β-mercaptoethanol, 1X penicillin/streptomycin, and human cytokines (100 ng/mL SCF, 10 ng/mL IL-3, 20 ng/mL IL-6, 10 ng/mL TPO, and 10 ng/mL FLT3L from Biolegend) for 2–4 days before lentiviral knockdown studies.
Method details
Plasmids and constructs
Retroviral constructs including pMYs-Setbp1-IRES-GFP, pMYs-Setbp1(D/N)-IRES-GFP, pMYs-3xFLAG-Setbp1-IRES-GFP, and pMYs-3xFLAG-Setbp1(D/N)-IRES-GFP were described previously (Nguyen et al., 2016). The pCMV-TNT-3xFLAG-Setbp1 and pCMV-TNT-3xFLAG-Setbp1(D/N) plasmids were generated by subcloning the 3xFLAG-Setbp1 and 3xFLAG-Setbp1(D/N) cDNA from pMYs-3xFLAG-Setbp1-IRES-GFP and pMYs-3xFLAG-Setbp1(D/N)-IRES-GFP into pCMV-TNT using XhoI and NotI sites. NH3-terminal 3xHA-tagged MLL1-N1 (a.a. 1–838) was amplified by PCR and cloned into pCMV-TNT using EcoRI and SalI sites. NH3-terminal 3xHA-tagged MLL1-N2 (a.a. 814–1874), MLL1-N3 (a.a. 1864–2658), MLL1-C1 (a.a. 2711–3485), and MLL1-C2 (a.a. 3454–3968) were similarly amplified by PCR and cloned into pCMV-TNT using XhoI and NotI sites. Primers used for generation of the MLL1 constructs are listed below. All constructs were confirmed by sequencing.
| Primers | Sequence |
|---|---|
| MLL1-N1 | 5′-CGC GGA ATT CGA ACA TGT ACC CAT ACG ATG TTC CAG ATT ACG CTG GCT ATC CCT ATG ACG TCC CGG ACT ATG CAG GAT CCT ATC CAT ATG ACG TTC CAG ATT ACG CTG CGC ACA GCT GTC GGT GGC GCT-3′ |
| MLL1-N2 | 5′-CGC GCT CGA GAT GTA CCC ATA CGA TGT TCC AGA TTA CGC TGG CTA TCC CTA TGA CGT CCC GGA CTA TGC AGG ATC CTA TCC ATA TGA CGT TCC AGA TTA CGC TGC AAG GAA GCA GAC TAG TG-3′ |
| MLL1-N3 | 5′-CGC GCT CGA GAT GTA CCC ATA CGA TGT TCC AGA TTA CGC TGG CTA TCC CTA TGA CGT CCC GGA CTA TGC AGG ATC CTA TCC ATA TGA CGT TCC AGA TTA CGC TGG CAT CGA TGA CAA CCG ACA G-3′ |
| MLL1-C1 | 5′-CGC GCT CGA GAT GTA CCC ATA CGA TGT TCC AGA TTA CGC TGG CTA TCC CTA TGA CGT CCC GGA CTA TGC AGG ATC CTA TCC ATA TGA CGT TCC AGA TTA CGC TGG TGT GGA TGA TGG GAC AGA G-3′ |
| MLL1-C2 | 5′-CGC GCT CGA GAT GTA CCC ATA CGA TGT TCC AGA TTA CGC TGG CTA TCC CTA TGA CGT CCC GGA CTA TGC AGG ATC CTA TCC ATA TGA CGT TCC AGA TTA CGC TGG CAA AAC CGG TAC CCT GAC-3′ |
Isolation and transduction of murine LSK cells
Bone marrow suspensions were harvested from female mice (2–4 months old), and mononuclear cells were isolated using LSM (MP Bio, Irvine, CA). Lineage positive (Lin+) cells were subsequently labeled with an antibody cocktail (Gr-1, CD4, CD8, B220, Ter119, Mac-1, and CD127) and depleted with Dynabeads covalently conjugated with sheep anti-rat IgG (Invitrogen, Carlsbad, CA, USA). Lineage negative (Lin−) cells were stained with Ly-6A/E (Sca-1) APC and CD117 (c-Kit) PE (BioLegend, San Diego, CA), and the LSK population was sorted using FACSAria (BD Biosciences, San Jose, CA). The purified LSK cells were initially cultured in DMEM media with 15% heat inactivated fetal bovine serum, and mouse cytokines (100 ng/mL SCF, 6 ng/mL IL-3, and 10 ng/mL IL-6) for 24 h to stimulate cell proliferation, and subsequently infected twice with high-titer pMYs retroviruses on retronectin-coated plates (Takara Bio, Mountain View, CA) for 48 h pMYs retroviruses were produced by transient transfection of Plat-E cells using Fugene-6 (Roche, Indianapolis, IN). Viral titer was assessed by serial dilution and infection of NIH-3T3 cells.
Co-immunoprecipitation and Western blotting
HEK293T cells were transfected with DNA construct expressing 3XFLAG-tagged SETBP1 (pMYs-3xFLAG-Setbp1-IRES-GFP) or 3XFLAG-tagged Setbp1(D/N) (pMYs-3XFLAG-Setbp1[D/N]-IRES-GFP), or control empty vector (pMYs-IRES-GFP) using LipoD293 (SignaGen, Rockville, MD) and nuclear extracts were isolated 36 h after transfection using the Nuclear Complex Co-IP kit (Active Motif, Carlsbad, CA). Nuclear lysates were incubated with either the anti-FLAG M2 (F1804, Sigma, St. Louis, MO) or an anti-MLL1 antibody (A300-086A, Bethyl Laboratories, Montgomery, TX) for 4 h at 4°C. Subsequently, pre-washed Dynabeads protein A or protein G (Invitrogen, Carlsbad, CA) were added to the nuclear lysate/antibody mixture and incubated for an additional 2 h at 4°C. Immunoprecipitated proteins were eluted and resolved on a 3–8% Tris-Acetate gel (NuPage, Invitrogen, Carlsbad, CA, USA). Proteins were transferred to nitrocellulose membrane using the IBlot2 system (Invitrogen, Carlsbad, CA, USA). For detection of the SETBP1/MLL1 complex components, the following antibodies were used: anti-SETBP1 (16841-1-AP; ProteinTech); anti-MLL1-N (14698S), anti-MLL1-C (14197S), anti-LEDGF (2088S) from Cell Signaling Technologies (Danvers, MA); anti-MEN1 (A300-105A), anti-WDR5 (A302-430A), and anti-ASH2 (300–489A) from Bethyl Laboratories (Montgomery, TX). Secondary antibodies used include goat anti-rabbit IgG-HRP (7074S, Cell Signaling Technologies, Danvers, MA) and anti-mouse IgG-HRP (A-9044, Sigma, St. Louis, MO). Protein bands were developed by incubation with SuperSignal West chemiluminescent substrate (Pierce, Thermo Fisher Scientific, Rockford, IL) and visualized using Image Lab software (Bio-Rad, Hercules, CA).
For co-immunoprecipitation experiments using in vitro transcribed and translated proteins, all cDNAs used were cloned into pCMV-TNT as described above. SETBP1, SETBP1(D/N), MLL1-N1, MLL1-N2, MLL1-N3, MLL1-C1, and MLL1-C2 proteins were produced using the TnT T7 Coupled Wheat Germ Extract system (Promega, Madison, WI) and by incubating the mixture at 30°C for 1.5 h. Immunoprecipitation of 3xFLAG-tagged SETBP1 or SETBP1(D/N) was performed using the anti-Flag M2 antibody (F1804, Sigma, St. Louis, MO) and immunoprecipitation of 3xHA-tagged MLL1 fragments was performed using an anti-HA antibody (3724S, Cell Signaling Technologies, Danvers, MA).
Lentiviral production, infection, and analysis
pLKO.1 lentiviral constructs containing shRNAs for NC-sh, GFP-sh, MLL1-sh1, MLL1-sh2, Wdr5-sh1, and Wdr5-sh2 were purchased from Sigma Aldrich (NC-sh, SHC002; GFP-sh, SHC005; MLL1-sh1, TRCN0000005954; MLL1-sh2, TRCN0000005956; Wdr5-sh1, TRCN0000034415; Wdr5-sh2, TRCN0000034416; St. Louis, MO). Mll1-sh1 and –sh2 were cloned into pLKO.1 and their targeting sequences are 5′-CGC CTT CAC TTG ACC ATA ATT -3′ and 5′-GCA CTT GAA GAA GAC TTC TAA -3′, respectively. Infectious lentivirus were produced and tittered as described previously (Nguyen et al., 2019). Lentiviral transduction of these immortalized cells was carried out by spinoculation in which a mixture of lentivirus and target cells was spun at 2000 x g for 90 min at 37°C. Transduced cells were selected with puromycin (2 or 3 μg/mL) at 24 h after infection. Cells were maintained in liquid culture in the presence of puromycin for the indicated time. For colony formation assays, cells were collected at 48 h after infection using 1 × 104 puromycin-resistant cells and plated in IMDM methylcellulose medium supplemented with 20% heat inactivated horse serum, mouse SCF (50 ng/mL) and IL-3 (10 ng/mL), and puromycin (2 or 3 μg/mL). Colony numbers were counted after 5–7 days.
Primary human leukemia cells after briefly expansion on irradiated (4,000 rad from 137Cs source) monolayer of Hs27 cells were infected with lentivirus by spinoculation as described above. After recovering on Hs27 monolayers for 24hrs, cells were selected with puromycin (2 μg/mL) for an additional 24hrs. Cells were then collected, centrifuge, and replaced with new media. Cells were maintained for an additional 24hrs (72hrs total) before they were isolated for Western blot and qPCR.
RNA extraction, real-time RT-PCR, and RNA-sequencing
For real-time RT-PCR, total RNA was extracted from cells using Nucleospin RNA (Takara Bio, Mountain View, CA). Oligo-dT-primed cDNA samples were prepared using Superscript IV (Invitrogen, Carlsbad, CA), and real-time PCR analysis was performed in triplicates using SYBR green detection reagents (Applied Biosystem, Grand Island, NY) on a QuantStudio 6 Flex (Applied Biosystems, Grand Island, NY). Relative changes in expression were calculated according to the ΔΔCt method. The cycling conditions were 50°C for 2 min followed by 95°C for 2 min, and then 40 cycles of 95°C for 15 s and 60°C for 1 min. The gene-specific primer sequences used are listed in Table S6.
For RNA-seq, RNA was isolated from primary colonies (day 5) of C57BL/6 LSK cells transduced with pMYs-Setbp1-IRES-GFP, pMYs-Setbp1(D/N)-IRES-GFP, or control (pMYs-IRES-GFP) retrovirus and sorted based on GFP positivity from methylcellulose using the RNeasy Mini Kit (Qiagen, Germantown, MD). RNA quality was checked with the Agilent Bioanalyzer 2100 (Agilent, Santa Clara, CA) and sequencing libraries were generated using the TruSeq Stranded mRNA Library Prep Kit (Illumina, San Diego, CA). Samples were sequenced using NextSeq 500 (Illumina, San Diego, CA) with ∼25–45 million 75bp, paired-end reads.
Chromatin immunoprecipitation (ChIP-qPCR and ChIP-seq)
For analysis of transcription factors, mouse LSK cells transformed by the expression of 3xFLAG-tagged SETBP1 or Setbp1(D/N) were initially fixed with ChIP cross-link Gold (Diagenode, Denville, NJ) for 30 min at a cell density of 1 × 106 cells per mL. Cells were centrifuged, fixative removed, and washed one time with PBS. All subsequent steps were the same for both analysis of transcription factors and histone marks. Cell pellets were resuspended in 1% formaldehyde and incubated for 10 min at room temperature with continuous shaking. After centrifugation and an additional wash with PBS, 0.125 M glycine in PBS was used to quench the reaction. Fixed cell pellets were stored at −80°C until ready for further processing. Fragmentation of chromatin for transcription factors or histone marks were performed using the Chromatin Shearing Optimization Kit (C01020013 and C01020010, respectively; Diagenode, Denville, NJ). Chromatin was sonicated for 10 cycles (30s on, 30s off) at 4°C using the Bioruptor Pico (Diagenode, Denville, NJ). Sheared chromatin was used for ChIP according to the iDeal ChIP-qPCR kit (Diagenode, Denville, NJ) using the indicated antibodies, with the exception that chromatin DNA was purified using the iPure kit (Diagenode, Denville, NJ). Purified chromatin DNA was used for qPCR as described above using ChIP primers specific for the Hoxa9 promoter listed below (Kuo et al., 2013).
For ChIP-sequencing, primary colonies (day 5) from wild-type LSK cells transduced with pMYs retrovirus expressing 3xFLAG-tagged SETBP1 (pMYs-3xFLAG-Setbp1-IRES-GFP) or SETBP1(D/N) (pMYs-3xFLAG-Setbp1[D/N]-IRES-GFP) were isolated and processed as described above. ChIP reactions were performed using the iDeal ChIP-seq kit for transcription factors or histones (C01010054 and C01010051, respectively; Diagenode, Denville, NJ) with the indicated antibodies. Purified ChIP samples were quantified using the Qubit dsDNA HS Assay kit (Invitrogen, Carlsbad, CA, USA) and sequencing libraries were produced using DNA SMART ChIP-Seq kit (Takara Bio, Mountain View, CA) according to the manufacturer's instructions. The concentrations of individual libraries were determined using the KAPA Library Quantification kit (Roche, Indianapolis, IN) and 1.8 pM of each library was used for running next-generation multiplexed sequencing using the NextSeq 500 instrument (Illumina, San Diego, CA).
| Primers | Sequence |
|---|---|
| Hoxa9 (−2087) Forward | 5′-CTG TCT CTC CGC TTC CAT TC-3′ |
| Hoxa9 (−2087) Reverse | 5′-CTG ACC AGC TTC CCT TAT GC-3′ |
| Hoxa9 (−1473) Forward | 5′-CAA ACT GGG GCA CTG GTA CT-3′ |
| Hoxa9 (−1473) Reverse | 5′-TTC TGT GGT CTC AGG CAG TG-3′ |
| Hoxa9 (−536) Forward | 5′-TGT CAG AGC GTT GGA AAG TG-3′ |
| Hoxa9 (−536) Reverse | 5′-TGT GAA TTT TGT GCC TTC CA-3′ |
| Hoxa9 (+24) Forward | 5′-ACC AGA GCG GTT CAT ACA GG-3′ |
| Hoxa9 (+24) Reverse | 5′-CAG ACT GGA GATG GGG AAA A-3′ |
Immunofluorescence
Setbp1 or Setbp1(D/N) transformed mouse LSK cells were fixed with 2% paraformaldehyde on poly-L-lysine coated microscope slides (Electron Microscopy Sciences, Hatfield, PA). The fixed cells were permeabilized with 0.05% Triton X-100, blocked with 10% normal goat serum and 1% BSA for 30 min. Cells were then incubated with anti-Setbp1 (16841-1-AP, Proteintech, Rosemont, IL) and anti-MLL1-N (05–764; Millipore, Temecula, CA) or anti-MLL1-C (05–765; Millipore, Temecula, CA) at room temperature for 3 h. After washing with PBS, cells were incubated with Alexa -Fluor 488 goat anti-mouse secondary antibody and Alexa-Fluor 647 goat anti-rabbit secondary antibody (Invitrogen, Carlsbad, CA, USA) at room temperature for 1 h. The nucleus was counter stained using DAPI and mounted with Fluoromount G (Electron Microscopy Sciences, Hatfield, PA). Images were acquired using a LSM 710 scanning confocal microscope (Zeiss).
Drug treatment
For in vitro Mll1 deletion, Cre induction was initiated in culture medium supplemented with 100 nM 4-OHT dissolved in ethanol for 6 h. Deletion efficiency for all alleles was determined by semi-quantitative genomic PCR using primers described (Chen et al., 2017), at 24 h after deletion was initiated.
MENIN inhibitors MI-3454 and SNDX-5613 were purchased from MedChemExpress (Monmouth Junction, NJ) and dissolved in DMSO. WDR5 inhibitor OICR-9429 was purchased from Selleck Chemicals (Houston, TX). Primary mouse leukemia cells generated previously (Nguyen et al., 2016) were plated at 0.5 to 1 × 105 cells/mL in IMDM supplemented with 15% heat inactivated fetal bovine serum, 1x penicillin/streptomycin, mouse cytokines (100 ng/mL SCF, 6 ng/mL IL-3, and 10 ng/mL IL-6), and treated with the inhibitors at the indicated concentrations or control DMSO. Cell concentrations were restored to the starting concentrations and inhibitors replenished after 72 h.
Cell culture
HEK293T, Plat-E, and Hs27 cells were maintained in DMEM supplemented with 10% heat inactivated fetal bovine serum and 1X penicillin/streptomycin. Setbp1 or Setbp1(D/N) transformed mouse LSK cells were maintained in IMDM supplemented with 20% heat inactivated horse serum, 1X penicillin/streptomycin, murine SCF (50 ng/mL) and IL-3 (10 ng/mL). Cell cultures were incubated in 5% CO2 at 37°C.
In vivo experiments
For Mll1 excision experiments, lethally irradiated (2 × 550 rad from 137Cs source) 8–12 week old C57BL/6 female mice were transplanted with 5 × 105 frozen viable spleen cells from primary leukemias along with 7.5 × 105 unfractionated support bone marrow cells by tail vein injection. Mll1+/+;Cre+ or Mll1F/F;Cre+ primary leukemias derived from transduction of Setbp1 cDNA (pMYs-Setbp1-IRES-GFP) or Setbp1(D/N) cDNA (pMYs-Setbp1[D/N]-IRES-GFP) were transplanted with the same cohort of recipient mice. Mice were treated by injecting intraperitoneally with 2 mg of Tamoxifen (MP Bio, Solon, OH) or equal volume of corn oil (Sigma, St. Louis, MO) at day 10 and 12 post transplantation. Recipients were monitored daily and mice were euthanized when moribund to collect issues for determining the presence of Mll1 deletion in leukemia cells. Deletion efficiency of Mll1 in vivo was determined by genotyping bone marrow cells from recipient mice at 10 days after Tamoxifen or corn oil treatments.
Data analysis
For RNA-seq analysis, raw sequencing reads were demuxed using bcl2fastq2 (v2.20) and aligned to the mouse reference genome (mm10) with MapSplice (v2.2.1). Gene-level quantification was performed with HTSeq (v0.9.1) against UCSC gene annotations (obtained February 21, 2016). Read alignment statistics and sample quality features were calculated with SAMtools and RSeQC. Sequencing quality was verified by manual inspection of sample-wise characteristics: total reads, mapping percentages, pairing percentages, transcript integrity number (TIN), 5′ to 3′ gene body read coverage slopes, and ribosomal RNA content. DESeq2 (v1.16.1) was used to identify differentially expressed genes (DEGs) between pairwise conditions from raw gene read counts. Significant DEGs were identified as those with an FDR q-value <0.05, an absolute fold change ≥2 (i.e. |log2 [fold-change]| ≥ 1), and mean transcripts per million (TPM) ≥ 1 across samples. Gene set enrichment analysis (GSEA) was performed using the GSEA v4.0.3 application (www.gsea-msigdb.org/gsea/downloads.jsp).
For ChIP-seq analysis, raw sequencing reads were demuxed using bcl2fastq2 (v2.20) and aligned to the mouse reference genome (mm10) with the BWA MEM algorithm. Peak calling was performed with MACS2 (version 2.1.1.20160309) for all ChIP samples against input controls. For histone modifications (H3K4me3 and H3K16ac), the MACS2 callpeak function was used with the standard model building procedure and additional parameters: -g mm --call-summits -B. For M2 and MLL1 experiments, the MACS2 callpeak function was also used but with a fixed shift size (i.e. no model building) of 284 and: -g mm --call-summits -B --nomodel –extsize 284. For all samples, peaks with q-value < 0.05 were considered significant and retained for further analyses. Overlaps among peaks from different experiments were determined with BEDTools, considering any intersection between peak coordinates as an overlap. For peak-to-gene mappings, we classified peaks within 2 kilobases (kb) of annotated gene transcription start sites as promoter mapped, peaks mapping within gene bodies as intragenic, and all other peaks as intergenic. Hypergeometric tests were used to determine the significance of overlapping peak counts against a background of the union of peaks identified in the two experiments. HOMER (version 4.11) was used to identify de-novo motifs of length 8, 10, or 12 overrepresented around top (up to 10,000) ChIP-seq peak summits with the command: findMotifsGenome.pl peakfile mm10r -S 25 -len 8,10,12 -size 50, where peakfile is an input file containing MACS2 peak summit coordinates.
Quantification and statistical analysis
Data are represented as mean ± SD. Sample sizes and animal numbers were determined by previous experiences. No samples were excluded from analyses. All data were analyzed by two-tailed Student's t-test except that survival curves were compared by Log-rank test, using GraphPad Prism 9 software. The researchers were not blinded during sample collection and analysis.
Acknowledgments
This work was supported by the National Cancer Institute Grant CA143193 (Y.D.) and Uniformed Services University of the Health Sciences (USUHS) Grant RAMP310534 (Y.D.). The views presented in this article are those of the authors; no endorsement by USUHS or the Department of Defense has been given or should be inferred.
Author contributions
N.N. designed and performed most of the experiments and data analysis and wrote the manuscript; K.O.G., K.O., and K.R.R. performed experiments and contributed to data analysis; Y.H. performed experiments; A.R.S. and C.L.D. performed data analysis; C.G., J.P.M., G.C., and P.E. contributed vital reagents; Y.D. designed and performed research, analyzed data, and wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: January 21, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.103679.
Supplemental information
Data and code availability
-
•
The RNA-seq and ChIP-seq data reported in this paper have been deposited at NCBI Gene Expression Ominibus (GEO: GSE167301).
-
•
This paper does not report original code.
-
•
Any additional information reported in this paper is available from the lead contact upon request.
References
- Aho E.R., Wang J., Gogliotti R.D., Howard G.C., Phan J., Acharya P., Macdonald J.D., Cheng K., Lorey S.L., Lu B., et al. Displacement of WDR5 from chromatin by a WIN site inhibitor with picomolar affinity. Cell Rep. 2019;26:2916–2928 e2913. doi: 10.1016/j.celrep.2019.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong S.A., Staunton J.E., Silverman L.B., Pieters R., den Boer M.L., Minden M.D., Sallan S.E., Lander E.S., Golub T.R., Korsmeyer S.J. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 2002;30:41–47. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- Bach C., Buhl S., Mueller D., Garcia-Cuellar M.P., Maethner E., Slany R.K. Leukemogenic transformation by HOXA cluster genes. Blood. 2010;115:2910–2918. doi: 10.1182/blood-2009-04-216606. [DOI] [PubMed] [Google Scholar]
- Barna M., Pusic A., Zollo O., Costa M., Kondrashov N., Rego E., Rao P.H., Ruggero D. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature. 2008;456:971–975. doi: 10.1038/nature07449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindels E.M., Havermans M., Lugthart S., Erpelinck C., Wocjtowicz E., Krivtsov A.V., Rombouts E., Armstrong S.A., Taskesen E., Haanstra J.R., et al. EVI1 is critical for the pathogenesis of a subset of MLL-AF9-rearranged AMLs. Blood. 2012;119:5838–5849. doi: 10.1182/blood-2011-11-393827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan A.F., Wang J., Howard G.C., Guarnaccia A.D., Woodley C.M., Aho E.R., Rellinger E.J., Matlock B.K., Flaherty D.K., Lorey S.L., et al. WDR5 is a conserved regulator of protein synthesis gene expression. Nucleic Acids Res. 2020;48:2924–2941. doi: 10.1093/nar/gkaa051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler L.H., Slany R., Cui X., Cleary M.L., Mason D.Y. The HRX proto-oncogene product is widely expressed in human tissues and localizes to nuclear structures. Blood. 1997;89:3361–3370. [PubMed] [Google Scholar]
- Cao F., Chen Y., Cierpicki T., Liu Y., Basrur V., Lei M., Dou Y. An Ash2L/RbBP5 heterodimer stimulates the MLL1 methyltransferase activity through coordinated substrate interactions with the MLL1 SET domain. PLoS One. 2010;5 doi: 10.1371/journal.pone.0014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Anastassiadis K., Kranz A., Stewart A.F., Arndt K., Waskow C., Yokoyama A., Jones K., Neff T., Lee Y., Ernst P. MLL2, not MLL1, plays a major role in sustaining MLL-rearranged acute myeloid leukemia. Cancer cell. 2017;31:755–770 e756. doi: 10.1016/j.ccell.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzio A., Passegue E., Ayton P.M., Karsunky H., Cleary M.L., Weissman I.L. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes Dev. 2003;17:3029–3035. doi: 10.1101/gad.1143403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristobal I., Blanco F.J., Garcia-Orti L., Marcotegui N., Vicente C., Rifon J., Novo F.J., Bandres E., Calasanz M.J., Bernabeu C., Odero M.D. SETBP1 overexpression is a novel leukemogenic mechanism that predicts adverse outcome in elderly patients with acute myeloid leukemia. Blood. 2010;115:615–625. doi: 10.1182/blood-2009-06-227363. [DOI] [PubMed] [Google Scholar]
- Damm F., Itzykson R., Kosmider O., Droin N., Renneville A., Chesnais V., Gelsi-Boyer V., de Botton S., Vey N., Preudhomme C., et al. SETBP1 mutations in 658 patients with myelodysplastic syndromes, chronic myelomonocytic leukemia and secondary acute myeloid leukemias. Leukemia. 2013;27:1401–1403. doi: 10.1038/leu.2013.35. [DOI] [PubMed] [Google Scholar]
- Dou Y., Milne T.A., Ruthenburg A.J., Lee S., Lee J.W., Verdine G.L., Allis C.D., Roeder R.G. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 2006;13:713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- Fernandez-Mercado M., Pellagatti A., Di Genua C., Larrayoz M.J., Winkelmann N., Aranaz P., Burns A., Schuh A., Calasanz M.J., Cross N.C., Boultwood J. Mutations in SETBP1 are recurrent in myelodysplastic syndromes and often coexist with cytogenetic markers associated with disease progression. Br. J. Haematol. 2013;163:235–239. doi: 10.1111/bjh.12491. [DOI] [PubMed] [Google Scholar]
- Garcia-Cuellar M.P., Buttner C., Bartenhagen C., Dugas M., Slany R.K. Leukemogenic MLL-ENL fusions induce alternative chromatin states to drive a functionally dichotomous group of target genes. Cell Rep. 2016;15:310–322. doi: 10.1016/j.celrep.2016.03.018. [DOI] [PubMed] [Google Scholar]
- Gentles A.J., Plevritis S.K., Majeti R., Alizadeh A.A. Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA. 2010;304:2706–2715. doi: 10.1001/jama.2010.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grebien F., Vedadi M., Getlik M., Giambruno R., Grover A., Avellino R., Skucha A., Vittori S., Kuznetsova E., Smil D., et al. Pharmacological targeting of the Wdr5-MLL interaction in C/EBPalpha N-terminal leukemia. Nat. Chem. Biol. 2015;11:571–578. doi: 10.1038/nchembio.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y., Nakamura T., Alder H., Prasad R., Canaani O., Cimino G., Croce C.M., Canaani E. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell. 1992;71:701–708. doi: 10.1016/0092-8674(92)90603-a. [DOI] [PubMed] [Google Scholar]
- Hsieh J.J., Cheng E.H., Korsmeyer S.J. Taspase1: a threonine aspartase required for cleavage of MLL and proper HOX gene expression. Cell. 2003;115:293–303. doi: 10.1016/s0092-8674(03)00816-x. [DOI] [PubMed] [Google Scholar]
- Huntly B.J., Shigematsu H., Deguchi K., Lee B.H., Mizuno S., Duclos N., Rowan R., Amaral S., Curley D., Williams I.R., et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer cell. 2004;6:587–596. doi: 10.1016/j.ccr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Jiang H., Shukla A., Wang X., Chen W.Y., Bernstein B.E., Roeder R.G. Role for Dpy-30 in ES cell-fate specification by regulation of H3K4 methylation within bivalent domains. Cell. 2011;144:513–525. doi: 10.1016/j.cell.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jude C.D., Climer L., Xu D., Artinger E., Fisher J.K., Ernst P. Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell stem cell. 2007;1:324–337. doi: 10.1016/j.stem.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawagoe H., Humphries R.K., Blair A., Sutherland H.J., Hogge D.E. Expression of HOX genes, HOX cofactors, and MLL in phenotypically and functionally defined subpopulations of leukemic and normal human hematopoietic cells. Leukemia. 1999;13:687–698. doi: 10.1038/sj.leu.2401410. [DOI] [PubMed] [Google Scholar]
- Klossowski S., Miao H., Kempinska K., Wu T., Purohit T., Kim E., Linhares B.M., Chen D., Jih G., Perkey E., et al. Menin inhibitor MI-3454 induces remission in MLL1-rearranged and NPM1-mutated models of leukemia. J. Clin. Invest. 2020;130:981–997. doi: 10.1172/JCI129126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Krivtsov A.V., Evans K., Gadrey J.Y., Eschle B.K., Hatton C., Uckelmann H.J., Ross K.N., Perner F., Olsen S.N., Pritchard T., et al. A menin-MLL inhibitor induces specific chromatin changes and eradicates disease in models of MLL-rearranged leukemia. Cancer cell. 2019;36:660–673 e611. doi: 10.1016/j.ccell.2019.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov A.V., Twomey D., Feng Z., Stubbs M.C., Wang Y., Faber J., Levine J.E., Wang J., Hahn W.C., Gilliland D.G., et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- Kuhn M.W., Song E., Feng Z., Sinha A., Chen C.W., Deshpande A.J., Cusan M., Farnoud N., Mupo A., Grove C., et al. Targeting chromatin regulators inhibits leukemogenic gene expression in NPM1 mutant leukemia. Cancer Discov. 2016;6:1166–1181. doi: 10.1158/2159-8290.CD-16-0237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo H.P., Wang Z., Lee D.F., Iwasaki M., Duque-Afonso J., Wong S.H., Lin C.H., Figueroa M.E., Su J., Lemischka I.R., Cleary M.L. Epigenetic roles of MLL oncoproteins are dependent on NF-kappaB. Cancer cell. 2013;24:423–437. doi: 10.1016/j.ccr.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard J., Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–260. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- Libbrecht C., Xie H.M., Kingsley M.C., Haladyna J.N., Riedel S.S., Alikarami F., Lenard A., McGeehan G.M., Ernst P., Bernt K.M. Menin is necessary for long term maintenance of meningioma-1 driven leukemia. Leukemia. 2021 doi: 10.1038/s41375-021-01146-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas C.M., Scott L.J., Carmell N., Holcroft A.K., Hills R.K., Burnett A.K., Clark R.E. CIP2A- and SETBP1-mediated PP2A inhibition reveals AKT S473 phosphorylation to be a new biomarker in AML. Blood Adv. 2018;2:964–968. doi: 10.1182/bloodadvances.2017013615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima H., Yoshida K., Nguyen N., Przychodzen B., Sanada M., Okuno Y., Ng K.P., Gudmundsson K.O., Vishwakarma B.A., Jerez A., et al. Somatic SETBP1 mutations in myeloid malignancies. Nat. Genet. 2013;45:942–946. doi: 10.1038/ng.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon K.A., Hiew S.Y., Hadjur S., Veiga-Fernandes H., Menzel U., Price A.J., Kioussis D., Williams O., Brady H.J. Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal. Cell stem cell. 2007;1:338–345. doi: 10.1016/j.stem.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Meyer C., Burmeister T., Groger D., Tsaur G., Fechina L., Renneville A., Sutton R., Venn N.C., Emerenciano M., Pombo-de-Oliveira M.S., et al. The MLL recombinome of acute leukemias in 2017. Leukemia. 2018;32:273–284. doi: 10.1038/leu.2017.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne T.A., Briggs S.D., Brock H.W., Martin M.E., Gibbs D., Allis C.D., Hess J.L. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol. cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- Minakuchi M., Kakazu N., Gorrin-Rivas M.J., Abe T., Copeland T.D., Ueda K., Adachi Y. Identification and characterization of SEB, a novel protein that binds to the acute undifferentiated leukemia-associated protein SET. Eur. J. Biochem. 2001;268:1340–1351. doi: 10.1046/j.1432-1327.2001.02000.x. [DOI] [PubMed] [Google Scholar]
- Mishra B.P., Zaffuto K.M., Artinger E.L., Org T., Mikkola H.K., Cheng C., Djabali M., Ernst P. The histone methyltransferase activity of MLL1 is dispensable for hematopoiesis and leukemogenesis. Cell Rep. 2014;7:1239–1247. doi: 10.1016/j.celrep.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T., Mori T., Tada S., Krajewski W., Rozovskaia T., Wassell R., Dubois G., Mazo A., Croce C.M., Canaani E. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol. cell. 2002;10:1119–1128. doi: 10.1016/s1097-2765(02)00740-2. [DOI] [PubMed] [Google Scholar]
- Nayak A., Viale-Bouroncle S., Morsczeck C., Muller S. The SUMO-specific isopeptidase SENP3 regulates MLL1/MLL2 methyltransferase complexes and controls osteogenic differentiation. Mol. cell. 2014;55:47–58. doi: 10.1016/j.molcel.2014.05.011. [DOI] [PubMed] [Google Scholar]
- Nguyen N., Oakley K., Han Y., Kwok M., Crouch G., Du Y. Interaction with XPO1 is essential for SETBP1 to induce myeloid transformation. Leukemia. 2019 doi: 10.1038/s41375-019-0521-x. [DOI] [PubMed] [Google Scholar]
- Nguyen N., Vishwakarma B.A., Oakley K., Han Y., Przychodzen B., Maciejewski J.P., Du Y. Myb expression is critical for myeloid leukemia development induced by Setbp1 activation. Oncotarget. 2016;7:86300–86312. doi: 10.18632/oncotarget.13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley K., Han Y., Vishwakarma B.A., Chu S., Bhatia R., Gudmundsson K.O., Keller J., Chen X., Vasko V., Jenkins N.A., et al. Setbp1 promotes the self-renewal of murine myeloid progenitors via activation of Hoxa9 and Hoxa10. Blood. 2012;119:6099–6108. doi: 10.1182/blood-2011-10-388710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott M.G., Schmidt M., Schwarzwaelder K., Stein S., Siler U., Koehl U., Glimm H., Kuhlcke K., Schilz A., Kunkel H., et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 2006;12:401–409. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- Patel A., Dharmarajan V., Vought V.E., Cosgrove M.S. On the mechanism of multiple lysine methylation by the human mixed lineage leukemia protein-1 (MLL1) core complex. J. Biol. Chem. 2009;284:24242–24256. doi: 10.1074/jbc.M109.014498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel J.L., Pournazari P., Haggstrom S.J., Kosari F., Shabani-Rad M.T., Natkunam Y., Mansoor A. LMO2 (LIM domain only 2) is expressed in a subset of acute myeloid leukaemia and correlates with normal karyotype. Histopathology. 2014;64:226–233. doi: 10.1111/his.12242. [DOI] [PubMed] [Google Scholar]
- Pelletier J., Thomas G., Volarevic S. Ribosome biogenesis in cancer: new players and therapeutic avenues. Nat. Rev. Cancer. 2018;18:51–63. doi: 10.1038/nrc.2017.104. [DOI] [PubMed] [Google Scholar]
- Phillips R.L., Ernst R.E., Brunk B., Ivanova N., Mahan M.A., Deanehan J.K., Moore K.A., Overton G.C., Lemischka I.R. The genetic program of hematopoietic stem cells. Science. 2000;288:1635–1640. doi: 10.1126/science.288.5471.1635. [DOI] [PubMed] [Google Scholar]
- Piazza R., Magistroni V., Redaelli S., Mauri M., Massimino L., Sessa A., Peronaci M., Lalowski M., Soliymani R., Mezzatesta C., et al. SETBP1 induces transcription of a network of development genes by acting as an epigenetic hub. Nat. Commun. 2018;9:2192. doi: 10.1038/s41467-018-04462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza R., Valletta S., Winkelmann N., Redaelli S., Spinelli R., Pirola A., Antolini L., Mologni L., Donadoni C., Papaemmanuil E., et al. Recurrent SETBP1 mutations in atypical chronic myeloid leukemia. Nat. Genet. 2013;45:18–24. doi: 10.1038/ng.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg A.J., Wang W., Graybosch D.M., Li H., Allis C.D., Patel D.J., Verdine G.L. Histone H3 recognition and presentation by the WDR5 module of the MLL1 complex. Nat. Struct. Mol. Biol. 2006;13:704–712. doi: 10.1038/nsmb1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi H., Okuno Y., Muramatsu H., Yoshida K., Shiraishi Y., Takahashi M., Kon A., Sanada M., Chiba K., Tanaka H., et al. Exome sequencing identifies secondary mutations of SETBP1 and JAK3 in juvenile myelomonocytic leukemia. Nat. Genet. 2013;45:937–941. doi: 10.1038/ng.2698. [DOI] [PubMed] [Google Scholar]
- Steward M.M., Lee J.S., O'Donovan A., Wyatt M., Bernstein B.E., Shilatifard A. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat. Struct. Mol. Biol. 2006;13:852–854. doi: 10.1038/nsmb1131. [DOI] [PubMed] [Google Scholar]
- Thomas L.R., Adams C.M., Wang J., Weissmiller A.M., Creighton J., Lorey S.L., Liu Q., Fesik S.W., Eischen C.M., Tansey W.P. Interaction of the oncoprotein transcription factor MYC with its chromatin cofactor WDR5 is essential for tumor maintenance. Proc. Natl. Acad. Sci. U S A. 2019;116:25260–25268. doi: 10.1073/pnas.1910391116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkachuk D.C., Kohler S., Cleary M.L. Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell. 1992;71:691–700. doi: 10.1016/0092-8674(92)90602-9. [DOI] [PubMed] [Google Scholar]
- Truitt M.L., Ruggero D. New frontiers in translational control of the cancer genome. Nat. Rev. Cancer. 2016;16:288–304. doi: 10.1038/nrc.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishwakarma B.A., Nguyen N., Makishima H., Hosono N., Gudmundsson K.O., Negi V., Oakley K., Han Y., Przychodzen B., Maciejewski J.P., Du Y. Runx1 repression by histone deacetylation is critical for Setbp1-induced mouse myeloid leukemia development. Leukemia. 2016;30:200–208. doi: 10.1038/leu.2015.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.F., Wu G., Mi S., He F., Wu J., Dong J., Luo R.T., Mattison R., Kaberlein J.J., Prabhakar S., et al. MLL fusion proteins preferentially regulate a subset of wild-type MLL target genes in the leukemic genome. Blood. 2011;117:6895–6905. doi: 10.1182/blood-2010-12-324699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong P., Iwasaki M., Somervaille T.C., So C.W., Cleary M.L. Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes Dev. 2007;21:2762–2774. doi: 10.1101/gad.1602107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Valerio D.G., Eisold M.E., Sinha A., Koche R.P., Hu W., Chen C.W., Chu S.H., Brien G.L., Park C.Y., et al. NUP98 fusion proteins interact with the NSL and MLL1 complexes to drive leukemogenesis. Cancer cell. 2016;30:863–878. doi: 10.1016/j.ccell.2016.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A., Cleary M.L. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer cell. 2008;14:36–46. doi: 10.1016/j.ccr.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A., Kitabayashi I., Ayton P.M., Cleary M.L., Ohki M. Leukemia proto-oncoprotein MLL is proteolytically processed into 2 fragments with opposite transcriptional properties. Blood. 2002;100:3710–3718. doi: 10.1182/blood-2002-04-1015. [DOI] [PubMed] [Google Scholar]
- Yokoyama A., Wang Z., Wysocka J., Sanyal M., Aufiero D.J., Kitabayashi I., Herr W., Cleary M.L. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol. Cell. Biol. 2004;24:5639–5649. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Sammons M.A., Donahue G., Dou Z., Vedadi M., Getlik M., Barsyte-Lovejoy D., Al-awar R., Katona B.W., Shilatifard A., et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature. 2015;525:206–211. doi: 10.1038/nature15251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J., Shi J., Wang E., Rappaport A.R., Herrmann H., Sison E.A., Magoon D., Qi J., Blatt K., Wunderlich M., et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The RNA-seq and ChIP-seq data reported in this paper have been deposited at NCBI Gene Expression Ominibus (GEO: GSE167301).
-
•
This paper does not report original code.
-
•
Any additional information reported in this paper is available from the lead contact upon request.