

Abstract
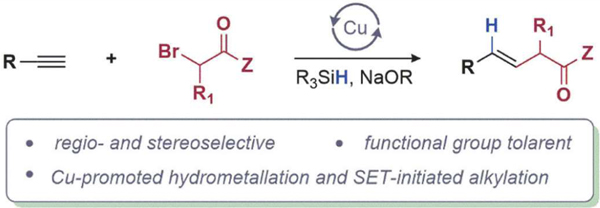
We have developed a method for the stereoselective coupling of terminal alkynes and α-bromo carbonyls to generate functionalized E-alkenes. The coupling is accomplished by merging the closed-shell hydrocupration of alkynes with the open-shell single electron transfer (SET) chemistry of the resulting alkenyl copper intermediate. We demonstrate that the reaction is compatible with various functional groups and can be performed in the presence of aryl bromides, alkyl chlorides, alkyl bromides, esters, nitriles, amides, and a wide range of nitrogen-containing heterocyclic compounds. Mechanistic studies provide evidence for SET oxidation of the alkenyl copper intermediate by an α -bromo ester as the key step that enables the cross coupling.
Graphical Abstract:
Selective and efficient synthesis of isomerically pure alkenes is challenging and has continuously inspired the development of new synthetic strategies. One recent approach is hydroalkylation of alkynes,1–9 which involves the direct coupling of unactivated alkyl electrophiles with alkynes. In the last several years, numerous hydroalkylation methods have been developed and now provide access to all three forms of disubstituted alkenes: 1,1 disubstiuted alkenes, Z- and E-alkenes (Scheme 1a). Furthermore, the high selectivity of these processes often enables the formation of a single regio- and stereoisomer of alkene products. Together, these methods have established hydroalkylation of alkynes as one of the most versatile and efficient strategies for selective alkene synthesis.
SCHEME 1.
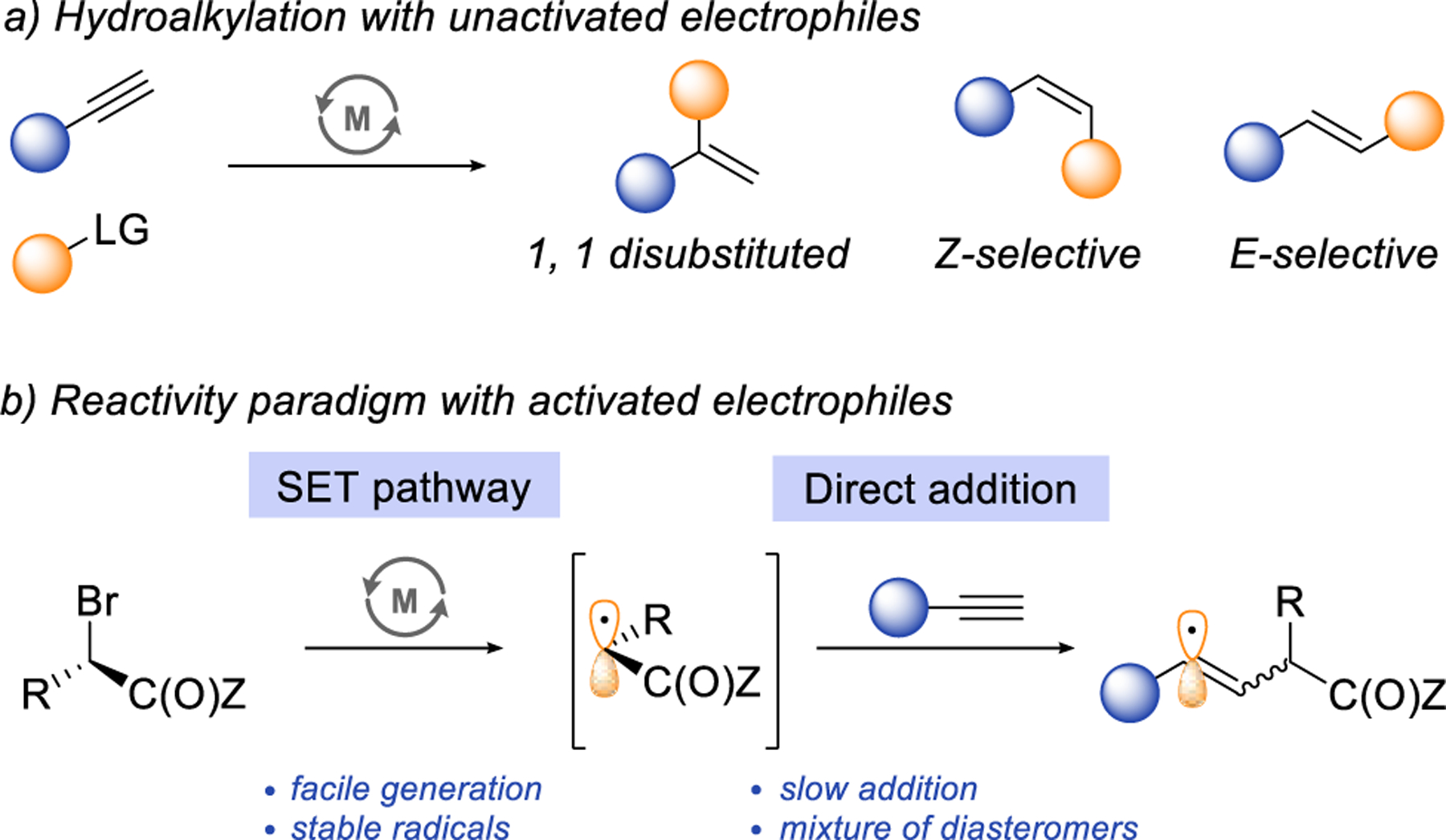
Despite these advancements, selective hydroalkylation remains difficult to accomplish with certain functionalized coupling partners. Particularly notable are α-halo carbonyls. These activated electrophiles have high redox potentials and readily generate alkyl radicals in the presence of first-row transition-metals through a single electron transfer (SET) process.10–11 However, leveraging this feature in selective hydroalkylation has proved to be challenging. So far, efforts have focused on the direct addition of these SET-generated alkyl radicals to the alkyne (Scheme 1b). Unfortunately, the addition step is slow12–13 and typically generates a mixture of E and Z isomers.12,14–16 As a result, the direct radical addition approach has found success only with activated aryl-substituted alkynes providing the alkene products with varying degrees of Z-diastereoselectivity.17–18
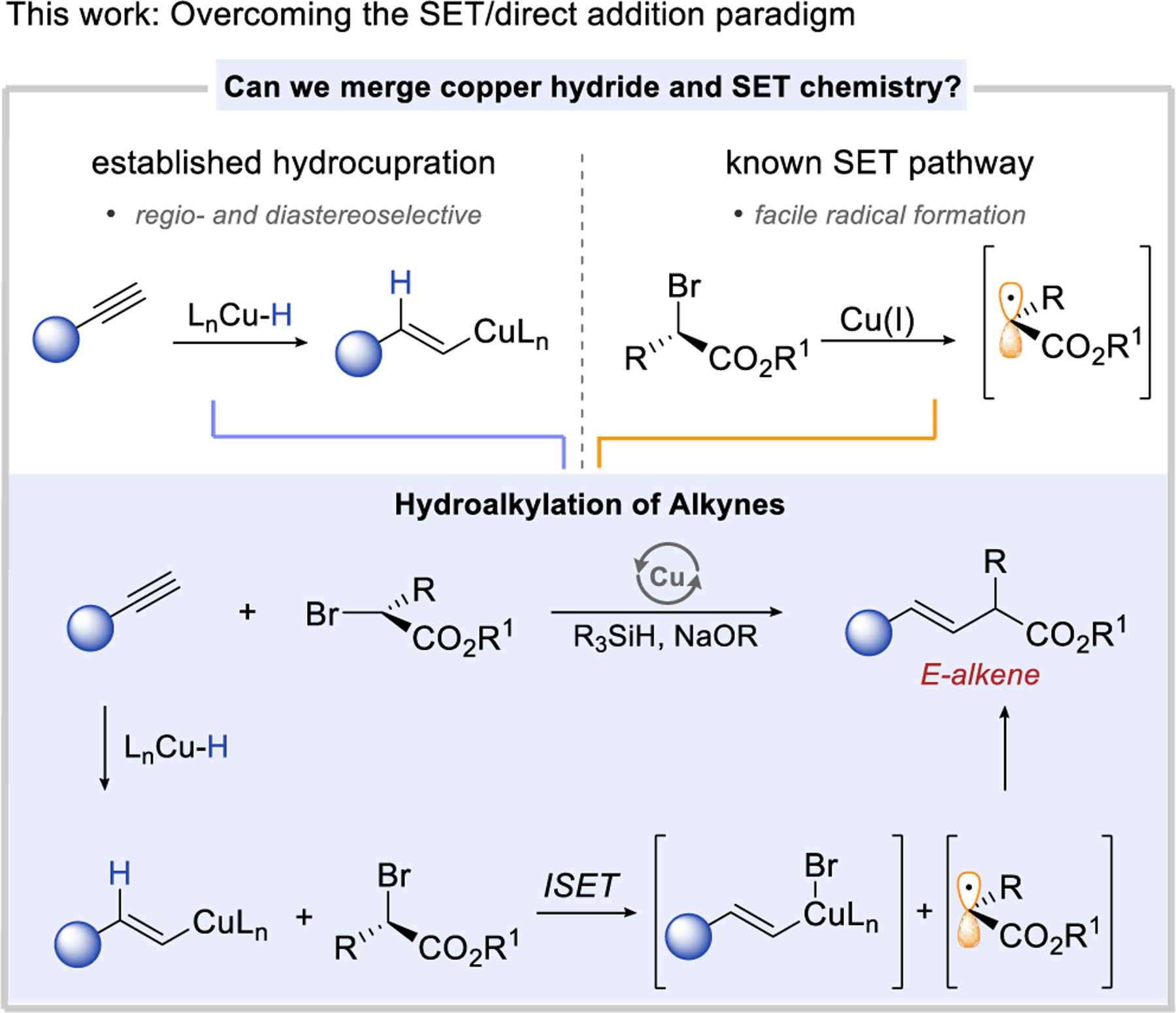
Recent developments in copper hydride chemistry have enabled a new approach to the hydrofunctionalization of alkynes based on the hydrocupration of the alkyne and subsequent functionalization of the reactive E-alkenyl copper intermediate.1,3,19–28 We propose to merge the hydrocupration of alkynes with the SET chemistry of α-halo carbonyls in order to achieve a selective hydroalkylation reaction (Scheme 2). The highly regio- and diastereoselective hysdrocupration step would ensure selective formation of the anti-Markovnikov addition product with excellent E selectivity. Subsequent halogen atom transfer via inner-sphere SET (ISET)10 would provide a facile alternative to the high barrier two-electron oxidative addition pathway29–30 and would lead to the alkylation of the alkenyl copper intermediate. Overall, the proposed combination of closed-shell copper-hydride addition to alkynes with open-shell SET chemistry provides a new strategy for hydroalkylation of alkynes.31
SCHEME 2.
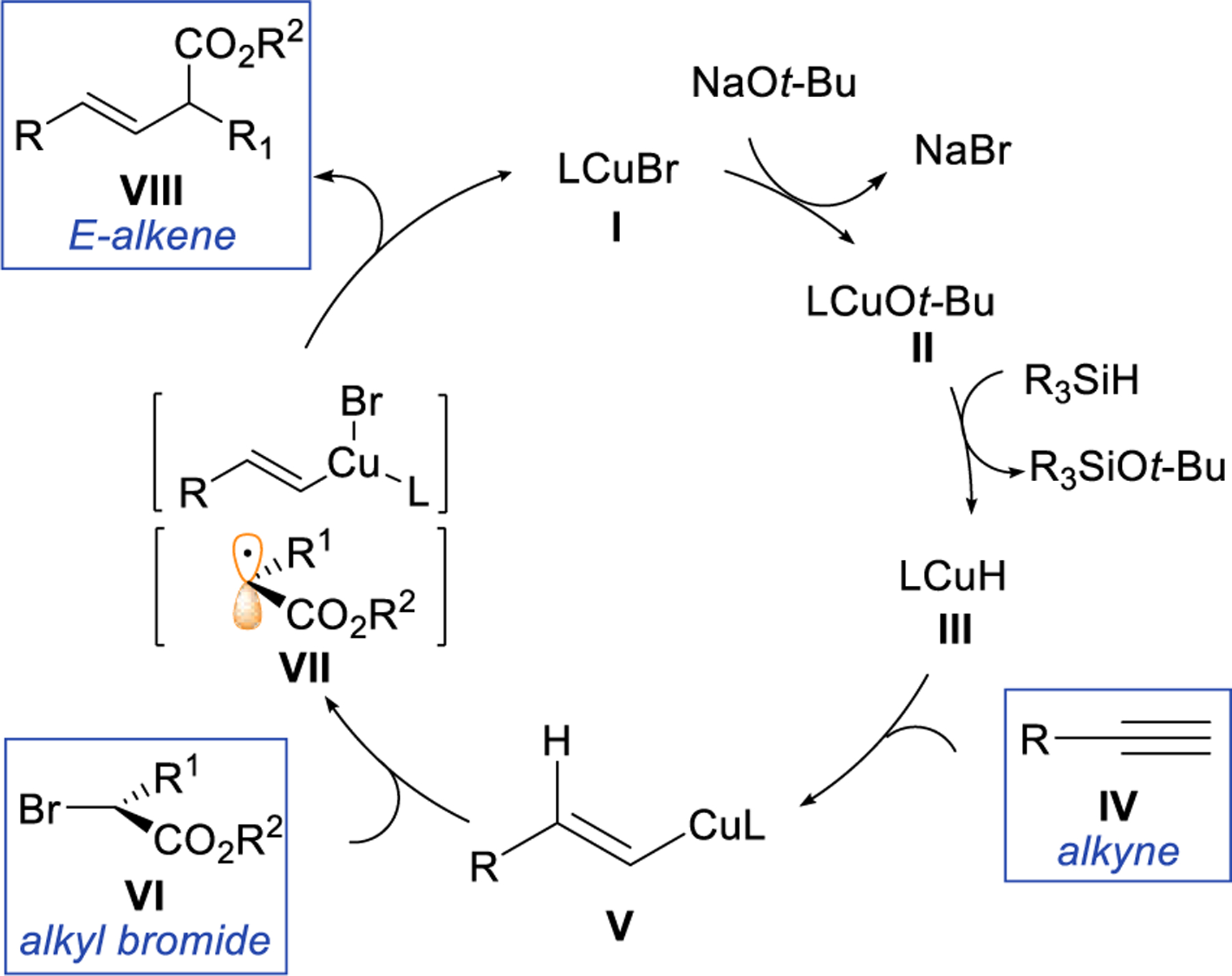
We envision the mechanism of hydroalkylation reaction as shown in Scheme 3. The alkenyl copper intermediate is formed according to the well-established mechanism (I-III).32 In a key step, the ISET involving alkenyl copper intermediate and an alkyl bromide (VI) would generate an alkenyl copper(II) species and a carbon-centered radical (VII).33 Finally, the reaction of the alkenyl copper(II) intermediate with the free radical delivers the product (VIII),34–36 possibly through radical capture followed by reductive elimination.
SCHEME 3.
From the outset, we recognized that a generally facile SET oxidation of copper(I) complexes by α-halo carbonyls could prevent the desired two-electron hydrocupration pathway. In effect, we had to suppress SET oxidation of all copper(I) complexes involved in forming the alkenyl copper intermediate (intermediates I-III).
We reasoned that a catalyst supported by an NHC ligand would allow us to achieve this goal by modulating the oxidation potential of these copper(I) intermediates. NHC ligands, such as IPr, favor the formation of linear two coordinate copper(I) complexes that are unusually resistant to oxidation.37–41 Hence, we expected that despite the σ-donating ability of NHC ligands, they would yield catalytic copper(I) intermediates with high reduction potentials red (E°1/2red [CuII/CuI]) and therefore relatively low rates in reaction with α-halo carbonyls. This correlation between the redox potentials of copper(I) complexes and rates of their reactions with α-halo carbonyls through ISET has been established in mechanistic studies of atom transfer radical polymerization (ATRP).33,42–43 We were also aware that IPr ligand promotes the rapid formation of the alkenyl copper intermediate through hydrocupration. Overall, we hoped that these features of IPr ligand will allow us to merge two electron hydrocupration pathway with the SET-based alkylation of the alkenyl copper intermediate.
We began our study by measuring the redox potential of IPrCuCl, a common catalyst in the hydrofunctionalization of alkynes. The anticipated effect of the IPr ligand was evident in the measured anodic peak potential (Ep,a= 1.68V vs SEC in MeCN; 1.58V vs SCE in DCM),44 which is high relative to the redox potentials for various copper(I) catalysts (E° ½ = 0.1V to −0.3V vs SCE in MeCN depending on the ligand) readily oxidized by α-halo carbonyls and used to promote ATRP33 or related ATR alkylation reactions.45
As expected, IPrCuCl did not readily react with an α-halo carbonyls. The exposure of secondary α-bromo ester 2 (E°1/2 = −0.43V vs. SCE in DMF)10 to IPrCuCl and TEMPO at 25 °C, resulted in full recovery of the α-bromo ester, indicating that SET did not occur (Scheme 4a). On the other hand, alkenyl copper complex 3 was significantly more easily oxidized (Ep,a = 0.90V vs. SCE in DCM) and, under similar reaction conditions, readily afforded cross-coupling product 5 (Scheme 4b). Less reactive electrophiles, such as α-chloro ester 4 did not provide the desired coupling product.
SCHEME 4.
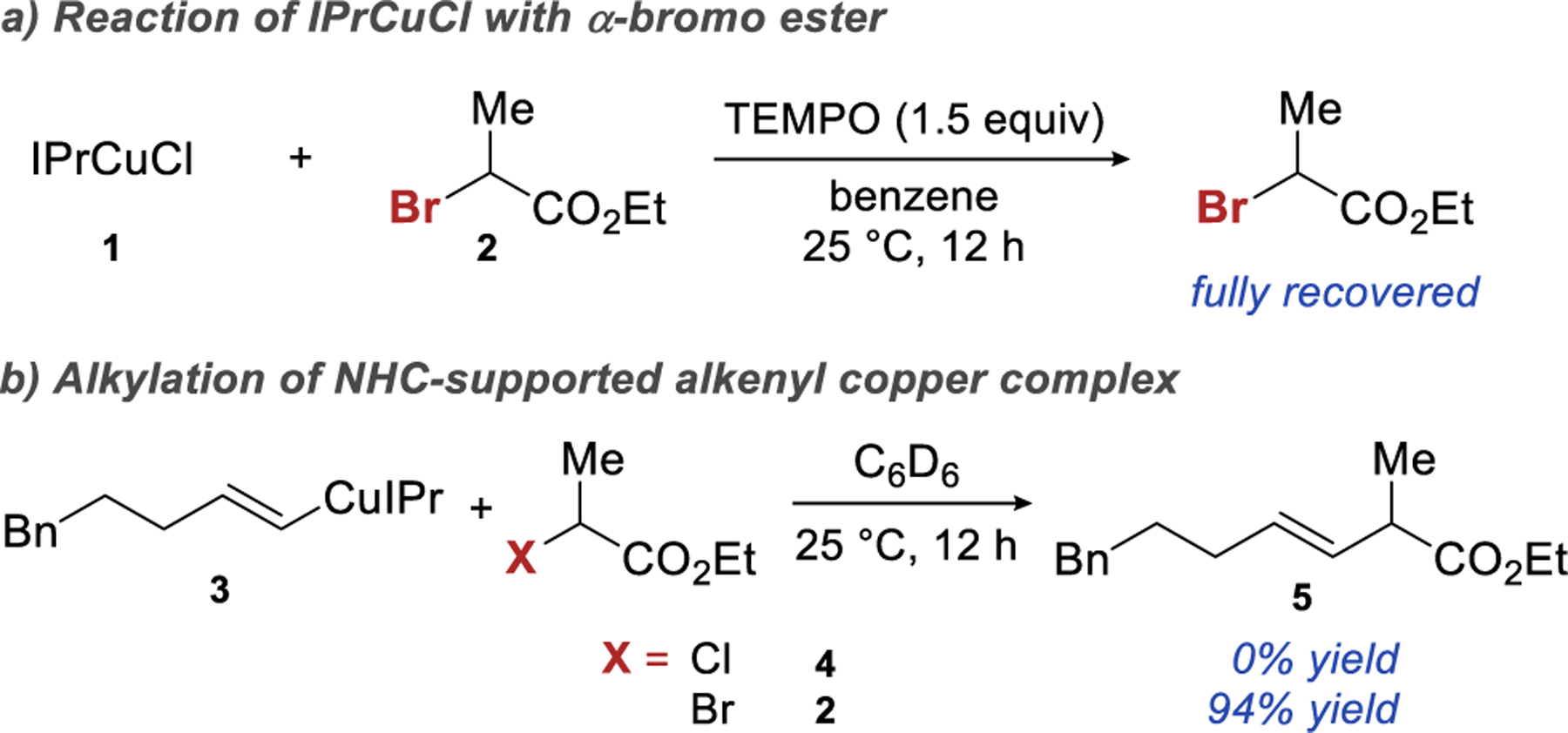
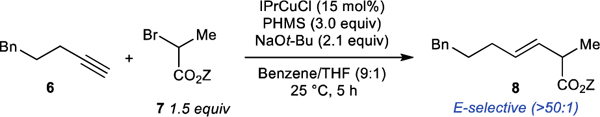
Encouraged by the results of our preliminary experiments and using them as a starting point, we developed the catalytic hydroalkylation of terminal alkynes with secondary α-bromo esters shown in Table 1. The best results were obtained using IPrCuCl as the catalyst, polymethylhydrosiloxane (PMHS) as the hydride source, and NaOt-Bu as the turnover reagent. The reaction is performed in a benzene/THF solvent combination and is complete in 5 hours at room temperature. During our efforts to identify the best conditions, we made several observations summarized in Table 1. In accordance with the results of the stoichiometric experiments shown in Scheme 4b, an α-cholo ester failed to provide any desired product in the catalytic reaction, while the corresponding α-iodo ester gave only 11% yield of the desired product. Copper complexes supported by IPr and SIPr ligand were the best catalysts. Even the closely related IMesCuCl catalyst provided less than 5% yield of the desired product. A catalyst prepared in situ from Cu(OAc)2 and (R)-DTBM-Segphos gave similarly low yield of the product.24 The choice of silane proved crucial to the success of the reaction. PMHS and structurally related tetrameric silicone hydride performed well. More reactive triethoxysilane or less reactive diphenylmethyl silane were ineffective. Sodium alkoxides were particularly successful in this transformation. Changing the alkoxide counterion from sodium to lithium or potassium led to significantly lower yields. While both sodium tert-butoxide and sodium tert-siloxide were effective turnover reagents, sodium isopropoxide was inferior and sodium methoxide failed to turn over the catalyst, presumably due to lower solubility. The highest yields of the desired product were obtained using a 9:1 benzene/THF solvent mix. Other ratios of these solvents and other aryl/etherial solvent mixtures resulted in depressed yields (see SI, Table S6) Benzene or toluene alone provided less of the desired product and a significant amount of the alkyne.
Table. 1.
Reaction development
| ||
|---|---|---|
| entry | change from standard conditions | yielda |
| 1. | none | 81% |
| 2. | Chloro ester instead of bromo ester | 0% |
| 3. | lodo ester instead of bromo ester | 11% |
|
| ||
| 4. | SIPrCuCI instead of IPrCuCI | 72% |
| 5. | IMesCuCI instead of IPrCuCI | 1% |
| 6. | (R)-DTBM-Segphos/Cu(OAc)2 instead of IPrCuCI | <5% |
|
| ||
| 7. | Ph2MeSiH instead of PMHS | 3% |
| 8. | Me(OEt)2SiH instead of PMHS | 0% |
| 9. | (MeOSiH)4 instead of PMHS | 46% |
|
| ||
| 10. | LiOt-Bu instead of NaOt-Bu | 37% |
| 11. | NaOi-Pr instead of NaOt-Bu | 22% |
| 12. | NaOSiMe3 instead of NaOt-Bu | 74% |
| 13. | KOt-Bu instead of NaOt-Bu | 0% |
|
| ||
| 14. | Benzene instead of Benzene/THF (9:1) | 58% |
| 15. | Benzene/THF (1:1) instead of Benzene/THF (9:1) | 66% |
| 16. | toluene instead of Benzene/THF (9:1) | 52% |
| 17. | THF instead of Benzene/THF (9:1) | 27% |
|
| ||
| 18. | Ethyl ester instead of pinacolyl ester | 61% |
Determined by GC using internal standard. Z stands for pinacolyl.

Using the standard conditions from Table 1, we found that a wide range of E-alkenes could be synthesized (Table 2) with E-selectivity greater than 50:1 (see SI). We also found that the reaction is compatible with many functional groups and can be accomplished in the presence of esters (11), epoxides (10), nitriles (17), alkyl chlorides (13), aryl bromides (24) and fluorides (9), acetals (20), and amides (22). Sterically demanding alkynes such as 15 and 25 also performed well under the reaction conditions. The reaction also tolerates several nitrogen-containing heteroarenes, such as halo pyridines (12, 26), indoles (21), quinoxalines (16), phenoxazines (19), and phenothiazines (14).
Table 2:
Substrate scopea

|
Yields of isolated are reported. Reactions performed on 0.5 mmol scale. Conditions A: IPrCuCI (15 mol%), NaOt-Bu (2.1 equiv), bromoester (1.5 equiv), PMHS (3 equiv), benzene/THF (9:1) 5 mL, 5–24 h.
NaOTMS was used as base instead of NaOt-Bu.
2 equiv of bromoester were used.
Conditions B: IPrCuCI (15 mol %), NaOt-Bu (2.1 equiv), bromoamide (2.0 equiv), PMHS (3 equiv), benzene/THF (7:3) 5 mL, 24h.
R = PhCH2CH2CH2, 1:1 benzene/THF was used as a solvent.
We also explored the reaction with different secondary α-bromo esters. In general, esters of sterically bulky alcohols afforded the desired product in good yield, presumably because of the increased stability of these esters under catalytic conditions. The presence of heteroatomic substituents, such as bromide (37) or thioether (38), at the γ position of the ester was well tolerated. Sterically demanding secondary bromides (36) also gave the product in good yield.
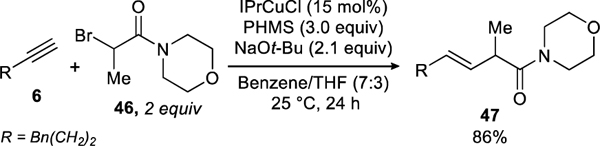
Our initial attempts to couple secondary α-bromo amides were unsuccessful. Under the reaction conditions used for coupling α-bromo esters, product 47 was formed in only 47% yield. The primary side reactions were the reductions of both the α-bromo amide and the alkyne. Eventually, we found that by subtly changing the reaction conditions, we could obtain product 47 in 86% yield.
 |
(eq 1) |
To achieve these results, we lowered the solvent ratio of benzene/THF from 9:1 to 7:3, adjusted the stoichiometry of the reactants, and increased the reaction time to 24 hours.
These modified conditions could be applied to the coupling of a variety of secondary α-bromo amides. These included amide derivatives of numerous biologically important amines, such as piperazine (34), pyrrolidine (32), indolone (33), and morpholine (31). This adaptation could also be used to couple cyclic tertiary α-bromo amides. Tertiary α-bromo-β-lactams (35), as well as secondary α-bromo-β-lactams (40) were found to provide the desired products in useful yields. Acyclic tertiary amides or esters failed to provide any product.
We also noted a few limitations of the present hydroalkylation reaction. Aryl acetylenes (41) and disubstituted internal alkynes (42) did not participate in the hydroalkylation reaction. Similarly, α-bromo ketones (43), α-bromo nitriles (45), aryl substituted α-bromo esters (44), and primary α-halo carbonyls were not viable substrates. Finally, protic functional groups, such as hydroxyl groups and unprotected amines, and reducible functional groups, like aldehydes and ketones were not tolerated.
The key feature of the mechanism proposed in Scheme 3 is the cross coupling of the alkenyl copper intermediate with the α-bromo carbonyl. Although stoichiometric experiments shown in Scheme 4b established the feasibility of this elementary step, the exact mechanism of this process was unclear. While we initially postulated a pathway initiated by SET, a two-electron oxidative addition/reductive elimination sequence is also plausible.
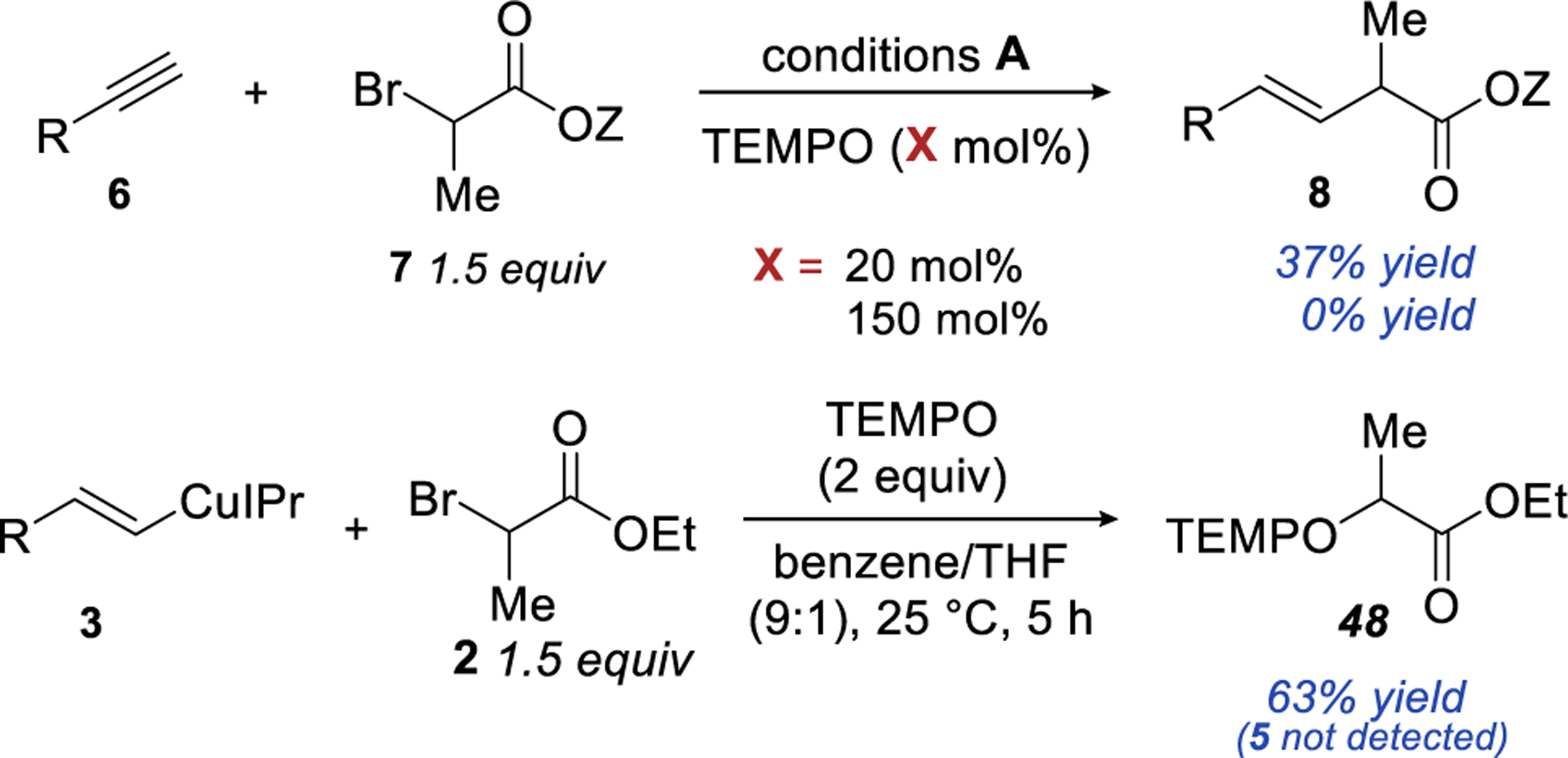
Support for a SET pathway came from radical trap experiments (Scheme 5). Our previous work has shown that the electrophilic functionalization of the alkenyl copper intermediate, is not affected by the addition of TEMPO.20 On the other hand, the SET pathway is expected to show sensitivity to TEMPO.43,46–47 We observed that as little as 20 mol% of TEMPO impacts the catalytic reaction and 1.5 equivalent of TEMPO completely prevents the formation of the alkene product (Scheme 5). Stochiometric experiment with alkenyl copper revealed that TEMPO inhibits the cross-coupling step of the reaction, producing TEMPO adduct 48 as the major product of the reaction (63% yield).
SCHEME 5. Radical Trap Probea.
a Z = pinacolyl, R = Ph(CH2)3; Conditions A (see Table 2)
The results of these experiments allow us to exclude the alkylation mechanism involving two electron processes. Distinguishing between different SET mechanisms is much harder. Extensive mechanistic investigations of similar processes involved in ATRP, suggest ISET as the most likely SET mechanism for the reaction of alkenyl copper with an α-bromo carbonyl.10,33,48 Furthermore, the relatively high redox potential of alkenyl copper intermediate makes the mechanisms involving outer-sphere SET processes unlikely, although we cannot completely exclude them.
We finished our preliminary investigation of the reaction mechanism by measuring redox potentials of other copper complexes involved in the proposed catalytic cycle. We found that IPrCuBr (Ep,a= 1.51V vs SCE in DCM) and IPrCuOt-Bu (Ep,a= 1.13V vs SCE in DCM) are both less reducing than the alkenyl copper intermediate 3. IPrCuH (Ep,a= −0.39V vs SCE in THF), on the other hand, is significantly more reducing than alkenyl copper intermediate 3, suggesting a potential for rapid SET with α-bromo carbonyls. Despite the highly reducing nature of IPrCuH, in a competition experiment, IPrCuH reacts significantly faster with a terminal alkyne than with α-bromo amide 46 (Scheme 6). Alkene 49, the nearly exclusive product of the competition experiment, is obtained through hydrocupration and protonation of the alkenyl copper intermediate (3) upon aqueous workup. Furthermore, a stoichiometric reaction of IPrCuH with α-bromo amide 46 is slow and not inhibited by TEMPO, suggesting that the mechanism of the reaction does not involve SET or radical intermediates (see the SI, Table S13). These results indicate surprising resistance of IPrCuH toward SET, despite its highly reducing nature.
SCHEME 6.

In conclusion, we have developed a hydroalkylation of alkynes using α-bromo carbonyls as alkylating reagents. The hydroalkylation reaction affords the E-alkene product with high selectivity and is compatible with several classes of alkynes and α-bromo carbonyls. Furthermore, the reaction can be accomplished in the presence of acetals, epoxides, aryl halides, and heteroaromatics. Mechanistic experiments reveal that the direct alkylation of the alkenyl copper intermediate obtained by hydrocupration of an alkyne is accomplished through a SET pathway.
Supplementary Material
ACKNOWLEDGMENT
M. T. Lee is acknowledged for help with preparation of the manuscript.
Funding Sources
NIH (1R01GM132200-01)
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website
Experimental procedure and product characterization (pdf)
The authors declare no competing financial interests.
REFERENCES
- (1).Hazra A; Chen J; Lalic G Stereospecific Synthesis of E-Alkenes through Anti-Markovnikov Hydroalkylation of Terminal Alkynes. J. Am. Chem. Soc 2019, 141, 12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lee MT; Goodstein MB; Lalic G Synthesis of Isomerically Pure (Z)-Alkenes from Terminal Alkynes and Terminal Alkenes: Silver-Catalyzed Hydroalkylation of Alkynes. J. Am. Chem. Soc 2019, 141, 17086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Uehling MR; Suess AM; Lalic G Copper-Catalyzed Hydroalkylation of Terminal Alkynes. J. Am. Chem. Soc 2015, 137, 1424. [DOI] [PubMed] [Google Scholar]
- (4).Suess AM; Uehling MR; Kaminsky W; Lalic G Mechanism of Copper-Catalyzed Hydroalkylation of Alkynes: An Unexpected Role of Dinuclear Copper Complexes. J. Am. Chem. Soc 2015, 137, 7747. [DOI] [PubMed] [Google Scholar]
- (5).Lu X-Y; Liu J-H; Lu X; Zhang Z-Q; Gong T-J; Xiao B; Fu Y 1,1-Disubstituted Olefin Synthesis Via Ni-Catalyzed Markovnikov Hydroalkylation of Alkynes with Alkyl Halides. Chem. Commun 2016, 52, 5324. [DOI] [PubMed] [Google Scholar]
- (6).Till NA; Smith RT; MacMillan DWC Decarboxylative Hydroalkylation of Alkynes. J. Am. Chem. Soc 2018, 140, 5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Yu L; Lv L; Qiu Z; Chen Z; Tan Z; Liang Y-F; Li C-J Palladium-Catalyzed Formal Hydroalkylation of Aryl-Substituted Alkynes with Hydrazones. Angew. Chem. Int. Ed 2020, 59, 14009. [DOI] [PubMed] [Google Scholar]
- (8).Cheung CW; Zhurkin FE; Hu X Z-Selective Olefin Synthesis Via Iron-Catalyzed Reductive Coupling of Alkyl Halides with Terminal Arylalkynes. J. Am. Chem. Soc 2015, 137, 4932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Barzanò G; Cheseaux A; Hu X Z-Selective Synthesis of Vinyl Boronates through Fe-Catalyzed Alkyl Radical Addition. Org. Lett 2019, 21, 490. [DOI] [PubMed] [Google Scholar]
- (10).Lin CY; Coote ML; Gennaro A; Matyjaszewski K Ab Initio Evaluation of the Thermodynamic and Electrochemical Properties of Alkyl Halides and Radicals and Their Mechanistic Implications for Atom Transfer Radical Polymerization. J. Am. Chem. Soc 2008, 130, 12762. [DOI] [PubMed] [Google Scholar]
- (11).Isse AA; Lin CY; Coote ML; Gennaro A Estimation of Standard Reduction Potentials of Halogen Atoms and Alkyl Halides. J. Phys. Chem. B 2011, 115, 678. [DOI] [PubMed] [Google Scholar]
- (12).Wille U Radical Cascades Initiated by Intermolecular Radical Addition to Alkynes and Related Triple Bond Systems. Chem. Rev 2013, 113, 813. [DOI] [PubMed] [Google Scholar]
- (13).Gómez-Balderas R; Coote ML; Henry DJ; Fischer H; Radom L What Is the Origin of the Contrathermodynamic Behavior in Methyl Radical Addition to Alkynes Versus Alkenes? J. Phys. Chem. A 2003, 107, 6082. [Google Scholar]
- (14).Jenkins PR; Symons MCR; Booth SE; Swain CJ Why Is Vinyl Anion Configurationally Stable but a Vinyl Radical Configurationally Unstable? Tetrahedron Lett 1992, 33, 3543. [Google Scholar]
- (15).Giese B; Lachhein S Addition of Alkyl Radicals to Alkynes: Distinction between Radical and Ionic Nucleophiles. Angew. Chem. Int. Ed 1982, 21, 768. [Google Scholar]
- (16).Mastandrea MM; Cañellas S; Caldentey X; Pericàs MA Decarboxylative Hydroalkylation of Alkynes Via Dual Copper-Photoredox Catalysis. ACS Catal 2020, 10, 6402. [Google Scholar]
- (17).Nakamura K; Nishikata T Tandem Reactions Enable Trans- and Cis-Hydro-Tertiary-Alkylations Catalyzed by a Copper Salt. ACS Catal 2017, 7, 1049. [Google Scholar]
- (18).For a different indirect approach to E-selective hydroalkylation involving hydroboration of alkynes followed by alkylation of the intermediate, see reference 17.
- (19).Mailig M; Hazra A; Armstrong MK; Lalic G Catalytic Anti-Markovnikov Hydroallylation of Terminal and Functionalized Internal Alkynes: Synthesis of Skipped Dienes and Trisubstituted Alkenes. J. Am. Chem. Soc 2017, 139, 6969. [DOI] [PubMed] [Google Scholar]
- (20).Uehling MR; Rucker RP; Lalic G Catalytic Anti-Markovnikov Hydrobromination of Alkynes. J. Am. Chem. Soc 2014, 136, 8799. [DOI] [PubMed] [Google Scholar]
- (21).Cheng L-J; Islam SM; Mankad NP Synthesis of Allylic Alcohols Via Cu-Catalyzed Hydrocarbonylative Coupling of Alkynes with Alkyl Halides. J. Am. Chem. Soc 2018, 140, 1159. [DOI] [PubMed] [Google Scholar]
- (22).Cheng L-J; Mankad NP Cu-Catalyzed Hydrocarbonylative C–C Coupling of Terminal Alkynes with Alkyl Iodides. J. Am. Chem. Soc 2017, 139, 10200. [DOI] [PubMed] [Google Scholar]
- (23).Cheng L-J; Mankad NP Heterobimetallic Control of Regioselectivity in Alkyne Hydrostannylation: Divergent Syntheses of α- and (E)-β-Vinylstannanes Via Cooperative Sn–H Bond Activation. J. Am. Chem. Soc 2019, 141, 3710. [DOI] [PubMed] [Google Scholar]
- (24).Shi S-L; Buchwald SL Copper-Catalysed Selective Hydroamination Reactions of Alkynes. Nat. Chem 2015, 7, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Das M; Kaicharla T; Teichert JF Stereoselective Alkyne Hydrohalogenation by Trapping of Transfer Hydrogenation Intermediates. Org. Lett 2018, 20, 4926. [DOI] [PubMed] [Google Scholar]
- (26).Fujihara T; Xu T; Semba K; Terao J; Tsuji Y Copper-Catalyzed Hydrocarboxylation of Alkynes Using Carbon Dioxide and Hydrosilanes. Angew. Chem. Int. Ed 2011, 50, 523. [DOI] [PubMed] [Google Scholar]
- (27).Semba K; Fujihara T; Terao J; Tsuji Y Copper-Catalyzed Highly Regio- and Stereoselective Directed Hydroboration of Unsymmetrical Internal Alkynes: Controlling Regioselectivity by Choice of Catalytic Species. Chem. Eur. J 2012, 18, 4179. [DOI] [PubMed] [Google Scholar]
- (28).Jang WJ; Lee WL; Moon JH; Lee JY; Yun J Copper-Catalyzed Trans-Hydroboration of Terminal Aryl Alkynes: Stereodivergent Synthesis of Alkenylboron Compounds. Org. Lett 2016, 18, 1390. [DOI] [PubMed] [Google Scholar]
- (29).Giri R; Brusoe A; Troshin K; Wang JY; Font M; Hartwig JF Mechanism of the Ullmann Biaryl Ether Synthesis Catalyzed by Complexes of Anionic Ligands: Evidence for the Reaction of Iodoarenes with Ligated Anionic Cu(I) Intermediates. J. Am. Chem. Soc 2018, 140, 793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhang S-L; Liu L; Fu Y; Guo Q-X Theoretical Study on Copper(I)-Catalyzed Cross-Coupling between Aryl Halides and Amides. Organometallics 2007, 26, 4546. [Google Scholar]
- (31). A closely related mechanism of carbonylative hydroalkylation of alkynes was proposed by Mankad et al. (reference 22). However, the proposed SET reduction of an alkyl iodide by alkenyl copper complex does not occur in the absence of CO.
- (32).Jordan AJ; Lalic G; Sadighi JP Coinage Metal Hydrides: Synthesis, Characterization, and Reactivity. Chem. Rev 2016, 116, 8318. [DOI] [PubMed] [Google Scholar]
- (33).Matyjaszewski K Atom Transfer Radical Polymerization (ATRP): Current Status and Future Perspectives. Macromolecules 2012, 45, 4015. [Google Scholar]
- (34).Bakhoda A; Okoromoba OE; Greene C; Boroujeni MR; Bertke JA; Warren TH Three-Coordinate Copper(II) Alkynyl Complex in C–C Bond Formation: The Sesquicentennial of the Glaser Coupling. J. Am. Chem. Soc 2020, 142, 18483. [DOI] [PubMed] [Google Scholar]
- (35).Salvador TK; Arnett CH; Kundu S; Sapiezynski NG; Bertke JA; Raghibi Boroujeni M; Warren TH Copper Catalyzed sp3 C–H Etherification with Acyl Protected Phenols. J. Am. Chem. Soc 2016, 138, 16580. [DOI] [PubMed] [Google Scholar]
- (36).Wiese S; Badiei YM; Gephart RT; Mossin S; Varonka MS; Melzer MM; Meyer K; Cundari TR; Warren TH Catalytic C-H Amination with Unactivated Amines through Copper(II) Amides. Angew. Chem. Int. Ed 2010, 49, 8850. [DOI] [PubMed] [Google Scholar]
- (37).IPrCuCl is air stable both in solid form and in solution. For examples of other two-coordinate copper(I) complexes that have high redox potentials and/or are resistant to oxidation, see references 38–41.
- (38).Sorrell TN; Jameson DL Synthesis, Structure, and Reactivity of Monomeric Two-Coordinate Copper(I) Complexes. J. Am. Chem. Soc 1983, 105, 6013. [Google Scholar]
- (39).Sanyal I; Karlin KD; Strange RW; Blackburn NJ Chemistry and Structural Studies on the Dioxygen-Binding Copper-1,2-Dimethylimidazole System. J. Am. Chem. Soc 1993, 115, 11259. [Google Scholar]
- (40).Banthia S; Samanta A Synthesis and Structure of Unusually Stable Linear Copper(I) Complexes with Blue Fluorescence. Polyhedron 2006, 25, 2269. [Google Scholar]
- (41).Le Clainche L; Giorgi M; Reinaud O Synthesis and Characterization of a Novel Calix[4]Arene-Based Two-Coordinate Copper(I) Complex That Is Unusually Resistant to Dioxygen. Eur. J. Inorg. Chem 2000, 2000, 1931. [Google Scholar]
- (42).Braunecker WA; Tsarevsky NV; Gennaro A; Matyjaszewski K Thermodynamic Components of the Atom Transfer Radical Polymerization Equilibrium: Quantifying Solvent Effects. Macromolecules 2009, 42, 6348. [Google Scholar]
- (43).Matyjaszewski K; Paik H.-j.; Zhou P; Diamanti SJ Determination of Activation and Deactivation Rate Constants of Model Compounds in Atom Transfer Radical Polymerization. Macromolecules 2001, 34, 5125. [Google Scholar]
- (44). For further details about the CV measurements, see the SI.
- (45).Shimkin KW; Watson DA Recent Developments in Copper-Catalyzed Radical Alkylations of Electron-Rich π-Systems. Beilstein J. Org. Chem 2015, 11, 2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Beckwith ALJ; Bowry VW; Ingold KU Kinetics of Nitroxide Radical Trapping. 1. Solvent Effects. J. Am. Chem. Soc 1992, 114, 4983. [Google Scholar]
- (47).Bowry VW; Ingold KU Kinetics of Nitroxide Radical Trapping. 2. Structural Effects. J. Am. Chem. Soc 1992, 114, 4992. [Google Scholar]
- (48).Fang C; Fantin M; Pan X; de Fiebre K; Coote ML; Matyjaszewski K; Liu P Mechanistically Guided Predictive Models for Ligand and Initiator Effects in Copper-Catalyzed Atom Transfer Radical Polymerization (Cu-ATRP). J. Am. Chem. Soc 2019, 141, 7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.