

Abstract
Ribosome profiling enables sequencing of ribosome-bound fragments of RNA, revealing which transcripts are being translated as well as the position of ribosomes along mRNAs. Although ribosome profiling has been applied to cultured bacterial isolates, its application to uncultured, mixed communities has been challenging. We present MetaRibo-Seq, a protocol that enables the application of ribosome profiling directly to the human fecal microbiome. MetaRibo-Seq is a benchmarked method that includes several modifications to existing ribosome profiling protocols, specifically addressing challenges involving fecal sample storage, purity and input requirements. We also provide a computational workflow to quality control and trim reads, de novo assemble a reference metagenome with metagenomic reads, align MetaRibo-Seq reads to the reference, and assess MetaRibo-Seq library quality (https://github.com/bhattlab/bhattlab_workflows/tree/master/metariboseq). This MetaRibo-Seq protocol enables researchers in standard molecular biology laboratories to study translation in the fecal microbiome in ~5 d.
Introduction
Regulation of gene expression at the level of translation is essential for the adaptability and survival of bacteria. For example, genes involved in translation itself are regulated at a translational level via feedback mechanisms1–5. Translational regulation is critical for generating proteins at the correct stoichiometry for many protein complexes6. This regulation even extends to pathway-specific enzyme stoichiometry in which protein synthesis levels across taxa remain correlated even as transcript abundance and architecture diverges over evolutionary time7. Transcript abundance and protein synthesis rates can differ by orders of magnitude6. Moreover, specific translational regulation has been extensively observed upon a variety of perturbations to bacteria8–14. Taken together, the ability to study translation in bacteria is critical for our understanding of their functions—yet, methods that enable the comprehensive study of translation in microbiomes have been lacking until now.
Existing methods for profiling bacterial translation
During translation, ribosomes protect small fragments of RNA from nuclease digestion. Capitalizing on this observation, ribosome profiling (Ribo-Seq) is an approach that sequences ribosome-protected fragments of mRNA after degrading unprotected mRNA with nuclease. First developed by Ingolia and Weissman for application in yeast, Ribo-Seq enables the study of transcriptome-wide translation and its regulation in vivo15. Many modifications and extensions to the protocol have been implemented since this initial application, enabling the application of this approach to a range of other cell types16.
Ribo-Seq was later applied to bacteria for the first time by Weissman and Bukau17–19. While the method is conceptually similar to the original Ribo-Seq protocol, unique challenges arose upon application of Ribo-Seq to bacteria, such as determining which methods to employ to stop elongation of ribosomes and choices of nucleases to generate ribosome-protected footprints17–19. New applications and modifications are being constantly implemented in bacterial Ribo-Seq, some of which are inspired by advances first discovered in eukaryotic Ribo-Seq. For example, while traditional Ribo-Seq involves enrichment of monosomes, which are single ribosomes bound to RNA, with a sucrose density gradient, protocols using size-exclusion columns have emerged as a replacement for sucrose density gradients. Size-exclusion columns are faster, require less equipment and produce comparable results20. Size-exclusion columns and monosome recovery were later implemented in bacterial Ribo-Seq protocols and demonstrated performance characteristics similar to those observed in eukaryotic Ribo-Seq21.
In addition to adaptations of methods for ribosome recovery, researchers have also experimented with various antibiotics and chemicals that inhibit translation by different mechanisms. Traditionally, bacterial Ribo-Seq was performed after the addition of chloramphenicol, a ribosome-targeting antibiotic that halts translation17–19. Subsequent adaptations have focused on enriching start sites and stop sites, and trying to improve single-codon resolution in bacterial Ribo-Seq signal. For example, antibiotics such as tetracycline22 and, more recently, retapamulin23 have been used to identify translation start sites; similarly, apidaecin, an antimicrobial peptide from bees, has been used to identify translation stop sites24. Unlike Ribo-Seq in eukaryotes, bacterial Ribo-Seq often struggles to achieve single-nucleotide resolution, in part due to the use of chloramphenicol to halt translation25,26. It has been shown that using the endonuclease RelE improves triplet periodicity, or the enrichment of in-frame signal, in bacterial Ribo-Seq25. More recently, high-salt buffers have been shown to halt translation better than chloramphenicol and improve resolution of triplet periodicity and pausing26. Overall, bacterial Ribo-Seq has a number of unique challenges that are continuing to be addressed by the research community.
Development of MetaRibo-Seq
Historically, Ribo-Seq has been performed on one cultured organism at a time. Consequently, our ability to study translation in high throughput has largely been limited to a small group of mostly model organisms in somewhat artificial culturing conditions. It would be advantageous to perform Ribo-Seq on a broader range of organisms, culture-free and on many organisms at once. By modifying existing Ribo-Seq protocols, we have enabled the direct study of translation in microbiomes. Previously, Ribo-Seq had never been implemented on uncultured mixtures of bacteria for three reasons: first, standard Ribo-Seq protocols are implemented on freshly grown bacteria in which culturing and growth conditions can be designed appropriately. It is often unrealistic to control the timing of fecal sample collection, and thus, storage methods for these fecal samples are needed; second, standard initial Ribo-Seq protocols require pure, concentrated bacterial cells. These protocols isolate bacterial cells by centrifugation and/or filtration of pure, cultured cells. Obtaining concentrated and pure bacteria from fecal samples is a challenge; third, standard Ribo-Seq protocols have high input requirements that require large quantities of bacteria. Culturing up to a liter of bacteria is typically necessary to satisfy input requirements.
We developed MetaRibo-Seq to solve these three problems. First, our method is compatible for use on frozen fecal samples stored in RNA preservative, not requiring fresh samples27. Second, we modified standard Ribo-Seq protocols to crudely isolate nucleic acids and ribonuclear complexes using ethanol precipitation instead of isolating bacterial cells. Thus, both the way we purify and the stage at which we purify ribonuclear complexes are different using MetaRibo-Seq. Third, we scaled down the input requirements and the micrococcal nuclease (MNase) reaction (to create ribosome-protected footprints). MetaRibo-Seq, with these modifications, demonstrates strong correlation with standard Ribo-Seq protocols and correlates more strongly to proteomics than RNA-Seq correlates to proteomics when benchmarked in a mock bacterial community27.
Of note, some of these revisions are conceptually similar to revised Ribo-Seq protocols that were independently developed simultaneously by Buskirk26. In their protocol, the authors freeze whole cultures of 100 mL of bacteria into pellets without centrifuging or filtering bacteria first28. These pellets are mechanically lysed, and MgCl2 is used to inhibit translation instead of chloramphenicol. Sucrose cushions are then used to purify ribosomes instead of purifying bacteria initially, which is conceptually similar to MetaRibo-Seq using an ethanol precipitation to more crudely purify nucleic acids and ribonuclear complexes. While Buskirk’s systematically revised protocol would not be directly applicable to fecal samples, the conceptual advances of freezing bacteria first and inhibiting translation and purifying ribosomes later are similar to those implemented in MetaRibo-Seq.
Advantages and applications of MetaRibo-Seq
Traditional Ribo-Seq is limited to studying translation only in organisms that are cultured and assayed in vitro. With MetaRibo-Seq, researchers can now study translation in fecal microbiomes. Beyond studying regulation of translation, there is substantial interest in detecting and measuring the proteome in microbiome research. As a surrogate for proteomics, MetaRibo-Seq can provide information on the open reading frames that are translated. Translational confirmation is particularly valuable for validating predicted small open reading frames, whose protein products are difficult to detect experimentally. Because proteomics methods are limited in their ability to detect small proteins and poorly abundant proteins, we rarely detect small proteins and often struggle to detect many larger proteins unless they are of relatively high abundance29. The ability to study protein synthesis is thus a great potential asset to microbiome researchers.
In addition to being a potential surrogate for proteomics, Ribo-Seq is a useful method to reannotate genes and identify new genes, especially small genes30,31. Ribo-Seq signal distribution across a genomic region can provide evidence that it likely encodes a protein. Previous work from our laboratory predicted thousands of novel families of small genes in microbiomes using comparative genomics32. Building on this framework, MetaRibo-Seq verified translation of hundreds of these novel families in the fecal microbiome and predicted thousands of additional small protein families in the human gut27. Many more unannotated small proteins likely exist in this space. MetaRibo-Seq has the potential to be instrumental in identifying additional small protein families in microbiomes. In fact, recent work used machine learning approaches to predict additional small genes and used MetaRibo-Seq to help validate predictions33. We anticipate more computational tools to predict small proteins by using MetaRibo-Seq signal distribution will be developed. The ability to study translation in microbiomes thus opens several new avenues of investigation for microbiome researchers.
MetaRibo-Seq can be used for several applications; therefore, the exact experimental design employed will vary based on the research objective of the study. If, for example, the goal is to study translational regulation in fecal samples over time, one could track specific strains of interest over time in people. This could be accomplished by performing metatranscriptomics and MetaRibo-Seq at time points of interest. In the design of such a study, we recommend careful planning and consideration of batch effects and replicates. Another potential study design of interest may be evaluating the impact of acute stresses, such as antibiotics, on a mixed microbial community. In this case, a researcher might choose to expose fecal samples to acute stresses ex vivo in short-term fecal cultures, and study translational regulation34. If the goal is to search for translation of genes for open reading frame prediction in individual samples, then metatranscriptomics may not be necessary and the relative timing of obtaining the different samples may require fewer considerations. However, it might be desirable to introduce an agent such as retapamulin, which enriches for start site positions and would thus enable accurate prediction of gene starts in bacteria whose ribosomes are sensitive to retapamulin-based inhibition. In addition to these applications, we expect that additional modifications will enable the study of translation in other types of microbiomes, such as environmental communities.
Overview of the procedure
We describe the MetaRibo-Seq protocol to perform Ribo-Seq on human fecal microbiomes (Fig. 1) and the computational workflow to create a de novo reference and assess library quality (Fig. 2). First, we describe lysis of microbes from fecal samples and the extraction of ribonuclear complexes (Steps 1–12), MNase digestion and purification of monosome footprints (Steps 13–28), and creation and sequencing of MetaRibo-Seq libraries (Steps 29–39). Second, we describe the computational workflow to assess MetaRibo-Seq library quality, including quality trimming of reads, metagenomic assembly, MetaRibo-Seq read alignment to the assembly, and visualizations (Steps 40–43).
Fig. 1 |. MetaRibo-Seq protocol workflow.
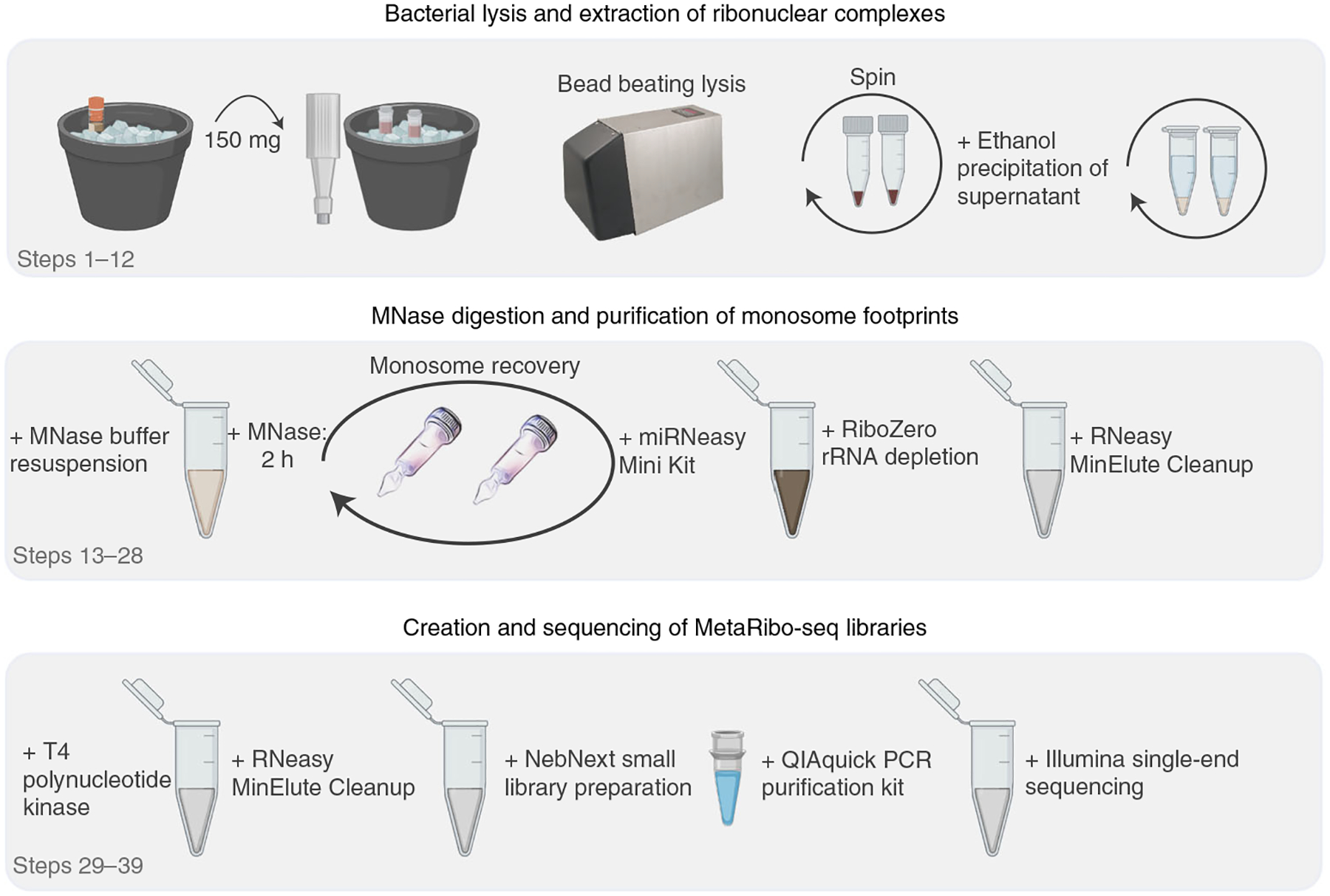
A biopsy punch with a plunger is used to prepare 300 mg aliquots of stool into two screw-top tubes (150 mg per tube) with lysis buffer (recipe 1). Bacterial cells are lysed using a bead beater and centrifuged to pellet fecal debris, and an ethanol precipitation is performed on the supernatant. Precipitate is resuspended in MNase buffer (recipe 2) and digested with MNase. Monosomes are recovered using size-exclusion columns, purified, and subjected to rRNA depletion. MetaRibo-Seq libraries are created from these footprints and sequenced.
Fig. 2 |. MetaRibo-Seq bioinformatic workflow.
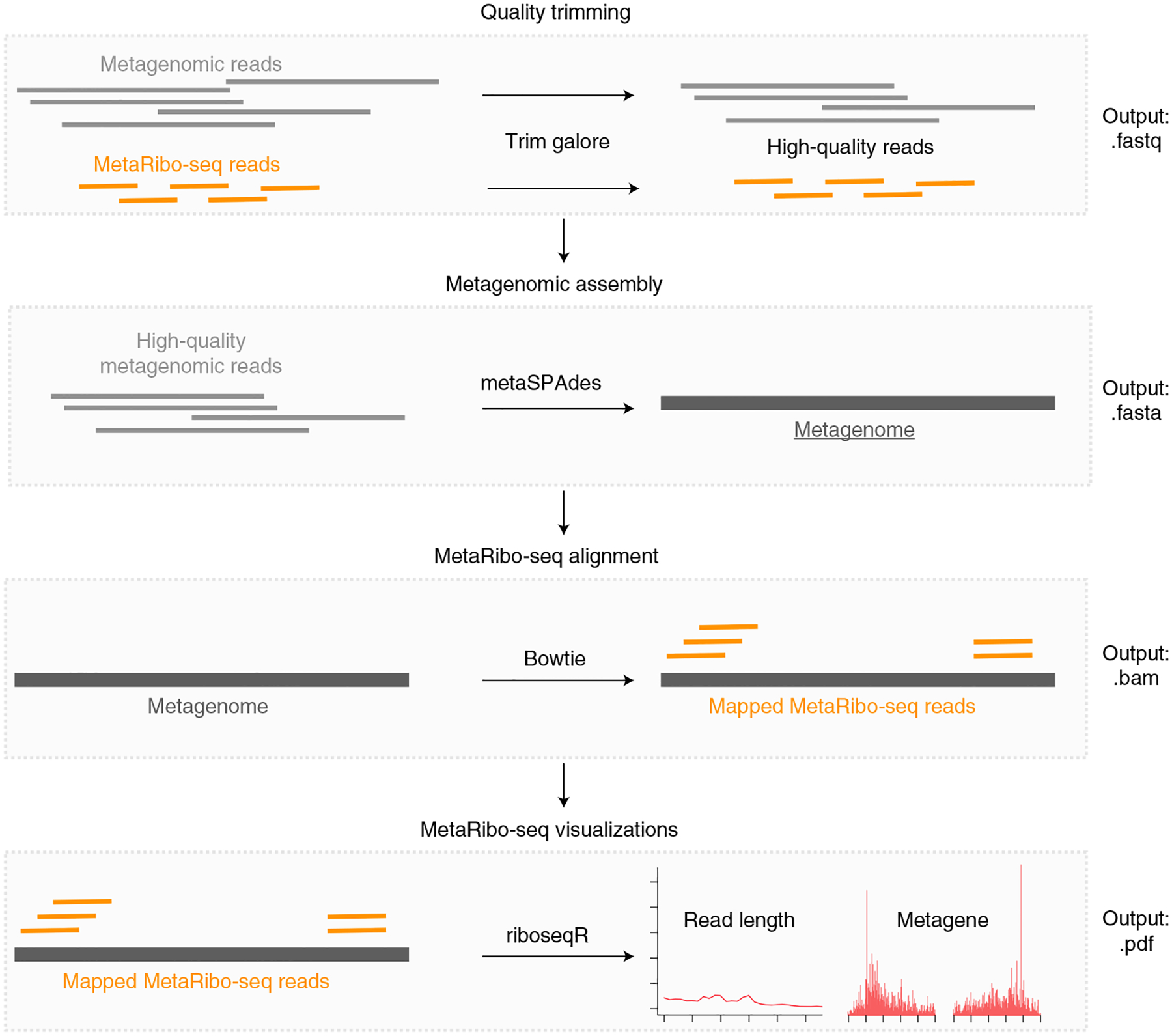
After sequencing, this computational workflow assesses MetaRibo-Seq library quality and serves as a starting point for downstream analyses. After quality trimming all relevant reads, this pipeline uses metagenomic reads for a given sample to create a de novo reference and then aligns MetaRibo-Seq reads corresponding to the sample to the assembly. These alignment files are used to create visualizations, including aligned footprint length distributions, footprint length specific triplet periodicity, and metagene plots for various footprint sizes.
Limitations of MetaRibo-Seq
While MetaRibo-Seq benchmarks well against gold-standard Ribo-Seq, there are several limitations to consider. Although MetaRibo-Seq correlates more strongly to protein abundance in mock communities than RNA-Seq, standard Ribo-Seq still correlates more strongly to protein abundance than MetaRibo-Seq. MetaRibo-Seq is also a snapshot of translation; therefore, the absence of signal does not imply that a protein is not real or that a protein does not exist in the human gut. MetaRibo-Seq must also be coupled with metagenomic assemblies to create a suitable reference for mapping MetaRibo-Seq reads, which requires additional bench and computational work, and also increases experimental costs.
MetaRibo-Seq also inherits many of the limitations of standard Ribo-Seq, such as contamination by other RNA–protein complexes or structured RNAs35. Because MetaRibo-Seq does not directly enrich for ribosomes (which could be achieved with sucrose density gradients), the potential for contaminating signal from nonribosomal complexes is higher. Additionally, the resolution of MetaRibo-Seq is compromised by several factors. First, using chloramphenicol to halt ribosomes can alter the ribosome pausing landscape36,37. Second, the resolution of ribosome pausing is also likely diminished by the broad distribution of footprint lengths and lack of stringent size selection (which could be achieved with PAGE gels, for example) in the MetaRibo-Seq protocol. Ribo-Seq footprint lengths are more variable in bacteria than in eukaryotes. It is not fully clear what causes this variability; however, this difference seems to be related to inherent properties of bacterial ribosomes as opposed to technical differences such as nucleases26. Third, nuclease-mediated depletion of ribosomal RNA (rRNA) (as performed in Ribo-Zero Plus) can obscure the position of ribosomes38. Fourth, ribosomes can continue to progress along a transcript during sample collection and processing, which takes longer for fecal samples than for laboratory cultured bacteria. While not yet rigorously evaluated, a future improvement may involve using a cocktail of RNAses to increase resolution of MetaRibo-Seq. Finally, we caution that rRNA depletion will not equally deplete rRNAs across different species.
Level of expertise needed to implement the protocol
This protocol can be performed by researchers with experience in molecular biology techniques and bioinformatics. Specifically, researchers must be familiar with fecal sample processing protocols and next-generation sequencing. Because some of the expensive equipment, such as a Bioanalyzer and Illumina sequencing platforms, can be outsourced, this protocol can be established in most laboratories.
Experimental design
Storage of fecal samples (Steps 1–5)
We recommend preparing fecal samples into aliquots and freezing them in RNAlater quickly after collection. We have not tried preservatives other than RNAlater, such as DNA/RNA Shield (Zymo Research, R1100) or ethanol; however, other preservatives may also be useful. Note that certain taxa may die, grow and alter their gene regulation in the time between collection and storage. We have successfully performed MetaRibo-Seq on samples that have been frozen for up to 1.5 years.
Obtaining monosome footprints from fecal samples (Steps 6–25)
We recommend using biopsy punches with plungers to divide fecal samples into aliquots in screw-top tubes for lysis. When handling biopsy punches, use twisting instead of jabbing motions and avoid holding the tube by hand. This minimizes the chances of injury. Biomass of fecal samples should also be a consideration. Samples of watery consistency and also those from patients being administered antibiotics typically require more total fecal mass to achieve the downstream RNA input requirements for MetaRibo-Seq.
Researchers may choose to perform RNA-Seq in addition to MetaRibo-Seq for comparison purposes, for example, to distinguish transcriptional changes from translational changes over time. We recommend performing the two methods at the same time to minimize batch effects. Additionally, we recommend performing technical duplicates within the same batch if the goal is to compare between technologies or monitor changes over time. MetaRibo-Seq was designed with timing in mind and contains modifications, such as using size-exclusion columns instead of sucrose density gradients, to simplify and shorten the protocol21,27. As a result, RNA-Seq and MetaRibo-Seq follow similar timelines and contain enough incubation periods in between steps to comfortably perform simultaneously27.
Bacterial cells are mechanically lysed using a bead beater. The samples are centrifuged to remove the fecal debris pellet. The supernatant is subjected to ethanol precipitation. After centrifugation, the pellet is a crude purification of nucleic acids and ribonuclear complexes. Sometimes, these pellets can be difficult to resuspend in MNase buffer. In such cases, gently breaking the pellet apart with a pipette tip is effective. Additionally, multiple extractions can be combined at this resuspension step. The target RNA input for MNase digestion is 80 μg, which is substantially lower than existing bacterial Ribo-Seq protocols. For this reason, we perform a scaled-down MNase reaction. It is also possible to perform this reaction with 40 μg of RNA and half the amount of MNase; however, this will make downstream steps more challenging as input will be limited. We highly recommend doing more extractions to achieve 80 μg if possible; however, we have also found that it can be very difficult to obtain 80 μg from samples collected from patients undergoing antibiotic treatment, even if many extractions are performed. In these scenarios, it is acceptable to carefully proceed with 40 μg. Size-exclusion columns are used for monosome recovery; however, the samples are split across two columns to avoid overloading. The samples are recombined upon purification with the miRNeasy Mini Kit.
rRNA depletion (Steps 26–28)
After the size-exclusion step, there should be between 1 and 5 μg of RNA. To deplete rRNA contaminants, we recommend using the Illumina Ribo-Zero Plus rRNA Depletion Kit and proceeding forward with depletion on 1 μg of RNA. Illumina continues to improve rRNA depletion from microbiome samples by adding additional rRNA probes to their kit. There are other options that we have not thoroughly compared with Ribo-Zero Plus for rRNA depletion, including NEBNext rRNA Depletion Kit (NEB, E7850) and MICROBExpress Bacterial mRNA Enrichment Kit (Invitrogen, AM1905). We prefer cleanup using the RNeasy MinElute Kit for greater purification through the silica membrane; however, ethanol precipitation has been a successful substitute for this step as a cheaper alternative.
Ligation preparation (Steps 29–34)
At this point, there should be at least 100 ng of RNA for downstream applications. To prepare samples for sequencing library construction, we subject them to T4 polynucleotide kinase to facilitate 5′ phosphorylation of RNA and 3′ phosphoryl removal. Again, we prefer cleanup using the RNeasy MinElute Kit; however, ethanol precipitation can be substituted as a cheaper alternative.
Library preparation (Steps 35–38)
Using 100 ng of input, we create MetaRibo-Seq libraries using the NEBNext Small RNA Library Prep. We recommend following the protocol at https://www.neb.com/protocols/2018/03/27/protocol-for-use-with-nebnext-small-rna-library-prep-set-for-illumina-e7300-e7580-e7560-e7330, with one exception: add 1.5 μL of SR Primer for Illumina and 1.5 μL Index Primer instead of the recommended 2.5 μL before library amplification. This modification was also recommended in a previous Ribo-Seq protocol21. We also found that using too much primer results in a substantial primer dimer peak in the final libraries. We have had limited success removing primer dimers using several rounds of a 1.3× ratio of AMPure XP Beads; however, the best solution is to avoid the formation of primer dimers. These primer dimers will not cluster on the flow cell when sequenced (unlike adapter dimers), meaning that libraries with primer dimers can still be sequenced effectively, but the primer dimers can make quantification of the library and pooling of samples more challenging than necessary. A peak around 150–155 bp, representing the MetaRibo-Seq library, should be present on a Bioanalyzer profile (Fig. 3a). For reference, the adapters within the fragments are 127 bp long; therefore, the position at 155 bp, for example, corresponds to a footprint of length of 28 bp.
Fig. 3 |. MetaRibo-Seq quality.
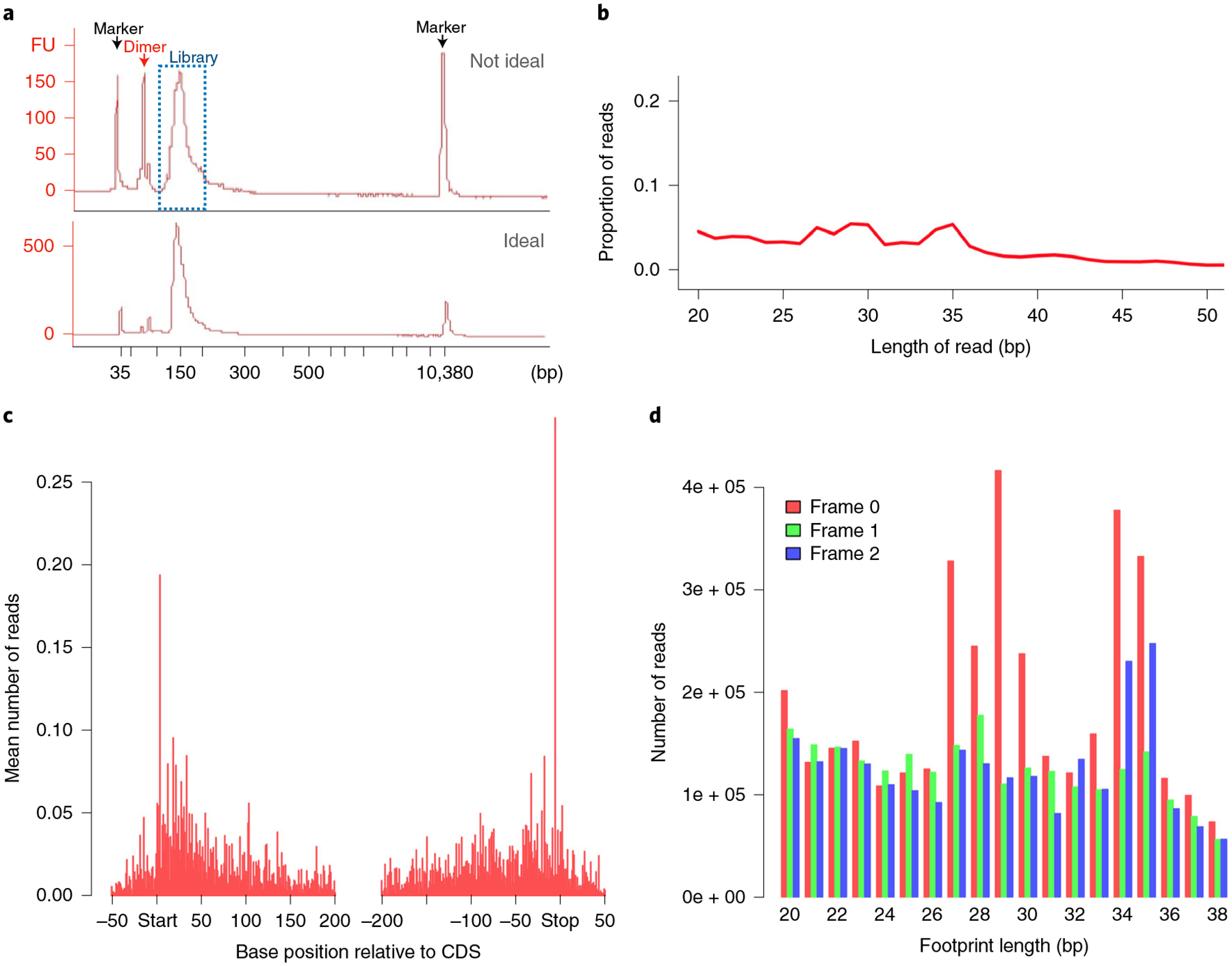
Assessing the quality of MetaRibo-Seq libraries experimentally and computationally. a, Bioanalyzer trace of a low-quality (top, with primer dimers) and high-quality (bottom, with minimal primer dimers) MetaRibo-Seq library. The bioanalyzer trace is labeled to indicate which peaks correspond to markers used by the bioanalyzer, primer dimer and library. b, Aligned read length distribution histogram of the MetaRibo-Seq library. c, Metagene plot of the MetaRibo-Seq library. d, Footprint length-specific triplet periodicity histogram of the MetaRibo-Seq library.
Sequencing (Step 39)
We recommend pooling all MetaRibo-Seq libraries to minimize batch effects and sequencing these libraries single-end. We sequence libraries using 50 bp reads. While the large majority of fragments will be <50 bp and require no more than 50 bp read lengths, some monosomes can reach a length >50 bp39, suggesting that 75 bp read lengths may be preferable. The sequencing depth depends on a number of factors including the diversity of the samples being tested and the effectiveness of rRNA depletion for those samples. To determine the sequencing depth needed and also get a sense of the quality of the libraries, it may be desirable to perform low-pass sequencing first by only sequencing several million reads per sample.
Processing and analysis of sequencing reads (Steps 40–43)
Many different analyses of the data can be performed depending on experimental design and research goals. The first computational challenge with MetaRibo-Seq analysis is that these very short reads can be difficult to analyze without representative reference genomes, and existing references are typically not specific to the strains present in a given fecal sample. Additionally, direct taxonomic classification of very short reads can be challenging without longer contigs to associate them to. To be confident in assessments of MetaRibo-Seq signal, it is important to first create a reference genome specific to the sample40. After metagenomic and MetaRibo-Seq reads are quality trimmed41, we use these high-quality metagenomic reads to create a de novo reference metagenome using metaSPAdes40. Depending on the sequencing depth of the samples, this assembly step can take up to 2 d to finish and is typically the rate-limiting step of the workflow (by default, the pipeline uses four threads to run metaSPAdes, though this can be increased to reduce time needed to run). The MetaRibo-Seq reads are aligned to this de novo reference using Bowtie42. Visualizations such as aligned footprint lengths, footprint length-specific triplet periodicity and footprint-specific metagene plots are created using riboseqR43 (Fig. 3). All of these steps are implemented by the provided pipeline: https://github.com/bhattlab/bhattlab_workflows/tree/master/metariboseq. This pipeline not only provides information on quality but also serves as a useful starting point for downstream analyses, by providing an assembly and MetaRibo-Seq read alignment files.
Materials
Biological samples
Stool sample, stored indefinitely at −80 °C in RNAlater ! CAUTION All samples should be obtained with informed consent and in accordance with relevant guidelines.
Reagents
RNeasy MinElute Cleanup Kit (Qiagen, cat. no. 74204)
Phenol/chloroform pH 8.0 (Sigma-Aldrich, cat. no. 77617–100ML) ! CAUTION Phenol and chloroform are toxic. Wear protective gloves and clothing to prevent contact.
3 M sodium acetate (Fisher Scientific, cat. no. BP333–500)
Absolute ethanol (Fisher Scientific, cat. no. BP2818500) ! CAUTION Ethanol is flammable. Store appropriately.
Nuclease-free water (Invitrogen, cat. no. AM9937)
1 M Tris-HCl pH 8.0 (Fisher Scientific, cat. no. 15-567-027)
0.5 M egtazic acid (EGTA; Fisher Scientific, cat. no. 50-255-956)
5 M sodium chloride (Fisher Scientific, cat. no. S640-3)
Superase-In 20 U/μL (Fisher Scientific, AM2694)
Calcium chloride, anhydrous (Fisher Scientific, cat. no. S25223)
Chloramphenicol (Fisher Scientific, cat. no. BP904-100)
Igepal CA-630 (Fisher Scientific, cat. no. ICN19859650)
MgCl2 (Fisher Scientific, cat. no. AC223211000)
Magnesium acetate (MgOAc), 1 M (Sigma-Aldrich, cat. no. 63052)
NH4Cl (Fisher Scientific, cat. no. A661-500)
RNase-free DNase I, 10 U/μL (Sigma-Aldrich, cat. no. 4716728001)
Qubit RNA HS Assay Kit (Life Technologies, cat. no. Q32852)
MNase, 500 Kunitz units/μL (High Concentration special order from New England Biolabs)
miRNeasy Mini Kit (Qiagen, cat. no. 217004)
Illumina Ribo-Zero Plus rRNA Depletion Kit (Illumina, cat. no. 20040526)
ATP, 10 mM (New England Biolabs, cat. no. P0756S)
T4 polynucleotide kinase (T4 PNK, New England Biolabs, cat. no. M0201S)
NEBNext Small RNA Library Prep Set for Illumina (New England Biolabs, cat. no. E7330S)
Qubit DNA HS Assay Kit (Life Technologies, cat. no. Q32851)
QIAquick PCR Purification Kit (Qiagen, cat. no. 28104)
Bioanalyzer High Sensitivity DNA Assay (Agilent, cat. no. 5067-4626)
Equipment
Class II A2 Biosafety Cabinets (Fisher Scientific, cat. no. 302610101)
1 mm zirconia/silica beads (Fisher Scientific, cat. no. NC9847287)
Biopsy punch (Fisher Scientific, cat. no. 12-460-410) ! CAUTION Biopsy punches are sharp and should be handled with care.
Sephacryl S400 MicroSpin Columns (Sigma-Aldrich, cat. no. GE27-5140-01)
LowBind tubes, 1.5 mL (Fisher Scientific, cat. no. 13-698-791)
LowBind tubes, 2 mL (Fisher Scientific, cat. no. 13-698-792)
PCR tubes, 0.2 mL (Fisher Scientific, cat. no. AM12230)
Pipettors (Fisher Scientific, cat. nos. 07-764-700, 07-764-701, 07-764-702, 07-764-704, 07-764-705)
Aerosol barrier pipette tips (Fisher Scientific, cat. nos. 02-707-439, 02-707-432, 02-707-430, 02-707-404)
Dry ice
Ice
Mini-beatbeater-16 (Biospec, cat. no. 607)
Analytical scale (Fisher Scientific, cat. no. S72710)
Microcentrifuge (Fisher Scientific, cat. no. 07-203-954)
Multi-head benchtop vortex (Benchmark Scientific, cat. no. BV1005)
Benchtop centrifuge (Beckman Coulter, cat. no. 392244)
Qubit fluorometer (Thermo Fisher Scientific, cat. no. Q33327)
Heat block (Fisher Scientific, cat. no. 88-870-001)
Thermal cycler (Thermo Fisher Scientific, cat. no. A37835)
Magnetic microcentrifuge tube rack (Thermo Fisher Scientific, cat. no. 12321D)
Bioanalyzer (Agilent, cat. no. G2939BA)
Software and hardware
Computer with Windows 7+; OSX Sierra, High Sierra or Mojave; or Linux Ubuntu 16.04 or 18.04; with 100 GB of storage
The only software that needs to be installed are NextFlow44 and Singularity45 with miniconda3 (https://docs.conda.io/en/latest/miniconda.html) being used to manage their installation. The Singularity image (https://github.com/orgs/bhattlab/packages/container/package/bhattlab-metariboseq) includes all other dependencies for the user, including metaSPAdes40, bowtie42 and riboseqR43
Reagent setup
! CAUTION Ensure that solutions are kept RNAse-free.
Stock solutions
5 M NaCl in RNase-free H2O. Store at 4 °C for up to 1 year
1 M pH 8.0 Tris-HCl in RNase-free H2O. Store at room temperature (RT, 20–25 °C) for up to 3 years
1 M MgCl2 in RNase-free H2O. Store at 4 °C for up to 1 year
0.5 M EGTA in RNase-free H2O. Store at RT for up to 3 years
500 mM CaCl2 in RNase-free H2O. Store at 4 °C for up to a few months ! CAUTION Calcium chloride releases heat when dissolved in water.
1 M MgOAc in RNase-free H2O. Store at RT for up to 1 year
1 M NH4Cl in RNase-free H2O. Store at RT for up to 3 years
50 mg/mL chloramphenicol in dimethyl sulfoxide. Make this fresh for every experiment
10 mM ATP in RNase-free H2O. Store at −20 °C for up to 1 year
Recipes ▲ CRITICAL All recipes should be freshly prepared.
Recipe 1:
lysis buffer
| Components | Volume |
|---|---|
| RLT buffer (Qiagen) | 965 μL |
| β-Mercaptoethanol | 10 μL |
| Superase-In, 20 U/uL | 15 μL |
| Chloramphenicol, 50 mg/mL | 10 μL |
| Total | 1 mL (scale as needed) |
Recipe 2:
MNase buffer
| Components | Volume |
|---|---|
| Tris-HCl pH 8.0, 1 M | 25 μL |
| NH4Cl, 1 M | 25 μL |
| MgOAc, 1 M | 10 μL |
| Chloramphenicol, 50 mg/mL | 10 μL |
| RNase-free water | To 1 mL (930 μL) |
Recipe 3:
polysome binding buffer
| Component | Volume |
|---|---|
| Igepal CA-630 | 100 μL |
| MgCl2, 1 M | 500 μL |
| EGTA, 0.5 M | 500 μL |
| NaCl, 5 M | 500 μL |
| Tris-HCl, pH 8.0, 1 M | 500 μL |
| RNase-free water | 7.9 mL |
| Total | 10 mL |
Note: this recipe is identical to the polysome buffer used in Latif et al.21.
Procedure
Storage of fecal samples ● Timing 15 min
-
1
Store collected fecal samples at 4 °C for up to 24 h before processing. It is preferable to process as soon as possible.
! CAUTION All samples are obtained with informed consent and in accordance with relevant guidelines.
! CAUTION All work with human stool samples is performed in a biosafety cabinet until bacteria are lysed. Protective laboratory coat, eyewear and gloves should be worn.
-
2
Prepare aliquots of 700 μL of RNAlater (Ambion) into 2 mL cryovials. It is helpful to store multiple tubes for each sample, especially if technical replicates are desirable.
-
3
Homogenize the fecal sample.
▲ CRITICAL STEP Try to avoid sampling from one specific region of the fecal sample as this may bias results and create technical differences. If the sample is liquid, pipette 700 μL into the vial containing RNAlater. Use a wide bore pipette tip or a pipette with trimmed tip to avoid difficulties with transferring the sample. If the sample is solid, transfer ~20 g of stool to one corner of a small plastic, zipper reclosable bag. Taking care to keep stool localized to a single corner of the bag, massage the contents to thoroughly homogenize the sample. Cut an opening in the clean corner of the bag ~0.5 mm wide. Transfer ~1.0–1.3 g of stool sample to the tube with RNAlater by squeezing the base of the bag gently.
-
4
Add ~1.0–1.3 g of fecal sample to cryovials with RNAlater. Mix by inverting and gently vortexing to ensure that the sample is submerged in RNAlater.
-
5
Freeze the samples at −80 °C, and store until ready to perform MetaRibo-Seq.
■ PAUSE POINT Successful libraries have been created from samples stored at −80 °C for as long as 1.5 years.
Obtaining monosome footprints from fecal samples ● Timing 5 h
-
6
Obtain two screw-top tubes for each sample.
▲ CRITICAL STEP For low microbial biomass samples with less extractable RNA (such as from patients on antibiotics), more tubes may be necessary. Additionally, technical replicates should be treated as different samples and would require two additional tubes.
-
7Add beads, lysis buffer and frozen stool to each screw-top tube. Keep the stool samples on dry ice the entire time they are out of the freezer. We recommend using a biopsy punch on frozen stool to prepare aliquots. Add the following to each screw-top tube:
Reagent Quantity 1.0 mm zirconia/silica beads ~20 beads Lysis buffer (recipe 1) 600 μL Frozen stool (in RNAlater) 150 mg ▲ CRITICAL STEP Prepare lysis buffer (recipe 1) and MNase buffer (recipe 2) ahead of time, and keep on ice until use.
! CAUTION Biopsy punches are sharp objects. Take precautions: avoid holding sample tubes by hand, and use twisting motions instead of forceful, jabbing motions to divide stool into aliquots.
-
8
Bead beat the tubes for 3 min at RT.
-
9
Centrifuge at RT for 3 min at 21,000g, and transfer 500 μL of supernatant to new 2 mL tubes; discard pellet.
-
10
Add 0.1 volumes 3 M sodium acetate and 2.5 volumes absolute ethanol.
-
11
Incubate for 30 min on ice.
-
12
Centrifuge at 21,000g for 30 min at 4 °C; discard supernatant.
-
13
Resuspend pellet in 100 μL MNase buffer (recipe 2), and combine the two resuspensions for each fecal sample. Keep on ice.
▲ CRITICAL STEP Recall that, for each fecal sample, two extractions were performed (starting with two screw-top tubes); therefore, two pellets are present, representing the same sample at this step. Resuspend these pellets in MNase buffer, and combine resuspensions into one sample with a combined volume of 200 μL. The resuspension volume may vary if more extractions were performed per sample. In the case of low-biomass fecal samples that require starting with four tubes, for example, resuspend each pellet in 50 μL and combine those four resuspensions.
-
14
Use Qubit RNA HS Assay Kit to assess RNA concentration. We recommend first taking 1 μL of sample and performing a ~1:50 dilution in nuclease-free water to bring it into detectable range for quantification. Then perform Qubit quantification on the diluted sample using fresh preparations of RNA standards, which are provided with the kit, to create the curve.
-
15Prepare MNase treatment reaction as follows:
Reagent Volume Lysate (from Step 13) X μL (to 80 μg RNA total) CaCl2, 500 mM 2 μL Superase-In, 20 U/μL 2 μL NEB MNase, 500 U/μL 1 μL MNase buffer To 200 μL -
16
Incubate for 2 h at RT.
-
17
Quench the reaction by adding 2.5 μL EGTA (500 mM stock).
-
18
Obtain two Sephacryl S400 MicroSpin columns for each sample.
-
19
Invert columns multiple times vigorously to resuspend resin.
-
20
Centrifuge columns at 600g for 1 min (all column centrifugation steps are performed at 4 °C); discard flow through.
-
21
Wash columns two times with 500 μL polysome binding buffer (recipe 3); centrifuge each time for 1 min at 600g.
-
22
Wash the column by adding 500 μL polysome binding buffer (recipe 3) and centrifuging for 4 min at 600g. Place each column into a clean collection tube.
-
23
Apply 100 μL of MNase Treatment reaction (from Step 17) to each column.
-
24
Centrifuge for 2 min at 600g to collect flow through.
-
25
Proceed using the flow through with miRNeasy Mini Kit (https://www.qiagen.com/us/resources/resourcedetail?id=da6c8d17-58c4-411c-a334-bc1754876db3&lang=en). Briefly, add 700 μL Qiazol and 140 μL chloroform to each flow through. Shake for 15 s. Incubate at RT for 3 min. Centrifuge at 21,000g for 15 min at 4 °C. Extract the aqueous (top) phase, and add 1.5× the volume of 100% ethanol and mix. Apply this to an RNeasy mini spin column (700 μL at a time), and centrifuge at 21,000g for 1 min at RT. Discard the flow through. Wash the column with RWT and RPE. Elute in 15 μL RNase-free water.
▲ CRITICAL STEP Please note that, at this step, the sample that was previously split into two upon loading onto Sephacryl S400 MicroSpin columns is combined. Apply both of these onto the same RNeasy column. Do not combine technical replicates if also being prepared, as these will be barcoded separately and pooled at the end of the protocol.
■ PAUSE POINT The purified RNA can now be stored at −80 °C for up to 30 d.
rRNA depletion ● Timing 2 h
-
26
Quantify RNA with Qubit RNA HS Assay Kit.
-
27
Treat 1 μg of RNA with Illumina Ribo-Zero Plus rRNA Depletion Kit using half reactions (https://www.illumina.com/content/dam/illumina-marketing/documents/products/technotes/ribo-zero-plus-data-concordance-tech-note-1270-2020-003.pdf).
-
28
Purify with RNeasy MinElute Cleanup, and elute in 19 μL RNase-free water.
▲ CRITICAL STEP 18 μL is needed for the downstream steps. For troubleshooting purposes, it can be helpful to save 1 μL of this elution in a separate tube for quantification later.
■ PAUSE POINT The rRNA-depleted RNA can be stored at −80 °C for up to 30 d.
Ligation preparation ● Timing 2 h
-
29Prepare the following reaction:
Reagent Volume RNA footprints (from Step 28) 18 μL 10X T4 PNK buffer 2.2 μL Superase-In, 20 U/μL 1 μL T4 PNK, 10 U/μL 1 μL Total volume 22.2 μL -
30
Incubate the reaction at 37 °C for 30 min.
-
31
Add 1 μL 10 mM ATP to the reaction, and incubate for 30 min at 37 °C.
-
32
Add 27 μL of RNase-free water, 350 μL RLT buffer and 600 μL absolute ethanol.
-
33
Apply to RNA MinElute column, wash with 500 μL of Buffer RPE, then wash with 500 μL of 80% (vol/vol) ethanol, and finally elute with 10 μL RNase-free water (https://www.qiagen.com/us/resources/resourcedetail?id=0acfc1c7-a1f8-4425-9aaf-b0d98b81bd1f&lang=en).
-
34
Check concentration with Qubit RNA HS Assay Kit.
Library preparation ● Timing 10 h
-
35
Perform NEBNext Small RNA Library Prep using recommendations for 100 ng input (https://www.neb.com/protocols/2018/03/27/protocol-for-use-with-nebnext-small-rna-library-prep-set-forillumina-e7300-e7580-e7560-e7330). The only exception to the manufacturer’s protocols is to add 1.5 μL of SR Primer for Illumina and the Index Primer instead of 2.5 μL before library amplification. This protocol involves the ligation of the 3′ adaptor, hybridization of the reverse transcription primer, ligation of the 5′ SR adaptor, reverse transcription, and library amplification.
-
36
Purify the reaction with QIAquick PCR purification kit (https://www.qiagen.com/us/resources/resourcedetail?id=95f10677-aa29-453d-a222-0e19f01ebe17&lang=en).
-
37
Quantify DNA with Qubit DNA HS Assay Kit.
-
38
Check distribution of the library on HS dsDNA Bioanalyzer (https://www.agilent.com/cs/library/usermanuals/public/G2938-90322_HighSensitivityDNAKit_QSG.pdf).
Sequencing ● Timing 1 d
-
39
Pool and sequence the libraries using an Illumina sequencing platform, for example, NextSeq or HiSeq, as single-end with at least 50 bp reads.
Processing and analysis of sequencing reads ● Timing 2 d
-
40
Install miniconda3 (https://docs.conda.io/en/latest/miniconda.html), Nextflow and Singularity. Clone the bhattlab_workflows repository from https://github.com/bhattlab/bhattlab_workflows, and navigate to the metariboseq subdirectory.
-
41Create a parameter file with the desired parameters using example settings found in the workflows/params subdirectory. This is described in detail within the GitHub repository, found at https://github.com/bhattlab/bhattlab_workflows/tree/master/metariboseq. An example can also be found below:
inputPrefix: /oak/stanford/scg/lab_asbhatt/cosn/metariboseq/samples/resultsPrefix: /oak/stanford/scg/lab_asbhatt/cosn/metariboseq/results/ trimGaloreOptions: “--cores 4 -q 30 --illumina” spadesOptions: “--threads=4 --memory=96” alignmentMemory: “96 GB” assemblyMemory: “96 GB” bowtieIndexOptions: “--threads 4” bowtieAlignmentOptions: “--threads 4” analysisScript: “/oak/stanford/scg/lab_asbhatt/cosn/bhattlab_workflows/metariboseq/workflows/analysis/plotVisualsRibo.R” sampleSpecs: - name: sample metagenomic: SampleBDNA metariboseq: SampleBRibo
Parameters that likely require changing include the inputPrefix, resultsPrefix, analysisScript and sampleSpecs because these all depend on the user’s specific data structure and sample names. The user should replace the example values with the file system paths that contain their input data (inputPrefix), the file system location where they wish their results to be written (outputPrefix), the location of the analysis script where they cloned the repository to (analysisScript) and, finally, the specification for the analysis they wish to run (sampleSpec), which specifies the original sample (the name field) and the analysis (metagenomic or metariboseq) to be performed on a given processed sample. The user can also control many aspects of the pipeline, including threads and memory usage of individual tools.
-
42
Run the Nextflow workflow (using nextflow run), and provide relevant parameters, including the parameter file you created and the Singularity image provided. More details are available on the GitHub repository, and NextFlow itself is also well documented44. The Github also includes an example script named-nextflow.sh, which can be used as a starting point for customization to match the local system configuration of the user.
-
43
Go to the results folder to view trimming statistics and alignments in BAM format. Assemblies can be found in the results/$SampleName-assembly folder in FASTA format. Visualizations of aligned footprint lengths, triplet periodicity and metagene plots at various footprint lengths can be found in the results/$SampleName-plots folder in PDF format.
Troubleshooting
Troubleshooting advice can be found in Table 1.
Table 1 |.
Troubleshooting table
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 13 | Difficult to resuspend pellet | Much variability exists in fecal samples. Some precipitations can be more crude than others | Use a pipette tip to gently break apart the pellet first, then gently vortex for a few minutes |
| 14 | Low yield (<80 μg total) | Low biomass of microbes in fecal sample. In this case, the RNA extractions will also be low yield. Alternatively, it could be an issue of RNA degradation during sample storage. For example, the RNAlater may not have been mixed well with the sample | Increase the number of extractions per sample. Also, it is acceptable to proceed with 40 μg, cutting the MNase treatment concentration in half as a last resort. Although the libraries will work, you will likely have just enough input for each downstream step |
| 34 | Not enough RNA for library preparation | RNA was degraded during rRNA depletion, from too many freeze-thaw cycles, or during the T4 PNK reaction | Backtrack and quantify how much RNA remained after rRNA depletion Try to avoid some pausing points to minimize freeze-thaws, and try to complete the entire protocol in 2 d Ensure that RNase inhibitors are being added diligently |
| 38 | Substantial amount of primer dimers | Too much primer was added | We recommend adding 1.5 instead of 2.5 μL of primers as the manufacturer recommends. If need be, this can be reduced to 1 μL Although less ideal, a few rounds of Ampure beads (1.3× ratio of beads to sample) can reduce the dimers in the library |
Timing
Steps 1–5, preparation of fecal samples for storage: 15 min
Steps 6–25, obtaining monosome footprints from fecal samples: 5 h
Steps 26–28, rRNA depletion: 2 h
Steps 29–34, ligation preparation: 2 h
Steps 35–38, library preparation: 10 h
Step 39, sequencing: 1 d
Steps 40–43, processing and analysis of sequencing reads: 2 d
Anticipated results
We find great variability in the extraction yield. From individuals who have not received antibiotics, we expect 40–200 μg of RNA from 150 mg of fecal sample. From lower-biomass samples, often from individuals receiving antibiotics, we expect 1–40 μg of RNA from 150 mg of fecal samples. Due to variability, these ranges may change as more samples are analyzed. These are just rough estimates to help inform how many extractions to perform. After MNase treatment and monosome recovery, before rRNA depletion, we expect 1–5 μg of RNA. After rRNA depletion, we expect 100–500 ng of RNA. The final library should contain a large peak in fragment length around 150–155 bp. Additionally, we expect to see local enrichment in MetaRibo-Seq signal within coding regions, relatively stronger signal across start and stop sites of genes on average, and, in many cases, weak triplet periodicity.
Data availability
The data presented in Fig. 3 were generated as part of ref.27 and can be accessed under BioProject accession PRJNA510123.
Code availability
The pipeline implemented in this paper is available at https://github.com/bhattlab/bhattlab_workflows/tree/master/metariboseq (https://zenodo.org/record/4638134#.YGDevkhKgcg)46. This pipeline creates a de novo assembly from metagenomic reads, maps MetaRibo-Seq reads to the assembly, calls open reading frames across the assembly and creates visualizations such as metagene plots, triplet periodicity histograms and fragment length distribution for aligned MetaRibo-Seq reads. This serves as a baseline assessment of library quality and a starting point for future analysis. Though specifically designed for MetaRibo-Seq, this pipeline would also be generally useful for any situation in which both DNA-sequencing and Ribo-Seq data for an organism with no existing reference genome are available.
Acknowledgements
We thank D. Maghini, E. Brooks and S. Vance for helpful comments on the manuscript. This work was funded by the Damon Runyon Clinical Investigator Award, grant nos. NIH R01AI148623 and NIH R01AI143757 as well as grant no. NIH P30 AG047366, which supports the Stanford ADRC. Computational work was supported by NIH S10 Shared Instrumentation grant no. 1S10OD02014101 and by NIH grant no. P30 CA124435.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Zengel JM & Lindahl L Diverse mechanisms for regulating ribosomal protein synthesis in Escherichia coli. Prog. Nucleic Acid Res. Mol. Biol 47, 331–370 (1994). [DOI] [PubMed] [Google Scholar]
- 2.Meyer MM The role of mRNA structure in bacterial translational regulation. Wiley Interdiscip. Rev. RNA 10.1002/wrna.1370 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Lindahl L, Jaskunas SR, Dennis PP & Nomura M Cluster of genes in Escherichia coli for ribosomal proteins, ribosomal RNA, and RNA polymerase subunits. Proc. Natl Acad. Sci. USA 72, 2743–2747 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fallon AM, Jinks CS, Strycharz GD & Nomura M Regulation of ribosomal protein synthesis in Escherichia coli by selective mRNA inactivation. Proc. Natl Acad. Sci. USA 76, 3411–3415 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dean D & Nomura M Feedback regulation of ribosomal protein gene expression in Escherichia coli. Proc. Natl Acad. Sci. USA 77, 3590–3594 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li G-W, Burkhardt D, Gross C & Weissman JS Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 157, 624–635 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lalanne J-B et al. evolutionary convergence of pathway-specific enzyme expression stoichiometry. Cell 173, 749–761.e38 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bucca G et al. Translational control plays an important role in the adaptive heat-shock response of Streptomyces coelicolor. Nucleic Acids Res. 46, 5692–5703 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sawyer EB, Grabowska AD & Cortes T Translational regulation in mycobacteria and its implications for pathogenicity. Nucleic Acids Res. 46, 6950–6961 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taylor RC et al. Changes in translational efficiency is a dominant regulatory mechanism in the environmental response of bacteria. Integr. Biol 5, 1393–1406 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morita Y, Gilmour C, Metcalf D & Poole K Translational control of the antibiotic inducibility of the PA5471 gene required for mexXY multidrug efflux gene expression in Pseudomonas aeruginosa. J. Bacteriol 191, 4966–4975 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Starosta AL, Lassak J, Jung K & Wilson DN The bacterial translation stress response. FEMS Microbiol. Rev 38, 1172–1201 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeong Y et al. The dynamic transcriptional and translational landscape of the model antibiotic producer Streptomyces coelicolor A3(2). Nat. Commun 7, 11605 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mathieu A et al. Discovery and function of a general core hormetic stress response in E. coli induced by sublethal concentrations of antibiotics. Cell Rep. 17, 46–57 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Ingolia NT, Ghaemmaghami S, Newman JRS & Weissman JS Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ingolia NT, Brar GA, Rouskin S, McGeachy AM & Weissman JS The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc 7, 1534–1550 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Becker AH, Oh E, Weissman JS, Kramer G & Bukau B Selective ribosome profiling as a tool for studying the interaction of chaperones and targeting factors with nascent polypeptide chains and ribosomes. Nat. Protoc 8, 2212–2239 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oh E et al. Selective ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo. Cell 147, 1295–1308 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li G-W, Oh E & Weissman JS The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature 484, 538–541 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Freeberg L, Kuersten S & Syed F Isolate and sequence ribosome-protected mRNA fragments using size-exclusion chromatography. Nat. Methods 10, i–ii (2013). [Google Scholar]
- 21.Latif H et al. A streamlined ribosome profiling protocol for the characterization of microorganisms. Bio-Techniques 58, 329–332 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Nakahigashi K et al. Comprehensive identification of translation start sites by tetracycline-inhibited ribosome profiling. DNA Res 23, 193–201 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meydan S et al. Retapamulin-assisted ribosome profiling reveals the alternative bacterial proteome. Mol. Cell 74, 481–493.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mangano K et al. Genome-wide effects of the antimicrobial peptide apidaecin on translation termination in bacteria. eLife 9, e62655 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hwang J-Y & Buskirk AR A ribosome profiling study of mRNA cleavage by the endonuclease RelE. Nucleic Acids Res. 45, 327–336 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mohammad F, Green R & Buskirk AR A systematically-revised ribosome profiling method for bacteria reveals pauses at single-codon resolution. eLife 8, e42591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fremin BJ, Sberro H & Bhatt AS MetaRibo-Seq measures translation in microbiomes. Nat. Commun 11, 3268 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mohammad F & Buskirk AR Protocol for ribosome profiling in bacteria. Bio Protoc. 9, e3468 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanca A et al. A straightforward and efficient analytical pipeline for metaproteome characterization. Microbiome 2, 49 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ndah E et al. REPARATION: ribosome profiling assisted (re-)annotation of bacterial genomes. Nucleic Acids Res. 45, e168 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clauwaerts J, Menschaert G & Waegeman W DeepRibo: a neural network for precise gene annotation of prokaryotes by combining ribosome profiling signal and binding site patterns. Nucleic Acids Res. 47, e36 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sberro H et al. Large-scale analyses of human microbiomes reveal thousands of small, novel genes. Cell 178, 1245–1259.e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Durrant MG & Bhatt AS Automated prediction and annotation of small open reading frames in microbial genomes. Cell Host Microbe 29, 121–131.e4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dar D et al. Term-seq reveals abundant ribo-regulation of antibiotics resistance in bacteria. Science 352, aad9822 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fremin BJ & Bhatt AS Structured RNA contaminants in bacterial Ribo-Seq. mSphere 5, e00855–20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mohammad F, Woolstenhulme CJ, Green R & Buskirk AR Clarifying the translational pausing landscape in bacteria by ribosome profiling. Cell Rep. 14, 686–694 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marks J et al. Context-specific inhibition of translation by ribosomal antibiotics targeting the peptidyl transferase center. Proc. Natl Acad. Sci. USA 113, 12150–12155 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zinshteyn B, Wangen JR, Hua B & Green R Nuclease-mediated depletion biases in ribosome footprint profiling libraries. RNA 26, 1481–1488 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Loughlin S et al. Polysomes bypass a 50-nucleotide coding gap less efficiently than monosomes due to attenuation of a 5′ mRNA stem-loop and enhanced drop-off. J. Mol. Biol 432, 4369–4387 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nurk S, Meleshko D, Korobeynikov A & Pevzner PA metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10 (2011). [Google Scholar]
- 42.Langmead B, Trapnell C, Pop M & Salzberg SL Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chung BY et al. The use of duplex-specific nuclease in ribosome profiling and a user-friendly software package for Ribo-seq data analysis. RNA 21, 1731–1745 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Di Tommasso P et al. Nextflow enables reproducible computational workflows. Nat. Biotechnol 35, 316–319 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Kurtzer GM, Sochat V & Bauer MW Scientific containers for mobility of compute. PLoS One 12, e0177459 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siranosian B bhattlab/bhattlab_workflows: metariboseq_v1.0.0 10.5281/zenodo.4638134 (2021). [DOI]
Related links
Key references using this protocol
- Fremin BJ et al. Nat. Commun 11, 3268 (2020): 10.1038/s41467-020-17081-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant MG & Bhatt AS Cell Host Microbe 29, 121–131.e4 (2021): 10.1016/j.chom.2020.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data presented in Fig. 3 were generated as part of ref.27 and can be accessed under BioProject accession PRJNA510123.