

Abstract
Proinflammatory activity of hepatic macrophages plays a key role during progression of alcoholic liver disease (ALD). Since mixed lineage kinase 3 (MLK3)-dependent phosphorylation of JNK is involved in the activation of macrophages, we tested the hypothesis that myeloid MLK3 contributes to chronic ethanol-induced inflammatory responses in liver, leading to hepatocyte injury and cell death. Primary cultures of Kupffer cells, as well in vivo chronic ethanol feeding, were used to interrogate the role of MLK3 in the progression of liver injury. Phosphorylation of MLK3 was increased in primary cultures of Kupffer cells isolated from ethanol-fed rats compared to cells from pair-fed rats. Kupffer cells from ethanol-fed rats were more sensitive to LPS-stimulated cytokine production; this sensitization was normalized by pharmacological inhibition of MLK3. Chronic ethanol feeding to mice increased MLK3 phosphorylation robustly in F4/80+ Kupffer cells, as well as in isolated nonparenchymal cells. MLK3−/− mice were protected from chronic ethanol-induced phosphorylation of MLK3 and JNK, as well as multiple indicators of liver injury, including increased ALT/AST, inflammatory cytokines, and induction of RIP3. However, ethanol-induced steatosis and hepatocyte apoptosis were not affected by MLK3. Finally, chimeric mice lacking MLK3 only in myeloid cells were also protected from chronic ethanol-induced phosphorylation of JNK, expression of inflammatory cytokines, and increased ALT/AST. MLK3 expression in myeloid cells contributes to phosphorylation of JNK, increased cytokine production, and hepatocyte injury in response to chronic ethanol. Our data suggest that myeloid MLK3 could be targeted for developing potential therapeutic strategies to suppress liver injury in ALD patients.
Key words: Alcoholic liver disease (ALD), Kupffer cells, Necroptosis, Toll-like receptor 4 (TLR4), Cytokines
INTRODUCTION
Alcoholic liver disease (ALD) develops in approximately 20% of all heavy drinkers with a higher prevalence in females. The development of ALD is a complex process involving both parenchymal and nonparenchymal cells resident in the liver, as well as the recruitment of other cell types to the liver in response to damage and inflammation1,2. Kupffer cells, the resident hepatic macrophages, are particularly critical to the onset of ethanol-induced liver injury1,2. Increased exposure of Kupffer cells to gut-derived lipopolysaccharide (LPS) during chronic ethanol feeding activates toll-like receptor 4 (TLR4)-dependent production of inflammatory mediators. Increased exposure to LPS results from an ethanol-induced loss of intestinal barrier function. In addition to increased exposure to LPS, chronic ethanol exposure sensitizes Kupffer cells to LPS, resulting in increased production of inflammatory mediators, associated with enhanced TLR4-mediated signaling and a predominantly M1 polarization1,2.
While activated macrophages release a variety of proinflammatory mediators, tumor necrosis factor-α (TNF-α) is of particular importance and is known to play a central role in the progression of hepatocyte injury3. In healthy hepatocytes, TNF-α serves a protective function via activation of NF-κB; however, in the context of ALD, the cellular environment shifts to enable TNF-α to drive hepatocellular cell death via necroptosis and/or apoptosis4. Ethanol feeding induces both apoptosis and receptor interacting protein kinase (RIP3)-dependent necroptosis in hepatocytes. RIP3-deficient mice are protected from chronic ethanol-induced hepatocyte injury and hepatic inflammation, suggesting that necroptosis plays a pivotal role during ethanol-induced hepatocyte injury5,6. RIP3-mediated necroptosis is activated by a number of upstream signaling molecules, including TNF-α7.
Despite the important role of the TLR4–Kupffer cell–TNF-α axis in effecting hepatocyte injury and death in response to chronic ethanol, we still do not have a complete understanding of the mechanisms by which ethanol sensitizes Kupffer cells to activation. Work over the last decade has found that overactivation of members of the mitogen-activated protein kinase (MAPK) family [e.g., p38, extracellular receptor kinase (ERK), and c-jun N-terminal kinase (JNK)] is critical for ethanol-induced sensitization of Kupffer cells to TLR4 ligation1,2. However, the upstream mechanisms involved in chronic ethanol-induced sensitization of Kupffer cells are not well understood.
MLKs, the mixed lineage kinase kinases (MAP3K), are a group of serine–threonine kinases that can activate MAP kinase pathways8. MLK3, a member of this MAP3K family (MAP3K11), can directly phosphorylate the MAPK family member JNK1/2 and also contribute to the activation of ERKs and p388,9. MLK3-induced JNK activation is involved in acetaminophen-driven liver injury10. Mice deficient in MLK3 are also protected from metabolic dysfunction and hepatic injury following exposure to high-fat diet11,12; phosphorylation of JNK and c-jun is reduced in both liver and adipose tissue of MLK3-deficent mice compared to wild-type mice fed high-fat diets11. MLK3 and JNK are central regulators in polarization of macrophages to a proinflammatory M1 phenotype13, and bone marrow-derived macrophages from MLK3-deficient mice are resistant to palmitic acid-induced macrophage M1 polarization14.
Since activation of Kupffer cells is crucial for ethanol-induced liver injury, here we tested the hypothesis that MLK3 serves as a pivotal link between ethanol-induced release of pro-death ligands (e.g. TNF-α) by activated macrophages and hepatocyte injury. Making use of both primary cultures of Kupffer cells and in vivo models of chronic ethanol exposure, we find that phosphorylation of MLK3 is increased by chronic ethanol in primary cultures of Kupffer cells, as well as in F4/80+ macrophages in mouse liver. Importantly, both global MLK3-deficient mice and chimeric mice lacking MLK3 only in myeloid cells were protected from multiple indicators of liver injury induced by chronic ethanol exposure. Collectively, these results suggest a novel link between MLK3-dependent sensitization of TLR4 signaling in Kupffer cells and hepatocyte injury following chronic ethanol feeding.
MATERIALS AND METHODS
Materials
MLK3−/− mice (generated by Roger J. Davis, University of Massachusetts Medical School, Worcester, MA) were a gift from Dr. Anja Jaeschke (University of Cincinnati). These mice were back-crossed for 10 generations to the C57BL/6J background. Lieber–DeCarli ethanol and control diets were purchased from Dyets (Bethlehem, PA, USA). Cytochrome P450 2E1 (CYP2E1)-deficient mice, generated by Dr. Frank Gonzalez, (National Institutes of Health, Bethesda, MD, USA), were a gift from Dr. Arthur Cederbaum (New York, NY, USA). RIP3−/− mice were a gift from Vishva Dixit (Genentech, San Fransisco, CA, USA). Knockout colonies were maintained at the Cleveland Clinic, and female mice at 8–10 weeks of age were used in all experiments. Female C57BL/6J mice and 129S1/SvlmJ mice (wild-type controls for CYP2E1−/− mice) (8–10 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Antibodies were purchased from the sources indicated in parentheses: 4-hydroxynonenal (Alpha Diagnostics, San Antonio, TX, USA), pSer345 MLKL (ab196436) and CYP2E1 (Abcam, Cambridge, MA, USA), RIP3 (ABGENT, San Diego, CA, USA), active-JNK (Promega, Madison, WI, USA), Ly6C (AbD Serotec, Raleigh, NC, USA), cytokeratin 18 fragment/M30 (Roche, Mannheim, Germany), Thr277/Ser281 phospho-MLK3 (Biorbyt, Cambridge, UK), RIP1 (BD Biosciences, San Jose, CA, USA), and TNF-α (Fitzgerald Inc., North Acton, MA, USA). Alexa Fluor 488- and Alexa Fluor 568-conjugated secondary antibodies were purchased from Life Technologies/Molecular Probes/Fisher. TUNEL assay kit ApopTag Plus In Situ Apoptosis Fluorescein Detection Kit was purchased from Millipore (S7111; Billerica, MA, USA). MLK3 inhibitor, URMC-099, was purchased from Adipogen (San Diego, CA, USA). 4-Methypyrazole and LPS were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Chronic Ethanol Feeding to Mice
All procedures using animals were approved by the Cleveland Clinic Institutional Animal Care and Use Committee. Female mice were housed in shoebox cages (two animals/cage) with microisolator lids. Standard microisolator handling procedures were used throughout the study. Age-matched, 8- to 10-week old female mice were randomized into ethanol-fed and pair-fed groups and then adapted to a control liquid diet for 2 days. Mice in the ethanol-fed groups were allowed free access to an ethanol diet containing 1% (v/v) ethanol for 2 days followed by 2% ethanol for 2 days, 4% ethanol for 1 week, 5% ethanol for 1 week followed by 6% ethanol for the final week (termed “25d, 32%”). In one experiment using RIP3−/− mice, mice were fed 1% ethanol for 2 days and then 6% ethanol for 2 days (termed “4d, 32%”). Control mice were pair-fed a control liquid diet, which isocalorically substituted maltose dextrins for ethanol15.
Bone Marrow Transplants: Generation of Myeloid MLK3−/− Mice
MLK3−/− chimeric mice were generated by transplantation of bone marrow from MLK3−/− mice to C57BL/6 wild-type mice. To create the bone marrow chimeras, 5-week-old female C57BL/6 mice were lethally irradiated, 2 h apart, with 600 RAD ×2. MLK3−/− mice (8 weeks old) were euthanized, and bone marrow cells were isolated from the donor mice by flushing the femur and tibia bones with sterile phosphate-buffered saline followed by lysis of the red blood cells using AKC lysis buffer. MLK3−/− bone marrow cells (at least 10 × 106 cells) were injected via the tail vein to irradiated C57BL/6 recipient mice. Control mice were generated by transplantation of bone marrow isolated from the C57BL/6 mice to C57BL/6 recipient mice. All chimeras were kept in sterile cages with irradiated food and autoclaved water. To deplete the resident macrophages in the liver, 4 weeks after bone marrow transplantation, MLK3 chimeras were injected via the tail vein with clodronate (200 μg/mouse) containing liposomes (SKU #8909; Encapsula Nanosciences, Nashville, TN, USA). One week after the clodronate injection, both the control and MLK3 chimeric mice were placed on the chronic ethanol feeding protocol, as described above.
Sample Collection
At the end of the feeding protocols, mice were anesthetized, blood samples were taken into nonheparinized syringes from the posterior vena cava, and livers were excised. Portions of each liver were then either fixed in formalin or frozen in optimal cutting temperature (OCT) compound (Sakura Finetek USA Inc., Torrance, CA, USA) for histology, frozen in RNAlater (Qiagen, Valencia, CA, USA), or flash frozen in liquid nitrogen and stored at −80°C until further analysis. Blood was transferred to EDTA-containing tubes for the isolation of plasma. Plasma was then stored at −80°C.
Rat Model of Chronic Ethanol Feeding and Isolation of Kupffer Cells
Chronic ethanol feeding to Wistar rats and isolation of Kupffer cells were performed, as previously described16. Isolated Kupffer cells were plated in 96-well plates (0.4 × 106 cells/ml) or chamber slides. Media were changed after 1 h to remove nonadherent cells. After 18 h in culture, cells were pretreated with MLK3 inhibitor (URMC-099; 100 nM), JNK inhibitor (SP600125; 30 μM), or vehicle for 2 h and then challenged with LPS (100 ng/ml) for 1 h. Kupffer cells were then collected for quantitative real-time polymerase chain reaction (qRT-PCR), Western blot analysis, or immunocytochemistry.
Immunohistochemistry
Apoptosis was detected using a TUNEL assay, as described previously17. Formalin-fixed paraffin-embedded liver sections were deparaffinized and stained for TNF-α, caspase-catalyzed cytokeratin-18 fragment (M30)5, phospho-JNK5, phospho-MLK3, or RIP35. Frozen liver sections were used for Ly6C staining5. All images presented in the results are representative of at least three images per liver and four to six mice per experimental condition. Images were analyzed and semiquantified using Image-Pro Plus software (Media Cybernetics, Bethesda, MD, USA).
Biochemical Assays
Activity of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) was measured in plasma samples using commercially available enzymatic assay kits (Sekisui Diagnostics, LLC, Lexington, MA, USA), as described in the manufacturer’s instructions. Total hepatic triglycerides were assayed using the Triglyceride Reagent Kit from Pointe Scientific Inc. (Lincoln Park, MI, USA)18.
Pieces of frozen liver (0.5–1.0 g) were homogenized in lysis buffer (10 ml/g tissue), and protein concentrations were measured by bicinchoninic acid assay using kit from Bio-Rad (Hercules, CA, USA). Liver lysates were used for Western blot analysis to detect CYP2E1 and RIP1. HSC70 was used as the loading control17. Nonparenchymal cells were isolated from ethanol-fed and pair-fed mice, as previously described15, and lysates were prepared and used in Western blot analysis to detect phospho-MLK3.
Isolation of RNA and qRT-PCR
Total RNA was isolated and reverse transcribed followed by amplification using qRT-PCR. The relative amount of target mRNA was determined using the comparative threshold (Ct) method by normalizing target mRNA Ct values to those of 18S19.
Statistical Analysis
Values shown in all figures represent means ± standard error of the mean (SEM), n = 4 for pair-fed and n = 6 for ethanol-fed. Data were analyzed by general linear models procedure (SAS, Carey, IN, USA). Data were log transformed, as needed, to obtain a normal distribution. Follow-up comparisons were made by least square means testing. If the data were not normally distributed, Kruskal–Wallis nonparametric statistical analysis was done using PRISM software.
RESULTS
Inhibition of MLK3 Attenuated Ethanol-Induced Sensitization of Kupffer Cells to LPS
Kupffer cell activation is an early event during progression of ALD3; chronic ethanol exposure sensitizes Kupffer cells to TLR4-dependent cytokine production. Activated Kupffer cells release a variety of proinflammatory mediators that contribute to hepatocyte injury following chronic ethanol feeding. Since MLK3 and its downstream target JNK are implicated in macrophage activation, here we tested the hypothesis that MLK3 is critical for ethanol-induced sensitization of Kupffer cells to LPS challenge. Phospho-MLK3 was detected in Kupffer cells isolated from ethanol-fed, but not pair-fed, rats at baseline (Fig. 1A). Phosphorylation was intermediate in Kupffer cells from pair-fed mice in response to LPS, but not further increased in cells from ethanol-fed rats (Fig. 1A). Challenge of Kupffer cells with LPS increased expression of TNF-α and interleukin-6 (IL-6) mRNA in Kupffer cells from ethanol- and pair-fed rats (Fig. 1B and C). While there was no effect of ethanol feeding on cytokine expression in the absence of LPS, Kupffer cells from ethanol-fed rats were more sensitive to LPS compared to cells from pair-fed rats (Fig. 1B and C). When Kupffer cells were pretreated with URMC-0999, an inhibitor of MLK3 (Fig. 1B and C), or SP0600125, an inhibitor of JNK (Fig. 1D), LPS-stimulated cytokine expression was reduced in both groups; expression in Kupffer cells from ethanol-fed rats was reduced to that of pair-fed controls not treated with inhibitor (Fig. 1B–D). In Kupffer cells from control rats, LPS activated both JNK1 and JNK2; pretreatment with URMC decreased activation of both isoforms (Fig. 1E). Taken together, these experiments suggest that ethanol enhances phosphorylation of MLK3; this activation alone is not sufficient to induce expression of cytokines, but does contribute to the increased sensitivity of Kupffer cells to LPS after chronic ethanol feeding.
Figure 1.

MLK3 contributed to sensitization of Kupffer cells from ethanol-fed rats to challenge with lipopolysaccharide (LPS). Kupffer cells isolated from ethanol- or pair-fed rats were cultured for 18 h. (A) Kupffer cells from pair- and ethanol-fed rats were challenged, or not, with 100 ng/ml LPS for 1 h. Kupffer cells were then fixed with 4% paraformaldehyde and stained for phospho-mixed lineage kinase 3 (MLK3). Images were acquired using 40× objective and semiquantified using Image-Pro Plus software. (B–E) Following pretreatment with (B, C, E) 100 nM URMC-099, a potent MLK3 inhibitor, or (D) 30 μM SP600125, a JNK inhibitor, for 2 h, cells were challenged with LPS (100 ng/ml) for 1 h. Expression of TNF-α (B, D) and IL-6 (C) mRNA was measured by quantitative real-time polymerase chain reaction (qRT-PCR). (E) Phosphorylation of JNK1 and JNK2 was assessed by Western blot and normalized to HSC70. Data are expressed relative phosphorylation in LPS-treated cells to better compare the effect of URMC between isoforms. Values represent means ± SEM, n = 4, except for (D) where n = 9. Values with different lower case letters are significantly different from each other, p < 0.05.
Chronic Ethanol Feeding Increased Phosphorylation of MLK3 in Mouse Liver
Since MLK3 phosphorylation was increased in primary Kupffer cells isolated from ethanol-fed rats, we next investigated the impact of chronic ethanol feeding on the phosphorylation of MLK3 in livers of wild-type and MLK3−/− mice. Chronic ethanol feeding increased phospho-MLK3 immunostaining in wild-type mouse liver; this signal was totally abrogated in livers of MLK3-deficient mice (Fig. 2A). Interestingly, morphological assessment of phospho-MLK3-positive staining indicated that phospho-MLK3 was robustly increased in nonparenchymal cells (Fig. 2A). Phospho-MLK3-positive staining in liver of ethanol-fed wild-type mice colocalized with F4/80, a marker of tissue-resident macrophages (Fig. 2B). Further, phospho-MLK3 immunoreactivity was also increased in nonparenchymal cells isolated from ethanol-fed compared to pair-fed wild-type mice in a Western blot analysis (Fig. 2C).
Figure 2.

Chronic ethanol feeding increased phosphorylation of MLK3 in mouse liver. C57BL/6 and MLK3−/− mice were allowed free access to diets with increasing concentrations of ethanol (final concentration 32% of kcal) or pair fed a control diet for 25 days. Paraffin-embedded livers were deparaffinized followed by immunohistochemistry. (A) Immunoreactive phospho-MLK3 was visualized, and nuclei were counterstained with hematoxylin. Images were acquired using 20× objective. Positive staining was quantified using Image-Pro Plus software and analyzed. Values represent means ± SEM, n = 4 pair-fed and 6 EtOH-fed mice. Values with different lower case letters are significantly different from each other, p < 0.05. (B) Immunoreactive phospho-MLK3 (green) and F4/80 (red) were visualized, and nuclei were labeled with DAPI. Images were acquired at 40×. (C) Nonparenchymal cells were isolated from livers of wild-type mice. Lysates were prepared and used for Western blot analysis of phospho-MLK3. GAPDH was used as a loading control. Values represent means ± SEM, n = 6–7 mice. *p < 0.05 compared to pair fed.
CYP2E1 Contributed to Ethanol-Induced Phosphorylation of MLK3
Increased oxidative stress is one important mechanism for the activation of MLK3; phosphorylation of MLK3 is increased in other systems in response to stress or excess reactive oxygen species (ROS)8,20. Since induction of CYP2E1 is a critical element in the generation of oxidative stress in response to ethanol exposure21,22, we next investigated the role of CYP2E1 in phosphorylation of MLK3. While CYP2E1 expression is primarily induced in hepatocytes after chronic ethanol, we and others have reported that chronic ethanol feeding also increases CYP2E1 expression in Kupffer cells23,24. When primary Kupffer cells isolated from ethanol-fed rats were preincubated with 4-methylpyrazole, an inhibitor of CYP2E1, the phosphorylation of phospho-MLK3 was reduced (Fig. 3A). Consistent with a role for CYP2E1 in the activation of MLK3, when CYP2E1−/− mice were exposed to chronic ethanol, phospho-MLK3 was undetectable in the liver (Fig. 3B). CYP2E1-deficient mice are known to be protected from chronic ethanol-induced liver injury21,22. Taken together, these data suggest that CYP2E1, an important source of reactive oxygen during chronic ethanol feeding, contributes to the activation of MLK3 in both primary cultures of Kupffer cells from ethanol-fed rats and in mice in response to chronic ethanol feeding.
Figure 3.

Ethanol-induced increases in phosphorylation of MLK3 in Kupffer cells and mouse liver were dependent on CYP2E1. (A) Kupffer cells isolated from ethanol- or pair-fed rats were cultured for 18 h. Following pretreatment with 5 nM 4-methyl pyrazole (4-MP) to inhibit CYP2E1 for 2 h, cells were challenged with LPS (100 ng/ml) for 1 h. Kupffer cells were then fixed with 4% paraformaldehyde and stained for phospho-MLK3. Values with different alphabetical superscripts were significantly different from each other, p < 0.05. (B) 129S1/SvlmJ (wild-type) and CYP2E1−/− mice were allowed free access to diets with increasing concentrations of ethanol (final concentration 32% of kcal) or pair-fed controls for 25 days. Paraffin-embedded livers were deparaffinized followed by immunohistochemistry for phospho-MLK3. Nuclei were counterstained with hematoxylin. All images were acquired using 20× objective. *p < 0.05 compared to pair fed within a genotype. Values represent means ± SEM, n = 4 pair-fed and 6 EtOH-fed mice.
MLK3-Deficiency Reduced the Ethanol-Induced Phosphorylation of JNK and Expression of TNF-α in Mouse Liver
Since MLK3 and JNK activity contributed to the chronic ethanol-induced sensitization of Kupffer cells to LPS (Fig. 1), we next investigated the impact of MLK3 deficiency on hepatic phosphorylation of JNK and expression of TNF-α after chronic ethanol feeding to mice. Chronic ethanol feeding increased the number of phospho-JNK+ cells in livers of wild-type mice (Fig. 4A). Similar to the distribution of phospho-MLK3, phospho-JNK+ cells were predominantly localized to cells of a nonparenchymal cell morphology (Fig. 4A, inset a), with some positive hepatocytes detected near portal veins (Fig. 4A, inset b). Consistent with the ability of MLK3 to activate JNK11, MLK3 deficiency attenuated the chronic ethanol-induced increase of phospho-JNK+ cells (Fig. 4A, inset c). Expression of TNF-α protein was also increased in livers of wild-type mice after chronic ethanol feeding, as evaluated by immunohistochemistry. Increased expression of TNF-α was predominantly detected in nonparenchymal cells (Fig. 4B, inset a), with some staining in hepatocytes, particularly near portal veins (Fig. 4B, inset b), as assessed by morphological examination. This expression pattern is similar to that previously reported by the Nieto group25. Chronic ethanol-induced expression of TNF-α protein was reduced in livers of MLK3-deficient mice (Fig. 4B, inset c).
Figure 4.

Chronic ethanol-induced phosphorylation of JNK and expression of TNF-α were dependent on MLK3. C57BL/6 and MLK3−/− mice were allowed free access to diets with increasing concentrations of ethanol (final concentration 32% of kcal) or pair fed a control diet for 25 days. Paraffin-embedded livers were deparaffinized followed by immunohistochemistry. (A) Immunoreactive phospho-JNK was visualized, and nuclei were counterstained with hematoxylin. Images were acquired using 20× objective, and positive staining was quantified using Image-Pro Plus software and analyzed. Red arrow indicates hepatocytes, and blue arrows indicate nonparenchymal cells. (B) Immunoreactive TNF-α was visualized, and nuclei were counterstained with hematoxylin. Images were acquired at 40× objective, and positive staining was quantified using Image-Pro Plus software and analyzed. Black arrows indicate hepatocytes, and white arrows indicate nonparenchymal cells. Values represent means ± SEM, n = 4 pair-fed and 6 EtOH-fed mice. Values with different lower case letters are significantly different from each other, p < 0.05.
MLK3 Deficiency Reduced the Development of Ethanol-Induced Liver Injury in Mice
Since expression of TNF-α by Kupffer cells is a known driver of chronic ethanol-induced liver injury, we next asked whether MLK3−/− mice would be protected from chronic ethanol-induced liver injury. Chronic ethanol feeding increased plasma concentrations of ALT and AST in wild-type mice; this response was reduced in MLK3-deficient mice (Fig. 5A). Chronic ethanol feeding also induced accumulation of lipids in the liver of wild-type mice (Fig. 5B). In contrast to circulating liver enzymes, MLK3-deficient mice were not protected from ethanol-induced hepatic lipid accumulation, as assessed by hepatic triglyceride content (Fig. 5B). Hematoxylin and eosin (H&E) sections illustrate the distribution of triglycerides in the liver of both wild-type and MLK3−/− mice (Fig. 5C).
Figure 5.

MLK3 deficiency reduced ethanol-induced liver injury but not steatosis in mice. C57BL/6 and MLK3−/− mice were allowed free access to diets with increasing concentrations of ethanol (final concentration 32% of kcal) or pair fed a control diet for 25 days. (A) Enzyme activities of ALT and AST were measured in plasma. (B) Hepatic triglyceride content was measured in whole-liver homogenate. (C) Liver histology was visualized in hematoxylin and eosin (H&E)-stained liver sections using a 10× objective. Values represent means ± SEM, n = 4 pair-fed and 6 EtOH-fed mice. Values with different lower case letters are significantly different from each other, p < 0.05.
Hepatocyte injury leads to hepatic inflammation, characterized by infiltration of immune cells in the liver and induction of proinflammatory mediators during progression of ALD1,2. Consistent with the increase in expression of TNF-α protein (Fig. 4B), chronic ethanol feeding also increased expression of mRNA for the proinflammatory mediators TNF-α and MCP-1 (Fig. 6A and B), as well as the number of infiltrating Ly6C+ immune cells in the livers of wild-type mice (Fig. 6C). In MLK3−/− mice, these inflammatory responses to chronic ethanol were ameliorated (Fig. 6A–C). Similarly, chronic ethanol feeding to wild-type mice, but not MLK3−/− mice, increased the accumulation of 4-hydroxynonenal (4-HNE), an indicator of oxidative stress (Fig. 6D).
Figure 6.

MLK3 deficiency reduced ethanol-induced inflammatory responses and oxidative stress in mice. C57BL/6 and MLK3−/− mice were allowed free access to diets with increasing concentrations of ethanol (final concentration 32% of kcal) or pair fed a control diet for 25 days. (A) MCP-1 and (B) TNF-α mRNA expression were detected in mouse livers using qRT-PCR measurement. (C) Ly6C+ cells were detected by immunostaining in frozen liver sections. Images for Ly6C+ cells were acquired using a 40× objective. The number of Ly6C+ cells was quantified using Image-Pro Plus software and analyzed. (D) Paraffin-embedded livers were deparaffinized followed by immunohistochemistry. Immunoreactive 4-hydroxynonenal (4-HNE) was visualized. Images were acquired using 20× objective, and positive staining was quantified using Image-Pro Plus software and analyzed. Values represent means ± SEM, n = 4 pair-fed and 6 EtOH-fed mice. Values with different lower case letters are significantly different from each other, p < 0.05.
Protection of MLK3-deficient mice from chronic ethanol-induced hepatocyte injury and inflammation was not due to differences in ethanol intake since the amount of ethanol consumed was comparable in both wild-type and MLK3-deficient mice (Table 1). Induction of CYP2E1 in the liver was also not affected by genotype (Table 1).
Table 1.
Body Weights, Food Intake, and CYP2E1 Expression in Pair-Fed and Chronic Ethanol-Fed (25d, 32%) Wild-Type and MLK3-Deficient Mice
| Wild Type | MLK3−/− | |||
|---|---|---|---|---|
| Pair Fed | EtOH Fed | Pair Fed | EtOH Fed | |
| Body weight (g) | ||||
| Initial | 18.5 ± 0.3 | 18.0 ± 0.09 | 18.9 ± 0.76 | 18.5 ± 0.31 |
| Final | 22.7 ± 0.52 | 19.3 ± 0.61 | 22.3 ± 1.06 | 20.2 ± 0.36 |
| Average daily food intake (ml/cage) | ↔ | 22.1 ± 0.44 | ↔ | 23.3 ± 0.24 |
| CYP2E1 expression/HSC70 expression | 0.37 ± 0.07a | 1.17 ± 0.08b | 0.43 ± 0.07a | 1.17 ± 0.13b |
↔: Pair-fed animals. Values with different alphabetical superscripts are significantly different from each other, p < 0.05.
Differential Contribution of MLK3 to Ethanol-Induced Hepatic Apoptosis and RIP3 Expression in Mouse Liver
Chronic ethanol feeding induces both apoptosis and necroptosis in hepatocytes15. Both pathways of cell death can be activated by TNF-α; therefore, we tested the hypothesis that MLK3 was upstream of both forms of hepatocellular death. Chronic ethanol feeding increased the number of TUNEL+ (Fig. 7A and C) and M30+ cells (Fig. 7B and D), markers of apoptosis, as well as indicators of necroptosis in livers of wild-type mice (Fig. 8). RIP3 expression was increased in response to 32% ethanol in the diet for 4 days (Fig. 8A) or 25 days (Fig. 8B). RIP3 immunoreactivity was absent in livers of RIP3−/− mice, demonstrating the specificity of the RIP3 immunohistochemistry (Fig. 8A). Chronic ethanol feeding also increased phosphorylation of MLKL in wild-type mice (Fig. 8C). Interestingly, MLK3 deficiency reduced ethanol-induced RIP3 and phospho-MLKL expression (Fig. 8B and C), consistent with a reduction in necroptosis, but ethanol-induced apoptosis was unaffected by genotype (Fig. 7A and B). RIP1 expression, measured by Western blot, was not different between diets or genotypes (Fig. 8D).
Figure 7.

MLK3-deficient mice were protected from chronic ethanol-induced apoptosis. C57BL/6 and MLK3−/− mice were allowed free access to diets with increasing concentrations of ethanol (final concentration 32% of kcal) or pair fed a control diet for 25 days. Paraffin-embedded livers were deparaffinized followed by (A) TUNEL or (B) M30 staining. Images were acquired using 40× for TUNEL and 20× objectives for M30. (C) TUNEL+ cells were counted and expressed as percent positive of total number of cells (detected by DAPI staining, not shown in the image). (D) M30+ cells per 20× frame were enumerated. Values represent means ± SEM, n = 4 pair-fed and 6 EtOH-fed mice. Values with different lower case letters are significantly different from each other, p < 0.05.
Figure 8.

Chronic ethanol feeding increased expression of RIP3 or phosphorylation of MLKL in both wild-type and MLK3−/− mice. (A) C57BL/6 and RIP3−/− mice were allowed free access to diets with increasing concentrations of ethanol (final concentration 32% of kcal) or pair fed a control diet for 4 days. (B–D) C57BL/6 and MLK3−/− mice were allowed free access to diets with increasing concentrations of ethanol (final concentration 32% of kcal) or pair fed a control diet for 25 days. Paraffin-embedded livers were deparaffinized followed by (A, B) RIP3 or (C) phospho-MLKL staining. Images were acquired using 20× objectives. The area positive for RIP3 or phospho-MLKL was quantified using Image-Pro Plus software and analyzed. (D) RIP1 protein was assessed via Western blot and quantified. Values represent means ± SEM, n = 4 pair-fed and 6 EtOH-fed mice. Values with different lower case letters are significantly different from each other, p < 0.05.
Myeloid-Specific MLK3−/− Mice Were Protected From Ethanol-Induced Hepatocyte Injury and Increased RIP3 Expression
Results from both primary Kupffer cells and in vivo chronic ethanol feeding suggested a critical role for MLK3 in myeloid cells. While phospho-MLK3, phospho-JNK, and TNF-α immunoreactivity was modestly increased in hepatocytes/parenchymal cells (Figs. 2 and 3) in response to chronic ethanol, here we have focused primarily on the role of MLK3 in nonparenchymal cells due both to the more robust activation of phospho-MLK3/phospho-JNK/TNF-α in nonparenchymal cells and to the well-known role of hepatic macrophages during the progression of chronic ethanol-induced liver injury. To further evaluate the contribution of myeloid-specific MLK3 to chronic ethanol-induced hepatic inflammation and liver injury, MLK3 chimera mice were generated by transplantation of bone marrow from MLK3−/− mice to wild-type recipients (myeloid-specific MLK3−/−). If MLK3 activity in Kupffer cells contributes to hepatocyte injury during ethanol exposure, myeloid-specific MLK3−/− mice should be protected from ethanol-induced liver injury. In C57BL/6 wild-type mice receiving the wild-type bone marrow, chronic ethanol feeding increased phospho-JNK immunoreactivity (Fig. 9A). In contrast, chronic ethanol-induced JNK phosphorylation in both nonparenchymal cells and hepatocytes was largely ameliorated in myeloid-specific MLK3−/− chimeric mice (Fig. 9A). C57BL/6 wild-type mice receiving the wild-type bone marrow exhibited typical increases in expression of mRNA for inflammatory cytokines (Fig. 9B) and ALT and AST (Fig. 9C) following chronic ethanol feeding. In contrast, ethanol-induced increases in expression of mRNA for proinflammatory mediators, including MCP-1, IL-6, and TNF-α (Fig. 9B) and ALT/AST (Fig. 9C), were reduced in myeloid MLK3−/− mice. These results further demonstrate that myeloid-specific MLK3 contributes to ethanol-induced hepatocyte injury, associated with a decrease in the activation of JNK and expression of inflammatory cytokines.
Figure 9.

Myeloid MLK3 contributed to chronic ethanol-induced liver injury. C57BL/6 received wild-type bone marrow or MLK3−/− bone marrow. Chimeric mice were allowed free access to diets with increasing concentrations of ethanol (final concentration 32% of kcal) or pair fed a control diet for 25 days. (A) JNK phosphorylation was evaluated by immunohistochemistry in paraffin-embedded livers. Red arrows indicate hepatocytes, and blue arrows indicates nonparenchymal cells. (B) MCP-1, interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) mRNA expression was detected in mouse livers using qRT-PCR measurement. *p < 0.05, +p < 0.07 compared to C57BL/6/C57BL/6 bone marrow-transplanted mice. (C) Enzyme activities of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured in plasma. Values with different lower case letters are significantly different from each other, p < 0.05. Values represent means ± SEM, n = 4 pair-fed and n = 6 EtOH-fed mice.
DISCUSSION
TLR4-mediated expression of TNF-α by Kupffer cells is a critical element in the development of ALD1,2. Chronic ethanol exposure enhances TLR4-mediated activation of NF-κB, as well as MAPK family members, leading to increased transcription of TNF-α and stabilization of TNF-α mRNA26. Recently, we identified RIP3-driven necroptotic hepatocyte cell death as an important downstream target of increased TNF-α expression in the context of chronic ethanol exposure5,6. Despite the key contributions of this TLR4–Kupffer cell–TNF-α axis to disease progression, the mechanisms by which ethanol sensitizes Kupffer cells to TLR4 ligands are not well understood. MLK3, a JNK-activating serine–threonine kinase, is a critical mediator of macrophage activation, as well as hepatocyte injury, in response to high-fat diet11,12,14 or acetaminophen toxicity10. Here we report that chronic ethanol feeding increased phosphorylation of MLK3 in a CYP2E1-dependent mechanism, in both primary cultures of Kupffer cells and in an in vivo mouse model of chronic ethanol exposure. Pharmacologic inhibition of MLK3 in Kupffer cells normalized TLR4-mediated cytokine expression and genetic deletion of MLK3 in mice reduced chronic ethanol-induced liver injury, decreasing phosphorylation of JNK and expression of TNF-α. These changes were associated with a reduction in ethanol-induced increases in ALT/AST concentrations in the plasma, numbers of infiltrating Ly6C+ cells, and accumulation of 4-HNE adducts. Importantly, protection was dependent on MLK3 expression in myeloid-derived cells, as chimeric mice lacking MLK3 only in myeloid cells were protected from chronic ethanol-induced phosphorylation of JNK, inflammatory cytokine expression, and increases to ALT and AST. Taken together, these data identify MLK3 as an upstream mediator contributing to chronic ethanol-induced inflammation and injury in the liver (Fig. 10).
Figure 10.
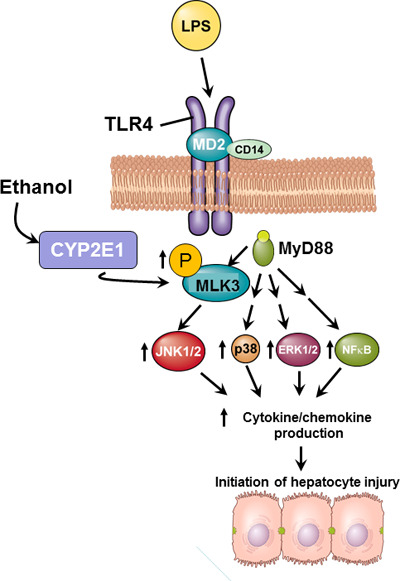
Schematic representation of the role of myeloid MLK3 in the progression of ethanol-induced liver injury. Ethanol feeding increases the phosphorylation of MLK3 in Kupffer cells; this increase is dependent on the activity of CYP2E1. Upon ligation of TLR4, MLK3 is an upstream activator of JNK and contributes, along with previously reported changes in p38, extracellular receptor kinase 1/2 (ERK1/2), and NF-κB signaling1,2, to the sensitization of TLR4-mediated cytokine and chemokine expression. Increased cytokine/chemokine expression contributes to ethanol-induced hepatocyte injury.
TLR4-mediated signaling in Kupffer cells is critical to progression of ALD1,2. Kupffer cells are activated by endotoxin in the portal circulation; translocation of endotoxin is increased after chronic ethanol due to impaired intestinal barrier function. Chronic ethanol feeding also sensitizes Kupffer cells to LPS; this sensitization is due, at least in part, to enhanced TLR4 signaling via both MyD88-dependent and -independent pathways and is associated with a predominant M1 polarization1,27,28. Activation of NF-κB, as well as multiple MAPK family members, contributes to increased cytokine and chemokine expression by Kupffer cells after chronic ethanol feeding (Fig. 10). Here we identify MLK3 as an upstream mediator of sensitization of Kupffer cells to activation by LPS. Chronic ethanol feeding increased the phosphorylation of MLK3, even under basal conditions, in rat Kupffer cells. Interestingly, this activation alone was not sufficient to induce expression of cytokines. Because the expression of cytokines is under the control of multiple mechanisms, both transcriptional and posttranscriptional, it is unlikely that a single signaling pathway would be sufficient to increase cytokine expression. This is also apparent from our studies using inhibitors, as either treatment with URMC to inhibit MLK3 (Fig. 1B and C) or with SP6 to inhibit JNK (Fig. 1D) only reduced, rather than ameliorated, cytokine expression. Taken together, these data illustrate that MLK3/JNK is a contributor to LPS-stimulated cytokine expression and not the sole driver of expression. Importantly, MLK3 contributed to the increased sensitivity of Kupffer cells to LPS after chronic ethanol feeding.
Phosphorylation of MLK3 was dependent on CYP2E1 activity in Kupffer cells; the contribution of CYP2E1 to activation of MLK3 was also confirmed in vivo in CYP2E1−/− mice. The role of CYP2E1 in the activation of MLK3 suggests an involvement of ROS generation, since CYP2E1 expression is associated with increased oxidative stress. This likely role of ROS in the activation of MLK3 is consistent with previous data demonstrating that CYP2E124 contributes to the increased activation of MAPK signaling in LPS-stimulated Kupffer cells isolated from ethanol-fed rats. While hepatocytes, where the majority of ethanol metabolism occurs, are typically thought of as the primary site of oxidative stress during ethanol exposure, these data highlight the importance of ethanol-induced oxidative stress in disrupting cellular function in Kupffer cells as well.
In other cell types, MLK3 provides a link between cellular ROS and MAPK activation, ultimately involved in controlling pathways of cell death8. These responses are dependent on the concentrations of ROS, with high ROS concentrations leading to MLK3-dependent cell death8. Our data suggest that MLK3 in myeloid cells is part of a complex network that links ethanol-induced oxidative stress and increased TLR4-mediated TNF-α expression in Kupffer cells to hepatocellular injury during ethanol exposure. It is interesting to note that phospho-MLK3 was more robustly expressed in F4/80+ macrophages in the liver in response to chronic ethanol feeding (Fig. 2). Similarly, chronic ethanol feeding increased phospho-JNK and TNF-α predominantly in nonparenchymal cells, although immunoreactive phospho-JNK and TNF-α were also observed in hepatocytes near the portal vein (Fig. 4). Phosphorylation of JNK and expression of TNF-α in hepatocytes were abrogated in chimeric mice lacking MLK3 only in myeloid cells (Fig. 9), thus placing myeloid MLK3 as a central driver for the activation of JNK and expression of TNF-α by both nonparenchymal cells and hepatocytes in wild-type mice.
It should be noted that our results do not preclude a contribution of MLK3 in hepatocytes to chronic ethanol-induced liver injury; a direct function of MLK3 in hepatocytes is currently under investigation. Recent studies from Ibrahim and colleagues12 found that MLK3 regulates the chemokine content of extracellular vesicles released from hepatocytes in response to challenge with toxic lipids. Future studies will be needed to determine whether MLK3 in hepatocytes also impacts the chemokine content of extracellular vesicles in response to challenge with ethanol. Indeed, it is possible that the decreased numbers of Ly6C+ cells infiltrating in the liver of MLK3−/− mice in response to chronic ethanol (Fig. 5) are at least partially dependent on changes in chemokine content of extracellular vesicles.
Ethanol feeding induces hepatocyte death via both apoptosis and necroptosis5,6. Importantly, RIP3-dependent necroptosis drives hepatocyte injury in response to chronic ethanol feeding6,29, while hepatocyte apoptosis tends to promote fibrogenesis in the liver30. We found that MLK3 deficiency prevents the induction of RIP3 protein expression and also decreased ALT/AST, an indication of leaky hepatocyte plasma membranes, in the circulation. Making use of global CYP2E1−/− mice, we previously reported that CYP2E1 is required for the induction of RIP3 in hepatocytes5; our current data suggest that MLK3 provides a link between CYP2E1 and RIP3 expression. It will be interesting to determine in future experiments if myeloid CYP2E1 is the essential cell type for promoting RIP3 expression in hepatocytes, since phospho-MLK3 is CYP2E1 dependent and myeloid MLK3 is required for chronic ethanol-induced increases in RIP3 expression.
Although MLK3 deficiency reduced chronic ethanol-induced increases in ALT/AST (indicators of hepatocyte injury) and hepatic RIP3 expression (an indirect indicator of necroptosis), chronic ethanol-induced hepatocyte apoptosis was not affected by MLK3 deficiency. In the model of chronic ethanol feeding used in the current studies, hepatocyte injury is predominantly driven by RIP3-dependent necroptosis with a concomitant rise in low-grade apoptosis (below 10%)5. Chronic ethanol-induced apoptosis is a direct result of ethanol metabolism; ethanol impairs mitochondrial function, leading to a release of cytochrome c and subsequent activation of the apoptotic cascade31,32. Since CYP2E1 was induced in both wild-type and MLK3−/− mice, ethanol metabolism via CYP2E1 was likely sufficient to activate caspase-dependent hepatocyte apoptosis, even in the absence of MLK3.
In summary, our studies demonstrate that myeloid-specific induction of the MLK3-JNK signaling axis contributes to ethanol-induced hepatocyte injury, associated with sensitization of Kupffer cells to activation by LPS and subsequent release of proinflammatory mediators, including TNF-α, in the hepatic milieu. Pharmacological intervention targeting the myeloid-specific MLK3-pathway could be beneficial for treatment of patients with ALD.
ACKNOWLEDGMENTS
This work was supported in part by National Institutes of Health (NIH) grants P20 AA17069, P50 AA024333, U01AA021890, and RO1 AA011975 (L.E.N.), and T32DK007319 (R.L.M.); ABMRF/The Foundation for Alcohol Research (S.R.); and NIH grant R21AA020941 (S.R.).
Footnotes
The authors declare no conflicts of interests.
REFERENCES
- 1. Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. Compr Physiol. 2013;3(2):785–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang HJ, Gao B, Zakhari S, Nagy LE. Inflammation in alcoholic liver disease. Annu Rev Nutr. 2012;32:343–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gao B, Seki E, Brenner DA, Friedman S, Cohen JI, Nagy L, Szabo G, Zakhari S. Innate immunity in alcoholic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2011;300(4):G516–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eguchi A, Wree A, Feldstein AE. Biomarkers of liver cell death. J Hepatol. 2014;60(5):1063–74. [DOI] [PubMed] [Google Scholar]
- 5. Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology 2013;57(5):1773–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang S, Ni HM, Dorko K, Kumer SC, Schmitt TM, Nawabi A, Komatsu M, Huang H, Ding WX. Increased hepatic receptor interacting protein kinase 3 expression due to impaired proteasomal functions contributes to alcohol-induced steatosis and liver injury. Oncotarget 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sun L, Wang X. A new kind of cell suicide: Mechanisms and functions of programmed necrosis. Trends Biochem Sci. 2014;39(12):587–93. [DOI] [PubMed] [Google Scholar]
- 8. Lee HS, Hwang CY, Shin SY, Kwon KS, Cho KH. MLK3 is part of a feedback mechanism that regulates different cellular responses to reactive oxygen species. Sci Signal. 2014;7(328):ra52. [DOI] [PubMed] [Google Scholar]
- 9. Handley ME, Rasaiyaah J, Barnett J, Thakker M, Pollara G, Katz DR, Chain BM. Expression and function of mixed lineage kinases in dendritic cells. Int Immunol. 2007;19(8):923–33. [DOI] [PubMed] [Google Scholar]
- 10. Sharma M, Gadang V, Jaeschke A. Critical role for mixed-lineage kinase 3 in acetaminophen-induced hepatotoxicity. Mol Pharmacol. 2012;82(5):1001–07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ibrahim SH, Gores GJ, Hirsova P, Kirby M, Miles L, Jaeschke A, Kohli R. Mixed lineage kinase 3 deficient mice are protected against the high fat high carbohydrate diet-induced steatohepatitis. Liver Int. 2014;34(3):427–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ibrahim SH, Hirsova P, Tomita K, Bronk SF, Werneburg NW, Harrison SA, Goodfellow VS, Malhi H, Gores GJ. Mixed lineage kinase 3 mediates release of C-X-C motif ligand 10-bearing chemotactic extracellular vesicles from lipotoxic hepatocytes. Hepatology 2016;63(3):731–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou D, Huang C, Lin Z, Zhan S, Kong L, Fang C, Li J. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal. 2014;26(2):192–97. [DOI] [PubMed] [Google Scholar]
- 14. Gadang V, Kohli R, Myronovych A, Hui DY, Perez-Tilve D, Jaeschke A. MLK3 promotes metabolic dysfunction induced by saturated fatty acid-enriched diet. Am J Physiol Endocrinol Metab. 2013;305(4):549–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Barnes MA, McMullen MR, Roychowdhury S, Pisano SG, Liu X, Stavitsky AB, Bucala R, Nagy LE. Macrophage migration inhibitory factor contributes to ethanol-induced liver injury by mediating cell injury, steatohepatitis, and steatosis. Hepatology 2013;57(5):1980–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mandal P, Roychowdhury S, Park PH, Pratt BT, Roger T, Nagy LE. Adiponectin and heme oxygenase-1 suppress TLR4/MyD88-independent signaling in rat Kupffer cells and in mice after chronic ethanol exposure. J Immunol. 2010;185(8):4928–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roychowdhury S, Chiang DJ, Mandal P, McMullen MR, Liu X, Cohen JI, Pollard J, Feldstein AE, Nagy LE. Inhibition of apoptosis protects mice from ethanol-mediated acceleration of early markers of CCl(4)-induced fibrosis but not steatosis or inflammation. Alcohol Clin Exp Res. 2012;36(7):1139–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roychowdhury S, McMullen MR, Pritchard MT, Hise AG, van Rooijen N, Medof ME, Stavitsky AB, Nagy LE. An early complement-dependent and TLR-4-independent phase in the pathogenesis of ethanol-induced liver injury in mice. Hepatology 2009;49(4):1326–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bakhautdin B, Das D, Mandal P, Roychowdhury S, Danner J, Bush K, Pollard K, Kaspar JW, Li W, Salomon RG, McMullen MR, Nagy LE. Protective role of HO-1 and carbon monoxide in ethanol-induced hepatocyte cell death and liver injury in mice. J Hepatol. 2014;61(5):1029–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van den Berg MC, van Gogh IJ, Smits AM, van Triest M, Dansen TB, Visscher M, Polderman PE, Vliem MJ, Rehmann H, Burgering BM. The small GTPase RALA controls c-Jun N-terminal kinase-mediated FOXO activation by regulation of a JIP1 scaffold complex. J Biol Chem. 2013;288(30):21729–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cederbaum AI. Role of CYP2E1 in ethanol-induced oxidant stress, fatty liver and hepatotoxicity. Dig Dis. 2010;28(6):802–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Roychowdhury S, McMullen MR, Pritchard MT, Li W, Salomon RG, Nagy LE. Formation of gamma-ketoaldehyde-protein adducts during ethanol-induced liver injury in mice. Free Radic Biol Med. 2009;47(11):1526–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thakur V, Pritchard MT, McMullen MR, Wang Q, Nagy LE. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: Role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. J Leukoc Biol. 2006;79(6):1348–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cao Q, Mak KM, Lieber CS. Cytochrome P4502E1 primes macrophages to increase TNF-alpha production in response to lipopolysaccharide. Am J Physiol Gastrointest Liver Physiol. 2005;289(1):G95–107. [DOI] [PubMed] [Google Scholar]
- 25. Ge X, Leung TM, Arriazu E, Lu Y, Urtasun R, Christensen B, Fiel MI, Mochida S, Sorensen ES, Nieto N. Osteopontin binding to lipopolysaccharide lowers tumor necrosis factor-alpha and prevents early alcohol-induced liver injury in mice. Hepatology 2014;59(4):1600–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nagy LE. Recent insights into the role of the innate immune system in the development of alcoholic liver disease. Exp Biol Med. (Maywood) 2003;228(8):882–90. [DOI] [PubMed] [Google Scholar]
- 27. Louvet A, Teixeira-Clerc F, Chobert MN, Deveaux V, Pavoine C, Zimmer A, Pecker F, Mallat A, Lotersztajn S. Cannabinoid CB2 receptors protect against alcoholic liver disease by regulating Kupffer cell polarization in mice. Hepatology 2011;54(4):1217–26. [DOI] [PubMed] [Google Scholar]
- 28. Mandal P, Pratt BT, Barnes M, McMullen MR, Nagy LE. Molecular mechanism for adiponectin-dependent M2 macrophage polarization: Link between the metabolic and innate immune activity of full-length adiponectin. J Biol Chem. 2011;286(15):13460–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Roychowdhury S, Chiang DJ, Mandal P, McMullen MR, Liu X, Cohen JI, Pollard J, Feldstein AE, Nagy LE. Inhibition of apoptosis protects mice from ethanol-mediated acceleration of early markers of CCl4-induced fibrosis but not steatosis or inflammation. Alcohol Clin Exp Res. 2012;36(7):1139–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fujii H, Kawada N. Fibrogenesis in alcoholic liver disease. World J Gastroenterol. 2014;20(25):8048–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cederbaum AI, Lu Y, Wang X, Wu D. Synergistic toxic interactions between CYP2E1, LPS/TNFalpha, and JNK/p38 MAP kinase and their implications in alcohol-induced liver injury. Adv Exp Med Biol. 2015;815:145–72. [DOI] [PubMed] [Google Scholar]
- 32. Yang L, Wu D, Wang X, Cederbaum AI. Cytochrome P4502E1, oxidative stress, JNK, and autophagy in acute alcohol-induced fatty liver. Free Radic Biol Med. 2012;53(5):1170–80. [DOI] [PMC free article] [PubMed] [Google Scholar]