

Abstract
Skyline Batch is a newly developed Windows forms application that enables the easy and consistent reprocessing of data with Skyline. Skyline has made previous advances in this direction; however, none enable seamless automated reprocessing of local and remote files. Skyline keeps a log of all of the steps that were taken in the document; however, reproducing these steps takes time and allows room for human error. Skyline also has a command-line interface, enabling it to be run from a batch script, but using the program in this way requires expertise in editing these scripts. By formalizing the workflow of a highly used set of batch scripts into an intuitive and powerful user interface, Skyline Batch can reprocess data stored in remote repositories just by opening and running a Skyline Batch configuration file. When run, a Skyline Batch configuration downloads all necessary remote files and then runs a four-step Skyline workflow. By condensing the steps needed to reprocess the data into one file, Skyline Batch gives researchers the opportunity to publish their processing along with their data and other analysis files. These easily run configuration files will greatly increase the transparency and reproducibility of published work. Skyline Batch is freely available at https://skyline.ms/batch.url.
Keywords: Skyline, workflow, reproducible, data analysis, reprocessing, software, mass spectrometry
1. Introduction
Skyline,1 a flexible Windows application with a highly informative user interface, is the leading tool for targeted mass spectrometry method development and quantitative data analysis. Skyline facilitates the processing and analysis of data from a range of mass spectrometry data acquisition techniques while supporting the instruments of six major vendors. With its comprehensive user interface and data visualization capabilities, Skyline enables complex analysis workflows for both proteomics and small-molecule data2 and can easily integrate external analysis tools.3 Whereas the convenience of a graphical user interface (GUI) tool makes it easy for any researcher to use Skyline, it also makes it challenging to document and reproduce the analysis steps for creating the final Skyline document that is the outcome of a study. The use of GUI-based tools has been noted as a limitation for the reproducible processing of proteomics data.4
Skyline introduced Audit Logs5 as a way of recording all of the operations that were performed by the user to transform an empty Skyline document into its current form. This log can be viewed on Panorama,6 a web application for storing and sharing Skyline documents, and assists in keeping a record of changes when multiple users are collaborating on a document. Audit logging brings Skyline a step closer to reproducible workflows, as the log offers the ability to replicate the exact steps that were taken to produce a Skyline document. However, audit logs cannot be used to automatically reconstruct a document given the original data and the log. A user must carry out each of the steps listed in the log which can be time-consuming and leaves room for human error.
Skyline also has an extensive and well-documented command-line interface that can be used to execute many of the actions that can be performed in the Skyline GUI. The command line executes each action without opening the Skyline window while also updating the document’s audit log. Actions can be combined into batch scripts, allowing the automation of multiple operations into complete workflows. Scripts can be shared to make it possible for others to independently reproduce an analysis. We have presented such scripts in multiple webinars (14 - https://skyline.ms/webinar14.url and 15 - https://skyline.ms/webinar15.url) and used them for our own data processing for several manuscripts7−9 and presentations.10,11 However, few bench scientists are comfortable writing their own scripts or editing the existing ones to use on their computers and data. Without an intuitive user interface, the potential of the Skyline command line is not widely utilized.
Generalized workflow managers such as Cromwell, Snakemake, and Nextflow have been created to improve research reproducibility.12 However, like the Skyline batch scripts, managing server-side workflows requires expertise that most Skyline users do not have. Whereas GUI tools facilitating the generation of these workflows do exist,13 they are still too flexible to be easily used by someone without IT experience.
User interfaces can be built on top of the Skyline command-line interface to make it easy for researchers to execute specific workflows. One example of a GUI tool that harnesses the power of the Skyline command-line interface is AutoQC Loader, a component of the Panorama AutoQC pipeline.14 While limited in its design and goals, AutoQC Loader addresses the need for automatic data import and file transfer from the mass spectrometer instrument computer to a Panorama server. AutoQC Loader has proven to be critical to convincing laboratories to adopt the Panorama pipeline for system suitability monitoring.
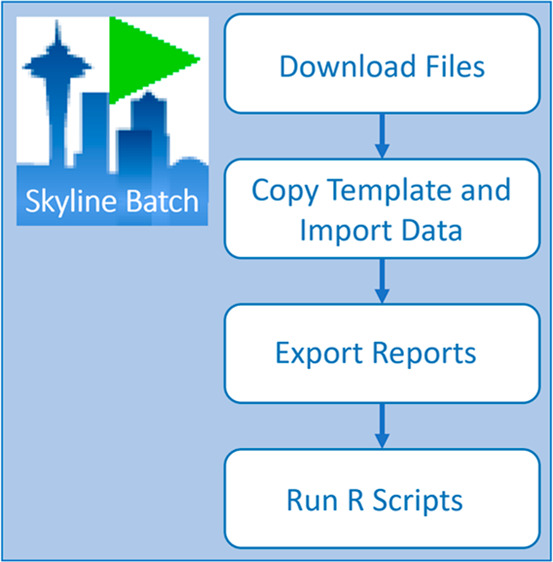
Here we present Skyline Batch (Figure 1), an intuitive, user-friendly interface that extends AutoQC Loader, leveraging the Skyline command-line interface to help users configure a well-defined, automated, reusable Skyline data processing workflow. A single declarative configuration file can fully capture the processing and analysis steps for an entire published experiment. These configuration files are well suited to sharing with others to allow reproducible data analysis and are easily adapted to reprocess the data with alternative parameters. Sharing is made easier by an intelligent file path replacement algorithm implemented in Skyline Batch for porting workflows across computers with similar file structures (Supplementary Figure 1). It also minimizes errors by remembering the Skyline and R versions that were used to run each individual configuration. Skyline Batch can be used with local input files and remote files stored on a Panorama server or any of the other ProteomeXchange15 repositories that provide a file transfer protocol (FTP) interface, making it possible to start with just a configuration file and have Skyline Batch automatically download everything else.
Figure 1.

Reprocessing data from a remote repository in Skyline Batch. The Skyline Batch configuration file is downloaded and opened in Skyline Batch for processing. Skyline Batch downloads the Skyline template document, data files, and R scripts. The processing is run and creates a Skyline results document, exported reports, and PDFs. R Logo courtesy of the R Foundation (https://www.r-project.org/logo) under the terms of the Creative Commons Attribution-ShareAlike 4.0 International license (CC-BY-SA 4.0) (https://creativecommons.org/licenses/by-sa/4.0/). Skyline logo reprinted with permission from Michael MacCoss.
The publicly available library of quantitative mass-spectrometry-based data sets has grown markedly in recent years. However, the information required to accurately reproduce published conclusions is often missing or incomplete.4 Skyline users can share documents as supporting information for published manuscripts via the Panorama Public16 repository, a member of the ProteomeXchange Consortium.15 By publishing the Skyline Batch configuration file, Skyline template file, data files, and R scripts they used, authors will enable reviewers, editors, and readers to re-execute the processing. The ability to easily reprocess these files will greatly improve the transparency and reproducibility of the data analysis of research published with data processed using Skyline.
2. Design and Implementation
The Skyline Batch application uses a similar interface design as the Panorama AutoQC Loader14 and shares a common code library. Both Skyline Batch and AutoQC contain a list of configurations on a main tab and the log output on a second tab. Analyses using a Skyline Batch configuration file can be run individually or in series, and all output appears in the log viewing and management tab.
Each configuration in Skyline Batch controls a five-step workflow that can be tailored to individual analysis needs by changing declarative properties. The steps are downloading files, copying the template document and importing data, optionally creating a new refined file, exporting reports, and running R scripts. To execute this workflow, a configuration requires the file locations of a Skyline template document, a folder containing data files, and any Skyline report files or R scripts that will be used for postimport results analysis. Skyline Batch also supports copying configurations, so many similar workflows can be easily created with different settings or with different Skyline templates and data. Skyline Batch can be downloaded from https://skyline.ms/batch.url.
2.1. Integration with Skyline, Panorama, and FTP Servers
Aside from storing file path locations, a Skyline Batch configuration also has options to change the settings inside the Skyline document. The settings specified by the user are then translated into Skyline commands that adjust the settings when the configuration is run. A Skyline Batch configuration starts with a Skyline template document. When it is run, it creates a new Skyline results document by altering the settings and then importing the data. This new document can be opened with a click of a button inside Skyline Batch. Optionally, a set of refinement options can be specified to control the creation of a new, refined Skyline document that can be used as the input document template to a subsequent configuration.
All of the required files for a Skyline Batch configuration can be either present on the local hard drive or automatically downloaded from a Panorama server such as PanoramaWeb (https://panoramaweb.org). Data files may also be downloaded from any FTP server. To specify a remote file in a configuration, the user supplies the Panorama URL, a folder path to store the downloaded file, and a username and password if the file is password protected. When the configuration is run, Skyline Batch downloads any missing files before beginning the analysis (Figure 2).
Figure 2.

Downloading data from a remote repository in Skyline Batch for the first case study, refinement configuration. A remote folder is specified with a URL, username, and password. Files are selected that match the data naming pattern regular expression. These files are downloaded to the computer when the configuration is run.
2.2. Skyline Batch Configuration Sharing
Skyline Batch was designed to make transferring batch workflows between computers as easy as possible. If a configuration downloads the Skyline template document, data files, and R scripts from a Panorama server, then all that the user must do to run the configuration on another computer is provide the path to a root folder in which downloads and processing will occur. In the scenario where some of the files were stored on disk on the original computer, Skyline Batch uses file path replacement to automatically guess where the same files will be stored on a different computer based on their location on the original computer (Supplementary Figure 1). If the folder containing the Skyline Batch configuration file has the same relative paths to the files as those on the computer where it was created, then the transfer is seamless.
Skyline Batch also takes into account software differences between computers. Storing the type of Skyline installation used (Skyline or Skyline-daily) and the version of R that the configuration was created with, Skyline Batch minimizes errors due to varying versions of software between machines. If a configuration is opened on a new computer without the correct software, then users are prompted to install the software or edit the configuration before running it.
2.3. Installation Options
Skyline Batch has both administrative and web installations. The web option is a stand-alone installation available on the Skyline Web site. It is self-updating and does not require administrative privileges on the computer to be installed. The Skyline Batch administrative installation is included with each Skyline administrative installer since Skyline version 20.2.1.454. The administratively installed Skyline Batch is almost identical to the web installation, with the key differences being that the administrative installation is not self-updating, and it automatically uses the version of Skyline it was installed with instead of letting the user select another version. The web installation allows each configuration to choose between a Skyline or Skyline-daily web installation or any folder containing SkylineCmd.exe.
3. Case Studies
We demonstrate the power of reprocessing with Skyline Batch using two data sets that require the processing of the proteome-wide data-independent acquisition (DIA) of a controlled mixture, which have been previously uploaded to PanoramaWeb (https://panoramaweb.org).
The first data set is based on the published work from Bruderer et al.,17 available on Peptide Atlas18 with the identifier PASS00589 (https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/PASS_View?identifier=PASS00589). The subset of the data used for processing, Skyline Template document, R script, and Skyline Batch configuration (.bcfg) file are available on ProteomeXchange (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD029663) via the Panorama Public partner repository at https://panoramaweb.org/dia-bruderer15.url. The R script used to analyze this data set utilizes the MSstats19 R package for statistical relative quantification of proteins and peptides to produce multiple plots for each individual protein. Processing the data entails importing it into the Skyline template document, exporting a report, and running the R script on the report. This workflow is described by a single Skyline Batch configuration (Supplementary Figure 2). The configuration file for this data set can be downloaded from Panorama, opened, and run after specifying a folder in which to perform the analysis. This reduces the time the user must spend monitoring the data processing from hours to minutes.
The second data set is based on the published work from Selevsek et al.,20 available with the identifier PXD001010 via the PRIDE partner repository21 of the ProteomeXchange Consortium. The subset of the data used for processing, Skyline Template documents, R scripts, and Skyline Batch configuration (.bcfg) file are available on ProteomeXchange (http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD029665) via the Panorama Public partner repository at https://panoramaweb.org/dia-selevsek15.url. This data set is processed with three separate workflows to explore the impact of using a tangential set of data to create a refined template file of high-quality targets versus analyzing all available targets through quantitative analysis without prior refinement. The first (refinement) workflow imports replicate runs, using them to select only targets detected (at q value ≤0.01) in at least 4 of 8 replicates with a low enough coefficient of variation (CV ≤ 20%), and then removes the data to create a new refined template document. The second (refined) workflow uses the refined template document, imports the osmotic shock data, exports a report, and runs an R script. The R script utilizes the MSstats19 R package for the statistical relative quantification of proteins and peptides to produce multiple plots for each individual protein. The third (unrefined) workflow uses the original template document, imports the osmotic shock data, exports a report, and runs the same R script for the relative quantification of all proteins and peptides. Before Skyline Batch, executing these three workflows could take hours of monitoring. Whether done through the Skyline GUI or the command-line interface, the user would have to download the data themselves. After that, processing the data through the GUI would require hours of manual monitoring to import the data, export reports, and run R scripts three times. Using batch scripts would require less monitoring, but the scripts would need to be written and updated by someone with scripting experience. Now, using Skyline Batch, these three workflows can be run by downloading the Skyline Batch configuration file, opening it, and specifying a base folder in which to store downloaded files and analysis results. This can be done in less than a minute.
Not only is Skyline Batch an important advancement in reproducibility and transparency for published analyses of quantitative mass spectrometry data but also it is useful for training scientists in complex analyses and aiding in the sharing/dissemination of Skyline workflows. The use of Skyline Batch in processing the Bruderer and Selevsek data sets17,20 enabled the instruction of these large-scale analyses in a way that was not possible before. During the 2021 May Institute (https://computationalproteomics.khoury.northeastern.edu), a 2 week long course on “Computation and statistics for mass spectrometry and proteomics”, some remote participants reported being entirely successful in processing overnight 187 GB of mass spectrometry data over three experiments, including 50 DIA runs, processed with whole proteome analysis, going far beyond anything ever tried, even in a week long instructional setting (Supplementary Figure 3). In past courses, data had to be greatly condensed to allow people to download the files manually, and the processing workflow was simply demonstrated for participants, as editing the batch scripts was outside the expertise of some (Supplementary Figure 4).
4. Conclusions
Skyline Batch fills the need for creating intuitive, automated Skyline workflows that can be easily executed by others. By connecting to Skyline and Panorama, Skyline Batch builds on top of tools that are already widely used in the proteomics community to make data processing easier and more transferrable. The GUI in Skyline Batch enables researchers, including those without scripting expertise, to configure reproducible Skyline workflows while capturing a record of the processing output in a log file. Skyline Batch configuration files can be shared to facilitate the sharing of quantitative analyses of proteomics data and data reprocessing. In future development, a Cromwell workflow will be designed to perform the processing specified by a Skyline Batch configuration file, enabling it to be run in a Docker Container on a server. Adding Skyline Batch to the Skyline software ecosystem fills a gap in scale and reproducibility for researchers lacking batch scripting and IT expertise.
Acknowledgments
Support for this research was provided in part by the National Institutes of Health grants R24 GM141156 and P41 GM103533.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.1c00749.
Supplementary Figure 1. Path replacement in Skyline Batch. Supplementary Figure 2. Screenshot of the Bruderer configuration in Skyline Batch. Supplementary Figure 3. Poll on the success of Skyline Batch use in an instructional setting. Supplementary Figure 4. Poll on Skyline users experience with scripting. Supplementary Note 1. Link to a webinar on using Skyline Batch for large-scale DIA. Supplementary Note 2. Link to a tutorial demonstrating how to use Skyline Batch instead of Skyline batch scripts for large-scale DIA. Supplementary Note 3. Link to a tutorial demonstrating how to use Skyline Batch instead of Skyline batch scripts for optimizing large-scale DIA. Supplementary Note 4. Skyline Batch Documentation. Supplementary Note 5. Tutorial on running a basic Skyline Batch workflow (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Pino L. K.; Searle B. C.; Bollinger J. G.; Nunn B.; MacLean B.; MacCoss M. J. The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass Spectrom. Rev. 2020, 39 (3), 229–244. 10.1002/mas.21540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams K. J.; Pratt B.; Bose N.; Dubois L. G.; St. John-Williams L.; Perrott K. M.; Ky K.; Kapahi P.; Sharma V.; MacCoss M. J.; et al. Skyline for Small Molecules: A Unifying Software Package for Quantitative Metabolomics. J. Proteome Res. 2020, 19 (4), 1447–1458. 10.1021/acs.jproteome.9b00640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broudy D.; Killeen T.; Choi M.; Shulman N.; Mani D. R.; Abbatiello S. E.; Mani D.; Ahmad R.; Sahu A. K.; Schilling B.; Tamura K.; Boss Y.; Sharma V.; Gibson B. W.; Carr S. A.; Vitek O.; MacCoss M. J.; MacLean B. A framework for installable external tools in Skyline. Bioinformatics 2014, 30 (17), 2521–2523. 10.1093/bioinformatics/btu148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M.; Carver J.; Chiva C.; Tzouros M.; Huang T.; Tsai T.; Pullman B.; Bernhardt O. M.; Hüttenhain R.; Teo G. C.; Perez-Riverol Y.; Muntel J.; Müller M.; Goetze S.; Pavlou M.; Verschueren E.; Wollscheid B.; Nesvizhskii A. I.; Reiter L.; Dunkley T.; Sabidó E.; Bandeira N.; Vitek O. MassIVE.quant: a community resource of quantitative mass spectrometry–based proteomics datasets. Nat. Methods 2020, 17 (10), 981–984. 10.1038/s41592-020-0955-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde T.; Chupalov R.; Shulman N.; Sharma V.; Eckels J.; Pratt B. S.; MacCoss M. J.; MacLean B. X. Audit logs to enforce document integrity in Skyline and Panorama. Bioinformatics 2020, 36 (15), 4366–4368. 10.1093/bioinformatics/btaa547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V.; Eckels J.; Taylor G. K.; Shulman N. J.; Stergachis A. B.; Joyner S. A.; Yan P.; Whiteaker J. R.; Halusa G. N.; Schilling B.; Gibson B. W.; Colangelo C. M.; Paulovich A. G.; Carr S. A.; Jaffe J. D.; MacCoss M. J.; MacLean B. Panorama: A Targeted Proteomics Knowledge Base. J. Proteome Res. 2014, 13 (9), 4205–4210. 10.1021/pr5006636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceja-Navarro J. A.; Vega F. E.; Karaoz U.; Hao Z.; Jenkins S.; Lim H. C.; Kosina P.; Infante F.; Northen T. R.; Brodie E. L. Gut microbiota mediate caffeine detoxification in the primary insect pest of coffee. Nat. Commun. 2015, 6 (1), 7618. 10.1038/ncomms8618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle B. C.; Pino L. K.; Egertson J. D.; Ting Y. S.; Lawrence R. T.; MacLean B. X.; Villén J.; MacCoss M. J. Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat. Commun. 2018, 9 (1), 5128. 10.1038/s41467-018-07454-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaca Jacome A. S.; Peckner R.; Shulman N.; Krug K.; DeRuff K. C.; Officer A.; Christianson K. E.; MacLean B.; MacCoss M. J.; Carr S. A.; Jaffe J. D. Avant-garde: an automated data-driven DIA data curation tool. Nat. Methods 2020, 17 (12), 1237–1244. 10.1038/s41592-020-00986-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde T.; Schmidt T.; Kuster B.; MacCoss M. J.; Wilhelm M.; MacLean B.. Integration of the Deep Learning Prediction Tool Prosit into Skyline for High-Accuracy, On-Demand Fragment Intensity and Irt Prediction. In Proceedings of the 68th ASMS Conference on Mass Spectrometry and Allied Topics, Online Meeting, 2020-06-03; ID: 305122. [Google Scholar]
- Pratt B. S.; Chambers M. C.; Kaspar-Schoenefeld S.; Brehmer S.; Lubeck M.; Distler U.; Tenzer S.; MacCoss M. J.; MacLean B. X. Skyline Support For Proteome-Wide Data Analysis Of Bruker TimsTOF DiaPASEF Acquisition. In Proceedings of the 68th ASMS Conference on Mass Spectrometry and Allied Topics, Online Meeting, 2020-06-01, ID: 305148. [Google Scholar]
- Larsonneur E.; Mercier J.; Wiart N.; Floch E. L.; Delhomme O.; Meyer V. Evaluating Workflow Management Systems: A Bioinformatics Use Case. 2018 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) 2018, 2773–2775. 10.1109/BIBM.2018.8621141. [DOI] [Google Scholar]
- Kaushik G.; Ivkovic S.; Simonovic J.; Tijanic N.; Davis-Dusenbery B.; Kural D. RABIX: an Open-Source Workflow Executor Supporting Recomputability and Interoperability of Workflow Descriptions. Biocomputing 2017 2017, 154–165. 10.1142/9789813207813_0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereman M. S.; Beri J.; Sharma V.; Nathe C.; Eckels J.; MacLean B.; MacCoss M. J. An Automated Pipeline to Monitor System Performance in Liquid Chromatography–Tandem Mass Spectrometry Proteomic Experiments. J. Proteome Res. 2016, 15 (12), 4763–4769. 10.1021/acs.jproteome.6b00744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch E. W.; Csordas A.; Sun Z.; Jarnuczak A.; Perez-Riverol Y.; Ternent T.; Campbell D. S.; Bernal-Llinares M.; Okuda S.; Kawano S.; Moritz R. L.; Carver J. J.; Wang M.; Ishihama Y.; Bandeira N.; Hermjakob H.; Vizcaíno J. A. The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res. 2017, 45 (D1), D1100–D1106. 10.1093/nar/gkw936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V.; Eckels J.; Schilling B.; Ludwig C.; Jaffe J. D.; MacCoss M. J.; MacLean B. Panorama Public: A Public Repository for Quantitative Data Sets Processed in Skyline. Molecular & cellular proteomics: MCP 2018, 17 (6), 1239–1244. 10.1074/mcp.RA117.000543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruderer R.; Bernhardt O. M.; Gandhi T.; Miladinović S. M.; Cheng L.; Messner S.; Ehrenberger T.; Zanotelli V.; Butscheid Y.; Escher C.; Vitek O.; Rinner O.; Reiter L. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Molecular & cellular proteomics: MCP 2015, 14 (5), 1400–1410. 10.1074/mcp.M114.044305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desiere F.; Deutsch E. W.; King N. L.; Nesvizhskii A. I.; Mallick P.; Eng J.; Chen S.; Eddes J.; Loevenich S. N.; Aebersold R. The PeptideAtlas project. Nucleic Acids Res. 2006, 34, D655–D658. 10.1093/nar/gkj040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M.; Chang C.; Clough T.; Broudy D.; Killeen T.; MacLean B.; Vitek O. MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 2014, 30 (17), 2524–2526. 10.1093/bioinformatics/btu305. [DOI] [PubMed] [Google Scholar]
- Selevsek N.; Chang C.; Gillet L. C.; Navarro P.; Bernhardt O. M.; Reiter L.; Cheng L.; Vitek O.; Aebersold R. Reproducible and consistent quantification of the Saccharomyces cerevisiae proteome by SWATH-mass spectrometry. Molecular & cellular proteomics: MCP 2015, 14 (3), 739–749. 10.1074/mcp.M113.035550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y.; Csordas A.; Bai J.; Bernal-Llinares M.; Hewapathirana S.; Kundu D. J.; Inuganti A.; Griss J.; Mayer G.; Eisenacher M.; et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019, 47 (D1), D442–D450. 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.