

Abstract
Simple Summary
Hematological malignancies are diseases involving the abnormal production of blood cells. The aim of the study is to collect comprehensive information on new drugs used in the treatment of blood cancers which have introduced into therapy in the last decade. The approved drugs were analyzed for their structures and their biological activity mechanisms.
Abstract
Hematological malignancies, also referred to as blood cancers, are a group of diseases involving abnormal cell growth and persisting in the blood, lymph nodes, or bone marrow. The development of new targeted therapies including small molecule inhibitors, monoclonal antibodies, bispecific T cell engagers, antibody-drug conjugates, recombinant immunotoxins, and, finally, Chimeric Antigen Receptor T (CAR-T) cells has improved the clinical outcomes for blood cancers. In this review, we summarized 52 drugs that were divided into small molecule and macromolecule agents, approved by the Food and Drug Administration (FDA) in the period between 2011 and 2021 for the treatment of hematological malignancies. Forty of them have also been approved by the European Medicines Agency (EMA). We analyzed the FDA-approved drugs by investigating both their structures and mechanisms of action. It should be emphasized that the number of targeted drugs was significantly higher (46 drugs) than chemotherapy agents (6 drugs). We highlight recent advances in the design of drugs that are used to treat hematological malignancies, which make them more effective and less toxic.
Keywords: small molecule agents, macromolecule agents, hematological malignancies, FDA, EMA
1. Introduction
Hematological malignancies, also known as blood cancers, are diseases characterized by the clonal proliferation of blood-forming cells, which occur in blood, bone marrow, or lymph nodes. Hematological malignancies include wild range types of leukemia, lymphoma, and myeloma, classified into two types: lymphoid and myeloid [1]. According to its mechanism of action, the drugs used for the treatment of hematological malignancies can historically be divided into the following groups: deoxyribonucleic acid (DNA)-interactive agents, antimetabolites, anti-tubulin agents, and molecular targeting agents such as highly specific small molecules and monoclonal antibodies. DNA interactive agents, the oldest group of anticancer medications, can be primarily categorized into alkylating agents, cross-linking agents, intercalating agents, topoisomerase inhibitors, and DNA-cleaving agents [2]. The first alkylating agent approved by the Food and Drug Administration (FDA) was chlormethine (mechlorethamine), also called nitrogen mustard. Goodman and coworkers described, in 1946, the pharmacological effect of mechlorethamine on Hodgkin’s lymphoma, lymphosarcoma, and leukemia [3], which led to this drug being registered in 1949 [4]. As a result of work on folic acid antagonists carried out by Farber, the next class of drug was developed, i.e., antifolate. In 1948, Farber reported the use of aminopterin, which was the 4-amino derivative of folic acid, to treat children with acute leukemia [5]. Methotrexate (amethopterin) replaced aminopterin in the treatment of patients in 1953 because it has a better therapy-versus-toxicity ratio [6,7]. Then, mercaptopurine and fluorouracil were discovered as the first structural analogs of purine and pyrimidine, respectively. Mercaptopurine was synthesized by Elion et al. in 1952 [8] and was first FDA-approved in 1953 [9], while fluorouracil was developed by Dushinsky et al. in 1957 [10] and received first approval in 1962 [11]. These drugs were widely used for the treatment of both solid and hematological malignancies [12]. Generally, folate, purine, and pyrimidine antagonists form one of the oldest classes of anticancer drugs, i.e., antimetabolites. The next discovered agents for the treatment of hematological malignancies were natural plant alkaloids with anti-tubulin activity. Noble and Beer isolated two first vinca alkaloids, i.e., vinblastine and vincristine, from Catharanthus roseus (L.) G. Don [13]. Both compounds received extensive clinical evaluation leading to the FDA approval of vincristine in 1963 as therapies for a variety of cancers [14]. Other natural products were cytotoxic antibiotics such as bleomycin and doxorubicin. Bleomycin was found in Streptomyces verticillus by Umezawa et al. in 1962. This antibiotic was the first DNA-cleaving agent to be registered, in 1973 [15], and can be used to treat malignant lymphoma as well as squamous cell carcinoma of the skin, head, and neck [16]. Doxorubicin was isolated from Streptomyces peucetius var. caesius in 1967 in Italy [17], and was first FDA-approved in 1974 [18]. The drug showed anticancer activity via multiple mechanisms including intercalation into DNA and inhibition of topoisomerase II activity. Doxorubicin was commonly used for the treatment of various hematological malignancies [19]. In 1965, Rosenberg and co-workers discovered that cisplatin, the platinum coordination complex synthesized by Peyrone for the first time in 1845 [20], caused inhibition of cellular division [21]. Then, cisplatin was entered in trials against a wide range of cancers where it showed potent anticancer activity through the cross-linking of DNA. The drug was approved by the FDA in 1978 and, since that time, has been used as a first-line treatment for patients with leukemia or lymphomas. Currently, it is still one of the most successful anticancer agents used in clinical practice [22]. A milestone for blood cancer treatment was the discovery of targeted therapy, consisting of the inhibition of molecular targets that are specific molecules involved in the growth, progression, and spread of cancer by monoclonal antibodies or small selective molecules. The first FDA-approved monoclonal antibody for the treatment of hematological malignancies, a genetically engineered chimeric anti-cluster of differentiation 20 (CD20) antibody, was rituximab. The drug was registered in 1997 for the treatment of relapsed or refractory, B-cell, low-grade, or follicular non-Hodgkin’s lymphoma (LG/F NHL) [23]. Imatinib was the first small molecule inhibitor (SMI) to be found to be selective against various protein tyrosine kinases. It was synthesized by Buchdunger in 1996 and approved by the FDA in 2001. The drug was indicated for patients with chronic myelogenous leukemia (CML) [24].
This article is an overview of drugs used in the treatment of hematological malignancies, which was approved by the FDA from 2011 until 2021. The most recent examples of small molecule and macromolecule drugs are detailed, focusing on the initial approval date, chemical structure, molecular target, route of administration, indication, and the most common adverse effects for each agent. Depending on the mechanism of action, the approved drugs are assigned to two categories: chemotherapy and targeted agents. In the present review, the medications containing a new molecular entity, or old active ingredient but in a new formulation, are summarized. The drugs referred to as biosimilars are also included. The biosimilars are an important group of biologic medicines which, although similar in structure, purity, and function to their reference products, with no meaningful differences in clinical efficacy and safety, increase access to hematologic malignancy therapies by mitigating the treatment costs [25]. Notably, the drugs received supplemental indications in the period from 2011 to 2021 but were originally approved before 2011, and drugs used to treat the side effects of cancer treatment are not included in this work.
2. Small Molecule Anticancer Drugs
2.1. Various Protein Kinase Inhibitors as Anticancer Agents
Protein kinases are enzymes, which catalyse the reversible phosphorylation of proteins. This reaction is one of the most important regulatory mechanisms and plays a crucial role in processes such as the transduction of external signals and the cell cycle regulation. Therefore, protein kinases inhibitors are an important group in need of new drugs, especially anticancer drugs. Protein kinase inhibitors are divided into three types. Type I inhibitors bind within and around the adenosine triphosphate (ATP) binding site of a catalytically active protein kinase, causing inhibition of its phosphorylation. Type II inhibitors bind to a hydrophobic pocket adjacent to the ATP binding site and are usually nonselective. In contrast, type III inhibitors bind to allosteric sites, remote from the ATP site, and are highly selective [26].
2.1.1. Tyrosine Kinase (TK) Inhibitors
Tyrosine kinases (TKs) are enzymes that selectively phosphorylate the hydroxyl groups of a tyrosine residue in different proteins using ATP. They have a share in the regulation of most fundamental cellular processes such as growth, differentiation, proliferation, survival, migration, and the metabolism of cells, as well as programmed cell death in response to extracellular and intracellular stimuli [27]. The human genome contains at least 90 tyrosine kinase genes, which codify 58 receptor tyrosine kinases (RTKs) and 32 nonreceptor tyrosine kinases (NRTKs) [28]. RTKs are surface transmembrane receptors with kinase activity. In the structure of the receptor tyrosine kinases, an extracellular ligand-binding domain occurs which is connected to an intracellular catalytic kinase domain by a single pass transmembrane hydrophobic helix [27]. RTKs are not phosphorylated and monomeric in an inactive state [29]. Activation by ligand binding to their extracellular domain results in receptors’ oligomerization and autophosphorylation of a tyrosine residue within the kinase domain. NRTKs are cytoplasmic proteins that have a kinase domain and various additional signaling or protein-protein interacting domains [27]. They are activated by intracellular signals through the dissociation of inhibitors, by recruitment to transmembrane receptors, and through trans-phosphorylation by other kinases [29]. A large number of RTKs and NRTKs are associated with cancers; thus, a significant number of tyrosine kinase inhibitors (TKIs) are currently in clinical development. In the last 10 years, the FDA has approved four new drugs for the treatment of hematological malignancies, which are tyrosine kinase inhibitors (Table 1). Among them, there are the agents that target non-receptor Bruton’s tyrosine kinase (BTK) or non-receptor Sarcoma (Src) and Abelson (Abl) kinases.
Table 1.
Features of the tyrosine kinase (TK) inhibitors approved by the Food and Drug Administration (FDA) from 2011 to 2021. The order of drugs is tabulated in order of most recent to oldest registration date.
| No. | Generic Name of Drug | Brand Name and Company |
First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
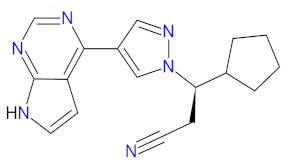
| 1 | Zanubrutinib | BRUKINSA BeiGene, Ltd., Beijing, China | FDA: 14 November 2019 EMA: 29 May 2019 |
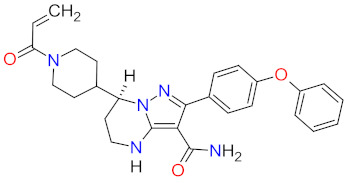
|
BTK 1 | Oral | Mantle Cell Lymphoma | Decreased neutrophil count, anemia, neutropenia, pneumonia, decreased platelet count, upper respiratory tract infection, rash, bruising, diarrhea, cough | [39,40] |
| 2 | Acalabrutinib | CALQUENCE AstraZeneca, Cambridge, UK | FDA: 31 October 2017 EMA: 5 November 2020 |
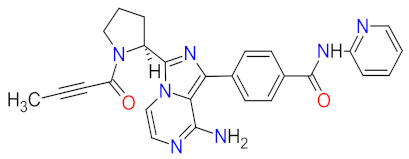
|
BTK 1 | Oral | Mantle Cell Lymphoma, Chronic Lymphocytic Leukemia, Small Lymphocytic Lymphoma |
Headache, diarrhea, fatigue, nausea, contusion, neutropenia, anemia, pneumonia, thrombocytopenia | [31,41,42] |
| 3 | Ibrutinib | IMBRUVICA AbbVie Inc., Lake Bluff, IL, USA | FDA: 13 November 2013 EMA: 21 October 2014 |
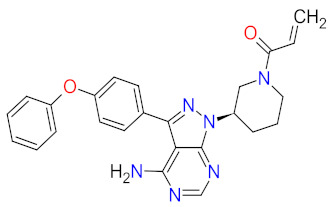
|
BTK 1 | Oral | Mantle Cell Lymphoma, Chronic Lymphocytic Leukemia, Waldenström’s Macroglobulinemia, Small Lymphocytic Lymphoma, Marginal Zone Lymphoma | Diarrhea, fatigue, nausea, dyspnea, constipation, peripheral edema, upper respiratory tract infection, rash, cough, arthralgia, vomiting, decreased appetite, thrombocytopenia, neutropenia, anemia, pneumonia, dehydratation | [30,43,44] |
| 4 | Bosutinib | BOSULIF Pfizer Inc., New York, NY, USA |
FDA: 4 September 2012 EMA: 27 March 2013 |
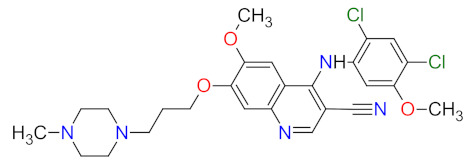
|
Src 2, Abl 3 |
Oral | Chronic Myelogenous Leukemia | Diarrhea, nausea, abdominal pain, vomiting, thrombocytopenia, anemia, neutropenia | [45,46,47] |
1 BTK: Bruton’s tyrosine kinase. 2 Src: non-receptor Sarcoma kinase. 3 Abl: Abelson kinase.
Ibrutinib, acalabrutinib, and zanubrutinib were originally developed as second-line therapy for the treatment of mantle cell lymphoma (MCL), a rare and aggressive type of blood cancer. To date, ibrutinib has received 11 FDA approvals since it was first registered in 2013, among others, as a breakthrough therapy for patients with Waldenström’s macroglobulinemia (WM) and chronic lymphocytic leukemia (CLL), who carry a deletion in chromosome 17 (17p deletion). In 2019, the FDA approved ibrutinib in combination with obinutuzumab, an anti-CD20 monoclonal antibody, as the first non-chemotherapy regimen for patients with previously untreated CLL [30]. In the same year, acalabrutinib received approval as the second BTK inhibitor to treat patients with CLL or small lymphocytic lymphoma (SLL). This drug can be used as monotherapy or in combination with obinutuzumab [31]. The mechanism of action of ibrutinib, acalabrutinib, and zanubrutinib is the irreversible inhibition of BTK activity by forming a covalent bond with a cysteine residue in the BTK active site. This results in blocking B cell antigen receptor signaling (i.e., nuclear factor of activated T-cells (NFAT) pathway, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway, and mitogen-activated protein kinase (ERK) pathway), thus inhibiting the malignant B cells’ proliferation and survival (Figure 1) [32,33,34].
Figure 1.

Mode of action of tyrosine kinase (TK) inhibitors such as non-receptor BTK and Src/Abl inhibitors. BCR: B-cell receptor. RTK: tyrosine kinase receptor. RAF: proto-oncogene serine/threonine-protein kinase. MEK: mitogen-activated protein kinase kinase. ERK: mitogen-activated protein kinase. Src: non-receptor Sarcoma kinase. Abl: Abelson kinase. Rac: Ras-related C3 botulinum toxin substrate. JNK: c-Jun N-terminal kinase. SYK: spleen tyrosine kinase. BCAP: B cell adapter for PI3K. DAG: diacylglycerol. PKC: protein kinase C. IKK: IκB kinase. NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells. Lyn: tyrosine-protein kinase Lyn. BTK: Bruton’s tyrosine kinase. PLC: phospholipase C. IP3: inositol trisphosphate. NFAT: nuclear factor of activated T-cells. Created with BioRender.com based on information in [37,38].
Bosutinib is a dual inhibitor of Src and Abl kinases that is used as a treatment for patients with Philadelphia chromosome positive (Ph+) chronic myeloid leukemia (CML), who show resistance or intolerance to previous therapy, including imatinib. The indication was extended in 2017 to include patients with newly diagnosed chronic phase Ph+ CML [35]. The drug shows activity against most imatinib-resistant mutants of BCR-ABL, which is a hybrid of a breakpoint cluster region protein (BCR) and Abelson tyrosine kinase (Abl), except the mutations T315I and V299. Bosutinib does not inhibit either the receptor tyrosine kinase c-Kit (known as mast/stem cell growth factor receptor or CD117) or platelet-derived growth factor receptor (PDGFR) [36]. The drug acts by binding to the active conformation of the kinase domain and inhibiting its autophosphorylation, resulting in a blockade of cancer cell growth (Figure 1) [37].
2.1.2. Multi Kinase Inhibitors
Multi kinase inhibitors are a group of ATP-competitive drugs that target a set of structurally related kinases. A single multi-inhibitor is preferred to two single inhibitors since drug-drug interactions might occur, changing the metabolism and activities against particular kinases. Multi kinase drugs become the second choice when their pharmacokinetic properties are worse. Besides, multi kinase inhibitors are less specific and might consequently lead to more side effects. A frequently observed disadvantage during treatment with multi kinase inhibitors is acquired resistance [48]. However, the inhibition of several kinases by one drug is useful in anticancer therapy, because oncogenesis and cancer growth have to be considered as multistep processes that are dependent on various signaling pathways (Figure 2) [49]. An overview of FDA-approved multi kinase inhibitors is presented in Table 2.
Figure 2.

Schematic representation of the signaling pathways that can potentially be inhibited by multi kinase inhibitors. BCR: B-cell receptor. PDGFR: platelet-derived growth factor receptor. FLT3: FMS-like tyrosine kinase-3. AXL: AXL receptor tyrosine kinase. ALK: anaplastic lymphoma kinase. VEGFR: vascular endothelial growth factor receptor. FGFR: fibroblast growth factor receptor. RET: receptor tyrosine kinase rearranged during transfection. c-Kit: mast/stem cell growth factor receptor. TIE2: tunica interna endothelial cell kinase 2. PI3K: phosphatidylinositol 3-kinase. PIP2: phosphatidylinositol 4,5-bisphosphate. PIP3: phosphatidylinositol-3,4,5-trisphosphate. PTEN: phosphatase and tensin homolog deleted on chromosome ten. PDK: 3-phosphoinositide-dependent protein kinase. AKT: protein kinase B. mTORC1: mammalian target of rapamycin complex 1. 4E-BP1: 4E-binding protein 1. eIF4E: eukaryotic translation initiation factor 4E. S6K: p70S6 kinase. S6: S6 protein. RAF: proto-oncogene serine/threonine-protein kinase. MEK: mitogen-activated protein kinase kinase. ERK: mitogen-activated protein kinase. Src: non-receptor Sarcoma kinase. Abl: Abelson kinase. Rac: Ras-related C3 botulinum toxin substrate. JNK: c-Jun N-terminal kinase. CDK: cyclin-dependent kinase. SYK: spleen tyrosine kinase. BCAP: B cell adapter for PI3K. DAG: diacylglycerol. PKC: protein kinase C. IKK: IκB kinase. NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells. Lyn: tyrosine-protein kinase Lyn. BTK: Bruton’s tyrosine kinase. PLC: phospholipase C. IP3: inositol trisphosphate. NFAT: nuclear factor of activated T-cells. Created with BioRender.com based on information in [37,38,50,51].
Table 2.
Features of the multi kinase inhibitors approved by the Food and Drug Administration (FDA) from 2011 to 2021. The order of drugs is tabulated in order of most recent to the oldest registration date.
| No. | Generic Name of Drug | Brand Name and Company |
First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Fedratinib | INREBIC Celgene Corporation, Summit, NJ, USA |
FDA: 16 August 2019 EMA: 8 February 2021 |
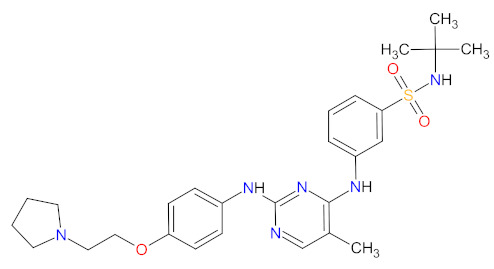
|
JAK2 2 | Oral | Myelofibrosis | Diarrhea, nausea, vomiting, constipation, anemia, thrombocytopenia | [66] 1, [67,68] |
| 2 | Gilteritinib | XOSPATA Astellas Pharma US, Inc., Northbrook, IL, USA |
FDA: 28 November 2018 EMA: 24 October 2019 |
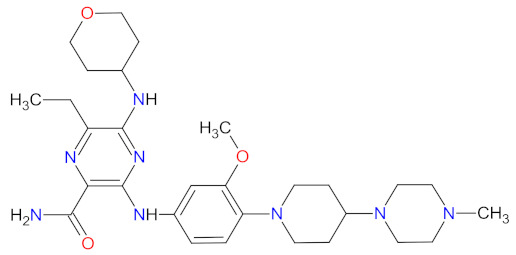
|
FLT3 3, AXL 4, ALK 5 |
Oral | Acute Myeloid Leukemia | Myalgia, arthralgia, increased levels of transaminases, fatigue, malaise, fever, diarrhea, dyspnea, edema, rash, pneumonia, sepsis, renal impairment | [69,70] |
| 3 | Midostaurin | RYDAPT Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA |
FDA: 28 April 2017 EMA: 18 September 2017 |
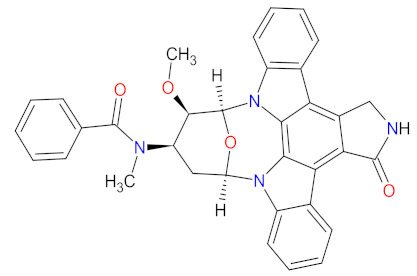
|
c-Kit 6, PDGFRA 7, PDGFRB 8, FLT3 3, PKC 9, CDK1 10, SYK 11, VEGFR-2 12 |
Oral | Acute Myeloid Leukemia, Cutaneous Mastocytosis | Febrile neutropenia, nausea, vomiting, diarrhea, edema, mucositis, headache, device-related infection, abdominal pain, fatigue, pyrexia, dyspnea, musculoskeletal pain, constipation, epistaxis, upper respiratory tract infection, petechial, hyperglycemia, | [71,72] |
| 4 | Ponatinib | ICLUSIG Ariad Pharmaceuticals, Inc., Cambridge, MA, USA |
FDA: 14 December 2012 EMA: 1 July 2013 |
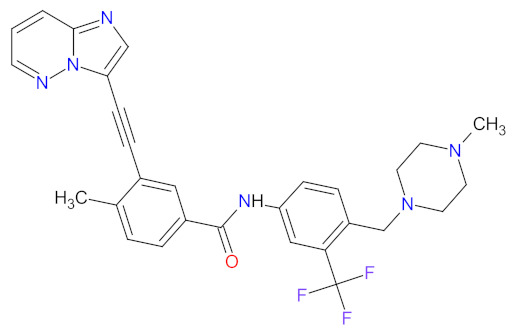
|
BCR-ABL 13, VEGFRs 14, FGFRs 15, PDGFRs 16, RET 17, c-Kit 6, TIE2 18, FLT3 3 |
Oral | Chronic Myelogenous Leukemia, Acute Lymphoblastic Leukemia |
Hypertension, cardiac failure, abdominal pain, constipation, diarrhea, oral mucositis, febrile neutropenia, fatigue, pneumonia, headache, peripheral neuropathy, dizziness, pleural effusion, cough, dyspnea, rush, dry skin, arthralgia, myalgia, spasms, decreased appetite, edema, weight loss, insomnia | [73,74] |
| 5 | Ruxolitinib | JAKAFI Incyte Corporation, Wilmington, DE, USA |
FDA: 16 November 2011 EMA: 23 August 2012 |
|
JAK1 19, JAK2 2 | Oral | Myelofibrosis, Polycythemia Vera, Graft-versus-host disease |
Anemia, thrombocytopenia, neutropenia | [75,76] |
1 Wrong chemical structure of the drug is given in the reference. 2 JAK2: Janus kinase 2. 3 FLT3: FMS-like tyrosine kinase-3. 4 AXL: AXL receptor tyrosine kinase. 5 ALK: anaplastic lymphoma kinase. 6 c-Kit: mast/stem cell growth factor receptor. 7 PDGFRA: platelet-derived growth factor receptor α. 8 PDGFRB: platelet-derived growth factor receptor β. 9 PKC: protein kinase C. 10 CDK1: cyclin-dependent kinase 1. 11 SYK: spleen tyrosine kinase. 12 VEGFR-2: vascular endothelial growth factor receptor-2. 13 BCR-ABL: BCR-ABL fusion protein. 14 VEGFRs: vascular endothelial growth factor receptors. 15 FGFRs: fibroblast growth factor receptors. 16 PDGFRs: platelet-derived growth factor receptors. 17 RET: receptor tyrosine kinase rearranged during transfection. 18 TIE2: tunica interna endothelial cell kinase 2. 19 JAK1: Janus kinase 1.
The kinase domain mutations are the reason for developing drug resistance during the treatment of various types of leukemia. One of the most common genetic alterations is the gatekeeper T315I substitution observed in chronic myeloid leukemia (CML) or Philadelphia chromosome positive (Ph+) acute lymphoblastic leukemia (ALL) and FMS-like tyrosine kinase-3 (FLT3)-activating mutations in acute myeloid leukemia (AML). Resistance to tyrosine kinase inhibitors has necessitated the designing of new mutation-resistant inhibitors, such as ponatinib, midostaurin, and gilteritinib. Ponatinib is a multitarget inhibitor characterized by high-affinity and optimized binding to the active site of the BCR-ABL kinase domain, in which the T315 can occur. This mutation is the major reason for inhibition access of the drug to the enzyme’s ATP-binding site, leading to resistance to first- and second-generation tyrosine kinase inhibitors [52]. Ponatinib is effective in the inhibition of native and mutant BCR-ABL, receptor tyrosine kinase rearranged during transfection (RET), FLT3, tunica interna endothelial cell kinase 2 (TIE2), mast/stem cell growth factor receptor (c-Kit), vascular endothelial growth factor receptors (VEGFRs), fibroblast growth factor receptors (FGFRs), and platelet-derived growth factor receptors (PDGFRs). Treatment with ponatinib shows substantial and durable clinical activity in patients with Ph+ leukemia with resistance or intolerance to all other approved tyrosine kinase inhibitors [53]. Adverse events of this therapy are defined as follows: nonhematologic toxic effects such as skin disorders (e.g., rash, acneiform dermatitis, and dry skin), constitutional symptoms (e.g., arthralgia, fatigue, and nausea), or hematologic—such as vascular occlusive—events, venous thromboembolic events, thrombocytopenia, and neutropenia [52]. The occurrence of vascular events during therapy was dependent on the dose of ponatinib, wherein lower doses have affected the improvement of the vascular safety profile [54]. Midostaurin and gilteritinib are approved drugs for patients with newly diagnosed FLT3-mutated AML. The clinical activity of midostaurin in combination with cytarabine and daunorubicin-based chemotherapy was positive for FLT3-activating mutations, such as, primarily, in-frame internal tandem duplications (ITD) and missense point mutations in the tyrosine kinase domain (TKD). Moreover, midostaurin inhibits c-Kit (wild type and D816V mutant) found in advanced systemic mastocytosis (SM), which includes aggressive systemic mastocytosis (ASM), systemic mastocytosis with associated hematological neoplasm (SM-AHN), and mast cell leukemia [55]. It was found to also be an inhibitor of protein kinase C (PKC), platelet-derived growth factor receptors (PDGFRs) alpha and beta, cyclin-dependent kinase 1 (CDK1), spleen tyrosine kinase (SYK), and vascular endothelial growth factor receptor-2 (VEGFR-2) [56]. Although midostaurin shows a broad spectrum of antikinase activity, it is characterized by lacked potency. Gilteritinib, on the other hand, is a selective, potent inhibitor of all FLT3-activating mutation types (e.g., ITC, TKD, D835Y, double ITD-D835Y) [57]. Furthermore, gilteritinib shows activity against c-Kit and the AXL receptor tyrosine kinase (AXL, also known as UFO), which is implicated in FLT3 inhibitor resistance [58]. The mechanism of action of gilteritinib involves binding to the active conformation of FLT3 at the ATP-binding site, resulting in reduced proliferation of cancer cells that overexpress the mutation [59].
The mutations that confer activation of the intracellular Janus kinase (JAK) signal transducer and activator of transcription (STAT) pathways (e.g., JAK2, V617F, and JAK2 exon 12) were identified as the most common in patients with myelofibrosis (MF). Only two drugs, namely ruxolitinib and fedratinib, are approved as JAK inhibitors for the treatment of MF [60]. Ruxolitinib is a JAK1/2 inhibitor that potently inhibits the proliferation of JAK2 V617F-driven Ba/F3 cells, resulting in decreased levels of phosphorylated JAK2 and signal transducer and activator of transcription 5 (STAT5) [61]. Ruxolitinib provides a rapid reduction in splenomegaly, ameliorating debilitating myelofibrosis-related symptoms and improving quality of life in patients with MF. The adverse events of ruxolitinib, like anemia and thrombocytopenia, were manageable and led to the discontinuation of therapy at a low rate [62]. Ruxolitinib is also an effective drug for patients with polycythemia vera, which allows for hematocrit control, reducing spleen size, and improving symptoms of disease [63]. However, some patients lose response to ruxolitinib and discontinue treatment over time because developing resistance or intolerance is associated with a substantially reduced life expectancy. Fedratinib, an alternative approved JAK inhibitor, is potent and selective for JAK2 regardless of its mutational status [64]. Compared with ruxolitinib, fedratinib causes a more effective reduction in spleen volume and disease-related symptoms [65].
2.2. Phosphatidylinositol 3-Kinase (PI3K) Inhibitors as Anticancer Agents
Phosphatidylinositol 3-kinases (PI3Ks) are a family of lipid kinases that phosphorylate phosphoinositides at the 3-hydroxyl group of the inositol ring that can be used to generate phosphatidylinositol 3,4,5-trisphosphate. Among these, several classes have been identified and characterized by different primary structures and substrate specificities. Class I PI3Ks are heterodimers and divide into two groups, IA and IB. Class IA PI3Ks are activated by a wide range of receptor tyrosine kinases (RTKs) and are frequently implicated in cancer, while class IB PI3Ks are activated by G-protein-coupled receptors [77]. Structurally, class IA PI3Ks exists in three isoforms (α, β, and δ) and class IB PI3Ks in one isoform (γ). Class II PI3Ks are monomeric proteins that consist of three isoforms (α, β, and δ), whereas class III PI3Ks are only one heterodimer composed of a catalytic (Vps34) and regulatory subunit [78]. PI3K-related kinases, which can be included as class IV of PI3K, are a group of protein kinases with structural similarity to PI3K, but without the lipid kinase activity. This group includes a mammalian target of rapamycin (mTOR), DNA-dependent protein kinase (DNA-PK), ataxia telangiectasia mutated gene product (ATM), and ataxia telangiectasia and Rad3-related gene product [79]. The dysregulation of the phosphatidylinositol-3 kinase pathway, especially abnormal activation, is one of the most frequently observed in blood cancers and an important target of selective anticancer therapies. The three inhibitors of PI3K have received market approval since 2011 (Table 3). Idelalisib, a first-in-class inhibitor of PI3K-δ, and the following inhibitors, i.e., copanlisib and duvelisib, directly reduce the proliferation and survival of malignant B-cell leukemia and lymphoma cells (Figure 3). Hence, they were approved by the FDA for the treatment of different types of leukemia and lymphoma [80,81,82]. Duvelisib is a first-in-class dual inhibitor of PI3Ks due to the fact that it also inhibits PI3K-γ activity, which leads to a reduction in the differentiation and migration of various components of the cancer microenvironment, such as T helper cells and M2 tumor-associated macrophages [82]. It is worth noting that the route of copanlisib administration as intermittent intravenous infusions lead to weaker gastrointestinal toxicity compared with the oral treatment of idelalisib [83].
Table 3.
Features of the phosphatidylinositol-3 kinase (PI3K) inhibitors approved by the Food and Drug Administration (FDA) from 2011 to 2021. The order of drugs is tabulated in order of most recent to oldest registration date.
| No. | Generic Name of Drug | Brand Name and Company |
First FDA/EMA Approved Date | Structure | Molecular Target |
Route of Administration |
Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Duvelisib | COPIKTRA Verastem, Inc. Needham, MA, USA | FDA: 24 September 2018 EMA: 19 May 2021 |
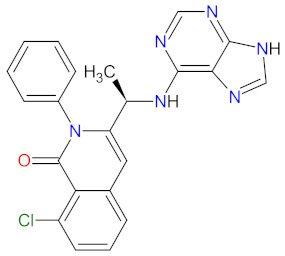
|
PI3K-δ 1, PI3K-γ 2 | Oral | Chronic Lymphocytic Leukemia, Follicular Lymphoma | Neutropenia, thrombocytopenia, anemia, diarrhea, pyrexia, nausea, vomiting, anorexia |
[84,85] |
| 2 | Copanlisib | ALIQOPA Bayer HealthCare Pharmaceuticals Inc., HanoverWhippany, NJ, USA |
FDA: 14 September 2017 EMA: Not approved |
|
PI3K-α 3, PI3K-δ 1 | Intravenous infusion | Follicular Lymphoma |
Hyperglycemia, hypertension, infections, neutropenia |
[86,87] |
| 3 | Idelalisib | ZYDELIG Gilead Sciences, Inc., Foster City, CA, USA |
FDA: 23 July 2014 EMA: 18 September 2014 |
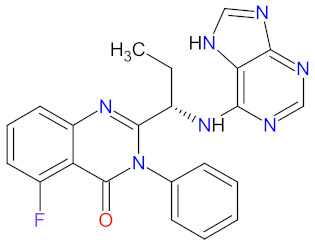
|
PI3K-δ 1 | Oral | Chronic Lymphocytic Leukemia, Follicular Lymphoma | Diarrhea, nausea, vomiting, fatigue, headache, pneumonia, chill, dyspnea, rash, neutropenia, pyrexia, sepsis, decreased neutrophil count, hypertriglyceridemia, hyperglycemia, elevated alanine and aspartate transaminases |
[88,89] |
1 PI3K-δ: phosphatidylinositol 3-kinase delta. 2 PI3K-γ: phosphatidylinositol 3-kinase gamma. 3 PI3K-α: phosphatidylinositol 3-kinase alpha.
Figure 3.

Mechanism of action of PI3K inhibitors. RTK: receptor tyrosine kinase. PI3K: phosphatidylinositol 3-kinase. PIP2: phosphatidylinositol 4,5-bisphosphate. PIP3: phosphatidylinositol-3,4,5-trisphosphate. PTEN: phosphatase and tensin homolog deleted on chromosome ten. PDK: 3-phosphoinositide-dependent protein kinase. AKT: protein kinase B. mTORC1: mammalian target of rapamycin complex 1. 4E-BP1: 4E-binding protein 1. eIF4E: eukaryotic translation initiation factor 4E. S6K: p70S6 kinase. S6: S6 protein. Created with BioRender.com based on information in [51,79].
2.3. Various Enzymes Inhibitors as Anticancer Agents
Enzymes are organic high-molecular-weight molecules that catalyze the synthesis or degradation reaction of a specific enzyme’s substrate. Enzyme inhibitors bind to the enzyme, resulting in disruption of the normal formation of an intermediate enzyme-substrate complex. Enzyme inhibitors, depending on their mechanism of action, cause the modification of enzyme’s activity in three ways, i.e., irreversible, competitive, and non-competitive [90]. The drugs that have received FDA approval for blood cancer treatment since 2011 inhibit the catalytic activity of such enzymes as histone deacetylase (HDAC), isocitrate dehydrogenases (IDHs), enhancers of zeste homolog 2 (EZH2), and deoxyribonucleic acid (DNA) or ribonucleic acid (RNA) methyltransferase (Figure 4). All new drugs are summarized in Table 4.
Figure 4.

Mode of action of various enzymes inhibitors. HDAC: histone deacetylase. RNMT: RNA methyltransferase. DNMT: DNA methyltransferase. EZH2: enhancer of zeste homolog 2. mIDH1: mutant isocitrate dehydrogenase 1. mIDH2: mutant isocitrate dehydrogenase 2. NADPH: nicotinamide-adenine dinucleotide phosphate (reduced form). NADP+: nicotinamide-adenine dinucleotide phosphate. αKG: α-ketoglutarate. D2-HG: D-2-hydroxyglutarate. Me: methyl group. Ac: acetyl group. DNA: deoxyribonucleic acid. tRNA: transfer ribonucleic acid. Created with BioRender.com.
Table 4.
Features of the other enzymes inhibitors approved by the Food and Drug Administration (FDA) from 2011 to 2021. The order of drugs is tabulated in order of most recent to oldest registration date.
| No. | Generic Name of Drug | Brand Name and Company |
First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Azacitidine | ONUREG Bristol-Myers Squibb Company, New York, NY, USA |
FDA: 1 September 2020 EMA: 17 June 2021 |
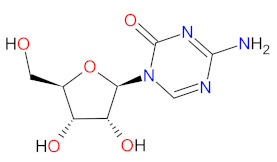
|
DNMTs 1, RNMTs 2 | Oral | Acute Myeloid Leukemia | Nausea, vomiting, diarrhea, fatigue, constipation, pneumonia, arthralgia, decreased appetite, febrile neutropenia, dizziness, abdominal pain. |
[112,114] |
| 2 | Tazemetostat | TAZVERIK Epizyme, Inc., Cambridge, MA, USA |
FDA: 23 January 2020 EMA: Not approved |
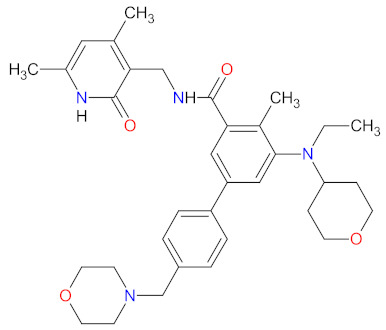
|
EZH2 3 | Oral | Epithelioid Sarcoma, Follicular Lymphoma |
Fatigue, nausea, decreased appetite, vomiting, constipation, upper respiratory tract infection, abdominal pain, musculoskeletal pain |
[106,115] |
| 3 | Ivosidenib | TIBSOVO Agios Pharmaceuticals, Inc., Cambridge, MA, USA |
FDA: 20 July 2018 EMA: Not approved |
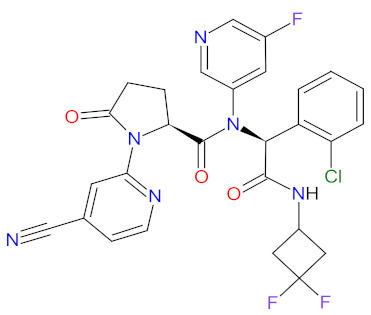
|
IDH1 4 | Oral | Acute Myeloid Leukemia | Diarrhea, leukocytosis, nausea, fatigue, dyspnea, electrocardiogram QT prolonged, edema, anemia, pyrexia, cough, febrile neutropenia, isocitrate dehydrogenase differentiation syndrome |
[116,117] |
| 4 | Enasidenib | IDHIFA Celgene Corporation, Summit, MA, USA |
FDA: 1 August 2017 EMA: Not approved |
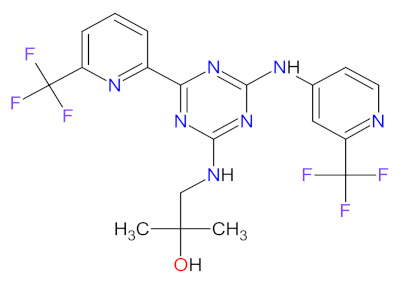
|
IDH2 5 | Oral | Acute Myeloid Leukemia | Elevated bilirubin, nausea, diarrhea, decreased appetite, vomiting |
[118,119] |
| 5 | Panobinostat | FARYDAK, Novartis Pharmaceuticals Corporation, East Hanover, NJ, USA |
FDA: 23 February 2015 EMA: 28 August 2015 |
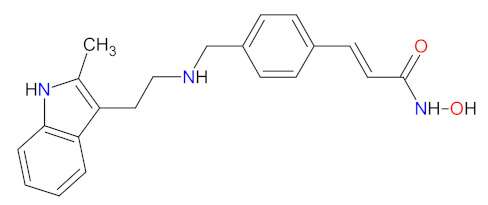
|
HDAC 6 | Oral | Multiple Myeloma | Diarrhea, fatigue, nausea, peripheral edema, decreased appetite, pyrexia, vomiting, thrombocytopenia, lymphopenia, leukopenia, neutropenia, anemia, hypophosphatemia, hypokalemia, hyponatremia, increased creatinine |
[120,121] |
| 6 | Belinostat | BELEODAQ, Spectrum Pharmaceuticals, Inc., Henderson, NV, USA | FDA: 3 July 2014 EMA: Not approved |
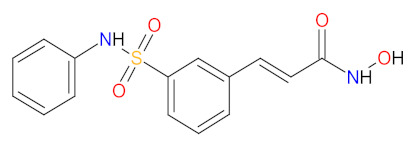
|
HDAC 6 | Intravenous infusion | Peripheral T-cell Lymphoma |
Nausea, vomiting, diarrhea, fatigue, pyrexia, anemia, constipation, dyspnea, rash, peripheral edema |
[122,123] |
1 DNMTs: DNA methyltransferases. 2 RNMTs: RNA methyltransferases. 3 EZH2: enhancer of zeste homolog 2. 4 IDH1: isocitrate dehydrogenase 1. 5 IDH2: isocitrate dehydrogenase 2. 6 HDAC: histone deacetylase.
2.3.1. Histone Deacetylase Inhibitors
Histone deacetylase (HDAC) enzymes catalyze the removal of an acetyl group from lysine residues of histone and non-histone (e.g., transcription factor p53) proteins. This process has a key role in modifying the structure of nucleosomes, the fundamental units of chromatin. Subsequently, the remodeling of chromatin form from open to closed is essential to the regulation of gene expression [91]. HDAC enzymes can be divided into four groups: class I (HDACs 1, 2, 3, and 8), class II a/b (HDACs 4, 5, 6, 7, 9, and 10), class III (sirtuin enzymes), and class IV (HDAC11). Class I, II, and IV HDACs are zinc-dependent enzymes [92]. The deregulation of HDACs activity has been reported in various cancer cell lines; therefore, their inhibition may be an attractive anticancer therapy. HDAC enzyme inhibitors (HDACi) induce rapid histone hyperacetylation and relaxation of chromatin, which results in cellular differentiation and apoptosis of cancer cells. Additionally, they promote cell cycle arrest, inhibit angiogenesis of cancer vasculature, and regulate host immune responses [91]. Since 2011, two HDACi are approved as a treatment for hematological malignancies, belinostat, for patients with relapsed or refractory peripheral T-cell lymphoma (PTCL), and panobinostat, for patients with recurrent multiple myeloma who have received at least two prior treatment regimens, including bortezomib and an immunomodulatory agent [93,94]. Both drugs are inhibitors of I, II, and IV HDAC isoforms. Their mechanism of action is the chelation of a zinc ion through the hydroxamate group region of their structure, resulting in the blocking of histone deacetylation [95].
2.3.2. Isocitrate Dehydrogenase Inhibitors
Isocitrate dehydrogenases (IDHs) are oxidoreductases presented as one of three isoforms, i.e., IDH1, IDH2, and IDH3, which are involved in the tricarboxylic acid (TCA) cycle, also called the Krebs cycle [96]. IDH1 and IDH2 are similar, homodimeric enzymes that catalyze the reversible conversion of isocitrate to α-ketoglutarate (α-KG), liberating CO2 and reducing nicotinamide adenine dinucleotide phosphate (NADP+) to NADPH. Although IDH1 and IDH2 catalyze identical reactions, these enzymes are situated in different localizations, namely, IDH1 in the cytoplasm and peroxisomes, and IDH2 in the mitochondrial matrix. Both IDHs play important roles in many cellular processes, including modulating the response to glucose by regulating insulin secretion, the metabolism of glutamine or synthesis, and the metabolism of lipids. What is more, the enzymes show antioxidant activity related to the regulation of cellular redox status, resulting in protection against lipid peroxidation and oxidative DNA damage [97]. A third IDH enzyme, IDH3, is a heterotetrameric protein that catalyzes the first oxidative reaction of the TCA cycle, which is the decarboxylation of isocitrate to αKG with a generation of reduced form of nicotinamide adenine dinucleotide (NADH) from nicotinamide adenine dinucleotide (NAD+) [96]. IDH1 and IDH2 mutations are found in multiple tumors, including glioma and acute myeloid leukemia (AML). They occur in a single arginine residue, i.e., Arg132 of IDH1 and Arg172 of IDH2, in the active catalytic sites of the enzyme. Further, IDHs acquire a neomorphic activity, resulting in a reduction in αKG into the oncometabolic product, D-2-hydroxyglutarate (D2-HG). D2-HG competitively inhibits α-KG-dependent dioxygenases resulting in DNA and histone hypermethylation, which leads to a block of normal differentiation processes via epigenetic and metabolic mechanisms and promotes oncogenic transformation [98]. Since 2011, the first-in-class mutant IDH (mIDH) inhibitors, such as enasidenib and ivosidenib, have been FDA-approved for relapsed or refractory AML. Enasidenib targets both IDH2 R140 and R172 isoforms, while ivosidenib targets a variety of IDH1 R132 mutants (R132C, R132G, R132H, R132S, and R132L) [99,100]. The drugs suppress the production of oncometabolite D2-HG from α-KG by binding to the allosteric site of the mutant enzyme, thus preventing a conformational change in its structure and inhibiting gain-of-function enzymatic activity [101]. Both of these induce terminal differentiation of leukemic bone marrow blasts, which is responsible for the clinical response and efficacy of treatment [100,102].
2.3.3. Enhancer of Zeste Homolog 2 Inhibitor
Enhancer of zeste homolog 2 (EZH2) is a histone methyltransferase that catalyzes trimethylation of histone H3 at Lys 27 (H3K27me3). EZH2 is also a catalytic component of polycomb repressive complex 2 (PRC2), which is a group of important epigenetic regulators of gene expression for regulating the differentiation of healthy cells. Mutation or overexpression of the EZH2 gene plays a critical role in the development of various cancers such as CRC, melanoma, ovarian cancer, and breast cancer. Dysregulation of EZH2 as a histone modifier causes the proliferation of cancer cells and promotes their survival and metastasis, resulting in invasion and progression of a malignant tumor. Moreover, EZH2 is involved in the regulation of immune cells (e.g., T cells, natural killer cells, dendritic cells, and macrophages), which are essential components in the cancer microenvironment [103]. Tazemetostat is a first-in-class, potent, and highly selective EZH2 inhibitor. The mechanism of its action is blocking EZH2 activity, thus the trimethylation of H3K27, which results in tumor regressions [104]. In clinical development, tazemetostat showed a favorable safety profile and responses in patients with either lymphoma, including both germinal center B-cell-like (GCB) and non-GCB subtypes of diffuse large B-cell lymphoma (DLBCL) [105], or advanced solid tumors such as epithelioid sarcoma [106]. The New Drug Application for tazemetostat as the treatment of relapsed or refractory follicular lymphoma (FL) was accepted by the FDA in June 2020 [107].
2.3.4. DNA and RNA Methyltransferases Inhibitor
DNA and RNA methyltransferases (DNMTs and RNMTs) are a family of enzymes that catalyze the methylation of DNA and RNA, respectively. The five human DNMTs, i.e., DNMT1, DNMT2, DNMT3A, DNMT3B, and DNMT3L, play important role in epigenetic gene regulation, including transcriptional silencing and transcriptional activation, whereas RNMTs are implicated in RNA stability, splicing, and epigenetic mechanisms [108,109]. Azacitidine is a cytidine nucleoside analog that is phosphorylated in cell to its active form, i.e., azacitidine triphosphate, and is incorporated into DNA and RNA. Subsequently, azacitidine reduces DNA and RNA methylation by noncompetitive inhibition of DNA methyltransferases and RNA methyltransferases, respectively. The resulting hypomethylation of DNA is involved in the activation and expression of genes which regulate cancer-suppressing functions and cell differentiation. The reduction in RNA methylation leads to decreased RNA stability and decreased protein synthesis. Azacitidine shows, also, direct cytotoxicity to abnormal hematopoietic cells in the bone marrow [110]. Azacitidine was originally approved in 2004 as an intravenous or subcutaneous treatment of myelodysplastic syndromes [111]. However, in 2020, ONUREG (a novel oral formulation of azacitidine as film-coated tablets) was registered as continued treatment of adult patients in the first remission with acute myeloid leukemia (AML) [112]. ONUREG is the first drug to receive FDA approval as maintenance therapy. An oral azacitidine formulation facilitates the drug administration, reduces the potential side effects associated with subcutaneous injection, and allows for the expansion of treatment regimens by searching for alternative doses or combinations of therapies [113].
2.4. Smoothed Receptor Antagonists as Anticancer Agents
The deregulation of the smoothened (SMO) receptor’s activity is frequent in human cancers and has great biomedical importance for the development of new anti-cancer agents. The SMO receptor belongs to the class Frizzled (class F) receptors, which are part of a G-protein-coupled receptor (GPCR) family. SMO mediates signal transduction in the Hedgehog (Hh) pathway, which plays a significant role in normal embryonic development and maintenance or repair of adult tissue [124]. Under physiological conditions, the activation of the SMO receptor is regulated by the binding of hedgehog signaling proteins to a transmembrane receptor called Patched (PTCH), and leads to the downstream activation of the Hh signaling cascade. The abnormal Hh signaling is implicated in carcinogenesis, most notably in basal cell carcinoma (BCC) or medulloblastoma, and supports the tumor microenvironment in disparate cancers [125]. The SMO receptor activity can be modulated by small-molecule inhibitors, some of which are currently FDA-approved anticancer drugs, including glasdegib, vismodegib, and sonidegib. Glasdegib, as the only one that has been registered in the last 10 years, is characterized in Table 5. This drug potently inhibits the Hedgehog signaling pathway with clinical activity in older patients with newly diagnosed acute myeloid leukemia (AML) (Figure 5) [126]. It is used in combination with chemotherapy, i.e., cytarabine, which is an attractive treatment approach for patients with AML, improving statistically significant overall survival (OS) [127]. Glasdegib is the next generation SMO inhibitor, and does not use the same binding site as vismodegib and sonidegib [128]. Therefore, it is not susceptible to the evolution of drug resistances, especially by acquired mutations in SMO receptors. However, vismodegib and sonidegib are under intensive investigation in ongoing clinical trials as a therapy for blood cancer types such as myelofibrosis, chronic myeloid leukemia, and myelodysplastic syndromes.
Table 5.
Features of the various receptor antagonists approved by the Food and Drug Administration (FDA) from 2011 to 2021.
| No. | Generic Name of Drug | Brand Name and Company |
First FDA/EMA Approved Date | Structure | Molecular Target |
Route of Administration |
Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Glasdegib | DAURISMO Pfizer Inc., New York, NY, USA | FDA: 21 November 2018 EMA: 26 June 2020 |
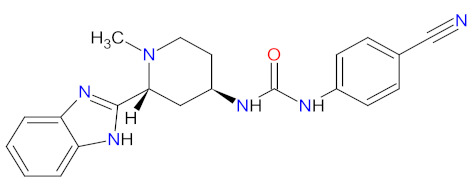
|
SMO receptor 1 | Oral | Acute Myeloid Leukemia |
Anemia, febrile neutropenia, thrombocytopenia |
[129,130] |
1 SMO receptor: smoothened receptor.
Figure 5.

Glasdegib inhibition of the Hedgehog signaling pathway. HH: Hedgehog. PTCH: Patched receptor. SMO: smoothened receptor. SUFU: suppressor of fused protein. GLI: glioma-associated oncogene protein. GLIr: repressor form of GLI. Created with BioRender.com based on information in [128].
2.5. Various Proteins Inhibitors as Anticancer Agents
Proteins have a key role in an array of biological processes, such as signal transduction pathways or programmed cell death, and, therefore, offer an attractive target for anticancer therapy. In the last 10 years, there has been important progress in developing small-molecule inhibitors (SMIs) targeting various types of proteins, including B-cell leukemia/lymphoma-2 (BCL-2), exportin-1 (XPO1), proteasome, and tubulin protein (Figure 6). The features of the FDA-approved protein inhibitors are presented in Table 6.
Figure 6.

The four types of protein inhibitors in the treatment of hematological malignancies and their mechanisms of action. BCL-2: B-cell leukemia/lymphoma-2. Bim: BCL-2-like protein 11. BAX: BCL-2-associated X protein. BAK: BCL-2 antagonist/killer 1. MOMP: mitochondrial outer membrane permeabilization. XPO1: exportin-1. TSP: tumor suppressor protein. Ub: ubiquitin. ADP: adenosine diphosphate. Created with BioRender.com.
Table 6.
Features of the various proteins inhibitors approved by the Food and Drug Administration (FDA) from 2011 to 2021. The order of drugs is tabulated in order of most recent to oldest registration date.
| No. | Generic Name of Drug | Brand Name and Company |
First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Selinexor | XPOVIO Karyopharm Therapeutics Inc., Newton Centre, MA, USA |
FDA: 3 July 2019 EMA: 26 March 2021 |
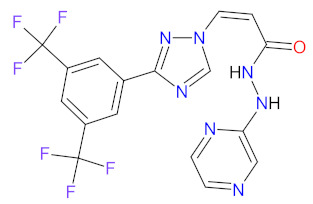
|
XPO1 1 | Oral | Multiple Myeloma, Diffuse Large B-cell Lymphoma | Thrombocytopenia, fatigue, nausea, anemia, decreased appetite, decreased weight, diarrhea, vomiting, hyponatremia, neutropenia, leukopenia, constipation, dyspnea and upper respiratory tract infection |
[149,150] |
| 2 | Venetoclax | VENCLEXTA AbbVie Inc., Lake Bluff, IL, USA | FDA: 11 April 2016 EMA: 5 December 2016 |
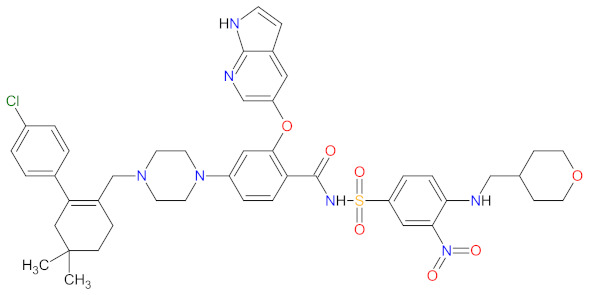
|
BCL-2 2 | Oral | Chronic Lymphocytic Leukemia, Acute Myeloid Leukemia |
Neutropenia, diarrhea, nausea, anemia, thrombocytopenia, upper respiratory tract infection, fatigue |
[151,152] |
| 3 | Ixazomib citrate | NINLARO Takeda Pharmaceuticals U.S.A., Inc., Deerfield, IL, usa |
FDA: 20 November 2015 EMA: 21 November 2016 |
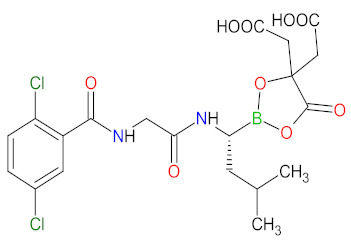
|
Proteasome | Oral | Multiple Myeloma | Diarrhea, constipation, thrombocytopenia, peripheral neuropathy, nausea, peripheral edema, vomiting, back pain, rash |
[153,154] |
| 4 | Vincristine sulfate | MARQIBO Talon Therapeutics Inc., Irvine, CA, USA |
FDA: 9 August 2012 EMA: Not approved |
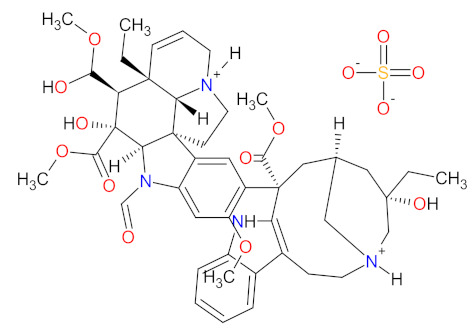
|
Tubulin | Intravenous | Acute Lymphoblastic Leukemia |
Constipation, nausea, pyrexia, fatigue, peripheral neuropathy, febrile neutropenia, diarrhea, anemia, decreased appetite, insomnia |
[155,156,157] |
| 5 | Carfilzomib | KYPROLIS Amgen Inc., Sauzend Oaks, CA, USA | FDA: 20 July 2012 EMA: 19 November 2015 |
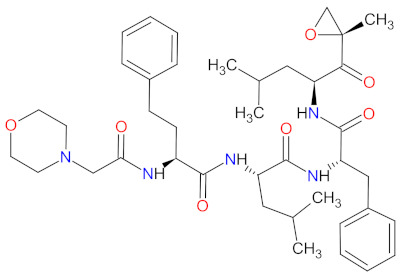
|
Proteasome | Intravenous | Multiple Myeloma | Fatigue, anemia, nausea, thrombocytopenia, dyspnea, diarrhea, pyrexia |
[158,159] |
1 XPO1: exportin-1. 2 BCL-2: B-cell leukemia/lymphoma-2 proteins.
2.5.1. B-Cell Leukemia/Lymphoma-2 Proteins Inhibitor
Proteins of the B-cell leukemia/lymphoma-2 (BCL-2) family are key regulators of the mitochondrial apoptotic pathway. They control cell death primarily by forming pores within the mitochondrial outer membrane which results in the release of intermembrane space proteins (e.g., cytochrome c), followed by the activation of a caspase cascade and apoptosis. The BCL-2 family of proteins is divided into three main groups, including anti-apoptotic members (BCL-2, B-cell lymphoma-extra large (BCL-XL), BCL-W, myeloid cell leukemia 1 (MCL1)), the pro-apoptotic pore-formers (BCL-2-associated X protein (BAX), BCL-2 antagonist/killer 1 (BAK), BCL-2 related ovarian killer (BOK)), and the pro-apoptotic BH3-only proteins (BCL-2 associated agonist of cell death (BAD), BH3 interacting domain death agonist (BID), BCL-2 interacting killer (BIK), BCL-2-like protein 11 (Bim), BCL-2 modifying factor (BMF), Harakiri (HRK), NOXA, PUMA, etc.). All members of the BCL-2 family contain at least one of the four BCL-2 homologies (BH) domains (BH1–4), wherein the anti-apoptotic and pore-forming proteins are multi-BH domain molecules and BH3-only proteins possess only one helical BH3 domain [131,132]. The small molecules mimicking the BH3 domain of BH3-only proteins can selectively bind to and antagonize anti-apoptotic members of the BCL-2 family, leading to apoptosis of various malignancies cells. Venetoclax is a first-in-class, highly selective BCL-2 inhibitor that is bound to the BH3-binding groove of BCL-2 and releases pro-apoptotic BH3-only proteins (e.g., Bim) from BCL-2 [133]. Consequently, free BH3-only proteins activate pro-apoptotic effectors such as BAX and BAK that induce permeabilization of the mitochondrial outer membrane and trigger cell death. Furthermore, venetoclax also inhibits anti-apoptotic proteins such as MCL1. The drug received first approval as monotherapy in patients with chronic lymphocytic leukemia (CLL) with the 17p deletion who have received at least one prior therapy. The indication was extended on June 2018 to include use as a combination therapy with rituximab for CLL or small lymphocytic lymphoma (SLL), and on May 2019 to include use in combination with obinutuzumab for previously untreated patients with CLL or SLL. Finally, in October 2020, the FDA was granted full approval of venetoclax in combination with azacitidine, decitabine, or cytarabine for acute myeloid leukemia (AML) patients [134].
2.5.2. Exportin-1 Inhibitors
Exportin-1 (XPO1), also known as chromosome maintenance protein 1 (CRM1), is a karyopherin involved in the export of various macromolecules from the nucleus to the cytosol, including tumor suppressor proteins (TSPs) (e.g., p53, p21, inhibitor of nuclear factor kappa B (IκB), p21), the glucocorticoid receptor, and messenger RNA (mRNA) for multiple oncoproteins (e.g., BCL-XL, mouse double minute 2 homolog (MDM2), cyclin D1). The export of mRNA to the cytoplasm is mediated exclusively by XPO1 after its binding to eukaryotic translation initiation factor 4E (eIF4e), which promotes the synthesis of cognate oncoproteins, cell survival, and proliferation [135]. Due to XPO1 being overexpressed in most cancer types, it is a promising target for anticancer therapy. Selinexor is a first-in-class selective inhibitor of nuclear export (SINE) compound. The drug forms a slowly reversible covalent bond to Cys528 in the cargo-binding pocket of XPO1, leading to inactivation of XPO1-mediated nuclear export [136]. The inhibition of XPO1 activity results in the accumulation of TSPs in the cell nucleus and the prevention of oncoprotein mRNA translation. It consequently leads to the induction of cell-cycle arrest and apoptosis [137]. Selinexor received first approval in combination with dexamethasone for the treatment of adult patients with relapsed or refractory multiple myeloma (MM), after at least four prior therapies. In June 2020, selinexor was registered, as well, for the treatment of adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL), who have previously attempted at least two lines of systemic therapy. The indication was then extended in December 2020 to include use in combination with bortezomib and dexamethasone for the treatment of adult patients with MM after at least one prior therapy [138].
2.5.3. Proteasome Inhibitors
The proteasome is a large protein complex responsible for the selective and efficient hydrolysis of proteins. The degradation of protein is initiated by the covalent attachment of chain consisting of several copies of ubiquitin (Ub), this is a process known as protein ubiquitylation. The polyubiquitin (poly-Ub) chain, the most abundant Lys48-linked poly-Ub (UbLys48) chain, is an intracellular signal to shuttle the target proteins to the proteasome, where they are proteolytically broken down [139]. The constitutive 26S proteasome, the main extralysosomal mediator of protein degradation, consists of two protein-recognizing 19S regulatory units and a 20S proteolytic core. The 20S core particle includes three protein-specific catalytic subunits, i.e., β1, β2, and β5, that are referred to as caspase-like, trypsin-like, and chymotrypsin-like, respectively [140]. The proteasome plays a key role in controlling the levels of disparate regulatory proteins and prevents the accumulation of mutant or damaged proteins in the cell. Moreover, proteasome activity exerts influence on the other cellular process, including proliferation, cell death, signal transduction, immune response or metabolism [141]. The malignant plasma cells, in particular, are degraded by the ubiquitin-proteasome pathway; thus, they are susceptible to the action of proteasome inhibitors (PI), which are a mainstay in the treatment of multiple myeloma (MM). Carfilzomib and ixazomib citrate are highly effective in the second-generation of PI registered in combination with dexamethasone and immunomodulatory drugs (lenalidomide) for patients with MM, who have received at least one previous therapy [142,143]. Carfilzomib is also used as monotherapy and in combination with dexamethasone and monoclonal antibody (daratumumab) [143]. Ixazomib citrate is the first orally administrated PI which contains a citrate-protected boric acid group. The drug hydrolyzes under physiological conditions (e.g., in the gastrointestinal tract and plasma) to its biologically active metabolite, i.e., ixazomib. The mechanism of action of carfilzomib and ixazomib is forming irreversible and reversible, respectively, adducts with a 20S unit of 26S proteasome, mainly in the chymotrypsin-like site. At high concentrations, both drugs show additional inhibitory effects on the trypsin-like and caspase-like sites [144]. Due to proteasome inhibition, the drugs cause an increase in ubiquitinated proteins in MM cells, resulting in activation of an unfolded protein stress response and, ultimately, cell-cycle arrest and apoptosis. Programmed cell death induced by carfilzomib is associated with the activation of caspase pathways and c-Jun-N-terminal kinase (JNK). Moreover, carfilzomib promotes mitochondrial membrane depolarization and the release of cytochrome c [145]. The anti-myeloma effect induced by ixazomib is mainly related to the blocking of the NF-κB pathway, which reduces the release of cytokines secreted by bone marrow stream cells, important for the proliferation of MM cells. This drug also causes activation of caspase pathways, an increase in cleavage of poly (ADPribose) polymerase, the overexpression of a tumor suppressor gene miR33b, and reduced angiogenesis by blocking expression of the VEGFR-2 and platelet endothelial cell adhesion molecule (PECAM). Furthermore, the use of ixazomib leads to upregulation of signaling pathways such as p53-p21, p53-NOXA-PUMA, and E2 factor (E2F) [146].
2.5.4. Tubulin Inhibitor
Tubulin is a protein which is essential for the normal polymerization of mitotic spindle microtubules, which play a pivotal role in cellular processes, including chromosome segregation during mitosis or the intracellular transport of organelles. Microtubules are also components of the cytoskeleton, determining cell shape. Structurally, they can be defined as cylindrical protofilaments composed of α/β-tubulin dimers. Microtubule-targeting agents (MTAs), e.g., vincristine (VCR), bind to tubulin proteins and, thus, inhibit their polymerization into microtubules. The effects of VCR on tubulin polymerization are dependent on the concentration of the dose. At low concentrations, VCR inhibits the formation of microtubules through the direct binding at α/β-tubulin dimers and indirect cross-linking of proteins which are responsible for the stabilization of protofilaments. This inhibition prevents chromosome segregation and results in arrest mitosis at the metaphase. At higher concentrations, VCR promotes the disruption and depolymerization of microtubules via binding tubules in a different conformation, leading to the formation of spiral aggregates and highly structured crystals. The alterations of the structure of the microtubules cause mitotic spindle disintegration and the accumulation of chromosomes, thus the activation of apoptotic pathways and cell death. Another mechanism of action of VCR includes the inhibition of axon transport, secretion processes, and impairment of platelet functions [147]. Vincristine is a vinca alkaloid that received initial approval in 1963, and, since then, has been used to treat different cancer types including leukemia, Hodgkin’s lymphoma, lung cancer, and breast cancer [14]. Vincristine sulfate encapsulated in sphingomyelin and cholesterol-based nanoparticles (VSLI; MARQIBO) is a novel formulation of VCR to treat adults with a rare type of leukemia called Philadelphia chromosome-negative (Ph-) acute lymphoblastic leukemia (ALL). VSLI consists of a uniquely slow-release liposomal system suited to deliver and intensify the dose of VCR. The liposomal carrier prolongs the circulation time of the drug, optimizes delivery to target tissues, and increases the therapeutic index. The long exposure time of cancer cells to VSLI leads to its therapeutic efficacy compared to standard VCR. These characteristics result in overcoming the dose-related neurotoxicity and pharmacokinetic limitations of free VCR [148].
2.6. Protein Translation Inhibitors as Anticancer Agents
Protein synthesis is the fundamental cellular process for living organisms, which can be defined as the transmitting of genetic information from DNA to proteins. First, the genetic code in the DNA is transcribed into messenger RNA (mRNA) in the nucleus. Next, information carried by the nucleotide sequence of mRNA is translated into the amino acid sequence of a polypeptide chain. The decoding of mRNA is performed after the transport of mRNA into the cytoplasm, where it becomes bound to the ribosome, and the placement of subsequent amino acid attached to transfer RNA (tRNA), i.e., aminoacyl-tRNA, in the A-site cleft of the ribosome. Finally, ribozyme, an integral part of the ribosome, catalyzes the formation of a peptide bond between amino acids [160]. Since components of protein translation machinery are often deregulated in cancer, inhibitors of protein synthesis are a promising group of anticancer drugs. Omacetaxine mepesuccinate is the first analog of cephalotaxine that has been approved by FDA for patients with chronic myeloid leukemia (CML) and who have demonstrated resistance or intolerance to two or more tyrosine kinase inhibitors (TKIs). The approval allows the home administration of subcutaneous injection using syringes prepared by a professional healthcare practicioner [161]. The drug is semisynthetic purified homoharringtonine, a natural alkaloid originally isolated from various Cephalotaxus species. The synthesis of omacetaxine mepesuccinate is based on direct esterification of cephalotaxine isolated from Cephalotaxus conifers [162]. Omacetaxine mepesuccinate acts by inhibiting protein synthesis during the G1 and G2 phases of the cell cycle (Figure 7). Its mechanism of its action is distinct from TKIs and includes binding to the A-site cleft of ribosomes that prevents the binding of incoming aminoacyl-tRNA to the ribosomal acceptor site and blocks peptide bond formation. In vitro studies have revealed that omacetaxine mepesuccinate selectively reduces levels of pro-oncogenic or anti-apoptotic proteins essential for leukemia cell survival, including both native and mutated forms of BCR-ABL and MCL1. The use of this drug leads to the degradation of BCR-ABL proteins by inhibiting heat shock protein 90 in a dose-dependent manner. The mutant BCR-ABL was more sensitive to inhibition by treatment with the drug compared to the native form of BCR-ABL. The results from in vitro studies have also demonstrated apoptotic activity of omacetaxine mepesuccinate related to activating caspase-9, caspase-8, caspase-3, and poly (ADP-ribose) polymerase (PARP) [163]. The overall characteristic of the drug is presented in Table 7.
Figure 7.

Schematic representation of major mechanisms of action for omacetaxine mepesuccinate. BCR-ABL: BCR-ABL fusion protein. MCL1: myeloid cell leukemia 1. mRNA: messenger ribonucleic acid. tRNA: transfer ribonucleic acid. Hsp90: heat shock protein 90. Created with BioRender.com.
Table 7.
Features of the protein translation inhibitor approved by the Food and Drug Administration (FDA) from 2011 to 2021.
| No. | Generic Name of Drug | Brand Name and Company |
First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Omacetaxine mepesuccinate | SYNRIBO Teva Pharmaceuticals, Tel Aviv, Israel |
FDA: 26 October 2012 EMA: Not approved |
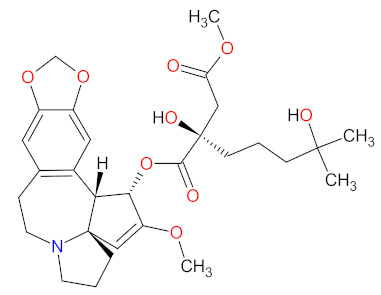
|
Protein level | Subcutaneous injection | Chronic Myelogenous Leukemia |
Thrombocytopenia, anemia, diarrhea, pyrexia, fatigue, nausea, neutropenia, injection site reaction, infection, lymphopenia |
[164,165] |
2.7. Purine Antagonist as Anticancer Agent
Purine antagonists, such as mercaptopurine (6-MP), belong to the class of drugs known as antimetabolites, which inhibit nucleic acid metabolism and are widely used in cancer chemotherapy. Mercaptopurine is a purine analog evaluated for the treatment of acute leukemias that act as antagonists to the endogenous purines [166]. It is a prodrug activated by hypoxanthine-guanine phosphoribosyltransferase (HGPRT), which converts it to thioinosine monophosphate (TIMP). TIMP is further methylated to 6-methylthioinosine monophosphate (Me-TIMP), a potent inhibitor of the purine de novo synthesis (Figure 8) [167]. Me-TIMP also reduces the amount of natural purines in lymphoblast, which results in the inhibition of DNA synthesis and early cytotoxicity in the S phase [168]. In addition, TIMP is metabolized to deoxy-6-thioguanine triphosphate (6-dTGTP) and 6-thioguanine triphosphate (6-TGTP), which are incorporated into DNA and RNA, respectively. This process causes inhibition of nucleotide and protein synthesis, thus inducing lymphocyte proliferation. 6-TGTP also inhibits Rac1, leading to apoptosis of T-lymphocytes via modulation of the Vav-Rac1 signaling pathway [169]. Mercaptopurine was approved for medical use in 1953 and was commercially available only as a 50 mg tablet until 2014, when the FDA approved a new liquid formulation of 6-MP (PURIXAN) [170]. PURIXAN is a 20 mg/mL oral suspension indicated for the treatment of patients with acute lymphoblastic leukemia (ALL) as part of a combination regimen. This drug provides flexibility and precision in terms of dosing based on the individual patient’s dosing needs. Especially, PURIXAN improves dose adjustments and accurately delivers the desired dose for children with a wide range of weights. Moreover, suspension as a route of administration is more convenient than tablets [171]. The overall features of this drug are presented in Table 8.
Figure 8.

Scheme of metabolism and mode of action of mercaptopurine (6-MP). SLC: solute carrier. 6-MP: mercaptopurine. XO: xanthine oxidase. TPMT: thiopurine methyltransferase. Me-MP: methylmercaptopurine. HPGRT: hypoxanthine-guanine phosphoribosyltransferase. TIMP: thioinosine monophosphate. Me-TIMP: methyl thioinosine monophosphate. IMPDH: inosine monophosphate dehydrogenase. 6-TGMP: 6-thioguanosine monophosphate. 6-TGDP: 6-thioguanosine diphosphate. 6-dTGTP: deoxy-6-thioguanine triphosphate. 6-TGTP: 6-thioguanosine triphosphate. Rac1: ras-related C3 botulinum toxin substrate 1. DNA: deoxyribonucleic acid. RNA: ribonucleic acid. Created with BioRender.com based on information in [167].
Table 8.
Features of the purine antagonist approved by the Food and Drug Administration (FDA) from 2011 to 2021.
| No. | Generic Name of Drug | Brand Name and Company |
First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Mercaptopurine | PURIXAN Nova Laboratories, Ltd., Leicester, UK |
FDA: 28 April 2014 EMA: 9 March 2012 |
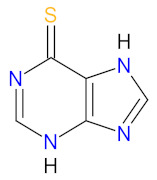
|
DNA 1 | Oral | Acute Lymphoblastic Leukemia |
Anemia, neutropenia, thrombocytopenia | [170,172,173] |
1 DNA: deoxyribonucleic acid.
2.8. Immunomodulators as Anticancer Agents
Immunomodulatory drugs (IMiDs) are a group of compounds that are structural and functional analogs of thalidomide, showing immunomodulatory, anti-angiogenic, and anti-inflammatory properties. IMiDs are currently under investigation and demonstrate activity against various hematologic malignancies, including multiple myeloma (MM), acute myeloid leukemia (AML), chronic lymphocytic leukemia (CLL), and myelofibrosis (MF), or solid tumors including lung, breast, renal, and colon cancer [174]. Pomalidomide, the newest IMiD characterized in Table 9, received approval in February 2013 as treatment for patients with multiple myeloma (MM) whose disease progressed after previous lines of therapy, including lenalidomide and bortezomib. On May 2020, the indication for treatment with pomalidomide was extended for patients with AIDS-related Kaposi sarcoma resistant to highly active antiretroviral therapy (HAART), or patients with HIV-negative Kaposi sarcoma [175]. Pomalidomide’s mechanism of action includes various molecular and cellular elements within the bone marrow microenvironment or cytokine production, as well as immunomodulation and direct antimyeloma activity. Pomalidomide causes a reduction in different proinflammatory cytokines, including tumor necrosis factor-alpha (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6). Furthermore, the anti-inflammatory effects of the drug are related to the inhibition of cyclooxygenase-2 (COX-2) and prostaglandin production by monocytes. Immunomodulatory mechanisms include enhanced T-cell proliferation leading to the increased production of interleukin-2 (IL-2) and interferon-γ (IFN-γ), in addition to amplified natural killer (NK) cells activity and antibody-dependent cell-mediated cytotoxicity (ADCC) in MM cells [176]. Pomalidomide also reduces the IL-2-mediated generation of T regulatory cells (FOXP3+CTLA-4+CD4+CD25high cells), which are important controllers of the immune response and suppressors of anti-tumor immunity [177]. Moreover, the drug shows antiangiogenic activity by inhibiting the secretion of vascular endothelial growth factor (VEGF) and hypoxia-inducible factor 1 alpha (HIF-1α). Pomalidomide achieves its antimyeloma action by causing both cell-cycle arrest and apoptosis. Specifically, the drug induces cell-cycle arrest via a Lysine-specific demethylase 1 (LSD1)-mediated epigenetic mechanism, resulting in p21 (WAF-1) activation independently of p53 signaling, apoptosis by activation of the caspase-8 pathway, increased sensitivity to Fas-mediated cell death, suppression of nuclear factor kappa-B (NF-κB) transcription, and inhibition of apoptosis 2 (IAP-2) [176]. The molecular target for pomalidomide is a highly conserved protein, cereblon (CRBN). The drug binds endogenous CRBN, resulting in the inhibition of its autoubiquitination and subsequent suppression of interferon regulatory factor 4 (IRF4), which is critical for MM cell survival [178]. Finally, clinical trials have proved that pomalidomide is the most potent IMiD [176].
Table 9.
Features of the immunomodulatory drug approved by the Food and Drug Administration (FDA) from 2011 to 2021.
| No. | Generic Name of Drug | Brand Name and Company |
First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Pomalidomide | POMALYST Bristol-Myers Squibb Company, New York, NY, USA |
FDA: 8 February 2013 EMA: 5 August 2013 |
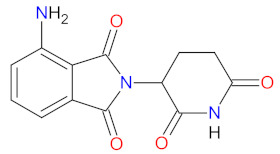
|
CRBN 1 | Oral | Multiple Myeloma, Kaposi’s Sarcoma |
Neutropenia, thrombocytopenia, anemia, fatigue |
[179,180] |
1 CRBN: protein cereblon.
2.9. Fixed-Dose Combination Drugs as Anticancer Agents
A fixed-dose combination (FDC) drug is a combination of two or more active agents within a single form of pharmaceutical administration. FDC drugs are a promising treatment strategy for anticancer therapy, due to that all of their ingredients reach a target cancer cell simultaneously and act by different mechanisms. The disruption of the cell cycle in different phases and the inhibition of various survival pathways results in better suppression of cancer chemoresistance, thereby synergistically inducing cellular apoptosis and inhibiting the growth of cancerous tissue. FDC drugs improve therapeutic efficiency and overcome potential adverse effects of treatment, as well as reduce the cost to patients compared to single drug therapies [181]. FDC drugs approved by the FDA since 2011 for treatment of certain hematological malignancies are shown in Table 10.
Table 10.
Features of the dual-drugs approved by the Food and Drug Administration (FDA) from 2011 to 2021. The order of drugs is tabulated in order of most recent to oldest registration date.
| No. | Generic Name of Drug | Brand Name and Company |
First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Decitabine | INQOVI Astex Pharmaceuticals, Taiho Oncology, and Otsuka Pharmaceutical, Tokyo, Japan |
FDA: 7 July 2020 EMA: Not approved |
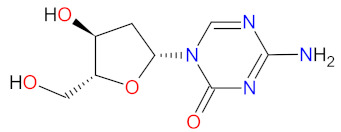
|
DNMTs 1 | Oral | Myelodysplastic Syndrome | Fatigue, constipation, hemorrhage, myalgia, nausea, arthralgia, pneumonia, sepsis, decreased leukocytes, decreased platelet count, decreased neutrophil count |
[193] |
| Cedazuridine |
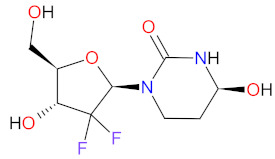
|
CDA 2 | |||||||
| 2 | Cytarabine | VYXEOS Jazz Pharmaceuticals plc, Dublin, Ireland |
FDA: 3 August 2017 EMA: 23 August 2018 |
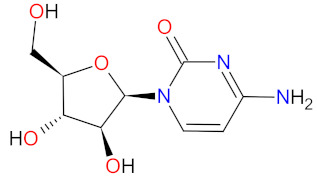
|
DNA 3 | Intravenous | Acute Myeloid Leukemia | Hemorrhagic events, febrile neutropenia, rash, edema, nausea, mucositis, diarrhea, constipation, musculoskeletal pain, fatigue, abdominal pain, dyspnea, headache, cough, decreased appetite, arrhythmia, pneumonia, bacteremia, chills, sleep disorders, vomiting |
[194,195] |
| Daunorubicin |
|
TOP2 4 |
1 DNMTs: DNA methyltransferases. 2 CDA: cytidine deaminase. 3 DNA: deoxyribonucleic acid. 4 TOP2: topoisomerase II.
VYXEOS is the first dual-drug liposomal encapsulation product registered by the FDA. It is composed of cytarabine and daunorubicin, in a molar ratio of 5:1, which are entrapped in liposomes. The liposome membrane, which contained distearoyl phosphatidylcholine, distearoyl phosphatidylglycerol, and cholesterol in a molar ratio of 7:2:1, is degraded upon internalization of the drug into target cells. Cytarabine and daunorubicin are then released within the intracellular environment [182]. Cytarabine, a pyrimidine nucleoside analog originally approved in 1969, is incorporated into DNA resulting in inhibition of DNA synthesis in the S-phase of the cell cycle and subsequent apoptotic cell death [183,184]. Daunorubicin, an anthracycline antibiotic approved for medical use in 1979, induces inhibition of DNA synthesis by the intercalation and blocking of activity of topoisomerase II [185,186]. This drug also generates free radicals, leading to DNA damage. Both chemotherapeutic agents in the indicated proportion have synergistic effects to induce leukemia cell death. VYXEOS has demonstrated a clinically significant improvement in overall survival compared with conventional cytarabine plus daunorubicin chemotherapy (7 + 3 regimen) in adults with newly diagnosed secondary acute myeloid leukemia (sAML) and therapy-related acute myeloid leukemia (t-AML), as well as AML with myelodysplasia-related changes (AML-MRC) [187,188]. By the end of 2020, the drug received approval as a treatment for adult patients with newly diagnosed t-AML or AML-MRC [189].
INQOVI is a fixed-dose combination of decitabine and cedazuridine. In 2006, decitabine was approved as a single drug that had to be administered by intravenous injection because of its degradation by cytidine deaminase in the gastrointestinal tract and liver [190]. INQOVI is designed to enable oral delivery of decitabine, due to the inhibition of cytidine deaminase activity by cedazuridine. The mechanism of action of decitabine, a cytosine analog, includes incorporation into replicating DNA and the inactivation of DNA methyltransferases (DNMTs). The subsequent hypomethylation of DNA is responsible for cancer cell death through a variety of mechanisms [191]. INQOVI is indicated for the treatment of adult patients with newly diagnosed and secondary myelodysplastic syndromes (MDS), a group of bone marrow diseases that change the production of functional blood cells, including refractory anemia, refractory anemia with ringed sideroblasts, refractory anemia with excess blasts, and chronic myelomonocytic leukemia (CMML) [192].
3. Macromolecule Anticancer Drugs
3.1. Monoclonal Antibodies as Anticancer Agents
Monoclonal antibodies (mAbs) represent a clonal version of a specified antibody isotype that can bind to antigens found on the surface of cancer cells. The activity of mAbs leads to normalizing growth rates and induces cancer cells’ death via various mechanisms (Figure 9). Furthermore, mAbs target the cancer microenvironment and inhibit processes such as angiogenesis. Monoclonal antibodies are composed of two heavy and two light chains, which form three functional protein domains: two identical fragments for antigen binding (Fab regions) and the constant or crystallizable fragment (Fc region). Depending on the type of Fab regions, mAbs can be divided into four classes: murine, chimeric, humanized, and human [196]. Immunoglobulin-type (IgG-type) monoclonal antibodies, an important class of therapeutic proteins which are the most frequently used for cancer immunotherapy, are classified into immunoglobulin G1 (IgG1), immunoglobulin G2 (IgG2), immunoglobulin G3 (IgG3), and immunoglobulin G4 (IgG4) [197]. Monoclonal antibodies approved by the FDA in the last decade for the treatment of hematological malignancies target cluster of differentiation 19 (CD19), cluster of differentiation 20 (CD20), cluster of differentiation 38 (CD38), signaling lymphocyte activation molecule family member 7 (SLAMF7), and programmed death receptor-1 (PD-1). The registered mAb drugs are summarized in Table 11.
Figure 9.

Mode of action of monoclonal antibodies. ADCC: antibody-dependent cellular cytotoxicity. NK: natural killer. ADCP: antibody-dependent cellular phagocytosis. CDC: complement-dependent cytotoxicity. mAb: monoclonal antibody. C1q: complement component 1q. PD-1: programmed death-1 protein. PD-L1: programmed death ligand-1. PD-1 Ab: monoclonal antibody directed against PD-1. TCR: T-cell receptor. MHC: major histocompatibility complex. Created with BioRender.com based on information in [197].
Table 11.
Features of the monoclonal antibody drugs approved by the Food and Drug Administration (FDA) from 2011 to 2021. The order of drugs is tabulated in order of most recent to oldest registration date.
| No. | Generic Name of Drug | Brand Name and Company |
First FDA/EMA Approved Date | Class | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Rituximab-arrx | RIABNI Amgen Inc., Sauzend Oaks, CA, USA |
FDA: 17 December 2020 EMA: Not approved |
Chimeric mouse/human immunoglobulin G1 kappa (IgG1 κ) monoclonal antibody | CD20 1 | Intravenous | Non-Hodgkin’s Lymphoma, Chronic Lymphocytic Leukemia | Infusion-related reactions, fever, lymphopenia, chills, infection, asthenia, neutropenia | [207] |
| 2 | Tafasitamab-cxix | MONJUVI MorphoSys AG, Planegg, Germany |
FDA: 31 July 2020 EMA: 26 August 2021 |
Humanized immunoglobulin G1/2 (IgG1/2) hybrid monoclonal antibody | CD19 2 | Intravenous | Diffuse Large B-Cell Lymphoma | Neutropenia, fatigue, anemia, diarrhea, thrombocytopenia, cough, pyrexia, peripheral edema, respiratory tract infection, decreased appetite |
[199,225] |
| 3 | Daratumumab and hyaluronidase-fihj | DARZALEX FASPRO Janssen Pharmaceuticals, Inc., Belce, Belgium |
FDA: 1 May 2020 EMA:Not approved |
Humanized immunoglobulin G1 kappa (IgG1 κ) monoclonal antibody and an endoglycosidase | CD38 3 | Intravenous | Multiple Myeloma | Upper respiratory tracts infection, constipation, nausea, fatigue, pyrexia, peripheral sensory neuropathy, diarrhea, cough, insomnia, vomiting, back pain, muscle spasms, pneumonia, dyspnea | [215,226] |
| 4 | Isatuximab | SARCLISA Sanofi, Paris, France | FDA: 2 March 2020 EMA: 30 May 2020 |
Chimeric mouse/human immunoglobulin G1 kappa (IgG1 κ) monoclonal antibody | CD38 3 | Intravenous | Multiple Myeloma | Infusion reactions, upper respiratory tract infections, bronchitis, pneumonia | [227,228] |
| 5 | Rituximab-pvvr | RUXIENCE Pfizer Inc., New York, NY, USA |
FDA: 23 July 2019 EMA: 1 April 2020 |
Chimeric mouse/human immunoglobulin G1 kappa (IgG1 κ) monoclonal antibody | CD20 1 | Intravenous | Non-Hodgkin’s Lymphoma, Chronic Lymphocytic Leukemia | Infusion-related reactions, fever, lymphopenia, chills, infection, asthenia, neutropenia | [206,229] |
| 6 | Rituximab-abbs | TRUXIMA Celltrion, Inc., Incheon, Korea | FDA: 28 November 2018 EMA: 17 February 2017 |
Chimeric mouse/human immunoglobulin G1 kappa (IgG1 κ) monoclonal antibody | CD20 1 | Intravenous | Non-Hodgkin’s Lymphoma | Infusion-related reactions, fever, lymphopenia, chills, infection, asthenia | [230,231] |
| 7 | Mogamulizumab-kpkc | POTELIGEO Kyowa Kirin, Inc., Bedminster, NJ, USA | FDA: 8 August 2018 EMA: 22 November 2018 |
Humanized immunoglobulin G1 kappa (IgG1 κ) monoclonal antibody | CCR4 4 | Intravenous | Mycosis Fungoides, Sézary syndrome | Rash, infusion related reactions, fatigue, diarrhea, musculoskeletal pain, and upper respiratory tract infection | [232,233] |
| 8 | Elotuzumab | EMPLICITI Bristol-Myers Squibb Company and AbbVie, Lake Bluff, IL, USA | FDA: 30 November 2015 EMA: 11 May 2016 |
Humanized immunoglobulin G1 (IgG1) monoclonal antibody | SLAMF7 5 | Intravenous | Multiple Myeloma | Fatigue, diarrhea, pyrexia, constipation, cough, peripheral neuropathy, nasopharyngitis, upper respiratory tract infection, decreased appetite, pneumonia | [234,235] |
| 9 | Daratumumab | DARZALEX Janssen Biotech, Inc., Horsham, PA, USA | FDA: 16 November 2015 EMA: 20 May 2016 |
Humanized immunoglobulin G1 kappa (IgG1 κ) monoclonal antibody | CD38 3 | Intravenous | Multiple Myeloma | Infusion-related reactions, lymphopenia, neutropenia, thrombocytopenia, anemia | [236,237] |
| 10 | Nivolumab | OPDIVO Bristol-Myers Squibb Company, New York, NY, USA |
FDA: 22 December 2014 EMA: 19 June 2015 |
Human immunoglobulin G4 kappa (IgG4 κ) monoclonal antibody | PD-1 6 | Intravenous | Hodgkin’s Lymphoma | Fatigue, upper respiratory tract infection, pyrexia, diarrhea, cough | [238,239,240] |
| 11 | Pembrolizumab | KEYTRUDA Merck, Kenilworth, NJ, USA | FDA: 4 September 2014 EMA: 17 July 2015 |
Humanized immunoglobulin G4 (IgG4) monoclonal antibody | PD-1 6 | Intravenous | Primary Mediastinal Large B-Cell Lymphoma, Hodgkin’s Lymphoma | Fatigue, cough, pruritus, nausea, rash, decreased appetite, constipation, arthralgia, diarrhea, anemia, hyperglycemia, hyponatremia, hypoalbuminemia, hypertriglyceridemia,, hypocalcaemia, elevated aspartate transaminase | [241,242,243,244] |
| 12 | Obinutuzumab | GAZYVA Genentech, Inc., South San Francisco, CA, USA | FDA: 1 November 2013 EMA: 23 July 2014. |
Humanized immunoglobulin G1 (IgG1) monoclonal antibody | CD20 1 | Intravenous | Chronic Lymphocytic Leukemia, Follicular Lymphoma | Infusion reactions, neutropenia | [245,246] |
1 CD20: cluster of differentiation 20. 2 CD19: cluster of differentiation 19. 3 CD38: Cluster of differentiation 38. 4 CCR4: CC chemokine receptor 4. 5 SLAMF7: Signaling Lymphocyte Activation Molecule Family member 7. 6 PD-1: programmed death receptor-1.
3.1.1. Anti-CD19 Monoclonal Antibody
Cluster of differentiation 19 (CD19) is a glycoprotein with a single transmembrane domain specifically expressed in B lymphocytes. CD19 is a cell surface response regulator that modulates B cell signaling. It plays a role in the development and immunoglobulin-induced activation of B cells [198]. Tafasitamab-cxix is an Fc-modified humanized monoclonal antibody that binds to the CD19 antigen expressed on the surface of B-cell malignancies, including diffuse large B-cell lymphoma (DLBCL). The drug induces B-cell lysis via apoptosis and immune effector mechanisms, i.e., antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP). In the structure of the Fc region of tafasitamab-cxix, two amino acids were substituted, resulting in increased Fcγ receptor affinity compared with a non-engineered anti-CD19 mAbs [199]. The drug is used in combination with lenalidomide as a treatment for adult patients with relapsed or refractory DLBCL, who are not eligible for autologous stem cell transplant (ASCT) [200].
3.1.2. Anti-CD20 Monoclonal Antibodies
Cluster of differentiation 20 (CD20) is a molecule with multiple membranes spanning domains expressed exclusively by B lymphocytes. The engagement of CD20 on antigen-presenting cells leads to the regulation of transmembrane transport of Ca2+ and cell-cycle progression during B cell activation and proliferation. Many hematological malignancies, including non-Hodgkin’s lymphomas, have been shown to express CD20; thus, anti-CD20 mAbs are an effective anticancer immunotherapy. Anti-CD20 mAbs bind to different epitopes on CD20, leading to the inhibition of B lymphocyte progression into the S/G2+M stages of the cell cycle [201]. There are two types of anti-CD20 mAbs based on different mechanisms causing malignant B cells death. Type I anti-CD20 antibodies, such as rituximab or its biosimilars rituximab-abbs, rituximab-pvvr, or rituximab-arrx, redistribute CD20 molecules into large insoluble lipid microdomains (lipid rafts) and exert potent complement-dependent cytotoxicity (CDC) activity. In contrast, type II antibodies such as obinutuzumab induce less accumulation of CD20 in lipid rafts and show weak CDC activity [202]. Obinutuzumab shows an increased ability to bind and recruit effector cells, thereby antibody-dependent cellular cytotoxicity (ADCC) activity, due to the glycoengineered the Fc region [203]. Obinutuzumab was more effective than rituximab as either first- or second-line therapy in mouse xenograft models of human lymphoma [204]. Rituximab-abbs received approval for the treatment of non-Hodgkin’s lymphoma (NHL), to be used as a single agent or in combination with chemotherapy, while rituximab-pvvr and rituximab-arrx were approved as single agents or in combination with first line chemotherapy to treat patients with NHL and chronic lymphocytic leukemia (CLL) [205,206,207]. Obinutuzumab was approved in combination with chlorambucil as a treatment for CLL and combination with bendamustine as a treatment for patients with follicular lymphoma (a type of non-Hodgkin lymphoma). This drug is also registered in combination with ibrutinib as a first non-chemotherapy treatment of patients with CLL [208].
3.1.3. Anti-CD38 Monoclonal Antibodies
A cluster of differentiation 38 (CD38) is a transmembrane glycoprotein expressed extensively on B and T lymphocytes. It shows an aplysia adenosine diphosphate (ADP)-ribosyl cyclase activity, and the ability to regulate intracellular Ca2+ release [209]. Anti-CD38 mAbs represent a promising therapy for patients with multiple myeloma (MM) since CD38 is expressed at relatively low levels on myeloid and lymphoid cells, and at high levels in various malignant plasma cells, including MM. Daratumumab and isatuximab, FDA-approved anti-CD38 antibodies, kill MM cancer cells via Fc-dependent immune effector mechanisms, i.e., complement-dependent cytotoxicity (CDC), antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), and apoptosis. Both drugs also show indirect anticancer activity consisting of the suppression of CD38+ T regulatory cells, thus restoring immune effector functions against MM [210,211]. On the other hand, daratumumab and isatuximab target a different amino acid sequence in CD38 molecules and exhibit several mechanistic differences. Daratumumab immediately induces redistribution of CD38 on the surface of MM cells and the formation of polar aggregates leading to the release of CD38 in microvesicles, while isatuximab does not result in a decrease in the CD38 level. Isatuximab induces direct apoptosis through the caspase-dependent apoptotic pathway and the lysosome-mediated cell death pathway, whereas daratumumab induces apoptosis only in the presence of cross-linking agents. Isatuximab strongly inhibits CD38 enzymatic activity, acting as an allosteric antagonist, while daratumumab, under the same experimental conditions, shows a more limited inhibition. Finally, isatuximab can regulate the cancer immunosuppressive environment of the bone marrow niche in MM patients via the modulation of adenosine levels [212]. Daratumumab was approved as monotherapy and in combination with various chemotherapy regiments (i.e., lenalidomide and dexamethasone, bortezomib, melphalan and prednisone, bortezomib, thalidomide and dexamethasone, bortezomib and dexamethasone, pomalidomide and dexamethasone, or carfilzomib and dexamethasone) to treat both relapsed or refractory multiple myeloma (RRMM) and newly diagnosed multiple myeloma (NDMM) [213]. Isatuximab was registered indicated with pomalidomide and dexamethasone for the treatment of adult patients with MM who have received at least two prior therapies and in combination with carfilzomib and dexamethasone to treat adult patients with RRMM who have received one to three prior lines of therapy [214]. DARZALEX FASPRO is a new subcutaneous formulation of daratumumab (DARZALEX) which is co-formulated with recombinant human hyaluronidase. The drug is administered in significantly less time (i.e., three to five minutes) compared to the hours of intravenous infusion required for DARZALEX. Hyaluronidase increases the permeability of the subcutaneous tissue. In the doses administered, hyaluronidase in DARZALEX FASPRO acts locally, and the effects of its action are reversible. The drug was approved as a monotherapy and in combination with four chemotherapy regimens across multiple myeloma, including newly diagnosed, transplant-ineligible patients as well as relapsed or refractory patients [215].
3.1.4. Anti-SLAMF7 Monoclonal Antibody
Signaling lymphocytic activation molecule family member 7 (SLAMF7, also known as CS1, CD319, and CRACC) is a transmembrane receptor that is expressed on hematopoietic cells such as natural killer (NK) cells, B cells, T cells, NK-T cells, dendritic cells, and monocytes. The receptor modulates the function of immune cells through immune-receptor tyrosine-based switch motifs, which mediates interaction with Ewing’s sarcoma-associated transcript 2 (EAT-2). SLAMF7 has both activating and inhibitory functions in NK cells, induces proliferation and the production of autocrine cytokines in B cells, and inhibits the production of proinflammatory cytokines by activated monocytes. Since high-level expression of SLAMF7 is retained in multiple myeloma (MM) cells, implicated in the uncontrolled proliferation of MM cells, the receptor is a promising target for immunotherapy [216]. Elotuzumab is a humanized IgG1 monoclonal antibody that binds to SLAMF7 on MM cells and natural killer (NK) cells. Elotuzumab exerts antimyeloma effects via direct NK cell activation and NK-mediated antibody-dependent cellular cytotoxicity (ADCC). The drug also prevents the adhesion of MM cells to bone marrow stromal cells, which may disrupt the growth and survival of MM cells [217]. Elotuzumab has no significant clinical efficacy as a single agent; however, it is effective in combination with lenalidomide and dexamethasone or pomalidomide and dexamethasone, and, thus, received approval in combination with these two chemotherapy regimens for the treatment of adult patients with relapsed or refractory multiple myeloma (RRMM) who have received prior therapies [218].
3.1.5. Anti-PD-1 Monoclonal Antibodies
Programmed death-1 (PD-1) is a cell surface protein belonging to the CD28 family, expressed on activated T cells, B cells, natural killer (NK) cells, and monocytes. Interaction between PD-1 and its ligands PD ligand-1 (PD-L1) and PD ligand-2 (PD-L2) plays an important role in the activation of inhibitory kinases involved in T-cell proliferation, adhesion, or cytokine production and secretion. This interaction causes the suppression of T cells’ response upon antigen exposure, which allows cancer cells to evade immune surveillance. PD-1 ligands are overexpressed in hematologic malignancies, such as multiple myeloma, lymphoma, and various leukemia types, which results in the inhibition of T-cell–mediated immune response in these diseases. Blocking the PD-1/PD-L1 signaling pathway with monoclonal antibodies (mAbs) has an important role in cancer treatment because it restores the efficacy of cancer-specific T cells, allowing them to recognize and counter cancer cells [219]. Nivolumab and pembrolizumab are the first two FDA-approved mAbs that target PD-1. These drugs selectively impede the interaction between PD-1 and PD-L1, and lead to the release of T cells from pathological immune suppression [220]. Nivolumab received accelerated approval for patients with classical Hodgkin lymphoma that has relapsed or progressed after autologous hematopoietic stem cell transplantation (HSCT) and posttransplantation treatment with brentuximab vedotin. Pembrolizumab was registered as monotherapy to treat pediatric and adult patients with relapsed or refractory classical Hodgkin lymphoma (cHL). Moreover, these drugs have earned a series of other FDA approvals, including for melanoma, non-small cell lung cancer (NSCLC), renal cell carcinoma, head and neck cancer, urothelial carcinoma, hepatocellular carcinoma, esophageal carcinoma, and gastric cancer [221,222].
3.1.6. Anti-CCR4 Monoclonal Antibody
C-C chemokine receptor 4 (CCR4) is a G protein-coupled receptor for small molecular weight chemotactic cytokines, i.e., chemokines, that is expressed on the surface of regulatory T cells (Treg) and type 2 T helper cells (Th2). CCR4 plays an important role in the migration of T cells to the skin during the response to skin inflammation. This receptor is also overexpressed in some subtypes of cutaneous T-cell lymphoma (CTCL), including mycosis fungoides (MF) and Sézary syndrome (SS) [223]. Mogamulizumab-kpkc is a first-in-class defucosylated, humanized mAb against CCR4, which is used to treat adult patients with relapsed or refractory MF and SS who received at least one prior systemic therapy. The drug, by selectively binding to CCR4, marks T cells for depletion through antibody-dependent cellular cytotoxicity (ADCC) and destroys target cancer cells [224].
3.2. Bispecific Antibody as Anticancer Agent
A bispecific antibody (bsAb) is a single, hybrid protein containing two different antibody-based binding specificities, thus limiting complement activation that is responsible for side effects of treatment. The use of bsAb profoundly improves the selectivity and efficacy of therapies of various diseases, including hematological malignancies. Bispecific antibodies differ in structure, size, and method of production [247]. Blinatumomab is the first-in-class bispecific T-cell-engager antibody (BiTE) approved to treat children and adults with relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL). The drug binds to both CD19 and CD3, which are expressed on the surface of malignant B cells and endogenous T cells, respectively (Figure 10). Subsequently, a cytolytic synapse between B cells and T cells is formed, resulting in the activation of cytotoxic T cells. This induces apoptosis and lysis of the CD19 positive B cells via the release of perforin and granzymes from cytotoxic granules in the T cells. Furthermore, inflammatory cytokines (i.e., interferon γ (IFNγ) and tumor necrosis factor α (TNFα)) are released and T cell proliferation is promoted [248]. The summary of blinatumomab characteristics is included in Table 12.
Figure 10.

Blinatumomab mechanism of action. CD19: cluster of differentiation 19. CD3: cluster of differentiation 3. Gzm: granzymes. PFN: perforin. IFNγ: interferon gamma. TNFα: tumor necrosis factor alpha. Created with BioRender.com.
Table 12.
Features of the bispecific monoclonal antibody drug approved by the Food and Drug Administration (FDA) from 2011 to 2021.
| No. | Generic Name of Drug | Brand Name and Company |
First FDA/EMA Approved Date | Class | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Blinatumomab | BLINCYTO Amgen Inc., Thousand Oaks, CA, USA | FDA: 3 December 2014 EMA: 23 November 2015 |
Bispecific T-cell engaging (BiTE) antibody | CD19 1, CD3 2 |
Intravenous | Acute Lymphoblastic Leukemia | Pyrexia, headache, peripheral edema, nausea, febrile neutropenia, hypokalemia, constipation, febrile neutropenia, pneumonia, device-related infection, tremor, encephalopathy, infection, overdose, confusion, Staphylococcal bacteremia, headache |
[249,250] |
1 CD19: cluster of differentiation 19. 2 CD3: cluster of differentiation 3.
3.3. Antibody-Drug Conjugates as Anticancer Agents
Antibody-drug conjugate (ADC) is an immunoconjugate consisting of an antibody, a highly potent cytotoxic agent (known as the payload), and a chemical linker. The antibody moiety enables selective delivery of the payload to the target cancer cells by directly binding to specified cancer-associated antigen. Upon the internalization of the ADC-antigen complex into the cancer cell, ADC degradation occurs in the lysosome by the cleavage of the linker, which permits the efficient release of the payload. Finally, the released payload induces target cell death (Figure 11). ADCs confer to reduce off-target toxicity and increase the efficacy of the payloads in patients by limiting their exposure to normal tissues [251]. By the end of 2020, four ADCs had received FDA approval. Among them were antibody-drug conjugate targeting cluster of differentiation 22 (CD22), cluster of differentiation 30 (CD30), cluster of differentiation 79b (CD79b), and B-cell maturation antigen (BCMA), which are summarized in Table 13.
Figure 11.

General mechanism of action of antibody-drug conjugates. ADC: antibody-drug conjugate. Adapted from “Intracellular layout—endocytosis pathway,” by BioRender.com (2021). Retrieved from https://app.BioRender.com/biorender-templates (accessed on: 8 December 2021).
Table 13.
Features of the antibody-drug conjugates approved by the Food and Drug Administration (FDA) from 2011 to 2021. The order of drugs is tabulated in order of most recent to oldest registration date.
| No. | Generic Name of Drug | Brand Name and Company |
First FDA/EMA Approved Date | Structure | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Belantamab mafodotin-blmf | BLENREP GlaxoSmithKline, Brentford, England |
FDA: 5 August 2020 EMA: 25 August 2020 |
|
BCMA 1 | Intravenous | Multiple Myeloma | Ocular toxicity, thrombocytopenia, infusion-related reactions, gastrointestinal disorders, pyrexia, fatigue | [265,266] |
| 2 | Polatuzumab vedotin-piiq | POLIVY Genentech, Inc., South San Francisco, CA, USA | FDA: 10 June 2019 EMA: 16 January 2020 |
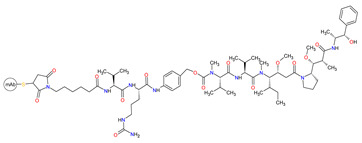
|
CD79b 2 | Intravenous | Diffuse Large B-Cell Lymphoma | Cytopenias | [262,267,268] |
| 3 | Inotuzumab ozogamicin | BESPONSA Pfizer Inc., New York, NY, USA | FDA: 17 August 2017 EMA: 29 June 2017 |
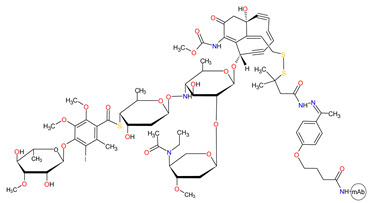
|
CD22 3 | Intravenous | Acute Lymphoblastic Leukemia | Cytopenias (including febrile neutropenia), infections, nausea, pyrexia, abnormal liver function and venoocclusive liver disease | [269,270] |
| 4 | Brentuximab vedotin | ADCETRIS Seattle Genetics, Inc., Bothell, WA, USA | FDA: 19 August 2011 EMA: 25 October 2012 |
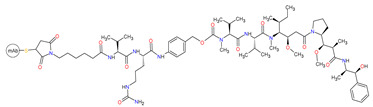
|
CD30 4 | Intravenous | Lymphoma, Hodgkin’s Lymphoma, Mycosis Fungoides | Neutropenia, anemia, peripheral sensory neuropathy, nausea, fatigue, constipation, diarrhea, vomiting, and pyrexia | [260,271,272] |
1 BCMA: B-cell maturation antigen. 2 CD79b: cluster of differentiation 79b. 3 CD22: cluster of differentiation 22. 4 CD30: cluster of differentiation 30.
3.3.1. Anti-CD22 Antibody-Drug Conjugate
A cluster of differentiation 22 (CD22) is a transmembrane sialoglycoprotein belonging to the immunoglobulin superfamily. CD22 expressed on mature B cells in a large variety of B-cell malignancies is involved in B cell activation through both a positive and negative regulation of B cell antigen receptor (BCR) signaling as well as in B cell migration and survival [252]. The expression of CD22 antigens is lineage-restricted, is generally not lost during neoplastic transformation, and is absent on normal hematopoietic stem cells, making them ideal targets for therapy of hematologic malignancies. Inotuzumab ozogamicin is a drug composed of a semisynthetic derivative of calecheamicin, a toxic product isolated from Micromonospora echinospora subsp. calichensis, and monoclonal IgG4 antibody targeting CD22. This antibody and calicheamicin derivative, i.e., N-acetyl-γ-calicheamicin dimethyl hydrazide (DMH), are covalently connected via a bifunctional acetophenone-butanoic acid linker. The linker forms an amide bond with lysine residues of the monoclonal antibody and a hydrazone linkage with N-acetyl-γ-calicheamicin DMH. The hydrolysis of the hydrazone functional group allows releasing of the cytotoxic component into the acidic environment of the lysosomes in cancer cells. Calecheamicin derivative is then activated by the reduction of a disulfide bond in the presence of intracellular glutathione (GSH), a cytoplasmic thiol cofactor. Calecheamicin shows a different mechanism of action from other tubulin-binding classes of cytotoxic agents. The enediyne moiety of calicheamicin, once reduced by cellular thiols, undergoes rearrangement, forming 1,4-benzenoid diradicals. Next, the diradical intermediates cause the abstraction of hydrogen atoms from the phosphodiester bonds of double-stranded DNA and their breaking. Subsequently, this leads to cell cycle arrest between the gap 2 phase and mitosis (G2/M) and apoptotic cell death. Overall, calecheamicin acts by binding to the minor DNA groove and causing double-strand DNA breaks in a manner independent of cell cycle progression, which induces apoptosis even in rapidly dividing cells [253,254]. Inotuzumab ozogamicin has been an approved drug since 2017 for the treatment of adults with relapsed or refractory B-cell precursor ALL [255].
3.3.2. Anti-CD30 and Anti-CD79b Antibody-Drug Conjugates
A cluster of differentiation 30 (CD30) is a transmembrane glycoprotein, a member of the tumor necrosis factor receptor (TNFR) superfamily, that is expressed on B or T lymphocytes in classical Hodgkin’s lymphoma (cHL) and anaplastic large cell lymphoma (ALCL). CD30 induces pleiotropic biological effects on cell growth and survival through activation of the NF-κB pathway [256]. A cluster of differentiation 79b (CD79b) is a component of the B cell receptor (BCR) complex that is expressed only on mature B cells and B cell malignancies. CD79b plays a key role in downstream BCR signaling [257]. Brentuximab vedotin and polatuzumab vedotin are drugs consisting of the monoclonal antibody directed against CD30 and CD79b, respectively, and monomethyl auristatin E (MMAE), a potent antimitotic agent, conjugated by a protease-cleavable maleimidocaproyl-valine-citrulline-p-aminobenzyloxycarbonyl linker. After internalization of these antibody-drug conjugates into a cancer cell, the linker is selectively cleaved by lysosomal enzymes and MMAE is released. The antimitotic agent then binds to tubulin and blocks their polymerization, leading to G2/M phase cell cycle arrest and cellular apoptosis [258,259]. Additionally, both drugs induce cancer cells’ death via antibody-mediated cellular phagocytosis (ADCP). Brentuximab vedotin is currently used as therapy for patients with systematic ALCL and cHL after the failure of prior combination chemotherapy regimens. The drug is also approved to treat adult patients with primary cutaneous anaplastic large cell lymphoma (pcALCL) and mycosis fungoides (MF) after the failure of prior systemic therapy. Moreover, the drug, in combination with chemotherapy, is the first-line treatment for adult patients with stage III or IV cHL who were previously untreated [260]. Clinical trials indicate that brentuximab vedotin followed by bendamustine supercharge is a promising treatment option for high-risk Hodgkin lymphoma. [261]. Polatuzumab vedotin is used in combination with bendamustine plus rituximab to treat adults with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) who have previously attempted two other therapies [262].
3.3.3. Anti-BCMA Antibody-Drug Conjugate
B cell maturation antigen (BCMA) is a transmembrane glycoprotein belonging to the tumor necrosis factor receptor superfamily 17 (TNFRSF17). The receptor is overexpressed on multiple myeloma (MM) cells as well as normal plasma cells, but not on other normal tissues. BCMA plays an important role in bone marrow plasma cell survival [263]. Belantamab mafodotin-blmf is a first-in-class anti-BCMA ADC indicated for the treatment of MM. The drug comprises an afucosylated IgG1 monoclonal antibody covalently conjugated to the cytotoxic microtubule inhibitor, i.e., monomethyl auristatin F (MMAF), through a maleimidocaproyl linker (mc). The antibody moiety delivers the MMAF to the BCMA-expressing cells. After the internalization of ADC and the release of MMAF via proteolytic degradation, the cytotoxic agent disrupts the formation of microtubules by inhibiting tubulin polymerization. Subsequently, this causes cell cycle arrest in the G2/M phase and apoptosis. Furthermore, belantamab mafodotin-blmf induces cancer cell lysis via antibody-dependent cellular cytotoxicity (ADCC) and phagocytosis (ADCP). The drug received FDA approval as a monotherapy for adults with relapsed or refractory MM after at least four prior treatments [264].
3.4. Recombinant Immunotoxin as Anticancer Agent
Recombinant immunotoxins (RITs) are fusion molecules that contain the Fv fragment of a monoclonal antibody (mAb) connected to the specified protein toxin. The toxin, produced by bacteria, fungi, or plants, is an extremely cytotoxic molecule that rapidly induces apoptotic cell death via inhibition of protein synthesis within the cell cytosol. Immunotoxins demonstrate potent clinical efficacy against various types of cancers, including hematological malignancies [273]. Moxetumomab pasudotox is the FDA-approved drug used to treat adult patients with relapsed or refractory hairy cell leukemia (HCL) after at least two prior systemic therapies [274]. Moxetumomab pasudotox is composed of the Fv fragment of an anti-CD22 monoclonal antibody genetically fused to PE38, which is a fragment of Pseudomonas exotoxin A. The Fv portion of this drug binds to cluster of differentiation 22 (CD22), a transmembrane sialoglycoprotein expressed on a variety of malignant B-cells, and thus delivers toxin moiety directly to target cells. After binding, the recombinant immunotoxin-CD22 complex is internalized into the cell, where the furin catalyzed cleavage of a fragment of PE38 occurs. The released PE38 then catalyzes the adenosine diphosphate (ADP)-ribosylation of the diphthamide residue in elongation factor-2 (EF-2), resulting in a decrease in protein myeloid cell leukemia 1 (MCL1) levels, and thereby cellular apoptosis (Figure 12) [275]. The overall features of the drug are summarized in Table 14.
Figure 12.

Intoxication of cell by moxetumomab pasudotox. ADP: adenosine diphosphate. ADPr: adenosine diphosphate ribose. EF-2: elongation factor-2. Figure based on the following reference [275]. Adapted from “Intracellular layout—endocytosis pathway,” by BioRender.com (2021). Retrieved from https://app.BioRender.com/biorender-templates (accessed on: 8 December 2021).
Table 14.
Features of the immunotoxin drug approved by the Food and Drug Administration (FDA) from 2011 to 2021.
| No. | Generic Name of Drug | Brand Name and Company |
First FDA/EMA Approved Date | Class | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Moxetumomab pasudotox-tdfk | LUMOXITI AstraZeneca, Cambridge, England |
FDA: 13 September 2018 EMA: Not approved |
Murine immunoglobulin and PE38 conjugate | CD22 1 | Intravenous | Hairy Cell Leukemia | Peripheral edema, nausea, fatigue, headache, pyrexia, decreased lymphocyte count, hemolytic uremic syndrome, | [276,277] |
1 CD22: cluster of differentiation 22.
3.5. Enzymes as Anticancer Agents
Enzymes used as anticancer therapy target amino acid metabolism. They are involved in the degrading of amino acids such as asparagine, arginine, and methionine, for which certain cancerous cells become auxotrophic. The amino acid deprivation leads to the inhibition and impairment of cancer growth [278]. The enzymes approved during the last decade for treatment of hematological malignancies are summarized in Table 15. Asparaginase Erwinia chrysanthemi is a novel formulation of L-asparaginase, which is the enzyme responsible for the metabolism of L-asparagine derived from the Gram-negative bacteria Erwinia chrysanthemi. The drug is immunologically distinct from Escherichia coli-derived L-asparaginase and demonstrates higher enzymatic activity. It was developed to avoid toxicities such as hypersensitivity, pancreatitis, and hyperglycemia associated with previously approved L-asparaginase therapies [279]. Calaspargase pegol-mknl is also an L-asparagine specific enzyme, but is connected with 31–39 molecules of monomethoxy-polyethylene glycol (mPEG) by a succinimidyl carbonate (SC) linker. This new drug is a long-acting product, which provides for less frequent dosing compared to other L-asparaginase therapies [280]. Asparaginase Erwinia chrysanthemi received approval to treat patients with acute lymphoblastic leukemia (ALL) who had to discontinue treatment with Escherichia coli-derived asparaginase because of hypersensitivity, while calaspargase pegol-mknl was approved to treat pediatric and young adult patients with ALL. Both drugs are used as components of a multi-agent chemotherapeutic regimen [281,282]. The mechanism of their action includes hydrolysis of L-asparagine to aspartic acid and ammonia resulting in depleting circulating levels of L-asparagine, which is essential for the protein metabolism of leukemic cells, and thereby their growth and survival (Figure 13). Subsequently, L-asparagine deprivation in leukemic cells induces cell death [282,283].
Table 15.
Features of the enzymes drugs approved by the Food and Drug Administration (FDA) from 2011 to 2021. The order of drugs is tabulated in order of most recent to oldest registration date.
| No. | Generic Name of Drug | Brand Name and Company |
First FDA/EMA Approved Date | Class | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Calaspargase pegol-mknl | ASPARLAS Servier Pharmaceuticals, Boston, MA, USA | FDA: 20 December 2018 EMA: Not approved |
Asparagine-specific enzyme | Asparagine level | Intravenous | Acute Lymphoblastic Leukemia | Elevated transaminase, bilirubin increased, pancreatitis, abnormal clotting studies | [280] |
| 2 | Asparaginase Erwinia chrysanthemi | ERWINAZE Jazz Pharmaceuticals plc, Dublin, Ireland |
FDA: 18 November 2011 EMA: nationally authorized |
Asparagine-specific enzyme | Asparagine level | Intravenous | Acute Lymphoblastic Leukemia | Anaphylaxis, pancreatitis, abnormal transaminases, thrombosis, hemorrhage, nausea, vomiting, hyperglycemia. | [283,284,285] |
Figure 13.

Schematic representation of mechanism of action for asparaginases such as asparaginase Erwinia chrysanthemi and calaspargase pegol-mknl. Created with BioRender.com.
3.6. Chimeric Antigen Receptor T (CAR-T) Cells as Anticancer Agents
Chimeric antigen receptor T (CAR-T) cell therapy is a novel, promising immunotherapy treatment of various hematological malignancies. In the CAR-T cell, T lymphocytes separated from the patient’s peripheral blood are engineered with genetic recombinant receptors known as chimeric antigen receptors (CAR). A CAR is a synthetic protein that consists of three parts, i.e., an extracellular antibody-derived domain, a transmembrane domain, and an intracellular T-cell signaling domain. The extracellular domain targets specific tumor-associated antigens (TAAs) while the intracellular domain enhances T-cell proliferation and differentiation. There are four generations of CAR proteins that differ in the structure of the intracellular signaling domain. CAR-T cells lead to cancer cell death mainly via the secretion of cytotoxic granules containing granzymes and perforin, and the release of cytokines (i.e., IFNγ and tumor necrosis factor (TNF)) (Figure 14). CAR-T cells provide antigen recognition in a major histocompatibility complex (MHC)-independent manner. For this reason, the immune response during CAR-T therapy may be out of control and cause some toxic side effects [286,287]. CAR-T therapies which are FDA-approved until 2021, i.e., tisagenlecleucel, axicabtagene ciloleucel, and brexucabtagene autoleucel, are second-generation CD19-directed CAR-T cells (Table 16). Tisagenlecleucel is the first genetically modified therapy, which is autologous T-cells modified through lentiviral transduction to express a CAR composed of a murine single-chain antibody fragment (scFv), a transmembrane domain, and an intracellular signaling domain (CD3ζ) fused to a costimulatory domain (4-1BB) [288]. It acts by binding to CD19-expressing cells, then initiating T-cell activation via the CD3ζ component. The 4-1BB domain provides enhancing of the expansion and persistence of tisagenlecleucel. This drug is now registered for two distinct indications in relapsed or refractory B-cell acute lymphoblastic leukemia (ALL) and certain types of relapsed or refractory large B-cell lymphomas, including diffuse large B-cell lymphoma (DLBCL), high-grade B-cell lymphoma, and DLBCL arising from follicular lymphoma, for which other therapies have failed [289]. Axicabtagene ciloleucel and brexucabtagene autoleucel are autologous T-cells that express a CAR, which contains a murine single-chain variable fragment (scFv), a transmembrane domain, and an intracellular signaling domain (CD3ζ) integrated with a costimulatory domain (CD28). The primary difference between these drugs is in their manufacturing processes. With brexucabtagene autoleucel specifically, there is an additional T-cell enrichment phase during production, which causes the removal of circulating CD19-expressing cancer cells in the leukapheresis material. This stage prevents potential manufacturing failure in the case of patients with mantle cell lymphoma (MCL), who may have a high number of cancer cells in their peripheral blood and relatively fewer T-cells [290,291]. Hence, axicabtagene ciloleucel and brexucabtagene autoleucel represent an effective treatment modality in adult patients with various large B-cell lymphomas, such as DLBCL, and relapsed or refractory MCL, respectively. These drugs are infused into the patient after conditioning with lymphodepleting chemotherapy. Their mechanism of action includes binding to CD19-expressing cancer cells, and subsequent activation of downstream signaling pathways through the CD28 and CD3ζ domains, leading to T-cell activation and proliferation, as well as the secretion of inflammatory cytokines and chemokines. This results in the effective eradication of cancer cells [292,293]. While tisagenlecleucel, axicabtagene ciloleucel, and brexucabtagene autoleucel are potent anticancer agents, they may cause severe adverse reactions, including cytokine release syndrome and neurologic toxicities. For this reason, CAR-T therapies are available only through restricted programs under a Risk Evaluation and Mitigation Strategy (REMS), i.e., the KYMRIAH REMS Program and the YESCARTA and TECARTUS REMS Programs [289,292,293].
Figure 14.

The immune-mediated cytotoxic mechanism of CAR-T cell. PFN: perforin. Gzm: granzymes. CD19: cluster of differentiation 19. IFNγ: interferon gamma. TNF: tumor necrosis factor. CAR-T: chimeric antigen receptor T. Created with BioRender.com.
Table 16.
Features of the chimeric antigen receptor T-cells (CAR-T cells) drugs approved by the Food and Drug Administration (FDA) from 2011 to 2021. The order of drugs is tabulated in order of most recent to oldest registration date.
| No. | Generic Name of Drug | Brand Name and Company |
First FDA/EMA Approved Date | Class | Molecular Target | Route of Administration | Indication | Adverse Effects | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Brexucabtagene autoleucel | TECARTUS Kite, a Gilead Company, Foster City, CA, USA |
FDA: 24 July 2020 EMA: 14 December 2020 |
Genetically modified autologous T cells |
CD19 1 | Intravenous | Mantle Cell Lymphoma | Cytokine release syndrome, cytopenias, hypotension, encephalopathy, fever, fatigue, tachycardia, arrhythmia, infection with pathogen unspecified, chills, hypoxia, cough, tremor, musculoskeletal pain, headache, nausea, edema, motor dysfunction, constipation, diarrhea, decreased appetite, dyspnea, rash, insomnia, pleural effusion, aphasia |
[290,294,295] |
| 2 | Axicabtagene ciloleucel | YESCARTA Kite Pharma, Inc., Los Angeles, CA, USA |
FDA: 18 October 2017 EMA: 23 August 2018 |
Genetically modified autologous T cells |
CD19 1 | Intravenous | Large B-Cell Lymphoma, Follicular Lymphoma | Cytokine release syndrome, fever, hypotension, encephalopathy, tachycardia, fatigue, headache, febrile neutropenia, nausea, infections with pathogen unspecified, decreased appetite, chills, diarrhea, tremor, musculoskeletal pain, cough, hypoxia, constipation, vomiting, arrhythmias, dizziness | [296,297] |
| 3 | Tisagenlecleucel | KYMRIAH Novartis Pharmaceuticals Corporation, Basel, Switzerland | FDA: 30 August 2017 EMA: 23 August 2018 |
Genetically modified autologous T cells |
CD19 1 | Intravenous | Acute Lymphoblastic Leukemia, Large B-Cell Lymphoma |
Cytokine release syndrome, infections-pathogen unspecified, pyrexia, decreased appetite, hypogammaglobinemia, headache, encephalopathy, hypotension, bleeding, episodes, tachycardia, nausea, diarrhea, vomiting, viral infectious disorders, hypoxia, fatigue, acute kidney injury, edema, cough, delirium | [298,299] |
1 CD19: cluster of differentiation 19.
4. Conclusions
Fifty two new drug registrations approved by the FDA were made in the period between 2011 and 2021 for use in therapies of hematological malignancies; 29 of them were for small molecule drugs and 23 of them were for macromolecules. Forty out of all these drugs (21 small molecule and 19 macromolecular drugs) were also approved by the EMA (European Medicines Agency). Depending on the mechanism of action, these drugs can be classified as chemotherapy agents, whose activity is due to disruption of the mitotic and/or DNA replication pathways, and targeted agents, which inhibit the molecular targets involved in cancer cell growth and spread. In the studied period, the number of FDA-approved chemotherapy drugs is significantly smaller (6 drugs) than ones containing targeted molecules (46 drugs) as defined above. The route of administration for these drugs is mainly intravenous (28 drugs) or oral (23 drugs), and only one (1) is subcutaneous injection, which may be administered at home by the patient or a caregiver. Six (6) of these registered drugs (i.e., ERWINAZE, PURIXAN, MARQIBO, INQOVI, ONUREG, and VYXEOS) are medications that contain anticancer active compounds (i.e., asparaginase Erwinia chrysanthemi, mercaptopurine, vincristine sulfate, decitabine, azacitidine, and a fixed-dose combination of cytarabine and daunorubicin, respectively) and that have been used for more than 10 years; however, their present forms have significantly better pharmacological parameters. In the case of MARQIBO and VYXEOS, which are liposome-encapsulated chemotherapy agents, delivery efficiency, biocompatibility, and pharmacokinetics are improved. In addition, VYXEOS consists of two active compounds in the optimal molar ratio, of which simultaneous delivery to cancer cells improves drug pharmacologic action and reduces the occurrence of resistance. ONUREG and INQOVI are formulated as film-coated tablets and fixed-dose combination with cedazuridine, respectively, that enable their oral administration due to avoiding degradation by cytidine deaminase. A new oral formulation of these drugs induces facilitated dosing, reduces administration side effects, and potentially increases efficacy. In contrast, PURIXAN is developed as an oral suspension to the accuracy of drug dose for individual patients. For ERWINAZE, there is increased enzymatic activity and reduced adverse events of asparaginase therapy.
All of the 29 presented new small molecule drugs are based on aromatic organic compounds, which, apart from two (2) drugs (i.e., ixazomib citrate (NINLARO) and belinostat (BELEODAQ)), contain a heterocyclic system in their structure. Among them, there is a large number of compounds bearing a nitrogen heterocycle ring (28 drugs) and a much smaller number with an oxygen heterocycle ring (6 drugs). A deeper analysis of pharmaceuticals containing nitrogen heterocycles indicates that bicyclic compounds, such as indole, purines, and pyrrolopyrimidines, and six-membered mono ring heterocycles, including pyridine, pyrimidine, pyrazine, and triazine, are the most common in structures of small molecule drugs. Only three (3) medicaments, i.e., omacetaxine mepesuccinate (SYNRIBO), vincristine (MARQIBO), and midostaurin (RYDAPT), are heteropolicyclic compounds. These belong to the class of compounds known as cephalotaxus alkaloids, vinca alkaloids, and indolocarbazoles, respectively. Moreover, almost half (14 drugs) of the drugs approved in the last decade contain chiral active compounds. It is interesting to note that only 1 (1) drug consists of a boron-containing molecule, 5 (5 drugs) are sulfur-containing compounds (i.e., sulfate, sulfonamide, or thioketone), and 10 (10 drugs) contain one or more halogen atoms (specifically fluorine or chlorine) in the structure. The boronic acids group, which is present in a peptidomimetic drug—ixazomib, acts as a reversible covalent inhibitor of nucleophilic threonine residues in the active sites of the 20S unit of the 26S proteasome. Subsequently, it blocks the ubiquitin-mediated protein degradation pathway leading to cancer cell death [300]. Other scaffolds found in drugs approved in the studied period are amide bonds and hydroxamic acid moiety. Due to the crucial role of the amide group for the function of many biomolecules, it is often represented in the pharmacophore of biologically active compounds [301]. The authors have noticed that 18 registered small molecules contain the amide functional group. The hydroxamic acid group is found in two (2) drugs (belinostat and panobinostat) which are HDACs inhibitors. The coordination of the hydroxamic acid to zinc ion in the active site of HDACs is essential to the inhibitory effect of these drugs [302]. Despite the toxicity of the nitrile (CN) and nitro (NO2) compounds [303,304], four (4) of the small molecule inhibitors are nitrile-containing pharmaceuticals, and one (1) contains the nitroaromatic compound. These drugs induce anticancer effects by diverse mechanisms including Src and Abl kinases inhibition (bosutinib), JAK1 and JAK2 inhibition (ruxolitinib), IDH1 inhibition (ivosidenib), SMO receptor antagonism (glasdegib), and BCL-2 inhibition (venetoclax). Since CN and NO2 groups have an electron-withdrawing nature, their role is a creation of electron-deficient sites in drug structures and nonspecific dipole interaction with biological nucleophiles present in living organisms, e.g., amino acids, nucleic acids, and enzymes [305,306]. It is worth noting that eight (8) of the small molecule drugs are characterized by a completely new mechanism of action, such as BTK inhibition (ibrutinib), PI3K-δ inhibition (idelalisib), PI3K- δ/γ inhibition (dulvalisib), IDH2 inhibition (enasidenib), IDH1 inhibition (ivosidenib), EZH2 inhibition (tazemetostat), BCL-2 inhibition (venetoclax), and XPO1 inhibition (selinexor). Furthermore, two (2) drugs contain a fixed-dose combination of two anticancer active ingredients. VYXEOS consists of topoisomerase II inhibitor—daunorubicin, and pyrimidine base antagonist—cytarabine. INQOVI contains decitabine, which inhibits DNA methyltransferases, and cedazuridine, which protects decitabine from being broken down by cytidine deaminase.
At the same time, it should be emphasized that more and more drugs from the macromolecular group (such as antibodies, enzymes, and CAR-T cells) are starting to be applied in anticancer therapy and it is supposed that this tendency will continue to the advantage of this composite biomolecular group of drugs. Among the 23 FDA-approved macromolecule drugs, more than half (12 drugs) are monoclonal antibodies (mAbs). Only one (1 drug) monoclonal antibody, i.e., mogamulizumab-kpkc, is referred to as a first-in-class drug, characterized by a novel anti-CCR4 mechanism of action. In contrast, three (3) mAbs (i.e., TRUXIMA, RUXIENCE, and RIABNI) are biosimilars of rituximab (RITUXAN), which is the first monoclonal antibody used in clinics for the treatment of hematological malignancies. Biosimilars of rituximab provide more affordable monoclonal antibody therapy and increase access for patients to receive optimal care. To improve the therapeutic efficacy of mAbs, various modifications have been developed, including currently approved immunoconjugates (five (5) antibody-drug conjugates and one (1) recombinant immunotoxin) and one (1) bispecific monoclonal antibody. There are three main structural elements of immunoconjugates: antibody directed against different types of cancer cell antigens; linker and payload, which is either cytotoxic agent, i.e., microtubule inhibitor (monomethyl auristatin F), antimitotic agent (monomethyl auristatin E), or DNA-damaging antibiotic derivative (N-acetyl-γ-calicheamicin dimethyl hydrazide); toxin (Pseudomonas exotoxin A). The analysis of linker structures indicates that they consist of the following chemical motifs: acid cleavable hydrazone, reducible disulfide, enzyme cleavable dipeptide with spacer unit (p-aminobenzyloxycarbonyl group), and non-cleavable maleimidocaproyl group. It is worth noting that the FDA registered, for the first time, the bispecific T-cell-engager (BiTE) antibody blinatumomab in 2014, and, in 2020, the first anti-BCMA antibody-drug conjugate—belantamab mafodotin-blmf. In the last decade, the FDA also approved the first-ever genetically modified CAR-T therapy. By the end of 2020, three (3) CAT-T therapies (i.e., KYMRIAH, YESCARTA, and TECARTUS) were used to treat certain types of leukemia and lymphoma. CAR-T cells differ significantly from other anticancer drugs because of their ability to proliferate and expand. They can persist for months or even years in a patient’s blood and, for this reason, are called “living drugs” [288]. CAR-T cells are the most promising type of anticancer therapy, especially for patients with relapsed or refractory hematological malignancies who do not respond to standard treatments.
Author Contributions
Conceptualization, A.S.-Ć., A.R. and M.M.; writing—original draft preparation, A.S.-Ć.; writing—review and editing, A.S.-Ć., A.R. and M.M.; visualization, A.S.-Ć.; supervision, A.R. and M.M.; funding acquisition, A.S.-Ć. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Wroclaw Medical University, grant number STM.D090.20.122.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Huang L., Huang J., Huang J., Xue H., Liang Z., Wu J., Chen C. Nanomedicine—A promising therapy for hematological malignancies. Biomater Sci. 2020;8:2376–2393. doi: 10.1039/d0bm00129e. [DOI] [PubMed] [Google Scholar]
- 2.Nussbaumer S., Bonnabry P., Veuthey J.-L., Fleury-Souverain S. Analysis of anticancer drugs: A review. Talanta. 2011;85:2265–2289. doi: 10.1016/j.talanta.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 3.Goodman L.S., Wintrobe M.M., Huang L., Huang J., Huang J., Xue H., Liang Z., Wu J., Chen C. Nitrogen mustard therapy; use of methyl-bis (beta-chloroethyl) amine hydrochloride and tris (beta-chloroethyl) amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. J. Am. Med. Assoc. 1946;132:126–132. doi: 10.1001/jama.1946.02870380008004. [DOI] [PubMed] [Google Scholar]
- 4.Mustargen NDA #006695—Drugs@FDA: FDA-Approved Drugs. [(accessed on 23 August 2021)]; Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=006695.
- 5.Farber S., Diamond L.K., Mercer R.D., Sylvester R.F., Wolff J.A. Temporary remissions in acute leukemia in children produced by folic acid antagonist, 4-Aminopteroyl-glutamic acid (Aminopterin) N. Engl. J. Med. 1948;238:787–793. doi: 10.1056/NEJM194806032382301. [DOI] [PubMed] [Google Scholar]
- 6.Methotrexate Sodium NDA #008085—Drugs@FDA: FDA-Approved Drugs. [(accessed on 23 August 2021)]; Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=008085.
- 7.Wright J.C., Prigot A., Wright B., Weintraub S., Wright L.T. An evaluation of folic acid antagonists in adults with neoplastic diseases: A study of 93 patients with incurable neoplasms. J. Natl. Med. Assoc. 1951;43:211–240. [PMC free article] [PubMed] [Google Scholar]
- 8.Elion G.B., Burgi E., Hitchings G.H. Studies on condensed pyrimidine systems. IX. The synthesis of some 6-substituted purines. J. Am. Chem. Soc. 1952;74:411–414. doi: 10.1021/ja01122a037. [DOI] [Google Scholar]
- 9.Purinethol NDA #009053—Drugs@FDA: FDA-Approved Drugs. [(accessed on 24 August 2021)]; Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=009053.
- 10.Dushinsky R., Pleven E., Heidelberger C. The synthesis of 5-fluoropyrimidines. J. Am. Chem. Soc. 1957;79:4559–4560. doi: 10.1021/ja01573a087. [DOI] [Google Scholar]
- 11.Fluorouracil NDA 012209—Drugs@FDA: FDA-Approved Drugs. [(accessed on 24 August 2021)]; Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=012209.
- 12.Hurley J.D. Chemotherapy of solid carcinoma. JAMA. 1960;174:1696–1701. doi: 10.1001/jama.1960.03030130024007. [DOI] [PubMed] [Google Scholar]
- 13.Noble R.L. The discovery of the vinca alkaloids—Chemotherapeutic agents against cancer. Biochem. Cell Biol. 1990;68:1344–1351. doi: 10.1139/o90-197. [DOI] [PubMed] [Google Scholar]
- 14.Oncovin NDA #014103—Drugs@FDA: FDA-Approved Drugs. [(accessed on 25 August 2021)]; Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=014103.
- 15.Watson R.A., De La Peña H., Tsakok M.T., Joseph J., Stoneham S., Shamash J., Joffe J., Mazhar D., Traill Z., Ho L.P., et al. Development of a best-practice clinical guideline for the use of bleomycin in the treatment of germ cell tumours in the UK. Br. J. Cancer. 2018;119:1044–1051. doi: 10.1038/s41416-018-0300-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takeuchi T. Antitumor antibiotics discovered and studied at the Institute of Microbial Chemistry. J. Cancer Res. Clin. Oncol. 1995;121:505–510. doi: 10.1007/BF01197761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonadonna G., Monfardini S., De Lena M., Fossati-Bellani F. Clinical evaluation of adriamycin, a new antitumour antibiotic. Br. Med. J. 1969;3:503–506. doi: 10.1136/bmj.3.5669.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doxorubicin Hydrochloride NDA #050467—Drugs@FDA: FDA-Approved Drugs. [(accessed on 25 August 2021)]; Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=050467.
- 19.Thorn C.F., Oshiro C., Marsh S., Hernandez-Boussard T., McLeod H., Klein T.E., Altman R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet. Genom. 2011;21:440–446. doi: 10.1097/FPC.0b013e32833ffb56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peyrone M. Ueber die einwirkung von ammoniak auf platinchlorü: Zweite abhandlung. Ann. Chem. Pharm. 1845;55:205–213. doi: 10.1002/jlac.18450550206. [DOI] [Google Scholar]
- 21.Rosenberg B., Vancamp L., Krigas T. Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature. 1965;205:698–699. doi: 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- 22.Ghosh S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019;88:102925. doi: 10.1016/j.bioorg.2019.102925. [DOI] [PubMed] [Google Scholar]
- 23.Grillo-López A.J., White C.A., Dallaire B.K., Varns C.L., Shen C.D., Wei A., Leonard J.E., McClure A., Weaver R., Cairelli S., et al. Rituximab: The first monoclonal antibody approved for the treatment of lymphoma. Curr. Pharm. Biotechnol. 2000;1:1–9. doi: 10.2174/1389201003379059. [DOI] [PubMed] [Google Scholar]
- 24.Cohen M.H., Williams G., Johnson J.R., Duan J., Gobburu J., Rahman A., Benson K., Leighton J., Kim S.K., Wood R., et al. Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin. Cancer Res. 2002;8:935–942. [PubMed] [Google Scholar]
- 25.Nava-Parada P., Shelbaya A., Nabhan C. Rituximab biosimilars in hematologic malignancies: The need for a real-world approach. Future Oncol. 2020;16:2017–2027. doi: 10.2217/fon-2020-0131. [DOI] [PubMed] [Google Scholar]
- 26.Pottier C., Fresnais M., Gilon M., Jérusalem G., Longuespée R., Sounni N.E. Tyrosine kinase inhibitors in cancer: Breakthrough and challenges of targeted therapy. Cancers. 2020;12:731. doi: 10.3390/cancers12030731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paul M.K., Mukhopadhyay A.K. Tyrosine kinase—Role and significance in cancer. Int. J. Med. Sci. 2004;1:101–115. doi: 10.7150/ijms.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Madhusudan S., Ganesan T.S. Tyrosine kinase inhibitors in cancer therapy. Clin. Biochem. 2004;37:618–635. doi: 10.1016/j.clinbiochem.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 29.Krause D.S., Van Etten R.A. Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med. 2005;353:172–187. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- 30.IMBRUVICA (Ibrutinib) Prescribing Information. [(accessed on 19 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/205552s030,210563s006lblPI.pdf.
- 31.CALQUENCE (Acalabrutinib) Prescribing Information. [(accessed on 19 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/210259s006s007lbl.pdf.
- 32.Wiestner A. Targeting B-Cell receptor signaling for anticancer therapy: The Bruton’s tyrosine kinase inhibitor ibrutinib induces impressive responses in B-cell malignancies. J. Clin. Oncol. 2013;31:128–130. doi: 10.1200/JCO.2012.44.4281. [DOI] [PubMed] [Google Scholar]
- 33.Byrd J.C., Harrington B., O’Brien S., Jones J.A., Schuh A., Devereux S., Chaves J., Wierda W.G., Awan F.T., Brown J.R., et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N. Engl. J. Med. 2016;374:323–332. doi: 10.1056/NEJMoa1509981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tam C.S., Trotman J., Opat S., Burger J.A., Cull G., Gottlieb D., Harrup R., Johnston P.B., Marlton P., Munoz J., et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B-cell malignancies and safety and efficacy evaluation in CLL. Blood. 2019;134:851–859. doi: 10.1182/blood.2019001160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.U.S. Food and Drug Administration FDA Grants Accelerated Approval to Bosutinib for Treatment of Newly-Diagnosed PH+ CML. [(accessed on 22 December 2021)]; Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-bosutinib-treatment-newly-diagnosed-ph-cml.
- 36.Cortes J.E., Kantarjian H.M., Brümmendorf T.H., Kim D.W., Turkina A.G., Shen Z.X., Pasquini R., Khoury H.J., Arkin S., Volkert A., et al. Safety and efficacy of bosutinib (SKI-606) in chronic phase Philadelphia chromosome-positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood. 2011;118:4567–4576. doi: 10.1182/blood-2011-05-355594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khatri A., Wang J., Pendergast A.M. Multifunctional Abl kinases in health and disease. J. Cell Sci. 2016;129:9–16. doi: 10.1242/jcs.175521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xue C., Wang X., Zhang L., Qu Q., Zhang Q., Jiang Y. Ibrutinib in B-cell lymphoma: Single fighter might be enough? Cancer Cell Int. 2020;20:467. doi: 10.1186/s12935-020-01518-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Syed Y.Y. Zanubrutinib: First approval. Drugs. 2020;80:91–97. doi: 10.1007/s40265-019-01252-4. [DOI] [PubMed] [Google Scholar]
- 40.European Medicines Agency BRUKINSA (Zanubrutinib) [(accessed on 1 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/19/2167-public-summary-opinion-orphan-designation-zanubrutinib-treatment-lymphoplasmatic-lymphoma_en.pdf.
- 41.Markham A., Dhillon S. Acalabrutinib: First global approval. Drugs. 2018;78:139–145. doi: 10.1007/s40265-017-0852-8. [DOI] [PubMed] [Google Scholar]
- 42.European Medicines Agency CALQUENCE (Acalabrutinib) [(accessed on 1 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/calquence-epar-medicine-overview_en.pdf.
- 43.Cameron F., Sanford M. Ibrutinib: First global approval. Drugs. 2014;74:263–271. doi: 10.1007/s40265-014-0178-8. [DOI] [PubMed] [Google Scholar]
- 44.European Medicines Agency IMBRUVICA (Ibrutinib) [(accessed on 1 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/imbruvica-epar-medicine-overview_en.pdf.
- 45.Bosulif NDA #203341—Drugs@FDA: FDA-Approved Drugs. [(accessed on 25 October 2021)]; Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&varApplNo=203341.
- 46.BOSULIF (Bosutinib) Prescribing Information. [(accessed on 7 September 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/203341s009lbl.pdf.
- 47.European Medicines Agency BOSULIF (Bosutinib) [(accessed on 1 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/bosulif-epar-medicine-overview_en.pdf.
- 48.Broekman F., Giovannetti E., Peters G.J. Tyrosine kinase inhibitors: Multi-targeted or single-targeted? World J. Clin. Oncol. 2011;2:80–93. doi: 10.5306/wjco.v2.i2.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krug M., Hilgeroth A. Recent advances in the development of multi-kinase inhibitors. Mini Rev. Med. Chem. 2008;8:1312–1327. doi: 10.2174/138955708786369591. [DOI] [PubMed] [Google Scholar]
- 50.Swords R., Freeman C., Giles F. Targeting the FMS-like tyrosine kinase 3 in acute myeloid leukemia. Leukemia. 2012;26:2176–2185. doi: 10.1038/leu.2012.114. [DOI] [PubMed] [Google Scholar]
- 51.Aydin E., Faehling S., Saleh M., Llaó Cid L., Seiffert M., Roessner P.M. Phosphoinositide 3-kinase signaling in the tumor microenvironment: What do we need to consider when treating chronic lymphocytic leukemia with PI3K inhibitors? Front. Immunol. 2021;11:595818. doi: 10.3389/fimmu.2020.595818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cortes J.E., Kantarjian H., Shah N.P., Bixby D., Mauro M.J., Flinn I., O’Hare T., Hu S., Narasimhan N.I., Rivera V.M., et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2012;367:2075–2088. doi: 10.1056/NEJMoa1205127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lipton J.H., Chuah C., Guerci-Bresler A., Rosti G., Simpson D., Assouline S., Etienne G., Nicolini F.E., le Coutre P., Clark R.E., et al. EPIC investigators. Ponatinib versus imatinib for newly diagnosed chronic myeloid leukaemia: An international, randomised, open-label, phase 3 trial. Lancet Oncol. 2016;17:612–621. doi: 10.1016/S1470-2045(16)00080-2. [DOI] [PubMed] [Google Scholar]
- 54.Gainor J.F., Chabner B.A. Ponatinib: Accelerated disapproval. Oncologist. 2015;20:847–848. doi: 10.1634/theoncologist.2015-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gotlib J., Kluin-Nelemans H.C., George T.I., Akin C., Sotlar K., Hermine O., Awan F.T., Hexner E., Mauro M.J., Sternberg D.W., et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N. Engl. J. Med. 2016;374:2530–2541. doi: 10.1056/NEJMoa1513098. [DOI] [PubMed] [Google Scholar]
- 56.Stone R.M., Mandrekar S.J., Sanford B.L., Laumann K., Geyer S., Bloomfield C.D., Thiede C., Prior T.W., Döhner K., Marcucci G., et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N. Engl. J. Med. 2017;377:454–464. doi: 10.1056/NEJMoa1614359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee L.Y., Hernandez D., Rajkhowa T., Smith S.C., Raman J.R., Nguyen B., Small D., Levis M. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood. 2017;129:257–260. doi: 10.1182/blood-2016-10-745133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perl A.E., Martinelli G., Cortes J.E., Neubauer A., Berman E., Paolini S., Montesinos P., Baer M.R., Larson R.A., Ustun C., et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N. Engl. J. Med. 2019;381:1728–1740. doi: 10.1056/NEJMoa1902688. [DOI] [PubMed] [Google Scholar]
- 59.Mori M., Kaneko N., Ueno Y., Yamada M., Tanaka R., Saito R., Shimada I., Mori K., Kuromitsu S. Gilteritinib, a FLT3/AXL inhibitor, shows antileukemic activity in mouse models of FLT3 mutated acute myeloid leukemia. Investig. Drugs. 2017;35:556–565. doi: 10.1007/s10637-017-0470-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bewersdorf J.P., Jaszczur S.M., Afifi S., Zhao J.C., Zeidan A.M. Beyond Ruxolitinib: Fedratinib and other emergent treatment options for myelofibrosis. Cancer Manag. Res. 2019;11:10777–10790. doi: 10.2147/CMAR.S212559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harrison C., Kiladjian J.J., Al-Ali H.K., Gisslinger H., Waltzman R., Stalbovskaya V., McQuitty M., Hunter D.S., Levy R., Knoops L., et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N. Engl. J. Med. 2012;366:787–798. doi: 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- 62.Verstovsek S., Mesa R.A., Gotlib J., Levy R.S., Gupta V., DiPersio J.F., Catalano J.V., Deininger M., Miller C., Silver R.T., et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N. Engl. J. Med. 2012;366:799–807. doi: 10.1056/NEJMoa1110557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vannucchi A.M., Kiladjian J.J., Griesshammer M., Masszi T., Durrant S., Passamonti F., Harrison C.N., Pane F., Zachee P., Mesa R., et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N. Engl. J. Med. 2015;372:426–435. doi: 10.1056/NEJMoa1409002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pardanani A., Tefferi A., Jamieson C., Gabrail N.Y., Lebedinsky C., Gao G., Liu F., Xu C., Cao H., Talpaz M. A phase 2 randomized dose-ranging study of the JAK2-selective inhibitor fedratinib (SAR302503) in patients with myelofibrosis. Blood Cancer J. 2015;5:e335. doi: 10.1038/bcj.2015.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pardanani A., Harrison C., Cortes J.E., Cervantes F., Mesa R.A., Milligan D., Masszi T., Mishchenko E., Jourdan E., Vannucchi A.M., et al. Safety and efficacy of Fedratinib in patients with primary or secondary myelofibrosis: A randomized clinical trial. JAMA Oncol. 2015;1:643–651. doi: 10.1001/jamaoncol.2015.1590. [DOI] [PubMed] [Google Scholar]
- 66.Blair H.A. Fedratinib: First approval. Drugs. 2019;79:1719–1725. doi: 10.1007/s40265-019-01205-x. [DOI] [PubMed] [Google Scholar]
- 67.INREBIC (Fedratinib) Prescribing Information. [(accessed on 5 December 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/212327s000lbl.pdf.
- 68.European Medicines Agency INREBIC (Fedratinib) [(accessed on 1 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/inrebic-epar-medicine-overview_en.pdf.
- 69.Dhillon S. Gilteritinib: First global approval. Drugs. 2019;79:331–339. doi: 10.1007/s40265-019-1062-3. [DOI] [PubMed] [Google Scholar]
- 70.European Medicines Agency XOSPATA (Gilteritinib) [(accessed on 1 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/xospata-epar-medicine-overview_en.pdf.
- 71.Kim E.S. Midostaurin: First global approval. Drugs. 2017;77:1251–1259. doi: 10.1007/s40265-017-0779-0. [DOI] [PubMed] [Google Scholar]
- 72.European Medicines Agency RYDAPT (Midostaurin) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/rydapt-epar-summary-public_en.pdf.
- 73.Jain H., Thorat J., Sengar M., Dubey A. Ponatinib: A drug review. Cancer Res. Stat. Treat. 2019;2:190–196. doi: 10.4103/CRST.CRST_98_19. [DOI] [Google Scholar]
- 74.European Medicines Agency ICLUSIG (Ponatinib) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/iclusig-epar-summary-public_en.pdf.
- 75.Deisseroth A., Kaminskas E., Grillo J., Chen W., Saber H., Lu H.L., Rothmann M.D., Brar S., Wang J., Garnett C., et al. Food and Drug Administration approval: Ruxolitinib for the treatment of patients with intermediate and high-risk myelofibrosis. Clin. Cancer Res. 2012;18:3212–3217. doi: 10.1158/1078-0432.CCR-12-0653. [DOI] [PubMed] [Google Scholar]
- 76.European Medicines Agency JAKAVI (Ruxolitinib) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/jakavi-epar-summary-public_en.pdf.
- 77.Verheijen J.C., Zask A. Phosphatidylinositol 3-kinase (PI3K) inhibitors as anticancer drugs. Drugs Future. 2007;32:537–547. doi: 10.1358/dof.2007.032.06.1107995. [DOI] [Google Scholar]
- 78.Falasca M. PI3K/Akt signalling pathway specific inhibitors: A novel strategy to sensitize cancer cells to anti-cancer drugs. Curr. Pharm. Des. 2010;16:1410–1416. doi: 10.2174/138161210791033950. [DOI] [PubMed] [Google Scholar]
- 79.Kong D., Yamori T. Phosphatidylinositol 3-kinase inhibitors: Promising drug candidates for cancer therapy. Cancer Sci. 2008;99:1734–1740. doi: 10.1111/j.1349-7006.2008.00891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Raedler L.A. Zydelig (Idelalisib): First-in-class PI3 kinase inhibitor approved for the treatment of 3 hematologic malignancies. Am. Health Drug Benefits. 2015;8:157–162. [PMC free article] [PubMed] [Google Scholar]
- 81.Liu N., Rowley B.R., Bull C.O., Schneider C., Haegebarth A., Schatz C.A., Fracasso P.R., Wilkie D.P., Hentemann M., Wilhelm S.M., et al. BAY 80-6946 is a highly selective intravenous pI3K inhibitor with potent p110α and p110δ activities in tumor cell lines and xenograft models. Mol. Cancer Ther. 2013;12:2319–2330. doi: 10.1158/1535-7163.MCT-12-0993-T. [DOI] [PubMed] [Google Scholar]
- 82.Flinn I.W., O’Brien S., Kahl B., Patel M., Oki Y., Foss F.F., Porcu P., Jones J., Burger J.A., Jain N., et al. Duvelisib, a novel oral dual inhibitor of PI3K-δ, γ, is clinically active in advanced hematologic malignancies. Blood. 2018;131:877–887. doi: 10.1182/blood-2017-05-786566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Krause G., Hassenrück F., Hallek M. Copanlisib for treatment of B-cell malignancies: The development of a PI3K inhibitor with considerable differences to idelalisib. Drug Des. Dev. Ther. 2018;12:2577–2590. doi: 10.2147/DDDT.S142406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Blair H.A. Duvelisib: First global approval. Drugs. 2018;78:1847–1853. doi: 10.1007/s40265-018-1013-4. [DOI] [PubMed] [Google Scholar]
- 85.European Medicines Agency COPIKTRA (Duvelisib) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/copiktra-epar-medicine-overview_en.pdf.
- 86.Markham A. Copanlisib: First global approval. Drugs. 2017;77:2057–2062. doi: 10.1007/s40265-017-0838-6. [DOI] [PubMed] [Google Scholar]
- 87.European Medicines Agency ALIQOPA (Copanlisib) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/18/2064-public-summary-opinion-orphan-designation-copanlisib-treatment-marginal-zone-lymphoma_en.pdf.
- 88.Markham A. Idelalisib: First global approval. Drugs. 2014;74:1701–1707. doi: 10.1007/s40265-014-0285-6. [DOI] [PubMed] [Google Scholar]
- 89.European Medicines Agency ZYDELIG (Idelalisib) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/zydelig-epar-medicine-overview_en.pdf.
- 90.Roberts S., Gibb A. Chapter 1–Introduction to enzymes, receptors and the action of small molecule drugs. In: Ganellin C., Roberts S., Jefferis R., editors. Introduction to Biological and Small Molecule Drug Research and Development. Elsevier; Amsterdam, The Netherlands: 2013. pp. 1–55. [DOI] [Google Scholar]
- 91.Bolden J.E., Peart M.J., Johnstone R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 92.Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589): Successes and challenges. Cancer Lett. 2009;280:233–241. doi: 10.1016/j.canlet.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 93.Lee H.Z., Kwitkowski V.E., Del Valle P.L., Ricci M.S., Saber H., Habtemariam B.A., Bullock J., Bloomquist E., Li Shen Y., Chen X.H., et al. FDA approval: Belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Clin. Cancer Res. 2015;21:2666–2670. doi: 10.1158/1078-0432.CCR-14-3119. [DOI] [PubMed] [Google Scholar]
- 94.Laubach J.P., Moreau P., San-Miguel J.F., Richardson P.G. Panobinostat for the treatment of multiple myeloma. Clin. Cancer Res. 2015;21:4767–4773. doi: 10.1158/1078-0432.CCR-15-0530. [DOI] [PubMed] [Google Scholar]
- 95.Zhang L., Zhang J., Jiang Q., Zhang L., Song W. Zinc binding groups for histone deacetylase inhibitors. J. Enzyme Inhib. Med. Chem. 2018;33:714–721. doi: 10.1080/14756366.2017.1417274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yen K., Bittinger M., Su S., Fantin V.R. Cancer-associated IDH mutations: Biomarker and therapeutic opportunities. Oncogene. 2010;29:6409–6417. doi: 10.1038/onc.2010.444. [DOI] [PubMed] [Google Scholar]
- 97.Waitkus M.S., Diplas B.H., Yan H. Isocitrate dehydrogenase mutations in gliomas. Neuro-Oncol. 2016;18:16–26. doi: 10.1093/neuonc/nov136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Golub D., Iyengar N., Dogra S., Wong T., Bready D., Tang K., Modrek A.S., Placantonakis D.G. Mutant isocitrate dehydrogenase inhibitors as targeted cancer therapeutics. Front Oncol. 2019;9:417. doi: 10.3389/fonc.2019.00417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Abou Dalle I., DiNardo C.D. The role of enasidenib in the treatment of mutant IDH2 acute myeloid leukemia. Ther. Adv. Hematol. 2018;9:163–173. doi: 10.1177/2040620718777467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Norsworthy K.J., Luo L., Hsu V., Gudi R., Dorff S.E., Przepiorka D., Deisseroth A., Shen Y.L., Sheth C.M., Charlab R., et al. FDA approval summary: Ivosidenib for relapsed or refractory acute myeloid leukemia with an isocitrate dehydrogenase-1 mutation. Clin. Cancer Res. 2019;25:3205–3209. doi: 10.1158/1078-0432.CCR-18-3749. [DOI] [PubMed] [Google Scholar]
- 101.Reed D.R., Elsarrag R.Z., Morris A.L., Keng M.K. Enasidenib in acute myeloid leukemia: Clinical development and perspectives on treatment. Cancer Manag. Res. 2019;11:8073–8080. doi: 10.2147/CMAR.S162784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Amatangelo M.D., Quek L., Shih A., Stein E.M., Roshal M., David M.D., Marteyn B., Farnoud N.R., de Botton S., Bernard O.A., et al. Enasidenib induces acute myeloid leukemia cell differentiation to promote clinical response. Blood. 2017;130:732–741. doi: 10.1182/blood-2017-04-779447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gan L., Yang Y., Li Q., Feng Y., Liu T., Guo W. Epigenetic regulation of cancer progression by EZH2: From biological insights to therapeutic potential. Biomark. Res. 2018;6:10. doi: 10.1186/s40364-018-0122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Italiano A., Soria J.C., Toulmonde M., Michot J.M., Lucchesi C., Varga A., Coindre J.M., Blakemore S.J., Clawson A., Suttle B., et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018;19:649–659. doi: 10.1016/S1470-2045(18)30145-1. [DOI] [PubMed] [Google Scholar]
- 105.Ribrag V., Soria J.-C., Michot J.-M., Schmitt A., Postel-Vinay S., Bijou F., Thomson B., Keilhack H., Blakemore S.J., Reyderman L., et al. Phase 1 study of Tazemetostat (EPZ-6438), an inhibitor of enhancer of Zeste-homolog 2 (EZH2): Preliminary safety and activity in relapsed or refractory non-hodgkin lymphoma (NHL) patients. Blood. 2015;126:473. doi: 10.1182/blood.V126.23.473.473. [DOI] [Google Scholar]
- 106.Hoy S.M. Tazemetostat: First approval. Drugs. 2020;80:513–521. doi: 10.1007/s40265-020-01288-x. [DOI] [PubMed] [Google Scholar]
- 107.TAZVERIK (Tazemetostat) Prescribing Information. [(accessed on 14 September 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/213400s000lbl.pdf.
- 108.Lyko F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2017;19:81–92. doi: 10.1038/nrg.2017.80. [DOI] [PubMed] [Google Scholar]
- 109.Schapira M. Structural chemistry of human RNA methyltransferases. ACS Chem. Biol. 2016;11:575–582. doi: 10.1021/acschembio.5b00781. [DOI] [PubMed] [Google Scholar]
- 110.Keating G.M. Azacitidine. Drugs. 2012;72:1111–1136. doi: 10.2165/11209430-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 111.VIDAZA (Azacitidine for Injection) Prescribing Information. [(accessed on 15 September 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/050794s011lbl.pdf.
- 112.ONUREG (Azacitidine) Prescribing Information. [(accessed on 15 September 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/214120s000lbl.pdf.
- 113.Garcia-Manero G., Stoltz M.L., Ward M.R., Kantarjian H., Sharma S. A pilot pharmacokinetic study of oral azacitidine. Leukemia. 2008;22:1680–1684. doi: 10.1038/leu.2008.145. [DOI] [PubMed] [Google Scholar]
- 114.European Medicines Agency ONUREG (Azacitidine) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/onureg-epar-medicine-overview_en.pdf.
- 115.European Medicines Agency TAZVERIK (Tazemetostat) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/18/2004-public-summary-opinion-orphan-designation-tazemetostat-treatment-diffuse-large-b-cell-lymphoma_en.pdf.
- 116.Dhillon S. Ivosidenib: First global approval. Drugs. 2018;78:1509–1516. doi: 10.1007/s40265-018-0978-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.European Medicines Agency TIBSOVO (Ivosidenib) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/medicine-qa/questions-answers-withdrawal-marketing-authorisation-tibsovo-ivosidenib_en.pdf.
- 118.Kim E.S. Enasidenib: First global approval. Drugs. 2017;77:1705–1711. doi: 10.1007/s40265-017-0813-2. [DOI] [PubMed] [Google Scholar]
- 119.European Medicines Agency IDHIFA (Enasidenib) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/medicine-qa/questions-answers-withdrawal-application-marketing-authorisation-idhifa-enasidenib_en.pdf.
- 120.Garnock-Jones K.P. Panobinostat: First global approval. Drugs. 2015;75:695–704. doi: 10.1007/s40265-015-0388-8. [DOI] [PubMed] [Google Scholar]
- 121.European Medicines Agency FARYDAK (Panobinostat) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/farydak-epar-summary-public_en.pdf.
- 122.Poole R.M. Belinostat: First global approval. Drugs. 2014;74:1543–1554. doi: 10.1007/s40265-014-0275-8. [DOI] [PubMed] [Google Scholar]
- 123.European Medicines Agency BELEODAQ (Belinostat) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/12/1055-public-summary-opinion-orphan-designation-belinostat-treatment-peripheral-t-cell-lymphoma-nodal/disseminated_en.pdf.
- 124.Scales S.J., de Sauvage F.J. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 2009;30:303–312. doi: 10.1016/j.tips.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 125.Wang C., Wu H., Evron T., Vardy E., Han G.W., Huang X.P., Hufeisen S.J., Mangano T.J., Urban D.J., Katritch V., et al. Structural basis for smoothened receptor modulation and chemoresistance to anticancer drugs. Nat. Commun. 2014;5:4355. doi: 10.1038/ncomms5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Norsworthy K.J., By K., Subramaniam S., Zhuang L., Del Valle P.L., Przepiorka D., Shen Y.L., Sheth C.M., Liu C., Leong R., et al. FDA approval summary: Glasdegib for newly diagnosed acute myeloid leukemia. Clin. Cancer Res. 2019;25:6021–6025. doi: 10.1158/1078-0432.CCR-19-0365. [DOI] [PubMed] [Google Scholar]
- 127.Cortes J.E., Heidel F.H., Hellmann A., Fiedler W., Smith B.D., Robak T., Montesinos P., Pollyea D.A., DesJardins P., Ottmann O., et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia. 2019;33:379–389. doi: 10.1038/s41375-018-0312-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Thomas X., Heiblig M. An evaluation of glasdegib for the treatment of acute myelogenous leukemia. Expert Opin. Pharmacother. 2020;21:523–530. doi: 10.1080/14656566.2020.1713094. [DOI] [PubMed] [Google Scholar]
- 129.Hoy S.M. Glasdegib: First global approval. Drugs. 2019;79:207–213. doi: 10.1007/s40265-018-1047-7. [DOI] [PubMed] [Google Scholar]
- 130.European Medicines Agency DAURISMO (Glasdegib) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/daurismo-epar-medicine-overview_en.pdf.
- 131.Kale J., Osterlund E., Andrews D. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018;25:65–80. doi: 10.1038/cdd.2017.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Besbes S., Mirshahi M., Pocard M., Billard C. New dimension in therapeutic targeting of BCL-2 family proteins. Oncotarget. 2015;6:12862–12871. doi: 10.18632/oncotarget.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mihalyova J., Jelinek T., Growkova K., Hrdinka M., Simicek M., Hajek R. Venetoclax: A new wave in hematooncology. Exp. Hematol. 2018;61:10–25. doi: 10.1016/j.exphem.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 134.VENCLEXTA (Venetoclax Tablets) Prescribing Information. [(accessed on 17 September 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208573s009lbl.pdf.
- 135.Abdul Razak A.R., Mau-Soerensen M., Gabrail N.Y., Gerecitano J.F., Shields A.F., Unger T.J., Saint-Martin J.R., Carlson R., Landesman Y., McCauley D., et al. First-in-class, first-in-human phase I study of Selinexor, a selective inhibitor of nuclear export, in patients with advanced solid tumors. J. Clin. Oncol. 2016;34:4142–4150. doi: 10.1200/JCO.2015.65.3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chari A., Vogl D.T., Gavriatopoulou M., Nooka A.K., Yee A.J., Huff C.A., Moreau P., Dingli D., Cole C., Lonial S., et al. Oral selinexor-dexamethasone for triple-class refractory multiple myeloma. N. Engl. J. Med. 2019;381:727–738. doi: 10.1056/NEJMoa1903455. [DOI] [PubMed] [Google Scholar]
- 137.Vogl D.T., Dingli D., Cornell R.F., Huff C.A., Jagannath S., Bhutani D., Zonder J., Baz R., Nooka A., Richter J., et al. Selective inhibition of nuclear export with oral selinexor for treatment of relapsed or refractory multiple myeloma. J. Clin. Oncol. 2018;36:859–866. doi: 10.1200/JCO.2017.75.5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.XPOVIO (Selinexor) Prescribing Information. [(accessed on 20 September 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212306s005lbl.pdf.
- 139.Grice G.L., Nathan J.A. The recognition of ubiquitinated proteins by the proteasome. Cell. Mol. Life Sci. 2016;73:3497–3506. doi: 10.1007/s00018-016-2255-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Orlowski M., Wilk S. Catalytic activities of the 20 S proteasome, a multicatalytic proteinase complex. Arch. Biochem. Biophys. 2000;383:1–16. doi: 10.1006/abbi.2000.2036. [DOI] [PubMed] [Google Scholar]
- 141.Tanaka K. The proteasome: Overview of structure and functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009;85:12–36. doi: 10.2183/pjab.85.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.NINLARO (Ixazomib) Prescribing Information. [(accessed on 21 September 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/208462lbl.pdf.
- 143.KYPROLIS (Carfilzomib) Prescribing Information. [(accessed on 21 September 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/202714s030lbl.pdf.
- 144.Muz B., Ghazarian R.N., Ou M., Luderer M.J., Kusdono H.D., Azab A.K. Spotlight on ixazomib: Potential in the treatment of multiple myeloma. Drug Des. Dev. Ther. 2016;10:217–226. doi: 10.2147/DDDT.S93602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kuhn D.J., Chen Q., Voorhees P.M., Strader J.S., Shenk K.D., Sun C.M., Demo S.D., Bennett M.K., van Leeuwen F.W., Chanan-Khan A.A., et al. Potent activity of carfilzomib, a novel, irreversible inhibitor of the ubiquitin-proteasome pathway, against preclinical models of multiple myeloma. Blood. 2007;110:3281–3290. doi: 10.1182/blood-2007-01-065888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gentile M., Offidani M., Vigna E., Corvatta L., Recchia A.G., Morabito L., Morabito F., Gentili S. Ixazomib for the treatment of multiple myeloma. Expert Opin. Investig. Drugs. 2015;24:1287–1298. doi: 10.1517/13543784.2015.1065250. [DOI] [PubMed] [Google Scholar]
- 147.Martino E., Casamassima G., Castiglione S., Cellupica E., Pantalone S., Papagni F., Rui M., Siciliano A.M., Collina S. Vinca alkaloids and analogues as anti-cancer agents: Looking back, peering ahead. Bioorg. Med. Chem. Lett. 2018;28:2816–2826. doi: 10.1016/j.bmcl.2018.06.044. [DOI] [PubMed] [Google Scholar]
- 148.Silverman J.A., Deitcher S.R. Marqibo® (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother. Pharmacol. 2013;71:555–564. doi: 10.1007/s00280-012-2042-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Syed Y.Y. Selinexor: First global approval. Drugs. 2019;79:1485–1494. doi: 10.1007/s40265-019-01188-9. [DOI] [PubMed] [Google Scholar]
- 150.European Medicines Agency NEXPOVIO (Selinexor) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/nexpovio-epar-medicine-overview_en-1.pdf.
- 151.Deeks E.D. Venetoclax: First global approval. Drugs. 2016;76:979–987. doi: 10.1007/s40265-016-0596-x. [DOI] [PubMed] [Google Scholar]
- 152.European Medicines Agency VENCLYXTO (Venetoclax) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/venclyxto-epar-medicine-overview_en.pdf.
- 153.Shirley M. Ixazomib: First global approval. Drugs. 2016;76:405–411. doi: 10.1007/s40265-016-0548-5. [DOI] [PubMed] [Google Scholar]
- 154.European Medicines Agency NINLARO (Ixazomib) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/ninlaro-epar-summary-public_en.pdf.
- 155.Waterhouse D.N., Madden T.D., Cullis P.R., Bally M.B., Mayer L.D., Webb M.S. Preparation, characterization, and biological analysis of liposomal formulations of vincristine. Methods Enzymol. 2005;391:40–57. doi: 10.1016/S0076-6879(05)91002-1. [DOI] [PubMed] [Google Scholar]
- 156.Davis T., Farag S.S. Treating relapsed or refractory Philadelphia chromosome-negative acute lymphoblastic leukemia: Liposome-encapsulated vincristine. Int. J. Nanomed. 2013;8:3479–3488. doi: 10.2147/IJN.S47037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.European Medicines Agency MARQIBO (Vincristine Sulfate LIPOSOME Injection) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/08/555-public-summary-positive-opinion-orphan-designation-vincristine-sulphate-liposomes-treatment_en.pdf.
- 158.Herndon T.M., Deisseroth A., Kaminskas E., Kane R.C., Koti K.M., Rothmann M.D., Habtemariam B., Bullock J., Bray J.D., Hawes J., et al. Food and drug administration approval: Carfilzomib for the treatment of multiple myeloma. Clin. Cancer Res. 2013;19:4559–4563. doi: 10.1158/1078-0432.CCR-13-0755. [DOI] [PubMed] [Google Scholar]
- 159.European Medicines Agency KYPROLIS (Carfilzomib) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/kyprolis-epar-medicine-overview_en.pdf.
- 160.Clark D., Pazdernik N., McGehee M. Chapter 13—Protein synthesis. In: Clark D., Pazdernik N., McGehee M., editors. Molecular Biology. 3rd ed. Elsevier; Amsterdam, The Netherlands: 2019. pp. 397–444. [DOI] [Google Scholar]
- 161.SYNRIBO (Omacetaxine Mepesuccinate) Prescribing Information. [(accessed on 24 September 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/203585s005lbl.pdf.
- 162.Kim T.D., Frick M., le Coutre P. Omacetaxine mepesuccinate for the treatment of leukemia. Expert Opin. Pharmacother. 2011;12:2381–2392. doi: 10.1517/14656566.2011.613378. [DOI] [PubMed] [Google Scholar]
- 163.Chen Y., Li S. Omacetaxine mepesuccinate in the treatment of intractable chronic myeloid leukemia. OncoTargets Ther. 2014;7:177–186. doi: 10.2147/OTT.S41786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Alvandi F., Kwitkowski V.E., Ko C.W., Rothmann M.D., Ricci S., Saber H., Ghosh D., Brown J., Pfeiler E., Chikhale E., et al. Food and Drug Administration approval summary: Omacetaxine mepesuccinate as treatment for chronic myeloid leukemia. Oncologist. 2014;19:94–99. doi: 10.1634/theoncologist.2013-0077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.U.S. Food and Drug Administration NDA 203585Orig1s000. [(accessed on 2 December 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203585Orig1s000SumR.pdf.
- 166.Liliemark J., Pettersson B., Engberg B., Lafolie P., Masquelier M., Peterson C. On the paradoxically concentration-dependent metabolism of 6-mercaptopurine in WEHI-3b murine leukemia cells. Cancer Res. 1990;50:108–112. [PubMed] [Google Scholar]
- 167.Gerbek T., Ebbesen M., Nersting J., Frandsen T.L., Appell M.L., Schmiegelow K. Role of TPMT and ITPA variants in mercaptopurine disposition. Cancer Chemother. Pharmacol. 2018;81:579–586. doi: 10.1007/s00280-018-3525-8. [DOI] [PubMed] [Google Scholar]
- 168.Bökkerink J.P., Stet E.H., De Abreu R.A., Damen F.J., Hulscher T.W., Bakker M.A., van Baal J.A. 6-Mercaptopurine: Cytotoxicity and biochemical pharmacology in human malignant T-lymphoblasts. Biochem. Pharmacol. 1993;45:1455–1463. doi: 10.1016/0006-2952(93)90045-X. [DOI] [PubMed] [Google Scholar]
- 169.Moon W., Loftus E.V. Review article: Recent advances in pharmacogenetics and pharmacokinetics for safe and effective thiopurine therapy in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2016;43:863–883. doi: 10.1111/apt.13559. [DOI] [PubMed] [Google Scholar]
- 170.PURIXAN (Mercaptopurine) Prescribing Information. [(accessed on 27 September 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/205919s000lbl.pdf.
- 171.Mulla H., Leary A., White P., Pandya H. A step toward more accurate dosing for mercaptopurine in childhood acute lymphoblastic leukemia. J. Clin. Pharmacol. 2012;52:1610–1613. doi: 10.1177/0091270011423663. [DOI] [PubMed] [Google Scholar]
- 172.Norman P. Orphan drug approvals of 2014: Europe and the United States. Expert Opin. Orphan Drugs. 2015;3:445–455. doi: 10.1517/21678707.2015.1022530. [DOI] [Google Scholar]
- 173.European Medicines Agency XALUPRINE (Mercaptopurine) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/xaluprine-epar-summary-public_en.pdf.
- 174.Raje N., Anderson K.C. Thalidomide and immunomodulatory drugs as cancer therapy. Curr. Opin. Oncol. 2002;14:635–640. doi: 10.1097/00001622-200211000-00008. [DOI] [PubMed] [Google Scholar]
- 175.POMALYST (Pomalidomide) Prescribing Information. [(accessed on 28 September 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/204026s024lbl.pdf.
- 176.Chanan-Khan A.A., Swaika A., Paulus A., Kumar S.K., Mikhael J.R., Rajkumar S.V., Dispenzieri A., Lacy M.Q. Pomalidomide: The new immunomodulatory agent for the treatment of multiple myeloma. Blood Cancer J. 2013;3:e143. doi: 10.1038/bcj.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Galustian C., Meyer B., Labarthe M.C., Dredge K., Klaschka D., Henry J., Todryk S., Chen R., Muller G., Stirling D., et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol. Immunother. 2009;58:1033–1045. doi: 10.1007/s00262-008-0620-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lopez-Girona A., Mendy D., Ito T., Miller K., Gandhi A.K., Kang J., Karasawa S., Carmel G., Jackson P., Abbasian M., et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. 2012;26:2326–2335. doi: 10.1038/leu.2012.119. Erratum in: Leukemia 2012, 26, 2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Elkinson S., McCormack P.L. Pomalidomide: First global approval. Drugs. 2013;73:595–604. doi: 10.1007/s40265-013-0047-x. [DOI] [PubMed] [Google Scholar]
- 180.European Medicines Agency IMNOVID (Pomalidomide) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/imnovid-epar-medicine-overview_en.pdf.
- 181.Moon C., Oh E. Rationale and strategies for formulation development of oral fixed dose combination drug products. J. Pharm. Investig. 2016;46:615–631. doi: 10.1007/s40005-016-0286-4. [DOI] [Google Scholar]
- 182.Pillai G. Chapter 9—Nanotechnology toward treating cancer: A comprehensive review. In: Mohapatra S., Ranjan S., Dasgupta N., Mishra R., Thomas S., editors. Micro and Nano Technologies, Applications of Targeted Nano Drugs and Delivery Systems. Elsevier; Amsterdam, The Netherlands: 2019. pp. 221–256. [DOI] [Google Scholar]
- 183.El-Subbagh H., Al-Badr A. Chapter 2—Cytarabine. In: Brittain H., editor. Profiles of Drug Substances, Excipients and Related Methodology. Volume 34. Academic Press; Cambridge, MA, USA: 2009. pp. 37–113. [DOI] [PubMed] [Google Scholar]
- 184.Cytarabine NDA #016793—Drugs@FDA: FDA-Approved Drugs. [(accessed on 29 September 2021)]; Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=016793.
- 185.Kwok K., Vincent E., Gibson J. 36—Antineoplastic drugs. In: Dowd F., Johnson B., Mariotti A., editors. Pharmacology and Therapeutics for Dentistry. 7th ed. Mosby; Maryland, MI, USA: 2017. pp. 530–562. [DOI] [Google Scholar]
- 186.Cerubidine NDA #050484—Drugs@FDA: FDA-Approved Drugs. [(accessed on 29 September 2021)]; Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=050484.
- 187.Lancet J.E., Uy G.L., Cortes J.E., Newell L.F., Lin T.L., Ritchie E.K., Stuart R.K., Strickland S.A., Hogge D., Solomon S.R., et al. CPX-351 (cytarabine and daunorubicin) liposome for injection versus conventional cytarabine plus daunorubicin in older patients with newly diagnosed secondary acute myeloid leukemia. J. Clin. Oncol. 2018;36:2684–2692. doi: 10.1200/JCO.2017.77.6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Tzogani K., Penttilä K., Lapveteläinen T., Hemmings R., Koenig J., Freire J., Márcia S., Cole S., Coppola P., Flores B., et al. EMA review of daunorubicin and cytarabine encapsulated in liposomes (Vyxeos, CPX-351) for the treatment of adults with newly diagnosed, therapy-related acute myeloid leukemia or acute myeloid leukemia with myelodysplasia-related changes. Oncologist. 2020;25:e1414–e1420. doi: 10.1634/theoncologist.2019-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.VYXEOS (Daunorubicin and Cytarabine) Prescribing Information. [(accessed on 4 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209401s000lbl.pdf.
- 190.Decitabine NDA #021790—Drugs@FDA: FDA-Approved Drugs. [(accessed on 4 October 2021)]; Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=021790.
- 191.Plimack E.R., Kantarjian H.M., Issa J.P. Decitabine and its role in the treatment of hematopoietic malignancies. Leuk. Lymphoma. 2007;48:1472–1481. doi: 10.1080/10428190701471981. [DOI] [PubMed] [Google Scholar]
- 192.INQOVI (Decitabine and Cedazuridine) Prescribing Information. [(accessed on 4 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212576s000lbl.pdf.
- 193.Dhillon S. Decitabine/Cedazuridine: First approval. Drugs. 2020;80:1373–1378. doi: 10.1007/s40265-020-01389-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Krauss A.C., Gao X., Li L., Manning M.L., Patel P., Fu W., Janoria K.G., Gieser G., Bateman D.A., Przepiorka D., et al. FDA approval summary: (Daunorubicin and Cytarabine) liposome for injection for the treatment of adults with high-risk acute myeloid leukemia. Clin. Cancer Res. 2019;25:2685–2690. doi: 10.1158/1078-0432.CCR-18-2990. [DOI] [PubMed] [Google Scholar]
- 195.European Medicines Agency VYXEOS (Daunorubicin and Cytarabine) [(accessed on 2 December 2021)]; Available online: https://www.ema.europa.eu/en/documents/overview/vyxeos-liposomal-epar-medicine-overview_en.pdf.
- 196.Bayer V. An overview of monoclonal antibodies. Semin. Oncol. Nurs. 2019;35:150927. doi: 10.1016/j.soncn.2019.08.006. [DOI] [PubMed] [Google Scholar]
- 197.Zahavi D., Weiner L. Monoclonal antibodies in cancer therapy. Antibodies. 2020;9:34. doi: 10.3390/antib9030034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Wang K., Wei G., Liu D. CD19: A biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012;1:36. doi: 10.1186/2162-3619-1-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Hoy S.M. Tafasitamab: First approval. Drugs. 2020;80:1731–1737. doi: 10.1007/s40265-020-01405-w. [DOI] [PubMed] [Google Scholar]
- 200.MONJUVI (Tafasitamab-cxix) Prescribing Information. [(accessed on 7 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761163s000lbl.pdf.
- 201.Uchida J., Lee Y., Hasegawa M., Liang Y., Bradney A., Oliver J.A., Bowen K., Steeber D.A., Haas K.M., Poe J.C., et al. Mouse CD20 expression and function. Int. Immunol. 2004;16:119–129. doi: 10.1093/intimm/dxh009. [DOI] [PubMed] [Google Scholar]
- 202.Niederfellner G., Lammens A., Mundigl O., Georges G.J., Schaefer W., Schwaiger M., Franke A., Wiechmann K., Jenewein S., Slootstra J.W., et al. Epitope characterization and crystal structure of GA101 provide insights into the molecular basis for type I/II distinction of CD20 antibodies. Blood. 2011;118:358–367. doi: 10.1182/blood-2010-09-305847. [DOI] [PubMed] [Google Scholar]
- 203.Mössner E., Brünker P., Moser S., Püntener U., Schmidt C., Herter S., Grau R., Gerdes C., Nopora A., van Puijenbroek E., et al. Increasing the efficacy of CD20 antibody therapy through the engineering of a new type II anti-CD20 antibody with enhanced direct and immune effector cell-mediated B-cell cytotoxicity. Blood. 2010;115:4393–4402. doi: 10.1182/blood-2009-06-225979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Herter S., Herting F., Mundigl O., Waldhauer I., Weinzierl T., Fauti T., Muth G., Ziegler-Landesberger D., Van Puijenbroek E., Lang S., et al. Preclinical activity of the type II CD20 antibody GA101 (obinutuzumab) compared with rituximab and ofatumumab in vitro and in xenograft models. Mol. Cancer Ther. 2013;12:2031–2042. doi: 10.1158/1535-7163.MCT-12-1182. [DOI] [PubMed] [Google Scholar]
- 205.TRUXIMA (Rituximab-abbs) Prescribing Information. [(accessed on 8 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761088s000lbl.pdf.
- 206.RUXIENCE (Rituximab-pvvr) Prescribing Information. [(accessed on 8 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761103s000lbl.pdf.
- 207.RIABNI (Rituximab-arrx) Prescribing Information. [(accessed on 8 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761140s000lbl.pdf.
- 208.GAZYVA (Obinutuzumab) Prescribing Information. [(accessed on 8 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125486s017s018lbl.pdf.
- 209.Partida-Sánchez S., Cockayne D., Monard S., Jacobson E.L., Oppenheimer N., Garvy B., Kusser K., Goodrich S., Howard M., Harmsen A., et al. Cyclic ADP-ribose production by CD38 regulates intracellular calcium release, extracellular calcium influx and chemotaxis in neutrophils and is required for bacterial clearance in vivo. Nat. Med. 2001;7:1209–1216. doi: 10.1038/nm1101-1209. [DOI] [PubMed] [Google Scholar]
- 210.Martin T.G., Corzo K., Chiron M., van de Velde H., Abbadessa G., Campana F., Solanki M., Meng R., Lee H., Wiederschain D., et al. Therapeutic opportunities with pharmacological inhibition of CD38 with Isatuximab. Cells. 2019;8:1522. doi: 10.3390/cells8121522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Van de Donk N.W.C.J., Usmani S.Z. CD38 antibodies in multiple myeloma: Mechanisms of action and modes of resistance. Front. Immunol. 2018;9:2134. doi: 10.3389/fimmu.2018.02134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Richardson P.G., Beksaç M., Špička I., Mikhael J. Isatuximab for the treatment of relapsed/refractory multiple myeloma. Expert Opin. Biol. Ther. 2020;20:1395–1404. doi: 10.1080/14712598.2021.1841747. [DOI] [PubMed] [Google Scholar]
- 213.DARZALEX (Daratumumab) Prescribing Information. [(accessed on 11 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761036s035lbl.pdf.
- 214.SARCLISA (Isatuximab-irfc) Prescribing Information. [(accessed on 11 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761113s003lbl.pdf.
- 215.DARZALEX FASPRO (Daratumumab and Hyaluronidase-fihj) Prescribing Information. [(accessed on 11 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761145s002lbl.pdf.
- 216.Malaer J.D., Mathew P.A. CS1 (SLAMF7, CD319) is an effective immunotherapeutic target for multiple myeloma. Am. J. Cancer Res. 2017;7:1637–1641. [PMC free article] [PubMed] [Google Scholar]
- 217.Magen H., Muchtar E. Elotuzumab: The first approved monoclonal antibody for multiple myeloma treatment. Ther. Adv. Hematol. 2016;7:187–195. doi: 10.1177/2040620716652862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.EMPLICITI (Elotuzumab) Prescribing Information. [(accessed on 12 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761035s008lbl.pdf.
- 219.Guan J., Lim K.S., Mekhail T., Chang C.C. Programmed death ligand-1 (PD-L1) expression in the programmed death receptor-1 (PD-1)/PD-L1 blockade: A key player against various cancers. Arch. Pathol. Lab. Med. 2017;141:851–861. doi: 10.5858/arpa.2016-0361-RA. [DOI] [PubMed] [Google Scholar]
- 220.Prasad V., Kaestner V. Nivolumab and pembrolizumab: Monoclonal antibodies against programmed cell death-1 (PD-1) that are interchangeable. Semin. Oncol. 2017;44:132–135. doi: 10.1053/j.seminoncol.2017.06.007. [DOI] [PubMed] [Google Scholar]
- 221.OPDIVO (Nivolumab) Prescribing Information. [(accessed on 15 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125554s058lbl.pdf.
- 222.KEYTRUDA (Pembrolizumab) Prescribing Information. [(accessed on 15 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/125514s096lbl.pdf.
- 223.Wu X.S., Lonsdorf A.S., Hwang S.T. Cutaneous T-cell lymphoma: Roles for chemokines and chemokine receptors. J. Investig. Dermatol. 2009;129:1115–1119. doi: 10.1038/jid.2009.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Watson S., Marx J.B. Mogamulizumab-kpkc: A novel therapy for the treatment of cutaneous T-cell lymphoma. J. Adv. Pract. Oncol. 2019;10:883–888. doi: 10.6004/jadpro.2019.10.8.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.European Medicines Agency. [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/minjuvi-epar-medicine-overview_en.pdf.
- 226.Cai H.H. Therapeutic monoclonal antibodies approved by FDA in 2020. Clin. Res. Immunol. 2021;4:1–2. [Google Scholar]
- 227.Dhillon S. Isatuximab: First approval. Drugs. 2020;80:905–912. doi: 10.1007/s40265-020-01311-1. [DOI] [PubMed] [Google Scholar]
- 228.European Medicines Agency SARCLISA (Isatuximab-irfc) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/sarclisa-epar-medicine-overview_en.pdf.
- 229.European Medicines Agency RUXIENCE (Rituximab-pvvr) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/ruxience-epar-medicine-overview_en.pdf.
- 230.U.S. Food and Drug Administration FDA Approves First Biosimilar for Treatment of Adult Patients with Non-Hodgkin’s Lymphoma. [(accessed on 10 September 2021)]; Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-biosimilar-treatment-adult-patients-non-hodgkins-lymphoma.
- 231.European Medicines Agency TRUXIMA (Rituximab-Abbs) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/truxima-epar-medicine-overview_en.pdf.
- 232.Kasamon Y.L., Chen H., de Claro R.A., Nie L., Ye J., Blumenthal G.M., Farrell A.T., Pazdur R. FDA approval summary: Mogamulizumab-kpkc for mycosis fungoides and Sézary syndrome. Clin. Cancer Res. 2019;25:7275–7280. doi: 10.1158/1078-0432.CCR-19-2030. [DOI] [PubMed] [Google Scholar]
- 233.European Medicines Agency POTELIGEO (Mogamulizumab-Kpkc) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/poteligeo-epar-medicine-overview_en.pdf.
- 234.Markham A. Elotuzumab: First global approval. Drugs. 2016;76:397–403. doi: 10.1007/s40265-016-0540-0. [DOI] [PubMed] [Google Scholar]
- 235.European Medicines Agency EMPLICITI (Elotuzumab) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/empliciti-epar-medicine-overview_en.pdf.
- 236.McKeage K. Daratumumab: First global approval. Drugs. 2016;76:275–281. doi: 10.1007/s40265-015-0536-1. [DOI] [PubMed] [Google Scholar]
- 237.European Medicines Agency DARZALEX (Daratumumab) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/darzalex-epar-medicine-overview_en.pdf.
- 238.Hazarika M., Chuk M.K., Theoret M.R., Mushti S., He K., Weis S.L., Putman A.H., Helms W.S., Cao X., Li H., et al. FDA approval summary: Nivolumab for treatment of unresectable or metastatic melanoma following progression on Ipilimumab. Clin. Cancer Res. 2017;23:3484–3488. doi: 10.1158/1078-0432.CCR-16-0712. [DOI] [PubMed] [Google Scholar]
- 239.OPDIVO (Nivolumab) Prescribing Information. [(accessed on 29 November 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/125554s019lbl.pdf.
- 240.European Medicines Agency OPDIVO (Nivolumab) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/opdivo-epar-medicine-overview_en.pdf.
- 241.Poole R.M. Pembrolizumab: First global approval. Drugs. 2014;74:1973–1981. doi: 10.1007/s40265-014-0314-5. [DOI] [PubMed] [Google Scholar]
- 242.U.S. Food and Drug Administration FDA Approves Pembrolizumab for Treatment of Relapsed or Refractory PMBCL. [(accessed on 10 September 2021)]; Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-pembrolizumab-treatment-relapsed-or-refractory-pmbcl.
- 243.U.S. Food and Drug Administration Pembrolizumab (KEYTRUDA) for Classical Hodgkin Lymphoma. [(accessed on 10 September 2021)]; Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/pembrolizumab-keytruda-classical-hodgkin-lymphoma.
- 244.European Medicines Agency KEYTRUDA (Pembrolizumab) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/keytruda-epar-medicine-overview_en.pdf.
- 245.Cameron F., McCormack P.L. Obinutuzumab: First global approval. Drugs. 2014;74:147–154. doi: 10.1007/s40265-013-0167-3. [DOI] [PubMed] [Google Scholar]
- 246.European Medicines Agency GAZYVARO (Obinutuzumab) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/gazyvaro-epar-summary-public_en.pdf.
- 247.Van Spriel A.B., van Ojik H.H., van de Winkel J.G.J. Immunotherapeutic perspective for bispecific antibodies. Immunol. Today. 2000;21:391–397. doi: 10.1016/S0167-5699(00)01659-5. [DOI] [PubMed] [Google Scholar]
- 248.Newman M.J., Benani D.J. A review of blinatumomab, a novel immunotherapy. J. Oncol. Pharm. Pract. 2016;22:639–645. doi: 10.1177/1078155215618770. [DOI] [PubMed] [Google Scholar]
- 249.Sanford M. Blinatumomab: First global approval. Drugs. 2015;75:321–327. doi: 10.1007/s40265-015-0356-3. [DOI] [PubMed] [Google Scholar]
- 250.European Medicines Agency BLINCYTO (Blinatumomab) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/blincyto-epar-medicine-overview_en.pdf.
- 251.Yaghoubi S., Karimi M.H., Lotfinia M., Gharibi T., Mahi-Birjand M., Kavi E., Hosseini F., Sineh Sepehr K., Khatami M., Bagheri N., et al. Potential drugs used in the antibody–drug conjugate (ADC) architecture for cancer therapy. J. Cell Physiol. 2019;235:31–64. doi: 10.1002/jcp.28967. [DOI] [PubMed] [Google Scholar]
- 252.Sato S., Miller A.S., Inaoki M., Bock C.B., Jansen P.J., Tang M.L., Tedder T.F. CD22 is both a positive and negative regulator of B lymphocyte antigen receptor signal transduction: Altered signaling in CD22-deficient mice. Immunity. 1996;5:551–562. doi: 10.1016/S1074-7613(00)80270-8. [DOI] [PubMed] [Google Scholar]
- 253.Shor B., Gerber H.P., Sapra P. Preclinical and clinical development of inotuzumab-ozogamicin in hematological malignancies. Mol. Immunol. 2015;67 Pt A:107–116. doi: 10.1016/j.molimm.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 254.Godwin C., Gale R., Walter R. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia. 2017;31:1855–1868. doi: 10.1038/leu.2017.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.BESPONSA (Inotuzumab Ozogamicin) Prescribing Information. [(accessed on 13 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761040s000lbl.pdf.
- 256.Sabattini E., Pizzi M., Tabanelli V., Baldin P., Sacchetti C.S., Agostinelli C., Zinzani P.L., Pileri S.A. CD30 expression in peripheral T-cell lymphomas. Haematologica. 2013;98:e81–e82. doi: 10.3324/haematol.2013.084913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Fichtner M., Dreyling M., Binder M., Trepel M. The role of B cell antigen receptors in mantle cell lymphoma. J. Hematol. Oncol. 2017;10:164. doi: 10.1186/s13045-017-0533-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Younes A., Bartlett N.L., Leonard J.P., Kennedy D.A., Lynch C.M., Sievers E.L., Forero-Torres A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N. Engl. J. Med. 2010;363:1812–1821. doi: 10.1056/NEJMoa1002965. [DOI] [PubMed] [Google Scholar]
- 259.Choi Y., Diefenbach C.S. Polatuzumab Vedotin: A new target for B cell malignancies. Curr. Hematol. Malig. Rep. 2020;15:125–129. doi: 10.1007/s11899-020-00572-7. [DOI] [PubMed] [Google Scholar]
- 260.ADCETRIS (Brentuximab Vedotin) Prescribing Information. [(accessed on 18 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/125388s097lbl.pdf.
- 261.Picardi M., Della Pepa R., Giordano C., Pugliese N., Mortaruolo C., Trastulli F., Rascato M.G., Cappuccio I., Raimondo M., Memoli M., et al. Brentuximab vedotin followed by bendamustine supercharge for refractory or relapsed Hodgkin lymphoma. Blood Adv. 2019;3:1546–1552. doi: 10.1182/bloodadvances.2019000123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.POLIVY (Polatuzumab Vedotin-Piiq) Prescribing Information. [(accessed on 18 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761121s000lbl.pdf.
- 263.Cho S.F., Anderson K.C., Tai Y.T. Targeting B cell maturation antigen (BCMA) in multiple myeloma: Potential uses of BCMA-based immunotherapy. Front. Immunol. 2018;9:1821. doi: 10.3389/fimmu.2018.01821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.BLENREP (Belantamab Mafodotin-Blmf) Prescribing Information. [(accessed on 18 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761158s000lbl.pdf.
- 265.Markham A. Belantamab Mafodotin: First approval. Drugs. 2020;80:1607–1613. doi: 10.1007/s40265-020-01404-x. [DOI] [PubMed] [Google Scholar]
- 266.European Medicines Agency BLENREP (Belantamab Mafodotin-Blmf) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/blenrep-epar-medicine-overview_en.pdf.
- 267.Deeks E.D. Polatuzumab Vedotin: First global approval. Drugs. 2019;79:1467–1475. doi: 10.1007/s40265-019-01175-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.European Medicines Agency POLIVY (Polatuzumab Vedotin-piiq) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/polivy-epar-medicine-overview_en.pdf.
- 269.Lamb Y.N. Inotuzumab Ozogamicin: First global approval. Drugs. 2017;77:1603–1610. doi: 10.1007/s40265-017-0802-5. [DOI] [PubMed] [Google Scholar]
- 270.European Medicines Agency BESPONSA (Inotuzumab Ozogamicin) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/besponsa-epar-summary-public_en.pdf.
- 271.De Claro R.A., McGinn K., Kwitkowski V., Bullock J., Khandelwal A., Habtemariam B., Ouyang Y., Saber H., Lee K., Koti K., et al. Food and Drug Administration approval summary: Brentuximab vedotin for the treatment of relapsed Hodgkin lymphoma or relapsed systemic anaplastic large-cell lymphoma. Clin. Cancer Res. 2012;18:5845–5849. doi: 10.1158/1078-0432.CCR-12-1803. [DOI] [PubMed] [Google Scholar]
- 272.European Medicines Agency ADCETRIS (Brentuximab Vedotin) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/adcetris-epar-medicine-overview_en.pdf.
- 273.Kreitman R.J. Immunotoxins in cancer therapy. Curr. Opin. Immunol. 1999;11:570–578. doi: 10.1016/S0952-7915(99)00005-9. [DOI] [PubMed] [Google Scholar]
- 274.LUMOXITI (Moxetumomab Pasudotox-Tdfk) Prescribing Information. [(accessed on 19 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761104s000lbl.pdf.
- 275.Kreitman R.J., Pastan I. Antibody fusion proteins: Anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin. Cancer Res. 2011;17:6398–6405. doi: 10.1158/1078-0432.CCR-11-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Dhillon S. Moxetumomab Pasudotox: First global approval. Drugs. 2018;78:1763–1767. doi: 10.1007/s40265-018-1000-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.European Medicines Agency LUMOXITI (Moxetumomab Pasudotox-tdfk) [(accessed on 2 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/lumoxiti-epar-medicine-overview_en.pdf.
- 278.Wang Z., Xie Q., Zhou H., Zhang M., Shen J., Ju D. Amino acid degrading enzymes and autophagy in cancer therapy. Front. Pharmacol. 2021;11:582587. doi: 10.3389/fphar.2020.582587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Salzer W.L., Asselin B.L., Plourde P.V., Corn T., Hunger S.P. Development of asparaginase Erwinia chrysanthemi for the treatment of acute lymphoblastic leukemia. Ann. N. Y. Acad. Sci. 2014;1329:81–92. doi: 10.1111/nyas.12496. [DOI] [PubMed] [Google Scholar]
- 280.Li R.J., Jin R., Liu C., Cao X., Manning M.L., Di X.M., Przepiorka D., Namuswe F., Deisseroth A., Goldberg K.B., et al. FDA approval summary: Calaspargase Pegol-mknl For treatment of acute lymphoblastic leukemia in children and young adults. Clin. Cancer Res. 2020;26:328–331. doi: 10.1158/1078-0432.CCR-19-1255. [DOI] [PubMed] [Google Scholar]
- 281.ERWINAZE (Asparaginase Erwinia Chrysanthemi) Prescribing Information. [(accessed on 20 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/125359lbl.pdf.
- 282.ASPARLAS (Calaspargase Pegol—mknl) Prescribing Information. [(accessed on 20 October 2021)]; Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/761102s000lbl.pdf.
- 283.Keating G.M. Asparaginase Erwinia chrysanthemi (Erwinaze®): A guide to its use in acute lymphoblastic leukemia in the USA. BioDrugs. 2013;27:413–418. doi: 10.1007/s40259-013-0051-4. [DOI] [PubMed] [Google Scholar]
- 284.Huang S.M., Abernethy D.R., Wang Y., Zhao P., Zineh I. The utility of modeling and simulation in drug development and regulatory review. J. Pharm. Sci. 2013;102:2912–2923. doi: 10.1002/jps.23570. [DOI] [PubMed] [Google Scholar]
- 285.European Medicines Agency List of Nationally Authorised Medicinal Products. [(accessed on 1 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/psusa/asparaginase-crisantaspase-pegaspargase-nationally-authorised-products-list-nationally-authorised/00003161/201808_en.pdf.
- 286.Yang X., Wang G.-X., Zhou J.-F. CAR T cell therapy for hematological malignancies. Curr. Med. Sci. 2019;39:874–882. doi: 10.1007/s11596-019-2118-z. [DOI] [PubMed] [Google Scholar]
- 287.Han D., Xu Z., Zhuang Y., Ye Z., Qian Q. Current progress in CAR-T cell therapy for hematological malignancies. J. Cancer. 2021;12:326–334. doi: 10.7150/jca.48976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Mueller K.T., Waldron E., Grupp S.A., Levine J.E., Laetsch T.W., Pulsipher M.A., Boyer M.W., August K.J., Hamilton J., Awasthi R., et al. Clinical pharmacology of Tisagenlecleucel in B-cell acute lymphoblastic leukemia. Clin. Cancer Res. 2018;24:6175–6184. doi: 10.1158/1078-0432.CCR-18-0758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.KYMRIAH (Tisagenlecleucel) Prescribing Information. [(accessed on 21 October 2021)]; Available online: https://www.fda.gov/media/107296/download.
- 290.Mian A., Hill B.T. Brexucabtagene autoleucel for the treatment of relapsed/refractory mantle cell lymphoma. Expert Opin. Biol. Ther. 2021;21:435–441. doi: 10.1080/14712598.2021.1889510. [DOI] [PubMed] [Google Scholar]
- 291.Riedell P.A., Bishop M.R. Safety and efficacy of axicabtagene ciloleucel in refractory large B-cell lymphomas. Ther. Adv. Hematol. 2020;11:2040620720902899. doi: 10.1177/2040620720902899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.YESCARTA (Axicabtagene Ciloleucel) Prescribing Information. [(accessed on 23 October 2021)]; Available online: https://www.fda.gov/media/108377/download.
- 293.TECARTUS (Brexucabtagene Autoleucel) Prescribing Information. [(accessed on 23 October 2021)]; Available online: https://www.fda.gov/media/140409/download.
- 294.U.S. Food and Drug Administration FDA Approves First Cell-Based Gene Therapy For Adult Patients with Relapsed or Refractory MCL. [(accessed on 25 October 2021)]; Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-cell-based-gene-therapy-adult-patients-relapsed-or-refractory-mcl.
- 295.European Medicines Agency TECARTUS (Brexucabtagene Autoleucel) [(accessed on 1 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/tecartus-epar-medicine-overview_en.pdf.
- 296.Bouchkouj N., Kasamon Y.L., de Claro R.A., George B., Lin X., Lee S., Blumenthal G.M., Bryan W., McKee A.E., Pazdur R. FDA approval summary: Axicabtagene Ciloleucel for relapsed or refractory large B-cell lymphoma. Clin. Cancer Res. 2019;25:1702–1708. doi: 10.1158/1078-0432.CCR-18-2743. [DOI] [PubMed] [Google Scholar]
- 297.European Medicines Agency YESCARTA (Axicabtagene Ciloleucel) [(accessed on 1 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/yescarta-epar-medicine-overview_en.pdf.
- 298.O’Leary M.C., Lu X., Huang Y., Lin X., Mahmood I., Przepiorka D., Gavin D., Lee S., Liu K., George B., et al. FDA approval summary: Tisagenlecleucel for treatment of patients with relapsed or refractory B-cell precursor acute lymphoblastic leukemia. Clin. Cancer Res. 2019;25:1142–1146. doi: 10.1158/1078-0432.CCR-18-2035. [DOI] [PubMed] [Google Scholar]
- 299.European Medicines Agency KYMRIAH (Tisagenlecleucel) [(accessed on 1 December 2021)]. Available online: https://www.ema.europa.eu/en/documents/overview/kymriah-epar-medicine-overview_en.pdf.
- 300.Plescia J., Moitessier N. Design and discovery of boronic acid drugs. Eur. J. Med. Chem. 2020;195:112270. doi: 10.1016/j.ejmech.2020.112270. [DOI] [PubMed] [Google Scholar]
- 301.Kumari S., Carmona A.V., Tiwari A.K., Trippier P.C. Amide bond bioisosteres: Strategies, synthesis, and successes. J. Med. Chem. 2020;63:12290–12358. doi: 10.1021/acs.jmedchem.0c00530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Liu W., Liang Y., Si X. Hydroxamic acid hybrids as the potential anticancer agents: An overview. Eur. J. Med. Chem. 2020;205:112679. doi: 10.1016/j.ejmech.2020.112679. [DOI] [PubMed] [Google Scholar]
- 303.Tanii H., Hashimoto K. Studies on the mechanism of acute toxicity of nitriles in mice. Arch. Toxicol. 1984;55:47–54. doi: 10.1007/BF00316585. [DOI] [PubMed] [Google Scholar]
- 304.Rosenkranz H.S., Mermelstein R. Mutagenicity and genotoxicity of nitroarenes. All nitro-containing chemicals were not created equal. Mutat. Res. 1983;114:217–267. doi: 10.1016/0165-1110(83)90034-9. [DOI] [PubMed] [Google Scholar]
- 305.Fleming F.F., Yao L., Ravikumar P.C., Funk L., Shook B.C. Nitrile-containing pharmaceuticals: Efficacious roles of the nitrile pharmacophore. J. Med. Chem. 2010;53:7902–7917. doi: 10.1021/jm100762r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Nepali K., Lee H.Y., Liou J.P. Nitro-Group-Containing Drugs. J. Med. Chem. 2019;62:2851–2893. doi: 10.1021/acs.jmedchem.8b00147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.