

Abstract
Crystallographic characterization of RuX(CO)(η3-C3H5)(JOSIPHOS), where X = Cl, Br, I, reveals a halide-dependent diastereomeric preference that defines metal-centered stereogenicity and, therefrom, the enantioselectivity of C-C coupling in ruthenium-catalyzed anti-diastereo- and enantioselective C-C couplings of primary alcohols with 1-aryl-1-propynes to form products of carbonyl anti-(α-aryl)allylation. Computational studies reveal a non-classical hydrogen bond between iodide and the aldehyde formyl CH bond stabilizes the favored transition state for carbonyl addition. An improved catalytic system enabling previously unattainable transformations was developed that employs an iodide-containing precatalyst RuI(CO)3(η3-C3H5) in combination with trifluoroethanol (TFE), as illustrated by the first enantioselective ruthenium-catalyzed C-C couplings of ethanol to form higher alcohols.
Introduction
The development of atom-efficient catalytic methods for the valorization of renewable feedstocks is a longstanding goal of chemical research.1 To this end, our laboratory has introduced a family of hydrogen auto-transfer (“borrowing hydrogen”) processes that convert lower alcohols to higher alcohols.2,3 These reactions affect carbinol C-H functionalization and, hence, differ from Guerbet-type reactions of alcohols, which result in hydroxyl substitution.3,4,5 This work encompasses the first catalytic enantioselective C-C couplings of methanol6b,c (>30 M tons/year) and ethanol7 (>80 M tons/year). Specifically, using iridium-catalysts, methanol was coupled to dienes6b and allenes6c to form primary neopentyl alcohols, and ethanol was coupled to allylic acetates7 to form branched secondary homoallylic alcohols. As reflected by annual production rates, ruthenium (30 tons/yr) is more abundant than iridium (3 tons/yr),8 yet use of ruthenium catalysts in asymmetric conversion of methanol or ethanol to higher alcohols is unknown.9 Recent advances in our laboratory on ruthenium-catalyzed alkyne-alcohol carbonyl reductive coupling via hydrogen auto-transfer support the feasibility of converting ethanol and 1-aryl-1-propynes to higher enantiomerically enriched alcohols.10,11e,i However, our previously reported catalytic system was inefficient, requiring high loadings of ruthenium (10 mol%).11i
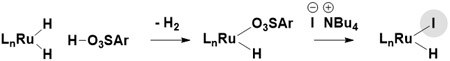
Here, by understanding halide counterion effectss12,13 and exploiting trifluoroethanol (TFE)-enhanced turnover,14 we report an improved catalytic system for alkyne-alcohol C-C coupling, as illustrated by the regio- and enantioselective conversion of ethanol (the most abundant renewable small molecule carbon source)15 to enantiomerically enriched homoallylic alcohols (Figure 1). Specifically, using a ruthenium catalyst modified by JOSIPHOS16 in the presence of TFE, diverse 1-aryl-1-propynes react with ethanol to form branched secondary homoallylic alcohols through a tandem catalytic cycle in which alkyne-to-allene internal redox isomerization10 is followed by allene-aldehyde reductive coupling via hydrogen auto-transfer.17,18,19 As corroborated by DFT calculations and crystallographic characterization of a series of halide-bound complexes RuX(CO)(η3-C3H5)(JOSIPHOS), where X = Cl, Br, I, there exists a halide-dependent diastereomeric preference that defines metal-centered stereogenicity and, therefrom, the absolute stereochemical course of C-C coupling. Whereas the chloride- and bromide-bound catalysts exist as stereoisomeric mixtures, the iodide-bound catalyst exists as a single stereoisomer, enforcing superior levels of enantioselectivity. These insights have led to a simplified catalyst system that exploits an iodide-containing precatalyst, RuI(CO)3(η3-C3H5) that operates at lower catalyst loadings and enables previously unattainable 1-aryl-1-propyne-mediated carbonyl anti-(α-aryl)allylations of alcohol reactants.20,21
Figure 1.

Halide effects in enantioselective ruthenium-catalyzed conversion of ethanol to higher alcohols.
Results and Discussion
In prior work on enantioselective ruthenium-JOSIPHOS-catalyzed couplings of alcohols and sec-alkyl-substituted propynes to form branched products of carbonyl allylation,11e iodide counterions were found to enforce high levels of enantioselectivity and suppress competing formation of (Z)-homoallylic alcohols that arise via allene-carbonyl oxidative coupling (for catalytic cycle, see Scheme 2).11c The iodide-bound ruthenium catalysts are generated in situ through an acid-base reaction of a ruthenium dihydride with an arylsulfonic acid followed by substitution of the resulting sulfonate counterion by iodide (eq. 1).22 The selectivity and productivity of the catalytic system is highly dependent upon the efficiency with which the iodide counterion is introduced. As illustrated in related reactions of 1-aryl-1-propynes,11i replacement of 2,4,6-(iPr)3PhSO3H with 4-NO2PhSO3H resulted in >50% increase in isolated yield, yet high catalyst loadings (10 mol%) remained necessary.
Scheme 2.

Proposed catalytic cycle and stereochemical model for the ruthenium-catalyzed C-C coupling of 1-aryl-1-propynes with ethanol to form products of carbonyl anti-(α-aryl)allylation.
 |
(eq.1) |
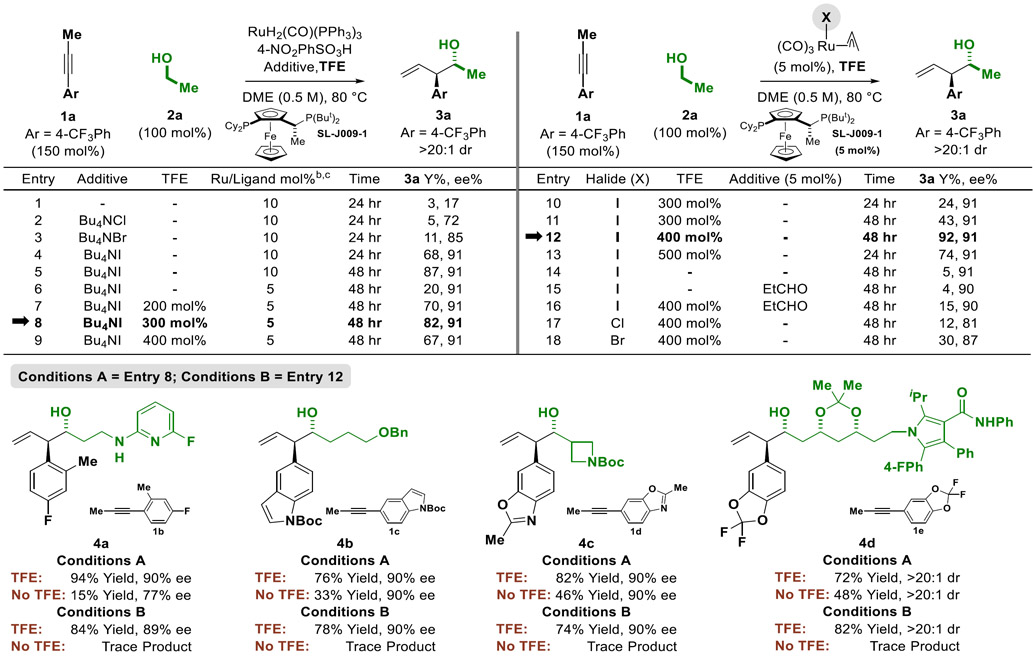
Given the low turnover numbers associated with the previously developed catalytic system additional optimization experiments were undertaken using 1-(4-CF3-phenyl)-1-propyne 1a (150 mol%) and ethanol 2a (100 mol%) (Table 1). Consistent with our previous observations,11i low conversion to the product of carbonyl anti-(α-aryl)allylation 3a is observed upon omission of halide additives or introduction of lower halides (Table 1, entries 1-3). Using the iodide-bound catalyst, adduct 3a is formed in 68% yield with a 91% enantiomeric enrichment (Table 1, entries 4). Extending reaction time to 48 hours, the yield of adduct 3a was increased to 87% (Table 1, entry 5). However, at 5 mol% loadings of RuH2(CO)(PPh3)3, SL-J009-1 and 4-NO2PhSO3H under otherwise identical conditions, the yield of 3a was substantially decreased. Low turnover was attributed to inefficient release of the homoallylic ruthenium alkoxide derived upon carbonyl addition. It was posited that TFE might facilitate exchange of the homoallylic ruthenium alkoxide with the primary alcohol reactant via protonolytic cleavage of the former species, or perhaps facilitate carbonyl addition via hydrogen bonding to the carbonyl ligand23 to enhance Lewis acidity at ruthenium. In the event, using TFE as an additive led to a pronounced increase in the yield of 3a without changing the degree of asymmetric induction (Table 1, entries 8-9). At optimal loadings of TFE (300 mol%), 3a was generated in 82% yield and 91% enantiomeric enrichment (Table 1, entry 8).
Table 1.
Top: Selected optimization experiments illustrating the influence of TFE and halide counterion in the enantioselective transfer hydrogenative coupling of alkyne 1a and ethanol 2a to form the product of carbonyl anti-(α-aryl)allylation 3a. Bottom: Effect of TFE on the ruthenium-JOSIPHOS catalyzed C-C coupling of alkynes 1b-1e with alcohols 2b-2e to form adducts 4a-4d.a
|
Yields of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. Diastereoselectivities were determined by 1H NMR analysis of crude reaction mixtures.
Entries 1-5: H2Ru(CO)(PPh3)3 (10 mol%), SL-J009-1 (10 mol%), Bu4NI (20 mol%) and 4-NO2PhSO3H (10 mol%).
Entries 6-9, H2Ru(CO)(PPh3)3 (5 mol%), SL-J009-1 (5 mol%), Bu4NI (10 mol%) and 4-NO2PhSO3H (5 mol%).
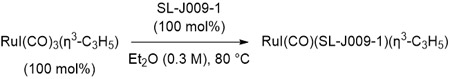
Use of the iodide-containing precatalyst, RuI(CO)3(η3-C3H5),24 should preclude the need for 4-NO2PhSO3H and Bu4NI, potentially simplifying the catalytic system. A series of experiments were conducted in which 1-(4-CF3-phenyl)-1-propyne 1a and ethanol 2a were exposed to RuI(CO)3(η3-C3H5) (5 mol%) and SL-J009-1 (5 mol%) in the presence of variable quantities of TFE (Table 1, entries 10-13). To our delight, reactions run for 48 hours using TFE (400 mol%) delivered adduct 3a in 92% yield and 91% enantiomeric enrichment (Table 1, entry 12). In the absence of TFE, a 5% yield of 3a was obtained (Table 1, entry 14). The increased sensitivity of reactions involving the iodide-bound precatalyst toward TFE may be due to the fact that 4-NO2PhSO3H is absent and an acid in the medium may be required to catalyze alkoxide exchange at the metal center. While it is possible that TFE initiates entry into the catalytic cycle through protonation of the π-allylruthenium precatalyst, a reaction conducted in the presence propanal (which can remove the π-allyl via carbonyl allylation) produced 3a in only 4% yield (Table 1, entry 15). Interestingly, in the presence propanal catalytic activity is not fully restored upon reintroduction of TFE (Table 1, entry 16). As will be discussed (vide infra), it appears pairwise generation of the aldehyde and π-allylruthenium intermediates is required for high catalytic turnover. Using the chloride- and bromide- bound precatalysts RuX(CO)3(η3-C3H5), X = Cl, Br, significantly lower yields and Reactions conducted using hexafluoroisopropanol HFIP in place of TFE (400 mol%) led to no conversion, however, lower loadings of HFIP were not explored.
To assess the generality of TFE-enhanced turnover in ruthenium-JOSIPHOS catalyzed C-C couplings beyond ethanol, alkynes 1b-1e were exposed to alcohols 2b-2e under the two optimal conditions identified for the formation of 3a in the presence and absence of TFE (Table 1). Specifically, “Conditions A” (Table 1, entry 8) utilizing the RuH2(CO)(PPh3)3 precatalyst in combination with 4-NO2PhSO3H and Bu4NI, and “Conditions B” (Table 1, entry 12) using the RuI(CO)3(η3-C3H5) precatalyst in the absence of 4-NO2PhSO3H and Bu4NI were employed. The products of carbonyl anti-(α-aryl)allylation 4a-4d were formed in substantially higher yields in the presence of TFE. Roughly equivalent enantioselectivities were obtained using either Conditions A or B. As demonstrated in the formation of 4a-4d (and in couplings to ethanol vide infra, Table 2), the superiority of Conditions A or B is case dependent and differences in yield typically do not diverge more than 10-20%.
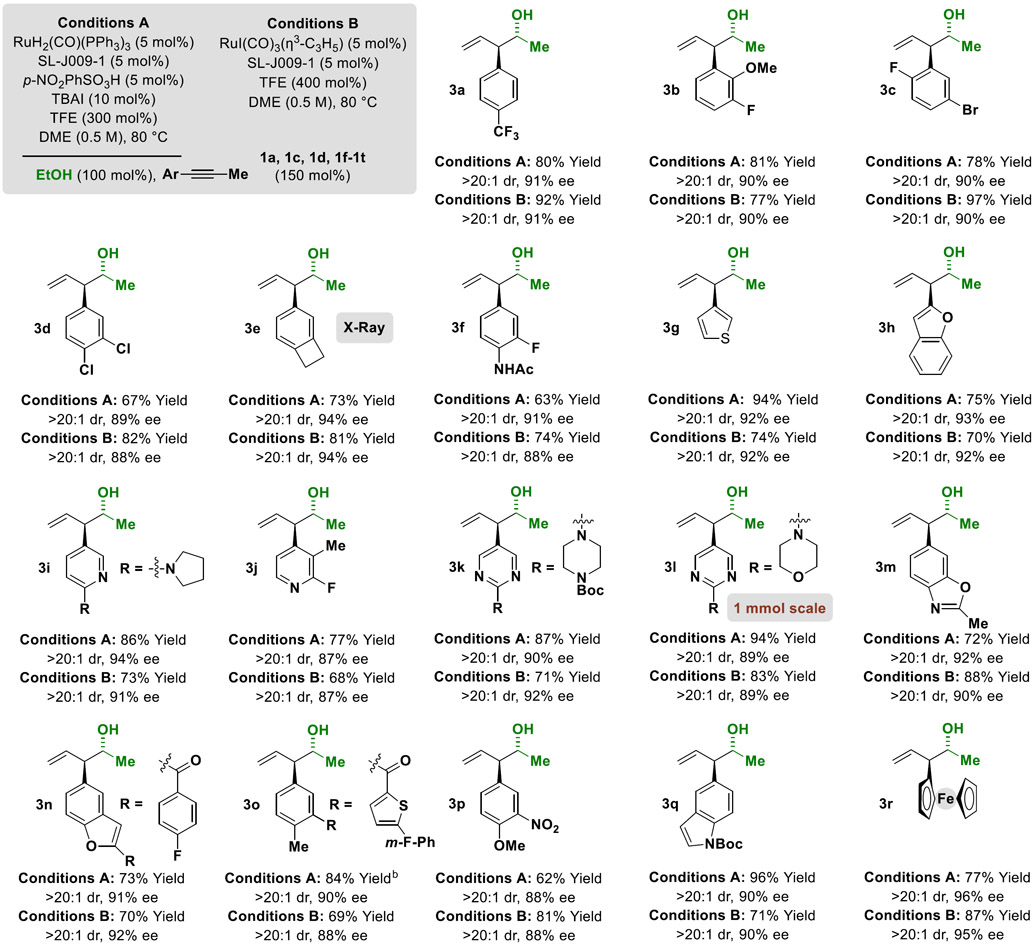
Table 2.
Ruthenium-catalyzed coupling of 1-aryl-1-propynes 1a, 1c, 1d, 1f-1t with ethanol 2a to form enantiomerically enriched homoallylic alcohols 3a-3r.a
|
Yields of material isolated by silica gel chromatography. Enantioselectivities were determined by chiral stationary phase HPLC analysis. Diastereoselectivities were determined via 1H NMR analysis of crude reaction mixtures.
DME (0.25 M). See Supporting Information for further experimental details.
 |
(eq.2) |
 |
(eq.3) |
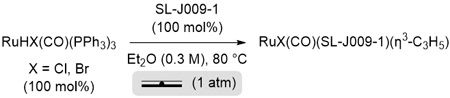
To gain insight into the pronounced effect of halide counterion on productivity and enantioselectivity in the present ruthenium-catalyzed (α-aryl)allylations, the complexes RuX(CO)(JOSIPHOS)(η3-C3H5), where X = Cl, Br, I, were prepared as shown (eq. 2, 3) and their solid state structures were characterized by single crystal X-ray diffraction (Figure 2). Comparison of these crystal structures reveals a halide-dependent selectivity for occupancy of the axial coordination sites to define metal-centered stereogenicity. For the chloride complex, a 3:1 ratio of isomers is observed with chloride predominantly occupying the “northern” coordination site. For bromide, a 1:1 ratio of isomers is observed. For iodide, the “southern” coordination site is exclusively occupied. An analogous trend is observed in relation to the degree of disorder the π-allyl moiety. For the chloride complex, there exists a 1:1 ratio of conformers in which the π-allyl is “up vs down.” For the bromide complex, the π-allyl is less disordered and simply tilts a small amount (a 3:1 ratio of conformers is observed). For the iodide complex, there is no discernable disorder associated with the π-allyl. We posit that steric interactions between the southern tert-butyl group guide site-occupancy. Although iodide has the largest Van der Waals radius (Cl = 1.75 Å, Br = 1.85 Å, I = 1.98 Å), the exceptionally long ruthenium-iodine bond (Ru-X, Cl = 2.57 Å, Br = 2.66 Å, I = 2.85 Å) alleviates steric interactions between iodide and the tert-butyl group relative to chloride, bromide and the carbonyl ligand. The long ruthenium-iodine bond also appears to mitigiate π-backbonding to the carbonyl ligand, as the C≡O bond length is shortest in the iodide complex. As will be discussed, diminished π-backbonding increases charge density on the iodide counterion, facilitating its role as a non-classical hydrogen-bond acceptor to the aldehyde formyl CH bond in the transition state for carbonyl addition (vide infra).
Figure 2.

Solid state structures of RuX(CO)(JOSIPHOS)(η3-C3H5), where X = Cl, Br, I, as determined by single crystal X-ray diffraction and selected bond distances. Disorder due to incomplete axial site selectivity between the CO ligand and halide counterion is evident for the complexes containing chloride and bromide but not iodide. Displacement ellipsoids are scaled to the 50% probability level. Hydrogen atoms have been omitted for clarity.a
a See Supporting Information for complete crystallographic data
To gain deeper insight into how the iodide counterion influences the transition state for enantioselective C-C bond formation, density functional theory (DFT) calculations were undertaken (Scheme 1).25 Based on the crystallographic data, these studies focused on complexes in which the iodide counterion resides in the “southern” coordination site. The JOSIPHOS ligand has one of the t-butyl groups positioned nearly on the P-Ru-P plane (with a P-Ru-P-C dihedral angle of 159°), which may introduce greater steric interactions with the allyl moiety to disfavor TS3 and TS4. Unlike TS2, TS1 has the iodide positioned in close proximity to the formyl C-H bond of acetaldehyde with an I···HCO distance of 2.845 Å, suggesting the existence of a non-classical hydrogen bond.26 Quantum theory of atoms in molecules (QTAIM)27 analysis identified the bond critical point (BCP) between the I···H region, which is in alignment with natural bond orbital (NBO)28 analysis (Figure 3 and SI). The fuzzy bond order (FBO)29 of 0.069 was computed, accounting for an overall stabilization energy of 4.44 kcal/mol (E(2)-NBO) in TS1 through the iodide’s three electron lone pairs with the σ* orbital of the formyl CH bond as determined by second-order perturbation treatments in NBO (See supporting information). In sharp contrast, no BCP was observed between the CO ligand and acetaldehyde in TS2 and E(2)-NBO from the overlap of the π orbitals of CO and the σ* of the CH group were found to be much smaller (0.53 kcal/mol). These results indicate that beyond steric effects, the formyl hydrogen bond to iodide contributes a critical stabilizing interaction in the carbonyl addition by way of TS1. Hence, TS1 is calculated to be more stable than TS2 and TS3 by 1.38 and 1.35 kcal/mol, respectively.
Scheme 1.

Chair-like transition structures leading to enantiomeric products of carbonyl anti-(α-aryl)allylation (ΔΔG‡ in kcal/mol at ωB97X-V).24
Figure 3.

Gradient trajectories mapped on a total electron density plot in the Ru-I-HCO plane showing bond critical points (BCP, blue circles).
Based on these data, a catalytic cycle for the ruthenium-catalyzed C-C coupling of 1-aryl-1-propynes with ethanol has been proposed in which stereogenicity at ruthenium is defined (Scheme 2). β-Hydride elimination from the indicated ethoxyruthenium species generates a ruthenium hydride with concomitant release of acetaldehyde. Hydrometalation of the allene,30 which is generated via alkyne isomerization, forms a π-allylruthenium complex that engages acetaldehyde in carbonyl addition by way of TS1 to form the indicated homoallylic ruthenium alkoxide. TFE-catalyzed exchange of the homoallylic alkoxide with ethanol liberates the product 3 and regenerates the ethoxyruthenium complex to close the catalytic cycle. Dehydrogenation of TFE vs ethanol are not competitive processes, as TFE dehydrogenation is prohibitively endothermic due to inductive destabilization of the transition state for hydride transfer to ruthenium.31
Conclusions
In summary, halide effects in transition metal catalysis are frequently observed, yet seldom understood.12 Here, we illuminate the origins of halide effects in ruthenium-JOSIPHOS-catalyzed anti-diastereo- and enantioselective C-C couplings of primary alcohols with 1-aryl-1-propynes through DFT calculations and crystallographic characterization of RuX(CO)(η3-C3H5)(JOSIPHOS), where X = Cl, Br, I. Notably, a “halide-dependent diastereomeric preference that defines metal-centered stereogenicity is observed. Whereas incomplete axial site-selectivity is observed for lower halides (Cl, Br), the iodide counterion exclusively occupies a single coordinate site. Additionally, the iodide counterion contributes a key stabilizing interaction, a non-classical CH···I hydrogen bond,26 in the preferred transition state for carbonyl addition. In this way, halide-directed metal-centered stereogenicity defines absolute stereochemical course of carbonyl addition. Based on these insights and exploiting trifluoroethanol (TFE)-enhanced turnover, a simplified catalyst system using an iodide-containing precatalyst, RuI(CO)3(η3-C3H5), that functions efficiently at lower catalyst loadings was designed that enables previously unattainable 1-aryl-1-propyne-mediated carbonyl anti-(α-aryl)allylations of alcohol reactants. Finally, the utility of this improved second-generation catalyst system was demonstrated in the first enantioselective ruthenium-catalyzed C-C couplings of ethanol, the most abundant renewable small molecule feedstock.15 These studies contribute to a growing class of hydrogen auto-transfer processes for catalytic carbonyl addition beyond premetalated reagents.1
Supplementary Material
ACKNOWLEDGMENT
The Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM069445) are acknowledged for partial support of this research. We are grateful for the assistance of Dr. Vincent Lynch for the acquisition and analysis of X-ray diffraction data. The service of Ibex, Shaheen 2 High Performance Computing Facilities was provided by King Abdullah University of Science and Technology (KAUST).
Footnotes
Supporting Information. Experimental procedures and spectroscopic data for all new compounds (1H NMR, 13C NMR, IR, HRMS), including images of NMR spectra and HPLC traces for racemic and enantiomerically enriched compounds. Single crystal X-ray diffraction data for RuX(CO)(JOSIPHOS)(η3-C3H5), where X = Cl, Br, I and compound 3e.
The authors declare no competing financial interest.
REFERENCES
- (1).For a recent review on metal-catalyzed C-C coupling of feedstocks, see: Doerksen RS; Meyer CC; Krische MJ Feedstock Reagents in Metal-Catalyzed Carbonyl Reductive Coupling: Minimizing Preactivation for Efficiency in Target-Oriented Synthesis. Angew. Chem., Int. Ed 2019, 58, 14055–14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) For selected reviews on metal-catalyzed carbonyl addition via hydrogen auto-transfer, see: Ketcham JM; Shin I; Montgomery TP; Krische MJ Catalytic Enantioselective C-H Functionalization of Alcohols by Redox-Triggered Carbonyl Addition: Borrowing Hydrogen, Returning Carbon. Angew. Chem. Int. Ed 2014, 53, 9142–9150. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim SW; Zhang W; Krische MJ Catalytic Enantioselective Carbonyl Allylation and Propargylation via Alcohol-Mediated Hydrogen Transfer: Merging the Chemistry of Grignard and Sabatier. Acc. Chem. Res 2017, 50, 2371–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Santana CG; Krische MJ From Hydrogenation to Transfer Hydrogenation to Hydrogen Auto-Transfer in Enantioselective Metal-Catalyzed Carbonyl Reductive Coupling: Past, Present and Future. ACS Catal. 2021, 11, 5572–5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) For selected reviews on metal-catalyzed hydroxyl substitution via hydrogen auto-transfer, see: Hamid MHSA; Slatford PA; Williams JMJ Borrowing Hydrogen in The Activation of Alcohols. Adv. Synth. Catal 2007, 349, 1555–1575. [Google Scholar]; (b) Guillena G; Ramón DJ; Yus M Alcohols as Electrophiles in C-C Bond-Forming Reactions: The Hydrogen Autotransfer Process. Angew. Chem. Int. Ed 2007, 46, 2358–2364. [Google Scholar]; (c) Dobereiner GE; Crabtree RH Dehydrogenation as a Substrate-Activating Strategy in Homogeneous Transition Metal Catalysis. Chem. Rev 2010, 110, 681–703.19938813 [Google Scholar]; (d) Bähn S; Imm S; Neubert L; Zhang M; Neumann H; Beller M The Catalytic Amination of Alcohols. ChemCatChem 2011, 3, 1853–1864. [Google Scholar]; (e) Yang Q; Wang Q; Yu Z Substitution of Alcohols by N-Nucleophiles via Transition Metal-Catalyzed Dehydrogenation. Chem. Soc. Rev 2015, 44, 2305–2329.25661436 [Google Scholar]; (f) Aitchison H; Wingad RL; Wass DF Homogeneous Ethanol to Butanol Catalysis - Guerbet Renewed. ACS Catal. 2016, 6, 7125–7132. [Google Scholar]; (g) Quintard A; Rodriguez J Catalytic Enantioselective OFF ↔ ON Activation Processes Initiated by Hydrogen Transfer: Concepts and Challenges. Chem. Commun 2016, 52, 10456–10473. [Google Scholar]; (h) Reed-Berendt BG; Polidano K; Morrill LC Recent Advances in Homogeneous Borrowing Hydrogen Catalysis Using Earth-Abundant First Row Transition Metals. Org. Biomol. Chem 2019, 17, 1595–1607.30222171 [Google Scholar]; (i) Kwok T; Hoff O; Armstrong RJ; Donohoe TJ Control of Absolute Stereochemistry in Transition-Metal-Catalysed Hydrogen-Borrowing Reactions. Chem. Eur. J 2020, 26, 12912–12926.32297370 [Google Scholar]; (j) Reed-Berendt BG; Latham DE; Dambatta MB; Morrill LC Borrowing Hydrogen for Organic Synthesis. ACS Cent. Sci 2021, 7, 570–585.34056087 [Google Scholar]
- (4).(a) For recent reports on the use of ethanol in Guerbet-type reactions, see: Mazzoni R; Cesari C; Zanotti V; Lucarelli C; Tabanelli T; Puzzo F; Passarini F; Neri E; Marani G; Prati R; Vigano F; Conversano A; Cavani F Catalytic Biorefining of Ethanol from Wine Waste to Butanol and Higher Alcohols: Modeling the Life Cycle Assessment and Process Design. ACS Sus. Chem. Eng 2019, 7, 224–237. [Google Scholar]; (b) Gawali SS; Pandia BK; Pal S; Gunanathan C Manganese(I)-Catalyzed Cross-Coupling of Ketones and Secondary Alcohols with Primary Alcohols. ACS Omega 2019, 4, 10741–10754.31460172 [Google Scholar]; (c) Kobayashi M; Itoh S; Yoshimura K; Tsukamoto Y; Obora Y Iridium Complex-Catalyzed C2-Extension of Primary Alcohols with Ethanol via a Hydrogen Autotransfer Reaction. J. Org. Chem 2020, 85, 11952–11958.32786619 [Google Scholar]
- (5).(a) For selected examples of enantioselective Guerbet-type reactions, see: Onodera G; Nishibayashi Y; Uemura S Ir- and Ru-Catalyzed Sequential Reactions: Asymmetric α-Alkylative Reduction of Ketones with Alcohols. Angew. Chem. Int. Ed 2006, 45, 3819–3822. [DOI] [PubMed] [Google Scholar]; (b) Armstrong RJ; Akhtar WM; Young TA; Duarte F; Donohoe TJ Catalytic Asymmetric Synthesis of Cyclohexanes by Hydrogen Borrowing Annulations. Angew. Chem. Int. Ed 2019, 58, 12558–12562. [DOI] [PubMed] [Google Scholar]; (c) Ng TW; Liao G; Lau KK; Pan H-J; Zhao Y Room-Temperature Guerbet Reaction with Unprecedented Catalytic Efficiency and Enantioselectivity. Angew. Chem. Int. Ed 2020, 59, 11384–11389. [DOI] [PubMed] [Google Scholar]
- (6).(a) Moran J; Preetz A; Mesch RA; Krische MJ Iridium Catalysed Direct C-C Coupling of Methanol and Allenes. Nature Chem. 2011, 3, 287–290. [DOI] [PubMed] [Google Scholar]; (b) Nguyen KD; Herkommer D; Krische MJ Enantioselective Formation of All-Carbon Quaternary Centers via C-H Functionalization of Methanol: Iridium-Catalyzed Diene Hydrohydroxymethylation. J. Am. Chem. Soc 2016, 138, 14210–14213. [DOI] [PubMed] [Google Scholar]; (c) Holmes M; Nguyen KD; Schwartz LA; Luong T; Krische MJ Enantioselective Formation of CF3-Bearing All-Carbon Quaternary Stereocenters via C-H Functionalization of Methanol: Iridium Catalyzed Allene Hydrohydroxymethylation. J. Am. Chem. Soc 2017, 139, 8114–8117. [DOI] [PubMed] [Google Scholar]
- (7).Meyer CC; Stafford NP; Cheng MJ; Krische MJ Ethanol: Unlocking an Abundant Renewable C2-Feedstock for Catalytic Enantioselective C-C Coupling. Angew. Chem. Int. Ed 2021, 60, 10542–10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).As indicated in the following review, the price per ounce for ruthenium vs iridium is 258 vs 1,480 USD/ounce, respectively (01/2019-01/2020): Doerksen RS; Hodík T; Hu G; Huynh NO; Shuler WG; Krische MJ Ruthenium-Catalyzed Cycloaddition to Form Five-, Six-, and Seven-Membered Rings. Chem. Rev 2021, 121, 4045–4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).A ruthenium-catalyzed C-C coupling of ethanol to form racemic products with incomplete regioselectivity has been described: Han H; Krische MJ Direct Ruthenium-Catalyzed C-C Coupling of Ethanol: Diene Hydro-Hydroxyethylation to Form All Carbon Quaternary Centers. Org. Lett 2010, 12, 2844–2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).For a review on the use of alkynes as π-allyl precursors in catalysis, see: Haydl AM; Breit B; Liang T; Krische MJ Alkynes as Electrophilic or Nucleophilic Allylmetal Precursors in Transition Metal Catalysis. Angew. Chem. Int. Ed 2017, 56, 11312–11325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) For alkynes as latent nucleophilic allylmetal equivalents in metal-catalyzed carbonyl reductive coupling and related redox-neutral processes, see: Obora Y; Hatanaka S; Ishii Y Iridium-Catalyzed Coupling Reaction of Primary Alcohols with 1-Aryl-1-propynes Leading to Secondary Homoallylic Alcohols. Org. Lett 2009, 11, 3510–3513. [DOI] [PubMed] [Google Scholar]; (b) Obora Y; Sawaguchi T; Tsubakimoto K; Yoshida H; Ogawa S; Hatanaka S Iridium-Catalyzed Synthesis of ω-Hydroxy Homoallylic Alcohols. Synthesis 2013, 2115–2119. [DOI] [PubMed] [Google Scholar]; (c) Park BY; Nguyen KD; Chaulagain MR; Komanduri V; Krische MJ Alkynes as Allylmetal Equivalents in Redox-Triggered C-C Couplings to Primary Alcohols: (Z)-Homoallylic Alcohols via Ruthenium-Catalyzed Propargyl C-H Activation. J. Am. Chem. Soc 2014, 136, 11902–11905. [DOI] [PubMed] [Google Scholar]; (d) Chen Q-A; Cruz FA; Dong VM Alkyne Hydroacylation: Switching Regioselectivity by Tandem Ruthenium Catalysis. J. Am. Chem. Soc 2015, 137, 3157–3160. [DOI] [PubMed] [Google Scholar]; (e) Liang T; Nguyen KD; Zhang W; Krische MJ Enantioselective Ruthenium Catalyzed Carbonyl Allylation via Alkyne-Alcohol C-C Bond Forming Transfer Hydrogenation: Allene Hydrometallation vs. Oxidative Coupling. J. Am. Chem. Soc 2015, 137, 3161–3164. [DOI] [PubMed] [Google Scholar]; (f) Liang T; Zhang W; Chen T-Y; Nguyen KD; Krische MJ Ruthenium Catalyzed Diastereo- and Enantioselective Coupling of Propargyl Ethers with Alcohols: Siloxy-Crotylation via Hydride Shift Enabled Conversion of Alkynes to π-Allyls. J. Am. Chem. Soc 2015, 137, 13066–13071. [DOI] [PubMed] [Google Scholar]; (g) Liang T; Zhang W; Krische MJ Iridium Catalyzed C-C Coupling of a Simple Propargyl Ether with Primary Alcohols: Enantioselective Homoaldol Addition via Redox-Triggered (Z)-Siloxyallylation. J. Am. Chem. Soc 2015, 137, 16024–16027. [DOI] [PubMed] [Google Scholar]; (h) Zhang W; Chen W; Xiao H; Krische MJ Carbonyl anti-(α-Amino)allylation via Ruthenium Catalyzed Hydrogen Autotransfer: Use of an Acetylenic Pyrrole as an Allylmetal Pronucleophile. Org. Lett 2017, 19, 4876–4879. [DOI] [PubMed] [Google Scholar]; (i) Xiang M; Ghosh A; Krische MJ Diastereo- and Enantioselective Ruthenium-Catalyzed C-C Coupling of 1-Arylpropynes and Alcohols: Alkynes as Chiral Allylmetal Precursors in Carbonyl anti-(α-Aryl)allylation. J. Am. Chem. Soc 2021, 143, 2838–2845. [DOI] [PubMed] [Google Scholar]
- (12).(a) For reviews on halide effects in transition metal catalysis, see: Maitlis PM; Haynes A; James BR; Catellani M; Chiusoli GP Iodide Effects in Transition Metal Catalyzed Reactions. Dalton Trans. 2004, 3409–3419. [DOI] [PubMed] [Google Scholar]; (b) Fagnou K; Lautens M Halide Effects in Transition Metal Catalysis. Angew. Chem. Int. Ed 2002, 41, 26–47. [DOI] [PubMed] [Google Scholar]
- (13).(a) The following studies suggest iodide is a stronger ligand for late transition metals than chloride or bromide: Pitteri B; Marangoni G; Cattalini L; Canovese L Nucleophilic Displacement of Halides from Dihalogeno-platinum(II) Complexes Containing a Chelating Thioether. Kinetics and Equilibria. J. Chem. Soc. Dalton Trans 1994, 169–174. [Google Scholar]; (b) Poulton JT; Sigalas MP; Folting K; Streib WE; Eisenstein O; Caulton KG RuHX(CO)(PR3)2: Can νCO Be a Probe for the Nature of the Ru-X Bond? Inorg. Chem 1994, 33, 1476–1485. [Google Scholar]
- (14).(a) For selected reviews on trifluoroethanol (and hexafluoroisopropanol) effects in metal-catalysis, see: Shuklov IA; Dubrovina NV; Boerner A Fluorinated Alcohols as Solvents, Cosolvents and Additives in Homogeneous Catalysis. Synthesis 2007, 2925–2943. [Google Scholar]; (b) Sugiishi T; Matsugi M; Hamamoto H; Amii H Enhancement of Stereoselectivities in Asymmetric Synthesis Using Fluorinated Solvents, Auxiliaries, and Catalysts. RSC Adv. 2015, 5, 17269–17282. [Google Scholar]; (c) Wencel-Delord J; Colobert F A Remarkable Solvent Effect of Fluorinated Alcohols on Transition Metal Catalyzed C-H Functionalizations. Org. Chem. Front 2016, 3, 394–400. [Google Scholar]; (d) Colomer I; Chamberlain AER; Haughey MB; Donohoe TJ Hexafluoroisopropanol as a Highly Versatile Solvent. Nat. Chem. Rev 2017, 1, 0088. [Google Scholar]
- (15).(a) For selected literature on ethanol production volumes, see: Kosaric N; Duvnjak Z; Farkas A; Sahm H; Bringer-Meyer S; Goebel O; Mayer D Ethanol. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2001. Vol. 13, pp. 333–403. [Google Scholar]; (b) Kennes D; Abubackar HN; Diaz M; Veiga MC; Kennes C Bioethanol Production from Biomass: Carbohydrate vs Syngas Fermentation. J. Chem. Tech. Biotech 2016, 91, 304–317 and references cited therein. [Google Scholar]
- (16).Blaser H-U; Brieden W; Pugin B; Spindler F; Studer M; Tognai A Solvias Josiphos Ligands: From Discovery to Technical Applications. Top. Catal 2002, 19, 3–16. [Google Scholar]
- (17).(a) For ruthenium-catalyzed allene-carbonyl reductive coupling via alcohol-mediated hydrogen transfer and hydrogen auto-transfer, see: Ngai M-Y; Skucas E; Krische MJ Ruthenium Catalyzed C-C Bond Formation via Transfer Hydrogenation: Branch-Selective Reductive Coupling of Allenes to Paraformaldehyde and Higher Aldehydes. Org. Lett 2008, 10, 2705–2708. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Skucas E; Zbieg JR; Krische MJ anti-Aminoallylation of Aldehydes via Ruthenium-Catalyzed Transfer Hydrogenative Coupling of Sulfonamido Allenes: 1,2-Aminoalcohols. J. Am. Chem. Soc 2009, 131, 5054–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zbieg JR; McInturff EL; Krische MJ Allenamide Hydro-Hydroxyalkylation: 1,2-Aminoalcohols via Ruthenium Catalyzed Carbonyl anti-Aminoallylation. Org. Lett 2010, 12, 2514–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zbieg JR; McInturff EL; Leung JC; Krische MJ Amplification of anti-Diastereoselectivity via Curtin-Hammett Effects in Ruthenium Catalyzed Hydrohydroxyalkylation of 1,1-Disubstituted Allenes: Diastereoselective Formation of All-Carbon Quaternary Centers. J. Am. Chem. Soc 2011, 133, 1141–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sam B; Montgomery TP; Krische MJ Ruthenium Catalyzed Reductive Coupling of Paraformaldehyde to Trifluoromethyl Allenes: CF3-Bearing All-Carbon Quaternary Centers. Org. Lett 2013, 15, 3790–3793. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Sam B; Luong T; Krische MJ Ruthenium-Catalyzed C-C Coupling of Fluorinated Alcohols with Allenes: Dehydrogenation at the Energetic Limit of β-Hydride Elimination. Angew. Chem. Int. Ed 2015, 54, 5465–5469. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Xiang M; Pfaffinger DE; Ortiz E; Brito GA; Krische MJ Enantioselective Ruthenium-BINAP-Catalyzed Carbonyl Reductive Coupling of Alkoxyallenes: Convergent Construction of syn-sec,tert-Diols via (Z)-σ-Allylmetal Intermediates. J. Am. Chem. Soc 2021, 143, 8849–8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).(a) For selected reviews on allene-mediated C-C coupling, see: Neff RK; Frantz DE Recent Applications of Chiral Allenes in Axial-to-Central Chirality Transfer Reactions. Tetrahedron 2015, 71, 7–18. [DOI] [Google Scholar]; (b) Pulis AP; Yeung K; Procter DJ Enantioselective Copper Catalysed, Direct Functionalisation of Allenes via Allyl Copper Intermediates. Chem. Sci 2017, 8, 5240–5247.28959423 [DOI] [Google Scholar]; (c) Holmes M; Schwartz LA; Krische MJ Intermolecular Metal-Catalyzed Reductive Coupling of Dienes, Allenes and Enynes with Carbonyl Compounds and Imines. Chem. Rev 2018, 118, 6026–6052.29897740 [DOI] [Google Scholar]; (d) Li G; Huo X; Jiang X; Zhang W Asymmetric Synthesis of Allylic Compounds via Hydrofunctionalisation and Difunctionalisation of Dienes, Allenes, and Alkynes. Chem. Soc. Rev 2020, 49, 2060–2118.32150186 [DOI] [Google Scholar]; (e) Blieck R; Taillefer M; Monnier F Metal-Catalyzed Intermolecular Hydrofunctionalization of Allenes: Easy Access to Allylic Structures via the Selective Formation of C-N, C-C and C-O Bonds. Chem. Rev 2020, 120, 13545–13598.33301308 [DOI] [Google Scholar]; (f) Talbot FJT; Dherbassy Q; Manna S; Shi C; Zhang S; Howell GP; Perry GJP; Procter DJ Copper-Catalyzed Borylative Couplings with C–N Electrophiles. Angew. Chem. Int. Ed 2020, 59, 20278–20289. [DOI] [Google Scholar]; (g) Xiang M; Pfaffinger DE; Krische MJ Allenes and Dienes as Chiral Allylmetal Pronucleophiles in Catalytic Enantioselective C=X Addition: Historical Perspective and State-of-The-Art Survey. Chem. Eur. J 2021, 27, DOI: 10.1002/chem.202101890. [DOI] [Google Scholar]
- (19).(a) For selected reviews on enantioselective carbonyl allylation, see: Denmark SE; Fu J Catalytic Enantioselective Addition of Allylic Organometallic Reagents to Aldehydes and Ketones. Chem. Rev 2003, 103, 2763–2794. [DOI] [PubMed] [Google Scholar]; (b) Hall DG Lewis and Brønsted Acid Catalyzed Allylboration of Carbonyl Compounds: From Discovery to Mechanism and Applications. Synlett 2007, 11, 1644–1655. [DOI] [PubMed] [Google Scholar]; (c) Hargaden GC; Guiry PJ The Development of the Asymmetric Nozaki–Hiyama–Kishi Reaction. Adv. Synth. Catal 2007, 349, 2407–2424. [DOI] [PubMed] [Google Scholar]; (d) Yus M; González-Gómez JC; Foubelo F Catalytic Enantioselective Allylation of Carbonyl Compounds and Imines. Chem. Rev 2011, 111, 7774–7854. [DOI] [PubMed] [Google Scholar]; (e) Huo H-X; Duvall JR; Huang M-Y; Hong R Catalytic Asymmetric Allylation of Carbonyl Compounds and Imines with Allylic Boronates. Org. Chem. Front 2014, 1, 303–320. [DOI] [PubMed] [Google Scholar]; (f) Spielmann K; Niel G; de Figueiredo RM; Campagne J-M Catalytic Nucleophilic ‘Umpoled’ π-Allyl Reagents. Chem. Soc. Rev 2018, 47, 1159–1173. [DOI] [PubMed] [Google Scholar]
- (20).(a) Prior to our recent work (ref. 11i) and prior studies involving activated aldehydes (ref. 19e, 20) only isolated examples of catalytic enantioselective carbonyl (α-aryl)allylations were reported, notwithstanding studies described in references 19i: Nakajima M; Saito M; Hashimoto S One-Pot Enantioselective Synthesis of Optically Active Homoallylic Alcohols from Allyl Halides. Chem. Pharm. Bull 2000, 48, 306–307. [DOI] [PubMed] [Google Scholar]; (b) Zanoni G; Gladiali S; Marchetti A; Piccinini P; Tredici I; Vidari G Enantioselective Catalytic Allylation of Carbonyl Groups by Umpolung of π-Allyl Palladium Complexes. Angew. Chem. Int. Ed 2004, 43, 846–849. [DOI] [PubMed] [Google Scholar]; (c) Zhu S-F; Yang Y; Wang L-X; Liu B; Zhou Q-L Synthesis and Application of Chiral Spiro Phospholane Ligand in Pd-Catalyzed Asymmetric Allylation of Aldehydes with Allylic Alcohols. Org. Lett 2005, 7, 2333–2335. [DOI] [PubMed] [Google Scholar]; (d) Hemelaere R; Carreaux F; Carboni B; Cross-Metathesis / Isomerization / Allylboration Sequence for a Diastereoselective Synthesis of Anti-Homoallylic Alcohols from Allylbenzene Derivatives and Aldehydes. Chem. Eur. J 2014, 20, 14518–14523. [DOI] [PubMed] [Google Scholar]; (e) Evans DA; Aye Y; Wu J Asymmetric, anti-Selective Scandium-Catalyzed Sakurai Additions to Glyoxyamide. Applications to The Syntheses of N-Boc d-Alloisoleucine and d-Isoleucine. Org. Lett 2006, 8, 2071–2073. [DOI] [PubMed] [Google Scholar]; (f) Bai B; Zhu H-J; Pan W Structure Influence of Chiral 1,1′-Biscarboline-N,N′-dioxide on The Enantioselective Allylation of Aldehydes with Allyltrichlorosilanes. Tetrahedron 2012, 68, 6829–6836. [DOI] [PubMed] [Google Scholar]; (g) Miura T; Nishida Y; Morimoto M; Murakami M Enantioselective Synthesis of Anti Homoallylic Alcohols from Terminal Alkynes and Aldehydes Based on Concomitant Use of a Cationic Iridium Complex and a Chiral Phosphoric Acid. J. Am. Chem. Soc 2013, 135, 11497–11500. [DOI] [PubMed] [Google Scholar]; (h) Tan Z; Wan X; Zang Z; Qian Q; Deng W; Gong H Ni-Catalyzed Asymmetric Reductive Allylation of Aldehydes with Allylic Carbonates. Chem. Commun 2014, 50, 3827–3830. [DOI] [PubMed] [Google Scholar]; (i) Xiong Y; Zhang G Enantioselective Synthesis of Quaternary Stereocenters via Chromium Catalysis. Org. Lett 2016, 18, 5094–5097. [DOI] [PubMed] [Google Scholar]
- (21).(a) For catalytic enantioselective carbonyl (α-aryl)allylations of formaldehyde (including use of methanol as a carbonyl proelectrophile) and fluoral, see: Garza VJ; Krische MJ Hydroxymethylation beyond Carbonylation: Enantioselective Iridium Catalyzed Reductive Coupling of Formaldehyde with Allylic Acetates via Enantiotopic π-Facial Discrimination. J. Am. Chem. Soc 2016, 138, 3655–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nguyen KD; Herkommer D; Krische MJ Enantioselective Formation of All-Carbon Quaternary Centers via C-H Functionalization of Methanol: Iridium-Catalyzed Diene Hydrohydroxymethylation. J. Am. Chem. Soc 2016, 138, 14210–14213. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Holmes M; Nguyen KD; Schwartz LA; Luong T; Krische MJ Enantioselective Formation of CF3-Bearing All-Carbon Quaternary Stereocenters via C-H Functionalization of Methanol: Iridium Catalyzed Allene Hydrohydroxymethylation. J. Am. Chem. Soc 2017, 139, 8114–8117. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cabrera JM; Tauber J; Zhang W; Xiang M; Krische MJ Selection between Diastereomeric Kinetic vs Thermodynamic Carbonyl Binding Modes Enables Enantioselective Iridium-Catalyzed anti-(α-Aryl)allylation of Aqueous Fluoral Hydrate and Difluoroacetaldehyde Ethyl Hemiacetal. J. Am. Chem. Soc 2018, 140, 9392–9395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).(a) For acid-base reactions of ruthenium dihydrides with Brønsted acids, see: Dobson A; Robinson SD; Uttley MF Complexes of the Platinum Metals. Part V. Perfluorocarboxylato Derivatives. J. Chem. Soc., Dalton Trans 1975, 370–377. [Google Scholar]; (b) McInturff EL; Yamaguchi E; Krische MJ Chiral-Anion-Dependent Inversion of Diastereo- and Enantioselectivity in Carbonyl Crotylation via Ruthenium Catalyzed Butadiene Hydrohydroxyalkylation. J. Am. Chem. Soc 2012, 134, 20628–20631.23234459 [Google Scholar]
- (23).(a) As addition of TFE does not influence enantiomeric excess, we believe hydrogen-bonding to the CO-ligand does not occur in the transition state for carbonyl addition. For selected examples of CH and OH hydrogen-bonding to carbonyl ligands, see: Kubas GJ; Burns CJ; Khalsa GRK; Van Der Sluys LS; Kiss G; Hoff CD Dihydrogen: A Better Ligand Than Water? IR and X-Ray Evidence for Aquo Coordination in W(CO)3(PR3)2(H2O), Thermodynamics of H2O Binding versus η2-H2 Binding and H2O/D2 Isotopic Exchange. Implications for The Biological Activation of Hydrogen. Organometallics 1992, 11, 3390–3404. [Google Scholar]; (b) Braga D; Grepioni F; Biradha K; Pedireddi VR; Desiraju GR Hydrogen Bonding in Organometallic Crystals. 2. C-H…O Hydrogen Bonds in Bridged and Terminal First-Row Metal Carbonyls. J. Am. Chem. Soc 1995, 117, 3156–3166. [Google Scholar]; (c) Braga D; Grepioni F; Wadepohl H; Gebert S; Calhorda MJ; Veiros LF Intramolecular and Intermolecular Bonding in Crystalline Clusters of the Type (CpR)3M3(CO)3 [M = Co, Rh, Ir; CpR = C5H5, C5Me5, C5H4Me]. Organometallics 1995, 14, 5350–5361. [Google Scholar]; (d) Wadepohl H; Braga D; Grepioni F Phosphine Derivatives of (μ-η-2-Methylidyne)(μ-hydrido)dodecacarbonyltetrairon. Organometallics 1995, 14, 24–33. [Google Scholar]; (e) Biradha K; Desiraju GR; Braga D; Grepioni F Hydrogen Bonding in Organometallic Crystals. 3.1 Transition-Metal Complexes Containing Amido Groups. Organometallics 1996, 15, 1284–1295. [Google Scholar]; (f) Brown M; Zubkowski JD; Valente EJ; Yang G-Z; Henry WP The Crystal Structure of The bis-Tricarbonylchromium Complex of Dibenzo[a,e]cyclooctatetraene: Cr(CO)3 Orientation Controlled by short C–H…O Hydrogen Bonding. J. Organomet. Chem 2000, 613, 111–118. [Google Scholar]; (g) Heinze K Patterns of Hydrogen Bonding in Crystals of Molybdenum Carbonyl Complexes. J. Chem. Soc., Dalton Trans 2002, 4, 540–547. [Google Scholar]; (h) Heinze K; Jacob V Hydrogen Bonds in Crystals of HO-(N∩N′)M(CO)3L Complexes: Chains, Dimers and an Unexpected Hexamer. J. Chem. Soc., Dalton Trans 2002, 11, 2379–2385. [Google Scholar]; (i) Braunstein P; Taquet J.-p.; Siri O; Welter R Supramolecular, Bifurcated N-H…OC-M Bonding Explains Unusually Low νCO Frequencies in Metal Carbonyl Compounds: A Case Study. Angew. Chem. Int. Ed 2004, 43, 5922–5925. [Google Scholar]
- (24).Sbrana G; Braca G; Piacenti F; Pino P Synthesis of π-Allylruthenium Tricarbonyl Halides. J. Organomet. Chem 1968, 13, 240–242. [Google Scholar]
- (25).Mardirossian N; Head-Gordon M ωB97X-V: A 10-Parameter, Range-Separated Hybrid, Generalized Gradient Approximation Density Functional with Nonlocal Correlation, Designed by a Survival-of-The-Fittest Strategy. Phys. Chem. Chem. Phys 2014, 16, 9904–9924. [DOI] [PubMed] [Google Scholar]
- (26).(a) For selected studies on aldehyde formyl CH hydrogen bonding, see: Corey EJ; Lee TW The Formyl C–H…O Hydrogen Bond as a Critical Factor in Enantioselective Lewis-Acid Catalyzed Reactions of Aldehydes. Chem. Commun 2001, 1321–1329. [Google Scholar]; (b) Thakur TS; Kirchner MT; Bläser D; Boese R; Desiraju GR Nature and Strength of C–H…O Interactions Involving Formyl Hydrogen Atoms: Computational and Experimental Studies of Small Aldehydes. Phys. Chem. Chem. Phys 2011, 13, 14076–14091.21475764 [Google Scholar]; (c) Grayson MN; Krische MJ; Houk KN Ruthenium-Catalyzed Asymmetric Hydrohydroxyalkylation of Butadiene: The Role of the Formyl Hydrogen Bond in Stereochemical Control. J. Am. Chem. Soc 2015, 137, 8838–8850.26107070 [Google Scholar]
- (27).Bader RF Atoms in Molecules: A Quantum Theory; Oxford University Press: New York, 1994. [Google Scholar]
- (28).Glendening ED; Landis CR; Weinhold F NBO 6.0: Natural Bond Orbital Analysis Program J. Comput. Chem 2013, 34, 1429–1437. [DOI] [PubMed] [Google Scholar]
- (29).Lu T; Chen F Bond Order Analysis Based on the Laplacian of Electron Density in Fuzzy Overlap Space. J. Phys. Chem. A 2013, 117, 3100–3108. [DOI] [PubMed] [Google Scholar]
- (30).(a) For the stoichiometric reaction of HXRu(CO)(PR3)3 (X = Cl, Br) with allenes and dienes to form isolable π-allylruthenium species, see: Hiraki K; Ochi N; Sasada Y; Hayashida H; Fuchita Y; Yamanaka S Organoruthenium(II) Complexes Formed by Insertion Reactions of Some Vinyl Compounds and Conjugated Dienes into a Hydrido–Ruthenium Bond. J. Chem. Soc., Dalton Trans 1985, 873–877. [Google Scholar]; (b) Hill AF; Ho CT; Wilton-Ely JDET The Coupling of Methylene and Vinyl Ligands at a Ruthenium(II) Centre. Chem. Commun 1997, 2207–2208. [Google Scholar]; (c) Xue P; Bi S; Sung HHY; Williams ID; Lin Z; Jia G Isomerism of [Ru(η3-allyl)Cl(CO)(PPh3)2] Organometallics 2004, 23, 4735–4743. [Google Scholar]
- (31).Sam B; Luong T; Krische MJ Ruthenium-Catalyzed C-C Coupling of Fluorinated Alcohols with Allenes: Dehydrogenation at the Energetic Limit of β-Hydride Elimination. Angew. Chem. Int. Ed 2015, 54, 5465–5469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.