

Abstract
To cope with unrepaired DNA lesions, cells are equipped with DNA damage tolerance mechanisms, including translesion synthesis (TLS). While TLS polymerases are well documented in facilitating replication across damaged DNA templates, it remains unknown whether TLS polymerases participate in transcriptional bypass of DNA lesions in cells. Herein, we employed the competitive transcription and adduct bypass assay to examine the efficiencies and fidelities of transcription across N2-alkyl-2′-deoxyguanosine (N2-alkyl-dG, alkyl = methyl, ethyl, n-propyl, or n-butyl) lesions in HEK293T cells. We found that N2-alkyl-dG lesions strongly blocked transcription and elicited CC → AA tandem mutations in nascent transcripts, where adenosines were misincorporated opposite the lesions and their adjacent 5′ nucleoside. Additionally, genetic ablation of Pol η, but not Pol κ, Pol ι, or Pol ζ, conferred marked diminutions in the transcriptional bypass efficiencies of the N2-alkyl-dG lesions, which is exacerbated by codepletion of Rev1 in Pol η-deficient background. We also observed that the repair of N2-nBu-dG was not pronouncedly affected by genetic depletion of Pol η or Rev1. Hence, our results provided insights into transcriptional perturbations induced by N2-alkyl-dG lesions and expanded the biological functions of TLS DNA polymerases.
Graphical Abstract
INTRODUCTION
The genomic integrity of human cells is constantly challenged by exposure to a variety of endogenous and exogenous DNA-damaging agents.1 Byproducts of endogenous metabolism, metabolic activation of some environmental chemicals, and cancer chemotherapeutic agents can lead to DNA alkylation, which represents one of the most common types of DNA damage.2–4
The N2 position of 2′-deoxyguanosine (dG) constitutes one of the major sites in DNA that can be modified by various alkylating agents. For example, benzo[a]pyrene-7,8-diol-9,10-epoxide (BPDE), a metabolite of benzo[a]pyrene, preferentially reacts with the N2 of dG to yield N2-BPDE-dG.5 In addition, formaldehyde and acetaldehyde can conjugate with dG, and the resulting products can undergo reduction to yield N2-methyl-dG (N2-Me-dG) and N2-ethyl-dG (N2-Et-dG), respectively, where N2-Et-dG is detected in blood DNA of aldehyde dehydrogenase 2-deficient individuals after drinking.6–9
Faithful and efficient propagation of genetic information is essential for all domains of life,10 and several studies have explored how N2-alkyl-dG lesions perturb the efficiency and accuracy of DNA replication and transcription.9,11,12 For instance, Wu et al.11 showed that replication across a series of N2-alkyl-dG lesions (with the alkyl group being a Me, Et, nPr, or nBu, Figure 1) is both accurate and efficient in human cells proficient in translesion synthesis (TLS). Depletion of polymerase (Pol) κ, Pol ι, or Rev1, however, results in G → A and G → T mutations at the lesion site.11
Figure 1.
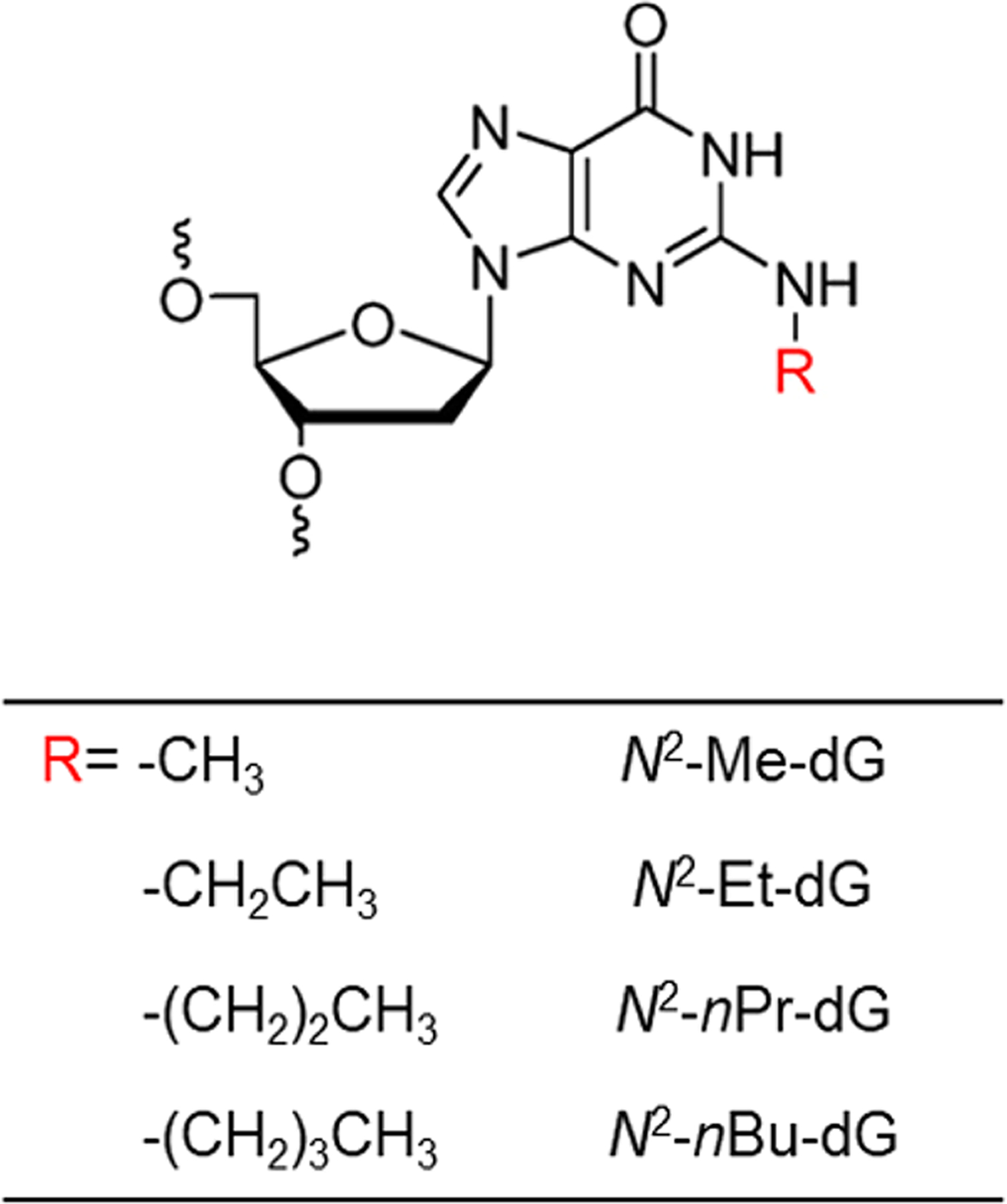
N2-Alkyl-dG lesions investigated in this study.
When located on the template strand of actively transcribed genes, many DNA lesions, including the methylglyoxal-induced N2-(1-carboxyethyl)-dG (N2-CE-dG),12 block transcription and trigger transcription-coupled nucleotide excision repair (TC-NER) in mammalian cells.12–15 In addition, N2-Et-dG impedes transcriptional elongation mediated by single and multisubunit RNA polymerases in vitro.9 Human RNA polymerase II (RNAPII), owing to its conformationally flexible active center, exhibits intrinsic translesion RNA synthesis ability, albeit with low efficiency and fidelity.16,17 The effects of N2-alkyl-dG lesions on perturbing the efficiencies and fidelities of transcription in human cells, however, have not yet been systematically examined.
TLS constitutes one of the major mechanisms of DNA damage tolerance, where specialized DNA polymerases insert a nucleotide opposite a DNA lesion and/or extend from the lesion site when replicative DNA polymerases are stalled.18,19 Recently, a growing number of studies revealed the versatile roles of TLS polymerases that are beyond their canonical functions in TLS.20–25 For instance, human DNA polymerase η can incorporate the correct ribonucleoside triphosphates (rNTPs) opposite undamaged and damaged nucleosides (e.g., 7,8-dihydro-8-oxo-dG and cyclobutane thymine dimer), albeit at rates that are markedly lower than those for inserting the corresponding dNTPs.22,26 Additionally, human Rev1 preferentially catalyzes rCTP incorporation directed by an arginine residue at the active site, with an efficiency that is approximately 280-fold lower than that of dCTP insertion.23 Moreover, human Pol ι exhibits intrinsic 5′-deoxyribose-5′-phosphate lyase activity and can function as a backup polymerase in base excision repair.24,25 It remains unclear if TLS polymerases can assist transcriptional bypass of DNA lesions in cells.
In the present study, we investigated systematically how N2-alkyl-dG lesions in template DNA strand affect transcription in human cells. We also assessed how transcription through these lesions is influenced by genetic ablation of TLS DNA polymerases.
RESULTS AND DISCUSSION
Transcriptional Perturbations of N2-Alkyl-dG Lesions in Human Cells.
The objectives of this study were to assess how N2-alkyl-dG lesions carrying various sizes of alkyl groups (Figure 1) perturb transcription and to investigate whether TLS DNA polymerases promote the transcriptional bypass of these lesions in human cells. To this end, we employed a previously established competitive transcription and adduct bypass (CTAB) assay,12,27 where we first constructed double-stranded, nonreplicative plasmids housing a site-specifically incorporated N2-alkyl-dG (Figures 1 and 2). In addition, the lesions are placed downstream of a CMV promoter to enable human RNAPII-mediated transcription.
Figure 2.
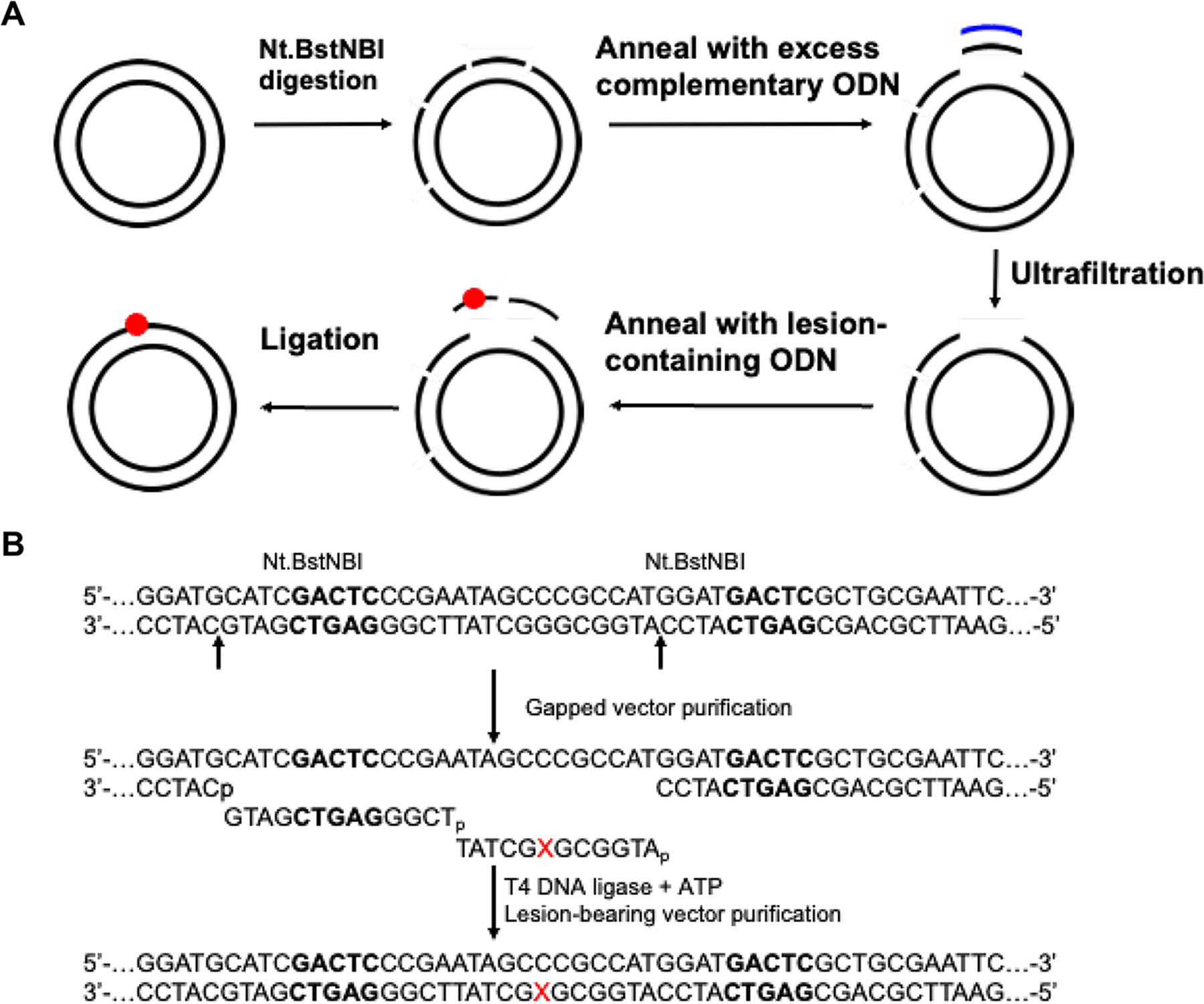
Construction of lesion-bearing plasmids. (a) Schematic diagram illustrating the procedures for the construction of plasmids harboring a site-specifically incorporated lesion. (b) Enzymatic digestion and ligation of the lesion-bearing ODNs into the gapped vector. “X” designates N2-alkyl-dG. The Nt.BstNBI recognition sequences are highlighted in bold.
The lesion-containing plasmid was transfected together with a lesion-free competitor plasmid into HEK293T cells. After cellular transcription, the runoff transcripts were reverse-transcribed with a gene-specific primer and the resulting cDNA amplified by PCR. The PCR products were digested with restriction enzymes, and the strand initially containing the lesion was selectively postlabeled on the 5′-terminus with 32P (Figures 3A and S1A). The resulting products were analyzed by native PAGE (Figures 3B and S1B–D) and LC–MS/MS (Figures S2–S3), where the 32P-labeling step was omitted in the sample preparation for LC–MS/MS experiments. The transcriptional bypass efficiency was calculated by dividing the band intensities of digestion products emanating from lesion-containing over competitor plasmids and then normalized against the ratio obtained from the parallel experiment conducted for the control lesion-free vector.
Figure 3.
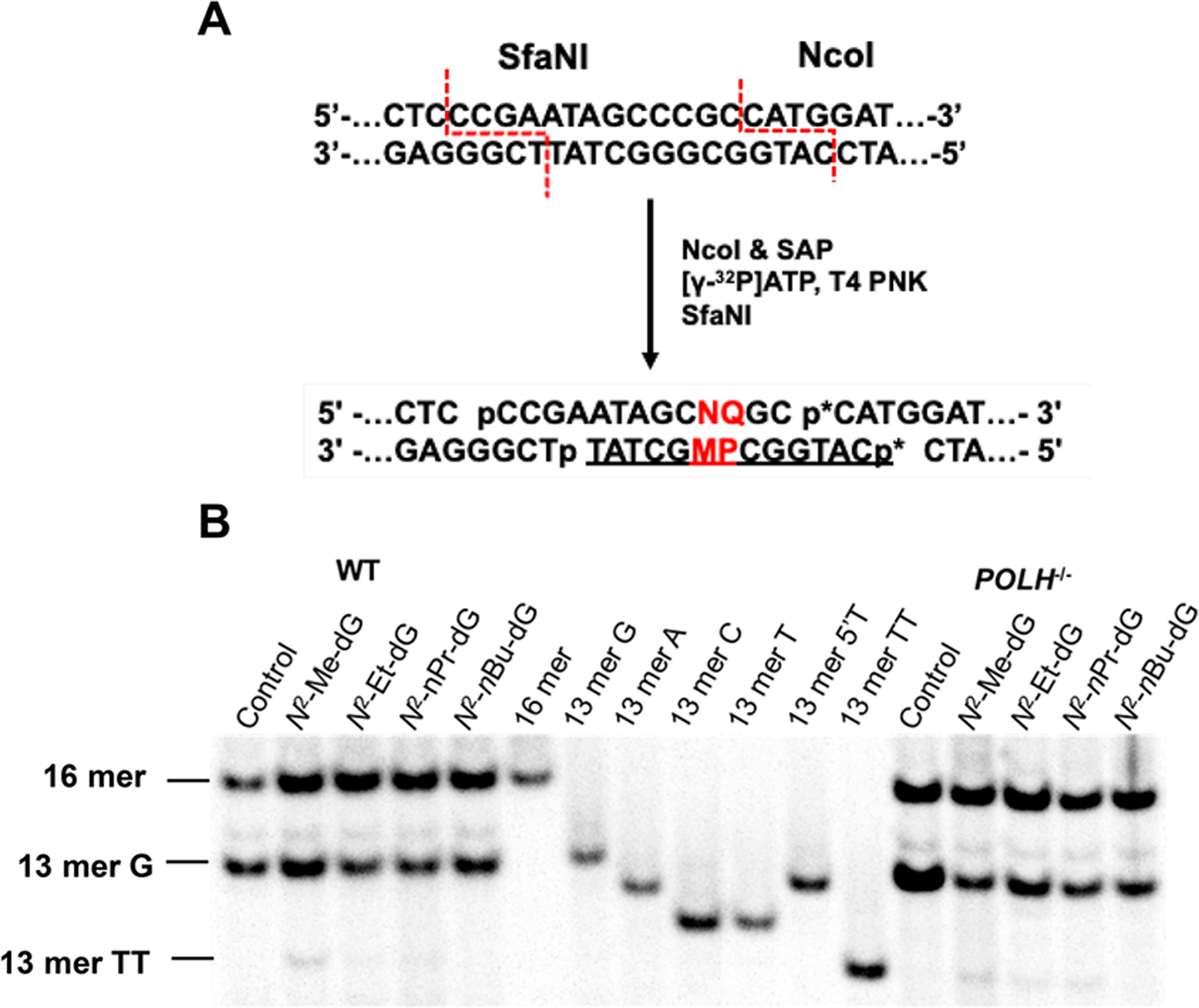
Restriction digestion and postlabeling method for determining the transcriptional bypass efficiencies and mutation frequencies of N2-alkyl-dG lesions in HEK293T cells and isogenic cells deficient of TLS polymerases. (a) A schematic diagram depicting the selective labeling of the template strand via sequential digestion of the RT-PCR products. “p*” denotes a 32P-labeled phosphate group. “M” represents the site where the lesion was initially situated. “P” indicates the nucleobase 5′ to the lesion site. “N” and “Q” are the complementary bases paired with “M” and “P”, respectively. Representative gel images for monitoring the restriction fragments of interest in wild-type or POLH-knockout cells (b). Lesion-containing plasmids were individually premixed with the competitor plasmid at a molar ratio of 2:1 (lesion/competitor) for transfection into HEK293T cells and 3:1 for POLH−/− cells, and the transcripts were isolated from cells at 24 h following transfection. The synthetic ODNs representing the restriction fragment arising from the competitor vector, i.e., d(CATGGCGATAGGCTAT), is designated as “16mer”; “13 mer G”, “13 mer A”, “13 mer C”, “13 mer T”, “13 mer 5′T”, and “13 mer TT” represent the standard synthetic ODNs d(CATGGCPMGCTAT), where “PM” is “GG”, “GA”, “GC”, “GT”, “TG”, and “TT”, respectively.
Our results showed that the presence of N2-alkyl-dG lesions on the transcribed strand led to pronouncedly diminished transcriptional bypass efficiencies in HEK293T cells, ranging from 27% to 35% relative to the corresponding dG-containing template (Figure 4A). In addition, increasing the size of the alkyl group elicited subtle differences in transcriptional bypass efficiency (Figure 4A). Together, N2-alkyl-dG lesions exert strong blockage effects on transcription machinery in human cells.
Figure 4.
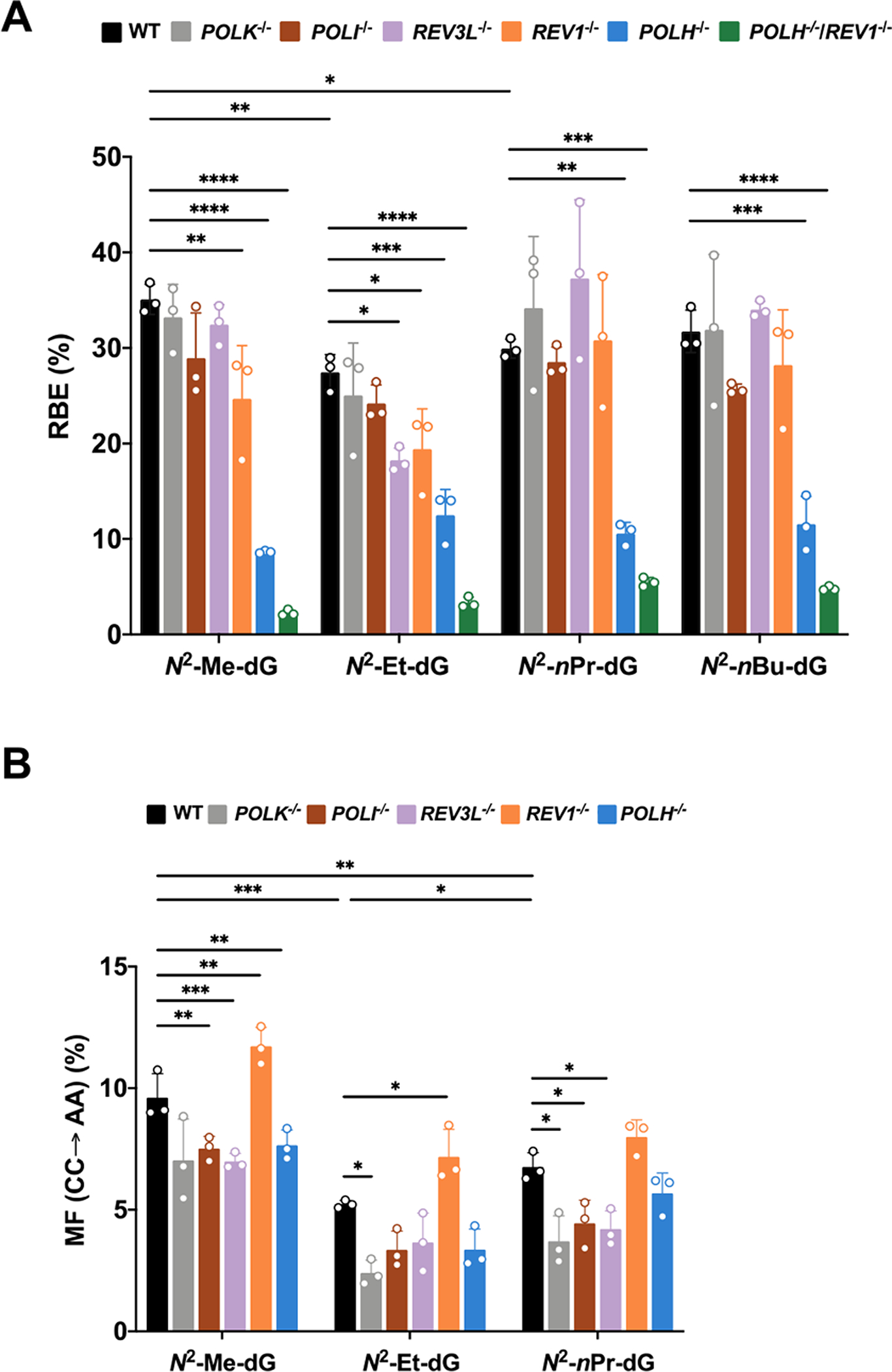
Relative transcriptional bypass efficiencies (RBEs) (A) and mutation frequencies (MFs) (B) of N2-alkyl-dG lesions in HEK293T cells and the isogenic cells where the indicated TLS polymerase genes were individually or simultaneously depleted by CRISPR/Cas9. POLH, POLI, POLK, REV1, and REV3L encode for DNA polymerases η, ι, κ, Rev1, and the catalytic subunit of Pol ξ, respectively. The transcripts were isolated at 24 h following plasmid transfection. The data represent the mean ± SD of results from three independent experiments. *, 0.01 < P < 0.05; **, 0.001 < p < 0.01; ***, 0.0001<p < 0.001; ****, p < 0.0001. The multiplicity-adjusted p values were calculated by using one-way ANOVA and Dunnett’s multiple comparisons test for the comparisons between parental HEK293T cells and isogenic TLS polymerase knockout cells and by one-way ANOVA and Tukey’s multiple comparisons test for the comparisons between different lesions in parental HEK293T cells.
Our PAGE and LC–MS and MS/MS analyses led to the identification of mutant transcripts arising from misinsertions of adenosines opposite both the site of N2-Me-dG, N2-Et-dG, and N2-nPr-dG as well as their neighboring 5′ nucleotide (CC → AA mutation) (Figures 3 and S1–S3), though this mutation was barely detectable for the N2-nBu-dG-containing substrate. We also detected a single 5′-A mutation based on LC–MS and MS/MS analyses (Figures S2 and S3), where RNAPII misincorporates an adenosine opposite the 5′-neighboring base of the lesions; the rate of the latter 5′-A mutation was, however, too low for robust quantification (<2%).
Roles of TLS Polymerases in Supporting the Transcriptional Bypass of N2-Alkyl-dG Lesions.
To explore whether TLS polymerases can assist the transcriptional bypass of these lesions, we conducted the CTAB experiments by employing isogenic HEK293T cells where TLS polymerases were individually or simultaneously knocked out by CRISPR-Cas9.11,28 We observed that individual ablation of Pol η conferred marked attenuations in transcriptional bypass efficiencies (to ~9–13%) for all four lesions (Figure 4A). In addition, depletion of Rev1 led to significant diminutions in bypass efficiencies of N2-Me-dG and N2-Et-dG. Interestingly, further depletion of Rev1 in Pol η-deficient background exacerbated the transcription blockage effects of all four lesions (with bypass efficiencies being reduced to 2–5%), suggesting that Pol η and Rev1 act independently to promote efficient transcriptional bypass of the N2-alkyl-dG lesions.
To further substantiate the role of Pol η in supporting the transcriptional bypass of the N2-alkyl-dG lesions, we conducted the CTAB assay by employing patient-derived Pol η-deficient cells and the isogenic cells complemented with wild-type human Pol η.29,30 Our results revealed that the diminished transcriptional bypass of N2-nPr-dG in patient-derived Pol η-deficient cells could be rescued by reconstituting the cells with wild-type human Pol η (Figure 5). In this vein, it is worth noting that the RBE value for N2-nPr-dG in XP30RO cells is substantially lower than what we observed for POLH−/− HEK293T cells. A previous study showed that the XP30RO cells carry an out-of-frame deletion in POLH gene, which gave rise to a truncated protein encompassing only the first 42 amino acids of Pol η (out of a total of 713 amino acids).31 Hence, the difference is unlikely attributable to the presence of mutant Pol η (if stable) in XP30RO cells; instead, it arises likely from the biological heterogeneities of the two cell lines.
Figure 5.

Transcriptional bypass efficiencies and mutation frequencies of N2-nPr-dG in Pol η-deficient XP30RO cells and the isogenic cells complemented with wild-type human Pol η. (A) Representative gel images for monitoring the restriction fragments of interest from the RT-PCR products of transcripts isolated from the transcription of a mixture of N2-nPr-dG- or dG-containing plasmid with competitor plasmid in XP30RO cells and the corresponding wild-type human Pol η-complemented human skin fibroblast cells at 24 h following transfection. The N2-nPr-dG-containing plasmid was premixed with the competitor plasmid at a molar ratio of 3:1 for XP30RO and 2:1 for XP30RO cells complemented with wild-type human Pol η. The synthetic ODNs representing the restriction fragment arising from the competitor vector, i.e., d(CATGGCGATAGGCTAT), are designated as “16mer”, “13 mer G”, “13 mer A”, “13 mer C”, “13 mer T”, “13 mer 5′T”, and “13 mer TT” represent the standard synthetic ODNs d(CATGGCPMGCTAT), where “PM” is “GG”, “GA”, “GC”, “GT”, “TG”, and “TT”, respectively. Shown in (B) and (C) are the relative transcriptional bypass efficiencies (RBE) and CC → AA mutation frequencies (MF) for N2-nPr-dG observed in the two cell lines. The data represent the mean ± SD of results obtained from three independent experiments. The p values were calculated by using unpaired two-tailed student’s t-test: **, 0.001< p < 0.01; ns, p > 0.05.
Depletion of Pol κ, Pol ι, or Pol ζ did not affect appreciably the efficiencies in transcriptional bypass of the N2-alkyl-dG lesions, except that genetic ablation of Pol ζ led to a significantly decreased transcriptional bypass efficiency for N2-Et-dG. On the other hand, the frequency of the aforementioned CC → AA mutation is the highest in REV1-knockout cells (Figure 4B), suggesting that Rev1 elicited error-free transcription across these lesions.
Pol η and Rev1 Do Not Substantially Modulate the Repair of N2-Alkyl-dG Lesions in Human Cells.
Our above results demonstrated that genetic depletion of Pol η led to pronounced drops in transcriptional bypass efficiencies of N2-alkyl-dG lesions, which are aggravated by further ablation of Rev1. In light of the previous observations that some TLS polymerases can function in DNA repair,20,21 we recognized that the reduced transcript yields may also arise from diminished repair of these lesions in the polymerase-deficient background. To examine this possibility, we conducted the CTAB assay by monitoring the time-dependent alterations in transcript yields, where we isolated nascent transcripts from the lesion-containing and control plasmids at different time points following transfection. Our results revealed a progressive increase in transcript yield for N2-nPr-dG in HEK293T cells. A similar increase in transcript yield was observed from 0 to 8 h for the isogenic Pol η-deficient cells, though no statistically significant elevation in transcript yield was observed for this lesion from 8 to 24 h in this genetic background. These results suggest that the repair of N2-nPr-dG in episomal plasmid is not pronouncedly compromised by the lack of Pol η (Figure S4).
We recognize that the repair of N2-alkyl-dG lesions located in the episomal vector employed for the CTAB assay may behave differently from the lesion situated in genomic DNA. To examine whether Pol η contributes to the repair of N2-alkyl-dG lesions in genomic DNA, we next assessed the roles of this and other TLS polymerases in the removal of N2-nBu-dG in HEK293T cells. A recent study by Spratt and co-workers32 showed that incubating cultured human cells with N2-substituted dG derivatives, including N2-nBu-dG, can allow for the facile incorporation of these modified nucleosides into genomic DNA, and Pol κ plays an important role in this process. We found that incubating HEK293T cells with 10 μM N2-nBu-dG for 3 h results in the incorporation of a substantial level of N2-nBu-dG into genomic DNA (Figure 6). While individual ablation of Pol ι, Pol ζ, or Rev1 did not appreciably affect the incorporation of N2-nBu-dG, losses of Pol κ and, to a lesser degree, Pol η gave rise to pronounced decreases in the incorporation of N2-nBu-dG into genomic DNA. This is in keeping with the results from in vitro biochemical assay, showing that Pol κ is highly efficient in inserting N2-nBu-dGTP opposite a cytosine base in template DNA; Pol η also exhibits such a function, albeit at a much lower efficiency.32
Figure 6.
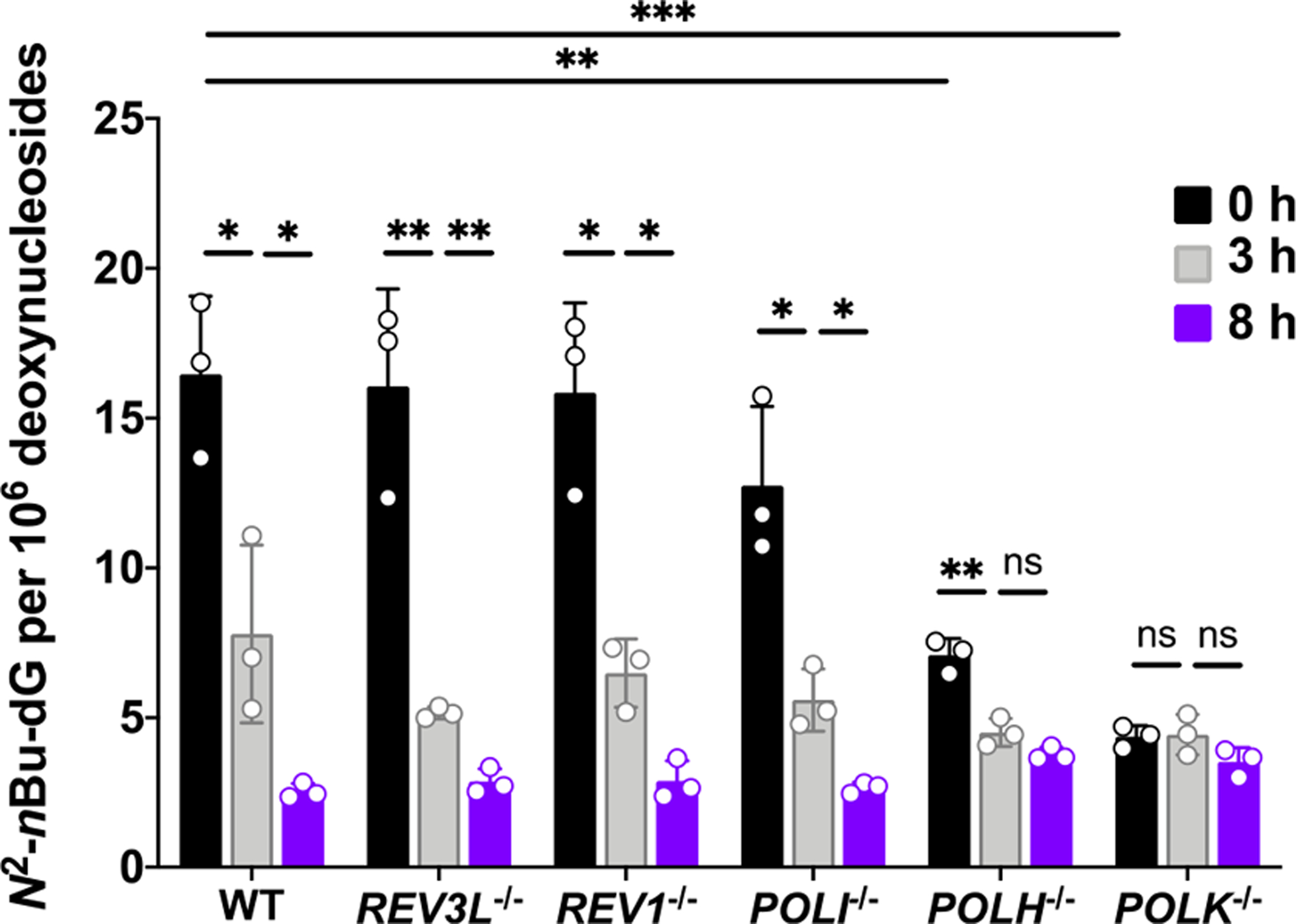
Frequencies of N2-nBu-dG in cellular DNA isolated from parental and TLS polymerase-depleted HEK 293T cells. All cells were exposed to 10 μM of N2-nBu-dG for 3 h. The cells were then harvested immediately or after incubation in fresh media for another 3 or 8 h. The data represent the mean ± SD of results obtained from three independent experiments. *, 0.01 < p < 0.05; **, 0.001 < p < 0.01; ns, p > 0.05. The multiplicity-adjusted p values were calculated by using multiple t-tests with a Holm–Sidak correction for comparisons between 0 and 3 h, and between 3 and 8 h, by using one-way ANOVA and Dunnett’s multiple comparisons test for the comparisons between parental HEK293T cells and the isogenic polymerase knockout cells at 0 h.
Our LC–MS/MS results also revealed that, after removal of medium containing the modified nucleoside, cellular DNA displays a time-dependent decrease in the level of N2-nBu-dG in parental HEK293T cells, or the isogenic cells depleted of Pol η, Pol ι, Rev1, or Pol ζ, though the difference in the levels of N2-nBu-dG in Pol η-deficient cells at 3 and 8 h was not statistically significant. The loss of Pol κ, however, abrogated such a progressive decrease in the level of N2-nBu-dG. These results, therefore, underscore that, among these TLS polymerases, Pol κ contributes to the removal of N2-nBu-dG from genomic DNA. In addition, our results suggest that Pol η may also assume a minor role in promoting the repair of N2-nBu-dG in cells. Together, the above results support that the reduced transcript yields of N2-alkyl-dG-containing template observed in cells depleted of Pol η arise mainly from its role in supporting the transcriptional bypass of these lesions, though we cannot exclude formally a minor role of Pol η in enhancing the repair of these lesions.
Previous studies showed that replication through N2-alkyl-dG and N2-carboxyalkyl-dG lesions in HEK293T cells was highly efficient, with the bypass efficiencies being 60–80% and 99–100%, respectively.11,33 Different from what were observed in replication studies, here we found that N2-alkyl-dG lesions conferred considerable impediments to transcription (with bypass efficiencies being 27–35%), which is consistent with what were previously reported for N2-CE-dG lesions in human skin fibroblast cells12 and for N2-Et-dG in RNAPII-mediated transcription in vitro.9
Recently, several studies showed that TLS polymerases have functions beyond their well-established roles in translesion DNA synthesis, where some of these polymerases are capable of incorporating ribonucleotides in vitro.22,23,26,34,35 By employing the CTAB assay, we examined systematically the involvement of different TLS polymerases in transcriptional output of N2-alkyl-dG lesions in human cells. Our results showed that single depletion of Pol η led to substantially decreased transcript yields of all four N2-alkyl-dG lesions (Figures 4 and 5). Dual ablation of Pol η and Rev1 conferred more pronounced attenuations in transcript yields of these lesions than depletion of Pol η alone (Figure 4). Our LC–MS/MS quantification results showed that N2-nBu-dG in genomic DNA can be repaired in Pol η-deficient HEK293T cells (Figure 6). In addition, we observed a time-dependent increase in transcript yield from an N2-nPr-dG-containing plasmid in HEK293T cells, and this increase was also observed in Pol η-deficient HEK293T cells (Figure S4). These results, therefore, support that the decreased transcript yields of N2-alkyl-dG-harboring template in the Pol η-deficient cells are attributed mainly to its role in supporting the transcriptional bypass of these lesions, though we cannot exclude completely a minor role of Pol η in promoting the repair of these lesions.
It is worth noting that the double-stranded plasmid that we employed in the current study does not carry any mammalian replication origin; hence, it is extremely unlikely that reduced transcript yield observed in Pol η-deficient background arises from its role in replicative bypass of N2-alkyl-dG lesions. This notion is corroborated by the lack of impact of Pol η depletion on the efficiency or fidelity of replication across the N2-alkyl-dG lesions in a similar, yet SV40 replication origin-carrying, plasmid,11 which is in stark contrast to Pol η’s prominent role in modulating the transcriptional bypass of these DNA adducts (Figures 4 and 5).
We postulate that the spacious active site of Pol η36 endows its ability in ribonucleotide incorporation. Additionally, recent studies revealed that human Pol η scaffolds the incoming ribonucleoside triphosphate to pair with the template base guanine or 7,8-dihydro-8-oxoguanine with a significant propeller twist.22,26 It will be important to examine, in the future, whether a similar alteration of active site structure occurs for Pol η during ribonucleotide incorporation opposite the N2-alkyl-dG lesions. Rev1 was found to function as a scaffold to assemble other TLS polymerases, including Pol κ, Pol η, Pol ζ, and Pol ι, during TLS across different DNA damage products.37,38 Rev1 itself could also direct dCTP insertion through an arginine residue (R357) near its active site, regardless of the template identity.39
Replicative bypass of N2-alkyl-dG lesions in HEK293T cells is error-free; however, G → T and G → A single-base substitutions were observed in cells deficient in Pol κ, Pol ι, or Rev1.11 In contrast, transcription across N2-alkyl-dG lesions (alkyl = Me, Et, and nPr) in HEK293T cells induced appreciable levels of CC → AA tandem mutation in nascent transcripts, where adenosine was inserted opposite both the lesion and its vicinal 5′ nucleoside, and this was accompanied by a much lower frequency of 5′-A mutation. The misincorporation of an adenosine opposite the neighboring 5′ nucleoside of N2-alkyl-dG is reminiscent of previous observations made for oxidatively induced purine cyclonucleo-sides12 and suggests that the lesion-induced distortion of the template strand renders the neighboring 5′ nucleoside inadequately recognized by polymerase(s) during ribonucleotide insertion. Although Pol ζ, Pol κ, and Pol ι contribute minimally to transcriptional bypass of N2-alkyl-dG lesions, their losses result in slightly elevated levels of CC → AA mutation in nascent transcripts. Additionally, genetic ablation of Rev1 results in pronounced increases in CC → AA mutation frequencies for these lesions, suggesting the role of this polymerase in promoting error-free transcription across these lesions. Primer extension assay showed that RNAPII preferentially inserts a rCTP opposite N2-Et-dG, but the accessory transcription factor TFIIS subsequently stimulates the backtrack of RNAPII and removes the rCTP.9 We reason that TLS polymerases are required to overcome the transcriptional blockage, where the involvement of TLS polymerases also elicits appreciable levels of mutant transcripts.
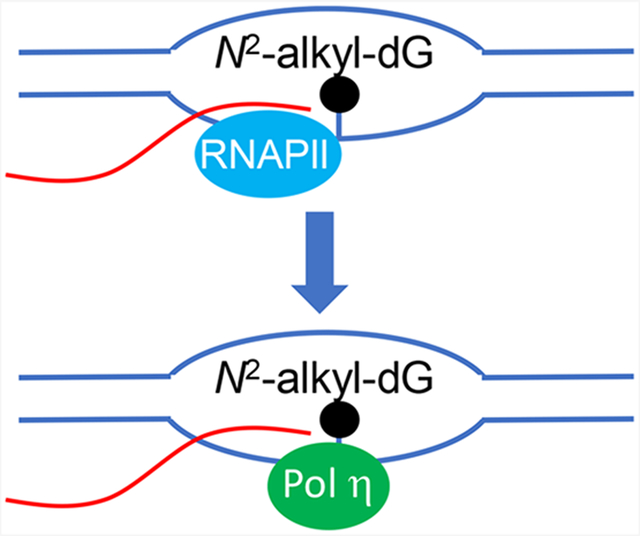
The mechanism through which Pol η is recruited to stalled RNAPII is unclear and warrants further investigation. Along this line, it was observed recently that ubiquitination of lysine 1268 in the largest subunit of RNAPII, RPB1, regulates transcription recovery and triggers TC-NER by stimulating the association of the core-TFIIH complex with stalled RNAPII and promoting its degradation.40,41 In addition, monoubiquitination of PCNA was shown to promote the recruitment of Pol η to stalled DNA replication machinery.42,43 It will be important to examine, in the future, whether a similar post-translational mechanism contributes to the recruitment of Pol η to stalled RNAP II.
CONCLUSIONS
In conclusion, we investigated systematically how minor-groove N2-alkyl-dG lesions impede transcription and induce transcriptional mutagenesis in human cells. We also unveiled novel biological functions of TLS polymerases in supporting transcriptional bypass of the minor-groove N2-alkyl-dG lesions in human cells. It will be important to investigate further the mechanism by which the TLS polymerases are recruited to transcription machinery during adduct bypass and to examine whether these functions of TLS polymerases can be extended to other types of DNA lesions.
EXPERIMENTAL SECTION
Unless otherwise stated, all chemicals were from Sigma-Aldrich or Thermo Fisher Scientific, and all enzymes were from New England Biolabs (Ipswich, MA). Unmodified ODNs were obtained from Integrated DNA Technologies (Coralville, IA). M-MLV reverse transcriptase was purchased from Promega (Madison, WI). [γ−32P]ATP was acquired from PerkinElmer Life Sciences (Waltham, MA).
The 12-mer ODNs carrying a site-specifically inserted N2-alkyl-dG were previously synthesized.11 HEK293T cells with single depletion of the POLH, POLI, POLK, REV3L, or REV1 gene were produced previously by the CRISPR-Cas9 genome editing method, where the successful depletion of these genes were confirmed by both Sanger sequencing and Western blot analysis.11,28 The isogenic cells with concurrent depletions of POLH and REV1 were generated from the POLH-knockout background using the same method. The successful depletion of REV1 gene was confirmed by Sanger sequencing and Western blot analysis (Figure S5). The SV40-transformed Pol η-deficient XP30RO fibroblasts and the corresponding cells reconstituted with wild-type human Pol η (XP30RO + Pol η) were kindly provided by Professor James E. Cleaver.29,30
Construction of Lesion-Containing Plasmids.
In-house synthesized 12mer N2-alkyl-dG-containing ODNs were incorporated into the transcribed strand of a double-stranded shuttle vector, i.e., pTGFP-T7-Hha10, following previously published procedures.27 This vector was constructed from pTurboGFPN,44 where the SV40 replication origin in the initial plasmid was removed. Briefly, the damage-free control vector was digested with Nt.BstNBI to nick the double-stranded parental vector. The 25mer ODN arising from Nt.BstNBI cleavage was subsequently removed by annealing with excess complementary 25mer ODN and by centrifugation using a 100 kDa cutoff centrifugal filter. The 12mer N2-alkyl-dG-containing ODN (5′-ATGGCGXGCTAT-3′, X= N2-alkyl-dG) was 5′-phosphorylated and annealed into the gap together with a 13mer 5′-phosphorylated lesion-free ODN (5′-TCGGGAGTCGATG-3′) (Figure 2). T4 DNA ligase was then added to seal the gap. The fully ligated, supercoiled plasmid was isolated from the ligation mixture by using agarose gel electrophoresis (Figure S6).
Cellular Transcription, RNA Isolation, and RT-PCR.
Cellular transcription experiments with the use of the above-constructed vectors were performed using the previously reported CTAB assay.27 The lesion-containing plasmids were individually premixed with the competitor plasmid at a molar ratio of 2:1 (lesion/competitor) for transfection into HEK293T, POLK−/−, POLI−/−, and REV3L−/− cells, 3:1 for POLH−/− cells, 5:1 for REV1−/− and POLH−/−/REV1−/− cells, 3:1 for XP30RO, and 2:1 for XP30RO cells complemented with wild-type human Pol η. The lesion-free control plasmid was premixed with the competitor vector at a molar ratio of 5:1 (control/competitor) for REV1−/− cells and 1:1 for all other cell lines. HEK293T cells and the isogenic TLS polymerase-deficient cells (1 × 105) were seeded in 24-well plates and cultured overnight at 37 °C in a 5% CO2 atmosphere, followed by transfection with 50 ng of the mixed plasmids and 450 ng of carrier plasmid (self-ligated pGEM-T, Promega) using TransIT-2020 (Mirus Bio) following the manufacturer’s recommended procedures. The cells were harvested at 24 h unless specified for the time-dependent experiments. After transfection, the transcripts of the mixed plasmids were isolated using Total RNA Kit I (Omega), and residual DNA in the mixture was removed with a DNA-free kit (Ambion). The transcripts of interest were reverse-transcribed and PCR-amplified, as described elsewhere.27 In this vein, we confirmed the lack of contamination of initial plasmid DNA in the RNA samples on the basis of absence of PCR products when the amplification was conducted directly for the RNA samples without the reverse transcription step, as described previously (Figure S7).27
Restriction Digestion and Polyacrylamide Gel Electrophoresis (PAGE) Analysis.
A NcoI/SfaNI-mediated restriction digestion and postlabeling method was employed for sample preparation prior to PAGE analysis. For each sample, 150 ng of the above-mentioned RT-PCR products was incubated with 5 U NcoI and 1 U shrimp alkaline phosphatase (rSAP) in 10 μL of NEB buffer 3.1 at 37 °C for 1 h. The enzymes were heat-inactivated by incubation at 80 °C for 20 min, and to the mixture were added with 5 U T4 polynucleotide kinase and 1.66 pmol [γ−32P]ATP to radiolabel the newly liberated 5′-termini in the template strand (shown as the bottom strand in Figure 3A). The resultant mixture was heated at 75 °C for 20 min and further digested with 2 U SfaNI in 20 μL of 1× NEB buffer 3.1 at 37 °C for 1.5 h (Figure 3). The reaction was terminated with 20 μL of formamide gel-loading buffer (2 ×), and the DNA mixture was resolved by using 30% native PAGE (acrylamide/bis-acrylamide = 19:1) and quantified by phosphor-imager analysis.27 The intensities of the radiolabeled DNA bands were used to calculate the relative bypass efficiency (RBE) with the following equation: RBE (%) = (lesion signal/competitor signal)/(control signal/competitor signal) × 100%, where the competitor signal was employed as the internal standard. The transcriptional mutation frequency (MF) was determined from the percentage of the amount of mutant transcript among the total amounts of all transcripts arising from the lesion-containing plasmid.
LC–MS/MS for the Identification of Mutant Transcripts.
LC–MS and MS/MS were used to identify unambiguously the transcription products arising from N2-alkyl-dG-containing templates, similar to those described elsewhere.12,27 RT-PCR products were treated with 50 U NcoI and 20 U rSAP in 250 μL of NEB buffer 3.1 at 37 °C for 2 h, followed by heating at 80 °C for 20 min. To the resulting solution was added 50 U SfaNI, and the reaction mixture was incubated at 37 °C for 2 h, followed by extraction with phenol/chloroform/isoamyl alcohol (25:24:1, v/v). The aqueous phase was collected, to which were added 0.1 volume of 3.0 M sodium acetate and 2.5 volumes of ethanol to precipitate the DNA. The DNA pellet was reconstituted in water and subjected to LC–MS/MS analysis. An LTQ linear ion trap mass spectrometer (Thermo Fisher Scientific) was set up for monitoring the fragmentations of the [M − 3H]3− ions of the 13-mer ODNs, 5′-CATGGCPMGCTAT-3′, where “PM” designates “GA”, “GT”, “GC”, “GG”, “TG”, or “TT”.
Incorporation of N2-nBu-dG into Cellular DNA and Extraction and Enzymatic Digestion of Genomic DNA.
HEK293T cells and the isogenic TLS polymerase-deficient cells were seeded in 6-well plates at 37 °C in a 5% CO2 atmosphere. N2-nBu-dG was added to the culture medium until its final concentration reached 10 μM. After incubation for 3 h, the cells were switched to fresh media without N2-nBu-dG and harvested 3 or 8 h later. Genomic DNA was extracted from HEK293T cells, the isogenic cells were depleted of TLS polymerases using Qiagen DNeasy Blood & Tissue Kit, and approximately 6 μg of DNA was recovered from a single well of cells.
We digested 1.0 μg of cellular DNA with 10 units of nuclease P1 (New England Biolabs) and 0.00125 unit of phosphodiesterase II in a buffer containing 30 mM sodium acetate (pH 5.6), 1 mM ZnCl2, and 2.5 nmol of EHNA. The above mixture was incubated at 37 °C for 24 h. To the mixture were then added 1.0 unit of alkaline phosphatase, 0.0025 unit of phosphodiesterase I, and one tenth volume of 0.5 M Tris–HCl (pH 8.9). After incubation at 37 °C for 4 h, the digestion mixture was neutralized with 1.0 M formic acid. The enzymes in the digestion mixture were subsequently removed by extraction with an equal volume of chloroform. The aqueous layer was dried in vacuo and reconstituted in water for LC–MS/MS analysis.
Online nLC–MS/MS Analysis of N2-nBu-dG in Cellular DNA.
HPLC separation was conducted on a Dionex Ultimate 3000 HPLC module (Thermo Fisher) with an in-house packed trapping column (150 μm × 40 mm) and an analytical column (75 μm × 200 mm), both packed with Magic C18 AQ (200 Å, 5 μm, Michrom BioResource, Auburn, CA) reversed-phase materials. Mobile phases A and B were composed of 0.1% formic acid in doubly distilled water and acetonitrile, respectively. The sample was loaded onto the trapping column with mobile phase A at a flow rate of 2.5 μL/min in 8 min, and the analyte (N2-nBu-dG) and its stable isotope-labeled standard (Figure S8) were then eluted from the column by using a 20 min linear gradient of 0–95% mobile phase B at a flow rate of 300 nL/min.
The LC effluent was directed to a TSQ-Altis mass spectrometer operated in the multiple-reaction monitoring (MRM) mode. The MRM transitions corresponding to the neutral loss of a 2-deoxyribose (116 Da) from the protonated ions ([M + H]+) of N2-nBu-dG (i.e., m/z 324 → 208) and [D9]-N2-nBu-dG (i.e., m/z 333 → 217) were monitored (Figure S9). The voltage for electrospray was set at 2.0 kV, and the temperature for the ion transport tube was maintained at 275 °C. The widths for precursor and fragment ion selection were both 0.7 m/z unit, and the collision energy was set at 20 V. A calibration curve was constructed by spiking 1.0 μg of calf thymus DNA with different amounts of an N2-nBu-dG-containing 12-mer ODN, and a fixed amount of [D9]-N2-nBu-dG; the samples were subjected to enzymatic digestion and LC–MS/MS analysis under the same conditions as described above for the cellular DNA samples (Figure S10).
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the National Institutes of Health for supporting this research (R35 ES031707) and Professor James E. Cleaver for providing the XP30RO and the corresponding Pol η-complemented cells.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c07374.
Synthetic schemes, ESI-MS, and MS/MS of the synthetic unlabeled and stable isotope-labeled N2-nBu-dG, PAGE, and LC–MS/MS characterizations of restriction fragments of RT-PCR products of transcripts from cellular transcription across the N2-alkyl-dG lesions) (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c07374
The authors declare no competing financial interest.
Contributor Information
Ying Tan, Environmental Toxicology Graduate Program, University of California, Riverside, California 92521-0403, United States.
Su Guo, Environmental Toxicology Graduate Program, University of California, Riverside, California 92521-0403, United States.
Jun Wu, Department of Chemistry, University of California, Riverside, California 92521-0403, United States.
Hua Du, Department of Chemistry, University of California, Riverside, California 92521-0403, United States.
Lin Li, Department of Chemistry, University of California, Riverside, California 92521-0403, United States.
Changjun You, Department of Chemistry, University of California, Riverside, California 92521-0403, United States.
Yinsheng Wang, Environmental Toxicology Graduate Program and Department of Chemistry, University of California, Riverside, California 92521-0403, United States;.
REFERENCES
- (1).Friedberg EC; Walker GC; Siede W; Wood RD; Schultz RA; Ellenberger T DNA Repair and Mutagenesis; ASM Press, 2006; pp 1–1118. [Google Scholar]
- (2).Fu D; Calvo JA; Samson LD Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat. Rev. Cancer 2012, 12, 104–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Helleday T; Petermann E; Lundin C; Hodgson B; Sharma RA DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [DOI] [PubMed] [Google Scholar]
- (4).Sedgwick B; Bates PA; Paik J; Jacobs SC; Lindahl T Repair of alkylated DNA: recent advances. DNA Repair 2007, 6, 429–42. [DOI] [PubMed] [Google Scholar]
- (5).Pfeifer GP; Denissenko MF; Olivier M; Tretyakova N; Hecht SS; Hainaut P Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 2002, 21, 7435–51. [DOI] [PubMed] [Google Scholar]
- (6).Cheng G; Shi Y; Sturla SJ; Jalas JR; McIntee EJ; Villalta PW; Wang M; Hecht SS Reactions of formaldehyde plus acetaldehyde with deoxyguanosine and DNA: formation of cyclic deoxyguanosine adducts and formaldehyde cross-links. Chem. Res. Toxicol 2003, 16, 145–52. [DOI] [PubMed] [Google Scholar]
- (7).Matsuda T; Yabushita H; Kanaly RA; Shibutani S; Yokoyama A Increased DNA damage in ALDH2-deficient alcoholics. Chem. Res. Toxicol 2006, 19, 1374–8. [DOI] [PubMed] [Google Scholar]
- (8).Fang JL; Vaca CE Detection of DNA adducts of acetaldehyde in peripheral white blood cells of alcohol abusers. Carcinogenesis 1997, 18, 627–32. [DOI] [PubMed] [Google Scholar]
- (9).Cheng T-F; Hu X; Gnatt A; Brooks PJ Differential blocking effects of the acetaldehyde-derived DNA lesion N2-ethyl-2′-deoxyguanosine on transcription by multisubunit and single subunit RNA polymerases. J. Biol. Chem 2008, 283, 27820–27828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lans H; Hoeijmakers JHJ; Vermeulen W; Marteijn JA The DNA damage response to transcription stress. Nat. Rev. Mol. Cell Biol 2019, 20, 766–784. [DOI] [PubMed] [Google Scholar]
- (11).Wu J; Du H; Li L; Price NE; Liu X; Wang Y The impact of minor-groove N2-alkyl-2′-deoxyguanosine lesions on DNA replication in human cells. ACS Chem. Biol 2019, 14, 1708–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).You C; Dai X; Yuan B; Wang J; Brooks PJ; Niedernhofer LJ; Wang Y A quantitative assay for assessing the effects of DNA lesions on transcription. Nat. Chem. Biol 2012, 8, 817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bregeon D; Doetsch PW Transcriptional mutagenesis: causes and involvement in tumour development. Nat. Rev. Cancer 2011, 11, 218–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Saxowsky TT; Doetsch PW RNA polymerase encounters with DNA damage: transcription-coupled repair or transcriptional mutagenesis? Chem. Rev 2006, 106, 474–88. [DOI] [PubMed] [Google Scholar]
- (15).Hanawalt PC; Spivak G Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol 2008, 9, 958–70. [DOI] [PubMed] [Google Scholar]
- (16).Walmacq C; Wang L; Chong J; Scibelli K; Lubkowska L; Gnatt A; Brooks PJ; Wang D; Kashlev M Mechanism of RNA polymerase II bypass of oxidative cyclopurine DNA lesions. Proc. Natl. Acad. Sci. U. S. A 2015, 112, E410–E419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Walmacq C; Cheung ACM; Kireeva ML; Lubkowska L; Ye C; Gotte D; Strathern JN; Carell T; Cramer P; Kashlev M Mechanism of translesion transcription by RNA polymerase II and its role in cellular resistance to DNA damage. Mol. Cell 2012, 46, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ghosal G; Chen J DNA damage tolerance: a double-edged sword guarding the genome. Transl Cancer Res. 2013, 2, 107–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ohmori H; Friedberg EC; Fuchs RP; Goodman MF; Hanaoka F; Hinkle D; Kunkel TA; Lawrence CW; Livneh Z; Nohmi T; Prakash L; Prakash S; Todo T; Walker GC; Wang Z; Woodgate R The Y-family of DNA polymerases. Mol. Cell 2001, 8, 7–8. [DOI] [PubMed] [Google Scholar]
- (20).Ogi T; Lehmann AR The Y-family DNA polymerase k (pol k) functions in mammalian nucleotide-excision repair. Nat. Cell Biol 2006, 8, 640–2. [DOI] [PubMed] [Google Scholar]
- (21).Ogi T; Limsirichaikul S; Overmeer RM; Volker M; Takenaka K; Cloney R; Nakazawa Y; Niimi A; Miki Y; Jaspers NG; Mullenders LH; Yamashita S; Fousteri MI; Lehmann AR Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol. Cell 2010, 37, 714–27. [DOI] [PubMed] [Google Scholar]
- (22).Su Y; Egli M; Guengerich FP Mechanism of Ribonucleotide Incorporation by Human DNA Polymerase eta. J. Biol. Chem 2016, 291, 3747–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Brown JA; Fowler JD; Suo Z Kinetic Basis of Nucleotide Selection Employed by a Protein Template-Dependent DNA Polymerase. Biochemistry 2010, 49, 5504–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Petta TB; Nakajima S; Zlatanou A; Despras E; Couve-Privat S; Ishchenko A; Sarasin A; Yasui A; Kannouche P Human DNA polymerase iota protects cells against oxidative stress. EMBO J. 2008, 27, 2883–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bebenek K; Tissier A; Frank EG; McDonald JP; Prasad R; Wilson SH; Woodgate R; Kunkel TA 5′-Deoxyribose phosphate lyase activity of human DNA polymerase iota in vitro. Science 2001, 291, 2156–9. [DOI] [PubMed] [Google Scholar]
- (26).Mentegari E; Crespan E; Bavagnoli L; Kissova M; Bertoletti F; Sabbioneda S; Imhof R; Sturla SJ; Nilforoushan A; Hubscher U; van Loon B; Maga G Ribonucleotide incorporation by human DNA polymerase h impacts translesion synthesis and RNase H2 activity. Nucleic Acids Res. 2016, 45, 2600–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).You C; Wang Y Quantitative measurement of transcriptional inhibition and mutagenesis induced by site-specifically incorporated DNA lesions in vitro and in vivo. Nat. Protoc 2015, 10, 1389–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wu J; Li L; Wang P; You C; Williams NL; Wang Y Translesion synthesis of O4-alkylthymidine lesions in human cells. Nucleic Acids Res. 2016, 44, 9256–9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Thakur M; Wernick M; Collins C; Limoli CL; Crowley E; Cleaver JE DNA polymerase h undergoes alternative splicing, protects against UV sensitivity and apoptosis, and suppresses Mre11-dependent recombination. Genes, Chromosomes Cancer 2001, 32, 222–35. [DOI] [PubMed] [Google Scholar]
- (30).de Feraudy S; Limoli CL; Giedzinski E; Karentz D; Marti TM; Feeney L; Cleaver JE Pol h is required for DNA replication during nucleotide deprivation by hydroxyurea. Oncogene 2007, 26, 5713–21. [DOI] [PubMed] [Google Scholar]
- (31).Masutani C; Kusumoto R; Yamada A; Dohmae N; Yokoi M; Yuasa M; Araki M; Iwai S; Takio K; Hanaoka F The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase h. Nature 1999, 399, 700–4. [DOI] [PubMed] [Google Scholar]
- (32).Gowda AS; Lee M; Spratt TE N2-substituted 2′-deoxyguanosine triphosphate derivatives as selective substrates for human DNA polymerase k. Angew. Chem., Int. Ed 2017, 56, 2628–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Yuan B; You C; Andersen N; Jiang Y; Moriya M; O’Connor TR; Wang Y The roles of DNA polymerases κ and ι in the error-free bypass of N2-carboxyalkyl-2′-deoxyguanosine lesions in mammalian cells. J. Biol. Chem 2011, 286, 17503–17511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Gali VK; Balint E; Serbyn N; Frittmann O; Stutz F; Unk I, Translesion synthesis DNA polymerase η exhibits a specific RNA extension activity and a transcription-associated function. Sci. Rep 2017, 7. DOI: 10.1038/s41598-017-12915-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Donigan KA; McLenigan MP; Yang W; Goodman MF; Woodgate R The steric gate of DNA polymerase ι regulates ribonucleotide incorporation and deoxyribonucleotide fidelity. J. Biol. Chem 2014, 289, 9136–9145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ling H; Boudsocq F; Plosky BS; Woodgate R; Yang W Replication of a cis–syn thymine dimer at atomic resolution. Nature 2003, 424, 1083–1087. [DOI] [PubMed] [Google Scholar]
- (37).Yoon J-H; Park J; Conde J; Wakamiya M; Prakash L; Prakash S Rev1 promotes replication through UV lesions in conjunction with DNA polymerases η, ι, and κ but not DNA polymerase ζ. Genes Dev. 2015, 29, 2588–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Guo C; Fischhaber PL; Luk-Paszyc MJ; Masuda Y; Zhou J; Kamiya K; Kisker C; Friedberg EC Mouse Rev1 protein interacts with multiple DNA polymerases involved in translesion DNA synthesis. EMBO J. 2003, 22, 6621–6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Swan MK; Johnson RE; Prakash L; Prakash S; Aggarwal AK Structure of the human Rev1-DNA-dNTP ternary complex. J. Mol. Biol 2009, 390, 699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Nakazawa Y; Hara Y; Oka Y; Komine O; van den Heuvel D; Guo C; Daigaku Y; Isono M; He Y; Shimada M; Kato K; Jia N; Hashimoto S; Kotani Y; Miyoshi Y; Tanaka M; Sobue A; Mitsutake N; Suganami T; Masuda A; Ohno K; Nakada S; Mashimo T; Yamanaka K; Luijsterburg MS; Ogi T Ubiquitination of DNA damage-stalled RNAPII promotes transcription-coupled repair. Cell 2020, 180, 1228–1244. [DOI] [PubMed] [Google Scholar]
- (41).Tufegdzic Vidakovic A; Mitter R; Kelly GP; Neumann M; Harreman M; Rodriguez-Martinez M; Herlihy A; Weems JC; Boeing S; Encheva V; Gaul L; Milligan L; Tollervey D; Conaway RC; Conaway JW; Snijders AP; Stewart A; Svejstrup JQ Regulation of the RNAPII pool is integral to the DNA damage response. Cell 2020, 180, 1245–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Bienko M; Green CM; Crosetto N; Rudolf F; Zapart G; Coull B; Kannouche P; Wider G; Peter M; Lehmann AR; Hofmann K; Dikic I Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science 2005, 310, 1821–4. [DOI] [PubMed] [Google Scholar]
- (43).Watanabe K; Tateishi S; Kawasuji M; Tsurimoto T; Inoue H; Yamaizumi M Rad18 guides polh to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004, 23, 3886–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Baker DJ; Wuenschell G; Xia L; Termini J; Bates SE; Riggs AD; O’Connor TR Nucleotide excision repair eliminates unique DNA-protein cross-links from mammalian cells. J. Biol. Chem 2007, 282, 22592–604. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.