

Abstract
Ischemia reperfusion injury contributes to adverse cardiovascular diseases in part by producing a burst of reactive oxygen species (ROS) that induce oxidations of many muscular proteins. Glutathionylation is one of the major protein cysteine oxidations that often serve as molecular mechanisms behind the pathophysiology associated with ischemic stress. Despite the biological significance of glutathionylation in ischemia reperfusion, identification of specific glutathionylated cysteines under ischemic stress has been limited. In this report, we have analyzed glutathionylation under oxygen-glucose-deprivation (OGD) or repletion of nutrients after OGD (OGD/R) by using a clickable glutathione approach that specifically detects glutathionylated proteins. Our data find that palmitate availability induces a global level of glutathionylation and decreases cell viability during OGD/R. We have then applied a clickable glutathione-based proteomic quantification strategy, which enabled the identification and quantification of 249 glutathionylated cysteines in response to palmitate during OGD/R in HL-1 cardiomyocyte cell line. The subsequent bioinformatic analysis found 18 glutathionylated cysteines whose genetic variants are associated with muscular disorders. Overall, our data report glutathionylated cysteines under ischemic stress that may contribute to adverse outcomes or muscular disorders.
Keywords: Glutathionylation, Ischemic stress, Clickable glutathione, Proteomics, Cardiomyocytes
Graphical Abstract
Introduction
Ischemia, restriction of arterial blood flow to tissues, is one of the major causes that lead to various diseases, including heart failure.1 Two main features of ischemia involve a limited supply of oxygen (hypoxia) and a shortage of nutrients, both of which are important for continuous ATP production and muscle contraction in the heart.2 Therefore, the length (or duration) and severity of the ischemic condition often determine the level of tissue damages,2 which should be resolved by restoration of a blood flow (reperfusion). However, it is well-known that ischemic reperfusion (I/R) causes an additional tissue injury that may not be manifested during ischemia, primarily resulting from sudden reintroduction of oxygen (re-oxygenation) to ischemic tissue.1
The underlying molecular and cellular events under ischemia and I/R are multifactorial, but in part consequential to changes in oxygen availability (hypoxia/reoxygenation) and nutrient availability (metabolic alteration).1–2 Cardiomyocytes produce necessary ATP primarily by fatty acid oxidation (60–90%) in mitochondria rather than glucose oxidation (10–40%), thus heavily relying on oxygen availability.3–4 Therefore, ischemia depletes an intracellular ATP level, which compromises ATP-dependent processes, such as calcium efflux, thus leading to intracellular and mitochondrial calcium overload.2, 5 Cardiomyocytes also turn to anaerobic glycolysis due to hypoxia, which acidifies intracellular environment.3, 5 These changes are associated with activation of proteases, including calpains,5 that degrade myofibrils and induce autophagy to reduce energy consumption, but eventually lead to cell death upon prolonged ischemia. Subsequently, reoxygenation during I/R causes a burst of reactive oxygen species (ROS) from mitochondria and others.1–2 Such burst of ROS overwhelms the detoxification capacity of redox enzymes and induces oxidation of proteins, lipids, and DNA, which is accompanied by a shrinkage of contractile cardiomyocytes (hypercontracture)5–6 and a reversible loss of contraction (cardiomyocyte stunning).2, 7 Importantly, many experiments demonstrated that metabolic activity or nutrient availability during reperfusion is crucial to determine the level of tissue damages.4, 8 For example, a plasma level of fatty acids was found elevated after ischemia.9 During reperfusion, the fatty acids are preferentially catabolized and inhibit glucose (pyruvate) oxidation,10–11 which is linked to increased cell death and diminished contractile activity of heart.9–10, 12 Indeed, metabolic modulations, such as a blockage of fatty acid oxidation or stimulation of glucose oxidation, have been evaluated or used as potential therapeutic interventions for I/R injury.13–16
The limited energy-production and the burst of ROS during ischemia and I/R consequently alter the redox homeostasis and readily induce reversible and irreversible oxidation of proteins, especially at cysteine residues.17–18 A large body of reports have demonstrated S-oxidation,19 disulfide formation,20–22 S-nitrosylation,23–24 4-hydroxynonenal (HNE) conjugation,25 and glutathionylation26–29 of individual proteins or global proteome in the heart subjected to I/R. Among these oxoforms, glutathionylation is one of the major protein cysteine oxidations that modulate the enzyme activity or protein-protein interactions, or confer protection from irreversible oxidations.30 Previously, glutathionylation of several individual contractile proteins, including actin, has been found during I/R, which correlated with compromised contractile activity.26 In addition, several metabolic and mitochondria enzymes, such as GAPDH and complex II, were found glutathionylated, which altered metabolic activity.28–29 Despite these analyses, global identification of specific glutathionylated cysteines under ischemic stress or upon metabolic alterations in cardiomyocytes has been limited.
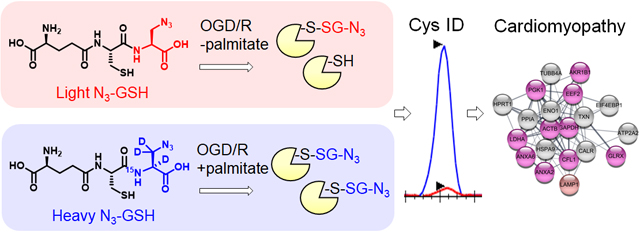
Previously, we have developed a chemoselective method, namely clickable glutathione, to detect glutathionylated proteins (Figure 1).31–32 In this approach, clickable glutathione (γGlu-Cys-azido-Ala, azido-glutathione, N3-GSH) is bio-synthesized in situ in cells with a mutant of glutathione synthetase (GS M4), which uses azido-Ala in place of Gly in glutathione biosynthesis. The azido-group on the clickable glutathione is used to identify glutathionylated proteins after click reaction with biotin- or fluorophore-alkyne. Recently, we have used this clickable glutathione approach with cleavable biotin-DADPS-alkyne and mass spectrometry to identify over 1,700 specific glutathionylated peptides in HL-1 cardiomyocytes upon production of hydrogen peroxide (H2O2).33 In addition, we have developed a quantification strategy with clickable glutathione by using isotopically labeled azido-Ala derivatives [heavy azido-Ala (+4 Da) and light azido-Ala (+0 Da)], which were used to quantify the levels of glutathionylation on proteins upon addition of H2O2.34 In this report, we have applied the clickable glutathione to HL-1 cardiomyocytes to investigate induction of glutathionylation under ischemic stress, especially with systemic alterations of metabolic nutrients and oxygen. Our analyses demonstrated that glutathionylation occurs at a proteome level under prolonged glucose depletion, but largely absent or weak under hypoxia or oxygen-glucose deprivation (OGD). However, during repletion of oxygen or nutrients after OGD (OGD/R), reoxygenation induces a high level of global glutathionylation where the level of glutathionylation is reduced by glucose availability but enhanced by fatty acid availability. The fatty acid availability during OGD/R correlated with decreased cellular viability. We further applied a quantification strategy with clickable glutathione, which resulted in the identification and quantification of 249 glutathionylated cysteines. Importantly, bioinformatic analysis was used to find 18 glutathionylated cysteines whose genetic variants are known and associated with muscular disorders, thus suggesting their potential functional outcomes resulting from ischemic stress.
Figure 1. Clickable glutathione approach for detection of glutathionylation.
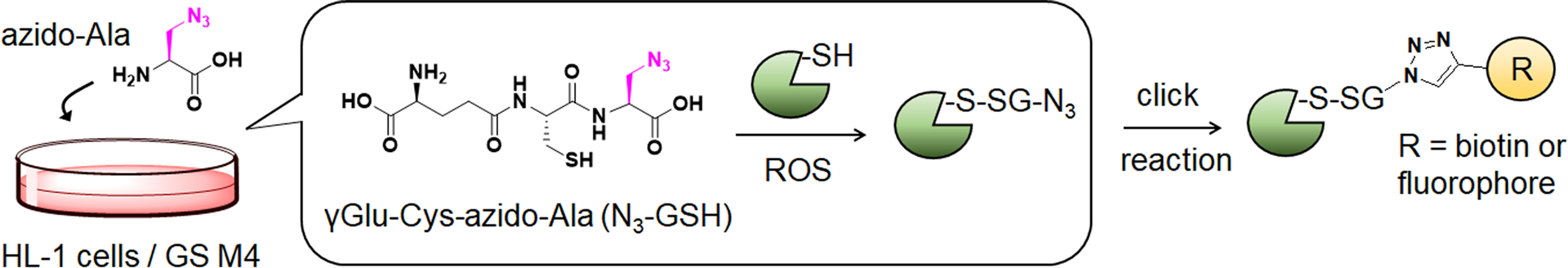
Glutathione synthetase mutant (GS M4) uses azido-Ala to synthesize azido-glutathione (γGlu-Cys-azido-Ala, N3-GSH). After incubation of azido-Ala to HL-1 cells expressing GS M4, azido-glutathione forms S-glutathionylation, which can be detected after the click reaction.
Experimental Procedures
Cell culture and induction of glutathionylation
HL-1 cells (Sigma) were cultured in Claycomb medium containing 10% FBS, penicillin (100 units/mL), streptomycin (100 μg/mL), norepinephrine (0.1 mM), and L-glutamine (2 mM) in fibronectin-gelatin coated flasks. HEK293/GS M4 stable cells35 were cultured in DMEM containing 10% FBS, penicillin (100 units/mL) streptomycin (100 units/mL). Cells were maintained in a humidified atmosphere containing 5% CO2 at 37°C. At 80% confluency, HL-1 cells were infected with adenovirus expressing GS M4. After 24 h, cells were incubated with a Claycomb medium containing L-azido-Ala (0.6 mM) for 20 h. Cells were then subjected to serum starvation for 12 h. Glutathionylation was induced using different stress conditions: Cells were subjected to glucose deprivation (GD, 0 mM glucose), hypoxia (1% oxygen), or oxygen-glucose-deprivation (OGD, 0 mM glucose and 1% oxygen) in a hypoxic chamber (Billups-Rothenberg, MIC-101) for indicated times. For OGD/R, after OGD, cells were repleted with glucose (25 mM glucose), oxygen (normoxia), or palmitate (1 mM) with BSA (0.17 mM). Cells were washed with cold PBS and lysed with a lysis buffer containing 1% SDS, 100 mM LiCl, 100 mM HEPES (pH 7.6), 50 mM N-ethylmaleimide (NEM), and protease inhibitor cocktail. Cell lysates were incubated for 30 min at 4°C, passed through 26-gauge needles ten times, and protein concentrations were determined using Bradford assay.
Detection of glutathionylation
Proteins (100 μg) were precipitated by adding acetone (4 times volume). After centrifugation at 13,000 rpm for 3 min, the supernatant was removed. Pellet was resuspended in a buffer (40 μL, 1× PBS, 0.1% SDS) by sonication. To the resuspended solution were added 10 mM Cy5-alkyne in DMSO (0.5 μL) and a click mix (10 μL) [20 mM CuBr in DMSO/tBuOH (3:1, v/v) (5 μL) and 20 mM THPTA (5 μL)]. The mixture was incubated for 1 h at room temperature in the dark. Proteins were separated by SDS-PAGE and analyzed by FluorChem Q imaging system (BioRad) or Coomassie stains.
ROS detection
Intracellular ROS levels were measured by using MitoSOX red (Sigma, M36008). HL-1 cells were uninduced or induced with different physiological conditions (GD, hypoxia, OGD, OGD/R). Cells were washed with PBS and incubated with MitoSOX red (5 μM) in DMEM without phenol red for 10 min in the dark. Cells were washed with PBS, and fluorescence values were monitored with a microplate reader (BioTek) with excitation (510 nm) and emission (595 nm) wavelengths.
NADPH quantification assay
The NADP+/NADPH levels were determined using the NADP+/NADPH quantification kit (Sigma, MAK038) according to the manufacturer’s instructions. HL-1 cells were uninduced or induced with different physiological conditions (GD, hypoxia, OGD, OGD/R). Cells were lysed using an NADP+/NADPH extraction buffer. Lysates (50 μL) were incubated with an NADP+ cycling enzyme mixture in an NADP+ cycling buffer (100 μL) and NADPH developer (10 μL) for 1 h at room temperature. Absorbance values were measured at 450 nm using a microplate reader (BioTek). The detection of NADPH was measured by heating lysates at 60°C for 30 min to decompose NADP+.
Intracellular glutathione assay
Reduced (GSH) and oxidized glutathione (GSSG) concentrations were determined by using a GSH colorimetric assay kit (BioVision, K261) according to the manufacturer’s instructions. HL-1 cells were uninduced or induced with different physiological stress conditions (GD, hypoxia, OGD, OGD/R). Lysates were incubated with glutathione reductase (20 μL), an NADPH generating solution (20 μL), and a glutathione reaction buffer (120 μL) for 10 min at room temperature. Absorbance values were obtained at 405 nm using a microplate reader (BioTek). Similarly, the reduced form of glutathione was measured without incubation of glutathione reductase in the lysates.
Cell viability assay
Trypan blue assay was used to determine cell viability. HL-1 cells were uninduced or induced with different physiological stress conditions (GD, OGD, OGD/R). Cells were detached by using 0.05% trypsin and diluted with a culture medium. The medium containing cells (100 μL) was mixed with trypan blue (100 μL, 0.4%). The mixture (20 μL) was loaded onto the slide and analyzed by TC 20 automated cell counter (Biorad) to determine the percentage of viable cells.
Pull down analysis for glutathionylated proteins
Proteins (1 mg) were subjected to click reaction with biotin-DADPS-alkyne, as described above. Proteins were precipitated by adding ice-cold acetone (4x volume). After centrifugation, the resulting pellet was resuspended in PBS containing 1.2% SDS (100 μL) by sonication. The resuspended proteins were added to streptavidin agarose beads (25 μL bead volume) (Pierce) in 1× PBS (500 μL). Beads were incubated overnight at 4°C and washed with 1× PBS containing 0.2% SDS, followed by 1× PBS. Proteins were eluted with SDS-loading buffer, resolved on SDS-PAGE, and transferred to the PVDF membrane. The membrane was blocked with 5% dry milk in TBST and incubated at 4°C overnight with primary antibody solution, including antibody against desmin (Abcam, ab32362, 1:1000), BAG3 (Novus Biologicals, NBP2–27398, 1:1000), and FLAG (Sigma, F1804, 1:1000). The membrane was then incubated with a secondary antibody conjugated with HRP, and proteins were visualized by chemiluminescence.
Proteomic sample preparation
Two cohorts of HL-1 cells expressing GS M4 were incubated with heavy or light azido-Ala (0.6 mM), and lysates were prepared after indicated stimulus, as described above. Two cohorts of lysates prepared with heavy or light azido-Ala (5 mg of proteins from each lysate) were combined and precipitated by adding cold acetone (4x volume). The supernatant was removed after centrifugation at 13,000 rpm for 10 min. Pellet was air-dried for 5 min and resuspended in a buffer (1440 μL) containing 1× PBS and 0.1% SDS by sonication. The resuspended solution was mixed with 10 mM biotin-DADPS-alkyne in DMSO (160 μL) and a click mix (400 μL) [20 mM CuBr in DMSO/tBuOH (3:1, v/v) (200 μL) and 20 mM THPTA (200 μL)]. The mixture was incubated for 1 h at room temperature in the dark. Proteins were precipitated by adding cold acetone (4x volume). The supernatant was removed after centrifugation at 14,000 rpm for 10 min. Pellet was resuspended in 1× PBS (1 mL) containing 1.2% SDS by sonication at room temperature. The resuspended solution was diluted with 1× PBS (5 mL) containing streptavidin-agarose beads (100 μL bead volume) and incubated overnight at 4°C. Proteins on beads were washed with 1× PBS containing 0.2% SDS, 1× PBS (3 times each), and incubated in a denaturation PBS solution (0.5 mL) containing 6 M urea for 45 min at 37°C. Proteins on beads were incubated with a digestion buffer (200 μL) containing 2 M urea, 1 mM CaCl2, and Trypsin/Lys-C (5 μg) overnight at 37°C. The beads were then washed with 1× PBS containing 0.2% SDS, 1× PBS, and water (3 times each). Peptides on beads were eluted by incubating with 10% formic acid (100 μL × 2) for 10 min, followed by a quick wash with 10% formic acid (100 μL). Eluates were combined, lyophilized, and subjected to LC-MS/MS analysis.
LC-MS/MS analysis
Lyophilized peptides were resuspended in 5% acetonitrile, 0.1% formic acid, and 0.005% trifluoroacetic acid. Peptides were then separated by UHPLC reverse phase chromatography using PepMap RSLC 18 columns and an EASY-nLC 1000 liquid chromatography system and introduced into an Orbitrap Fusion mass spectrometer (Thermo Fisher). MS1 scans were between 375–1600 m/z and at 240,000 orbitrap resolution. For MS2 scans, peptides with +2 and +3 charges were fragmented by collision-induced dissociation (CID) at 32% collision energy. Peptides with charges +3 to +7 were fragmented by electron transfer dissociation (ETD) with calibrated charge-dependent ETD parameters. The cycle time was set to 2.5 seconds over the 90 min gradient. Dynamic Exclusion was turned on.
Data analysis
Raw data files were analyzed in MaxQuant (version 1.6.2.10) against the Uniprot mouse database (downloaded on 2017-07-14, 16844 entries) and a contaminant database. N-terminal acetylation and methionine oxidation were set as variable modifications. Modifications included S-glutathionylation of cysteine with light azido-glutathione (addition of C16H24N6O7S, 444.14272 Da) or heavy azido-glutathione (addition of C16H212H3N515NO7S, 448.15858 Da). All other parameters were left at default values. As determined by reverse database search, peptide spectral matches were accepted at a 1% false discovery rate. Peptides were quantified using Skyline software (version 20.2). Spectral libraries were built by importing all msms.txt files to Skyline from MaxQuant. FASTA database (Uniprot mouse) and raw files were imported to Skyline for peak picking. In MS1-filtering, mass accuracy was set to 10 ppm. The retention time window (±2.0 min) was used to find the corresponding peptide peaks in all runs lacking MS/MS identification. Peptides were filtered to remove peptides without glutathionylation. Individual peptide peaks were inspected manually for accurate peak picking for the top three isotope peaks in the chromatographic traces. When necessary, manual integration was applied to have isotope dot product (idotp) scores higher than 0.8. Peptides with an idotp score lower than 0.8 were removed. The ratios of peptide areas of heavy to light labels were calculated automatically. From three biological replicate data, peptides identified more than two times with an idotp score higher than 0.8 were assigned with the median RH/L values.
Bioinformatic analysis
STRING analysis was performed using Cytoscape software. Identified mouse proteins containing glutathionylated cysteines with RH/L values above 2 were converted to human equivalents. The list of human proteins was loaded to STRING program as ‘glutathionylation’ network. For identification of cardiomyopathy-associated proteins, the ‘glutathionylation’ network was merged with the ‘cardiomyopathy’ network containing cardiomyopathy proteins (cutoff value of 0.4) loaded from the STRING disease search. The merged network was then subjected to CLUSTER MAKER analysis using MCL clustering with a granularity parameter of 4 and array sources from the score. For identification of mitochondrial and sarcomere-associated proteins, the list of ‘glutathionylation’ network was analyzed in DAVID GO analysis. Similarly, the selected ‘mitochondrial’ and ‘sarcomere associated’ proteins were clustered, as described above. The disease relevance was analyzed from the UniProt database (feature viewer and variants) and polyphen-2 program.
Results
Glutathionylation is significantly induced during glucose deprivation but weakly in hypoxia or OGD
In vitro models using specific cell types with hypoxia or OGD have proven useful for reproducing phenotype responses to ischemia reperfusion (I/R).1 To examine the response of glutathionylation in cardiomyocytes, we applied a clickable glutathione approach to the HL-1 mouse cardiomyocyte cell line, which retains contractility with the major cardiac phenotypes of adult cardiomyocytes.36 After the expression of GS M4, HL-1 cells were incubated with azido-Ala and subjected to various conditions that mimic glucose deprivation, hypoxia, or OGD. Notably, glucose depletion induced a global level of glutathionylation in concentration- and time-dependent manners (Figure 2A). Decreasing concentrations of glucose led to increasing levels of glutathionylation, where the longer depletion of glucose induced a higher level of global glutathionylation (Figure 2A). This observation was also seen in HEK293 cells (Supplementary Figure 1A) and our previous reports,35, 37 where glucose availability was important to maintain the redox homeostasis, potentially due to its role in NADPH production.38 Indeed, the ratio of NADP+/NADPH was significantly increased under glucose deprivation (GD) (Figure 2E). In addition, the level of ROS, detected by a mitochondrial ROS probe (MitoSox), and the ratio of GSSG/GSH were both increased (Figure 2D and 2F), supporting the induction of global glutathionylation under GD.
Figure 2. Global protein glutathionylation is significantly induced upon glucose deprivation (GD) but relatively weak in hypoxia or oxygen-glucose deprivation (OGD).
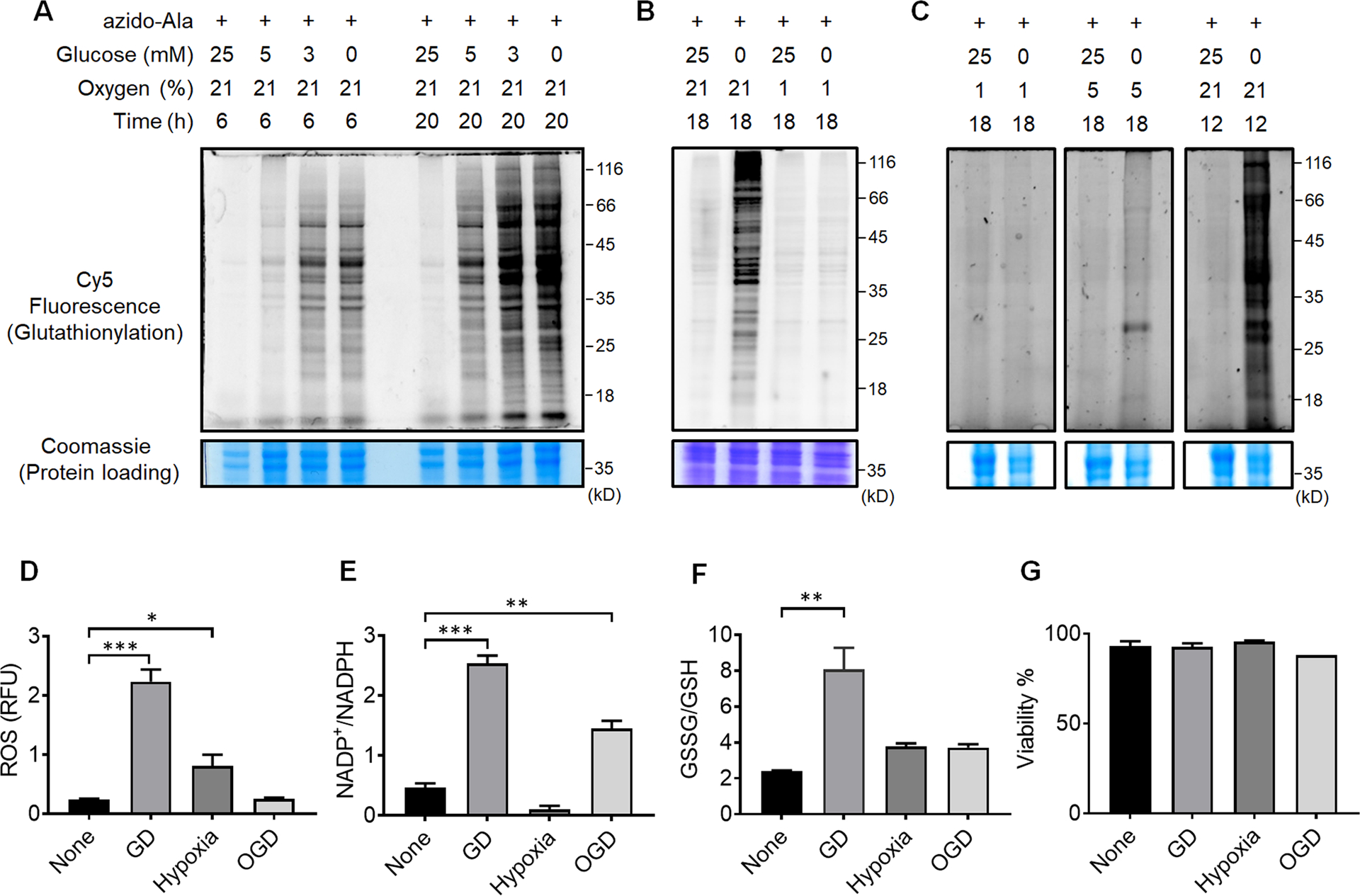
(A-C) Analysis of protein glutathionylation under GD, hypoxia, or OGD. HL-1 cells expressing GS M4 were incubated with azido-Ala. Cells were then subjected to decreasing concentrations of glucose (0–25 mM) under normoxia (A), glucose deprivation in comparison to hypoxia and OGD (B), or glucose deprivation in different percentages of oxygen (1, 5, 21%) (C). After the stimulus, cells were lysed and subjected to click reaction with Cy5-alkyne and analyzed for fluorescence (glutathionylation level) and Coomassie stains (protein loading control). Data are representative of at least 3 independent experiments. (D-G) Analysis of redox environment under GD, hypoxia, or OGD. HL-1 cells were independently analyzed, without using the clickable glutathione approach, for ROS (D), a ratio of NADP+ over NADPH (E), a ratio of oxidized glutathione (GSSG) over reduced glutathione (GSH) (F), and viability (G) after subjecting cells in GD, hypoxia, or OGD for 18 h. Data represent the mean ± SD, n = 3 independent experiments. Difference is significant by one-way ANOVA followed by Tukey’s post-hoc test, *p < 0.05, **p < 0.01, ***p < 0.001.
Next, HL-1 cells were subjected to hypoxia (1% O2) or OGD (1% O2 and no glucose) (Figure 2B). Interestingly, the level of glutathionylation was largely absent under hypoxia (Figure 2B, lane 1 vs. 3). Moreover, glutathionylation was significantly low or rarely observed under OGD (Figure 2B, lane 1 vs. 4) as opposed to under GD (Figure 2B, lane 2). A similar pattern was observed in HEK293 cells (Supplementary Figure 1A), supporting that the low level of oxygen (1% O2) in hypoxia or OGD induces no or low levels of glutathionylation, and the ambient level of oxygen is necessary for global induction of glutathionylation. To this agreement, in hypoxia, the ROS level was increased (Figure 2D), but the ratio of NADP+/NADPH was largely unchanged (Figure 2E), possibly due to glucose availability. In contrast, under OGD (1% O2), the ratio of NADP+/NADPH was increased (Figure 2E), but the ROS level was unchanged (Figure 2D). Consequently, neither hypoxia nor OGD (1% O2) was able to significantly alter the ratio of GSSG/GSH (Figure 2F), which supports no or low level of global glutathionylation under hypoxia and OGD (Figure 2B). Consistently, the increasing percentage of oxygen availability (1, 5, and 21%) during GD resulted in higher levels of glutathionylation (Figure 2C and Supplementary Figure 1B), further supporting the importance of oxygen availability during glutathionylation. Lastly, the cell viability was not significantly altered during the indicated conditions of glucose deprivation, hypoxia, or OGD (Figure 2G).
Glutathionylation is significantly induced by palmitate availability during OGD/R
With a low level of glutathionylation under OGD, we next examined glutathionylation under repletion of oxygen or glucose after OGD (OGD/R). After OGD (1% O2), HL-1 cells were re-oxygenated with or without glucose. Upon reoxygenation, the level of glutathionylation was significantly increased in the absence of glucose (OGD/O, Fig. 3A, lane 1 vs. 2–3, and Figure 3B, lane 1 vs. 2), but remained low with glucose repletion (OGD/OG, Fig. 3A, lane 1 vs. 4–5, and Figure 3B, lane 1 vs. 3). A similar pattern was observed in HEK293 cells during OGD/R (Supplementary Figure 2). These data further support that oxygen needs to be available to induce a high level of glutathionylation, whereas glucose metabolism is used to restore redox homeostasis. Consistently, the ROS level (Figure 3D), NADP+/NADPH (Figure 3E), and GSSG/GSH (Figure 3F) were all significantly elevated upon reoxygenation without glucose (OGD/O) versus with glucose repletion (OGD/OG).
Figure 3. Global protein glutathionylation increases upon the addition of palmitate during repletion of glucose and oxygen after OGD (OGD/R).
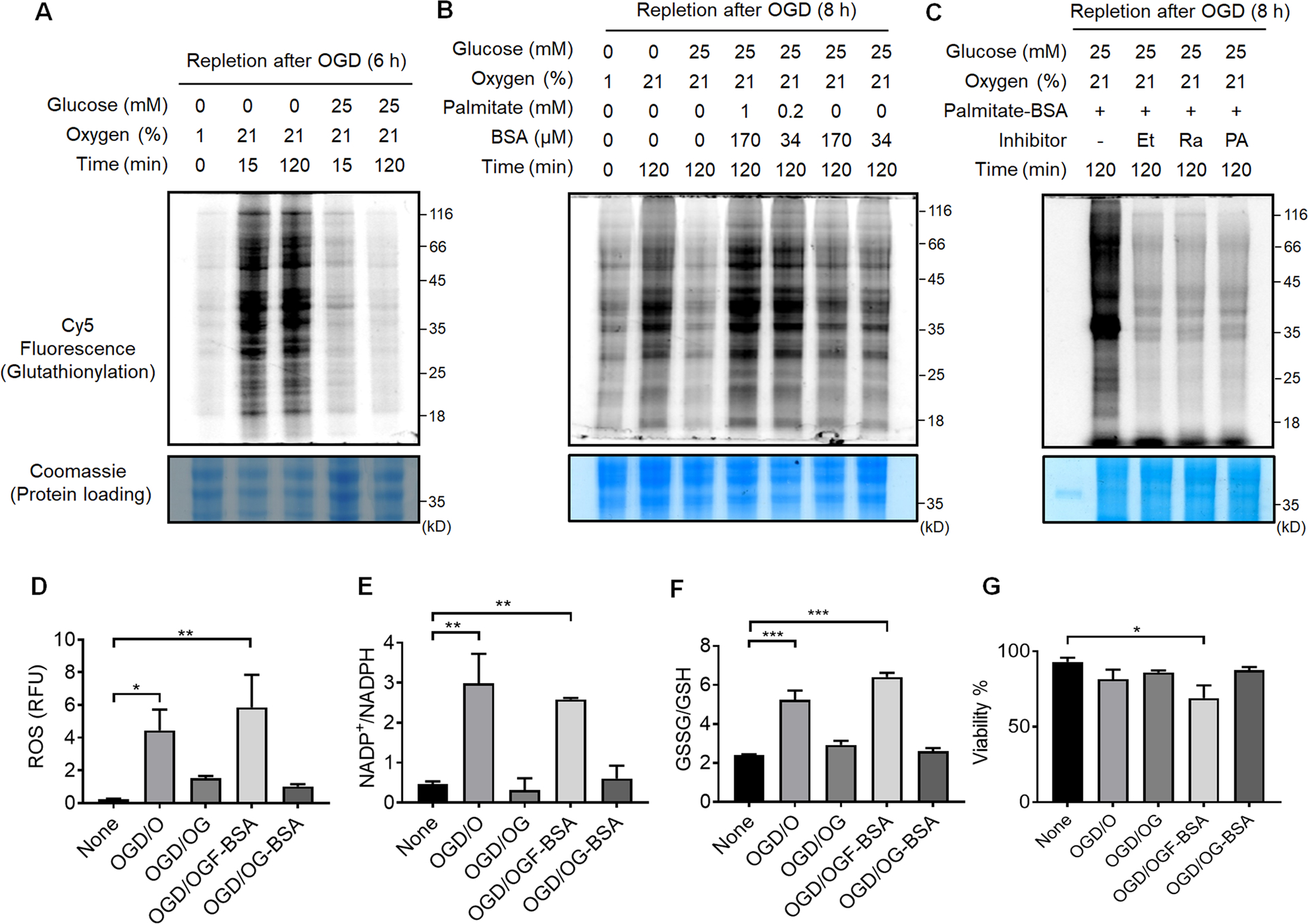
(A-C) In-gel fluorescence detection of global levels of glutathionylation during repletion of glucose or oxygen after OGD (A), repletion of palmitate after OGD (B), or co-addition of fatty acid oxidation inhibitors during OGD/R (C). HL-1/GS M4 cells incubated with azido-Ala were subjected to the indicated conditions after OGD (1% O2 and no glucose) in a hypoxic chamber, or co-treated with Etomoxir (Et), Ranolazine (Ra), or 4-pentenoic acid (PA) in the presence of palmitate (1 mM) and BSA (170 μM). Cells were then lysed and analyzed by in-gel fluorescence (glutathionylation level) or Coomassie stains (protein loading control) after click reaction with Cy5-alkyne. Data are representative of at least 3 independent experiments. (D-G) Analysis of redox environment during OGD/R. HL-1 cells were analyzed for ROS (D), a ratio of NADP+ over NADPH (E), a ratio of oxidized glutathione (GSSG) over reduced glutathione (GSH) (F), and viability (G) after repletion of glucose, oxygen, or palmitate for 2 h, following OGD for 8 h. Data represent the mean ± SD, n = 3 independent experiments. Difference is significant by one-way ANOVA followed by Tukey’s post-hoc test, *p < 0.05, **p < 0.01, ***p < 0.001.
It has been previously shown that the plasma level of fatty acids is elevated to high concentrations (e.g., 1–3 mM) after ischemia,9 and the fatty acids in the perfused heart inhibit glucose (pyruvate) oxidation and increase I/R injury.10–12 Therefore, HL-1 cells were subjected to the addition of palmitate, a major fatty acid, with its carrier protein BSA, during OGD/R. The level of glutathionylation was significantly increased upon addition of palmitate, together with reoxygenation and glucose repletion (OGD/OGF-BSA) (Figure 3B, lane 4–5) versus without palmitate (OGD/OG or OGD/OG-BSA) (Figure 3B, lane 3 and 6–7). Consistently, the ROS level (Figure 3D), NADP+/NADPH (Figure 3E), and GSSG/GSH (Figure 3F) were significantly higher upon addition of palmitate (OGD/OGF-BSA) versus without palmitate (OGD/OG-BSA or OGD/OG). Notably, the viability was reduced upon the addition of palmitate versus all other conditions (Figure 3G). These data are reminiscent of the adverse effect of fatty acids upon addition to perfused heart,12–13 while showing that glutathionylation is intensified upon the addition of fatty acid during OGD/R. To support the significance of fatty acid availability or oxidation for induction of glutathionylation, several fatty acid oxidation inhibitors were introduced together with palmitate during OGD/R, including Etomoxir (Et, carnitine palmitoyl-transferase inhibitor39), Ranolazine (Ra, Na-K pump inhibitor40–41), and 4-pentenoic acid (PA, thiolase inhibitor42). Notably, the level of glutathionylation was significantly reduced upon the addition of individual inhibitors (Figure 3C), supporting that fatty acid oxidation or availability is a crucial factor that contributes to the induction of glutathionylation during OGD/R.
Identification and quantitative analysis of glutathionylated cysteines upon addition of palmitate
Having seen that palmitate availability or oxidation is an important factor that induces a global level of glutathionylation, we sought to identify specific cysteines susceptible to glutathionylation upon addition of palmitate during OGD/R by using an isotopically labeled clickable glutathione approach.34 In this approach, an isotopically labeled light or heavy derivative of azido-Ala was introduced to HL-1 cells expressing a mutant of glutathione synthetase, GS M4 (Figure 4A and Supplementary Figure 3). The GS M4 uses light or heavy azido-Ala as a substrate, which results in the biosynthesis of isotopically labeled clickable glutathione with +4 Da mass difference (Supplementary Figure 3).34 After being subjected to OGD/R with and without palmitate (steps 1 & 2 in Figure 4A), two cohorts of cells labeled with heavy or light azido-Ala were lysed and combined. Glutathionylated proteins in lysates were conjugated with biotin-DADPS-alkyne43 by click reaction and bound to streptavidin beads. After on-bead trypsin digestion, glutathionylated peptides were eluted by the acidic cleavage of the DADPS linker and analyzed by LC-MS/MS (Figure 4A). Individual pairs of glutathionylated peptides labeled with light or heavy derivatives of azido-Ala (+4 Da mass difference) were identified (minimum 2 out of 3 biological replicates). Subsequently, MS1 peak intensity ratios of heavy- and light-labeled peptides were calculated to determine RH/L values, which represent the relative levels of glutathionylation on cysteines with versus without the addition of palmitate during OGD/R (Figure 4A).
Figure 4. Identification and quantification of glutathionylated cysteines upon addition of palmitate.
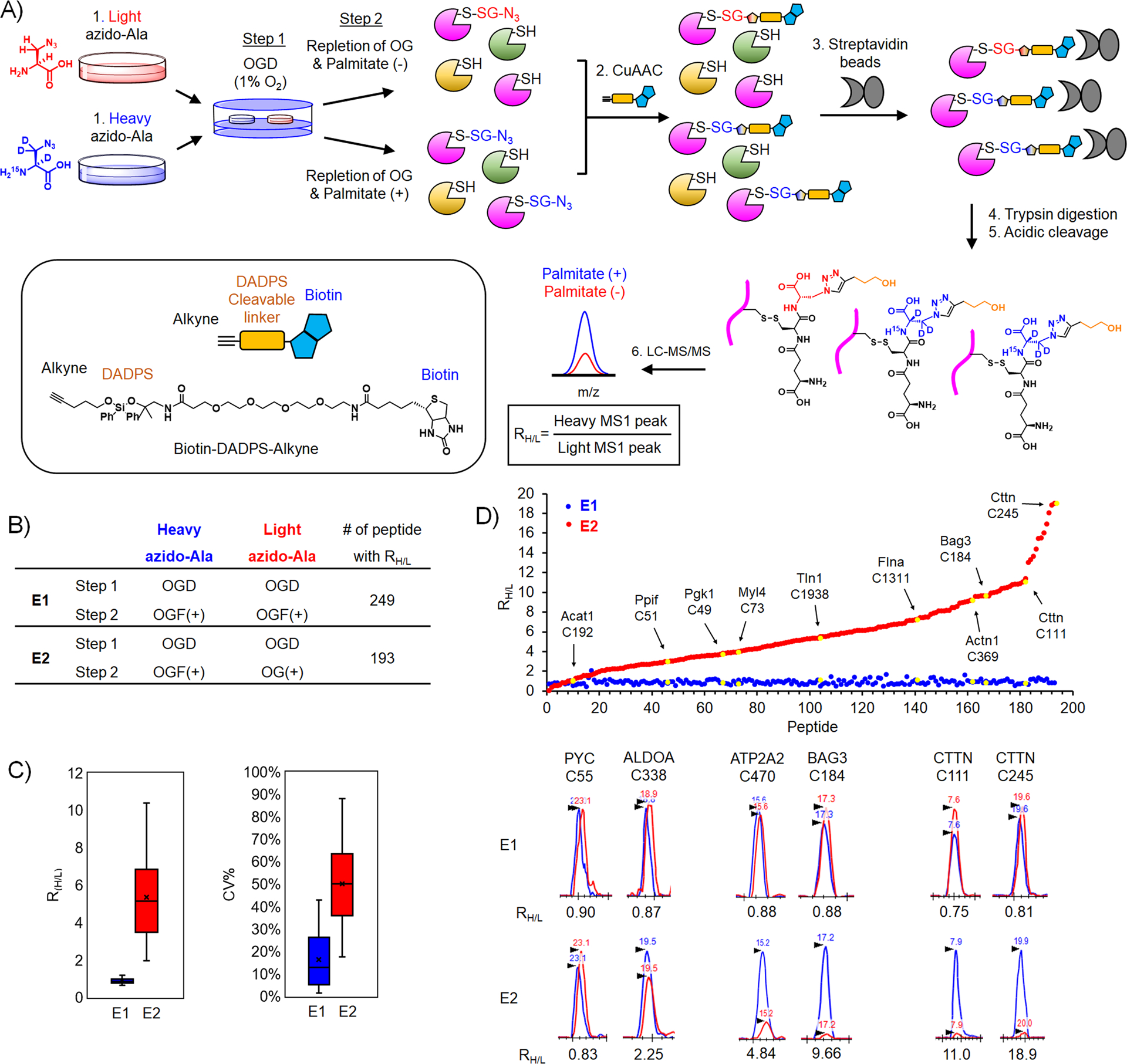
(A) Isotopically labeled clickable glutathione approach to identify and quantify levels of glutathionylation. After incubation of light or heavy azido-Ala, two cohorts of HL-1 cells expressing GS M4 were subjected to OGD for 8 h (step 1), followed by repletion of glucose and oxygen (OG) without or with palmitate for 2 h (step 2). Lysates collected were combined and processed for click reactions with biotin-DADPS-alkyne, pull-down with streptavidin beads, on-bead tryptic digestion, and elution of glutathionylated peptides by acidic cleavage of DADPS linker, followed by LC-MS/MS to determine an MS1-peak area ratio (RH/L) of heavy- to light-labeled peptides. (B) Two experimental conditions [E1: OGD, followed by repletion of oxygen, glucose, and palmitate (OGF) for both heavy and light isotopes. E2: OGD, followed by OGF for heavy isotope or repletion of oxygen and glucose only (OG) for light isotope] and the number of quantified glutathionylated peptides. (C) Plots of RH/L values and the coefficient of variation (CV) of glutathionylated peptides. Box plots are shown with the median value (line), box (25–75%), and whiskers (10–90%). (D) RH/L values of individual glutathionylated peptides. Distribution of RH/L values (top). Examples of identified proteins with their cysteine residues are shown by the arrow and yellow color. Graphical display of MS1-peaks showing relative quantification of heavy (blue)- to light (red)-labeled peptides (bottom).
To demonstrate validation of our quantitative strategy, we have analyzed two sets of experiments. In the E1 experiment, two cohorts of HL-1 cells were subjected to an identical OGD/R condition with the addition of palmitate (Figure 4B, E1 experiment = OGD/OGF-BSA for both light and heavy), which is expected to induce a global level of glutathionylation and give RH/L values close to 1. On the other hand, in the E2 experiment, two cohorts of cells were subjected to two different OGD/R conditions (E2 experiment = OGD/OGF-BSA and OGD/OG for heavy and light, respectively), which could be compared to quantify glutathionylated cysteines resulting from palmitate availability. In the E1 and E2 experiments, 249 and 194 glutathionylated peptides labeled with both heavy and light isotopes were identified with their RH/L values (Supplementary Table 1). Notably, the median RH/L values in E1 is 0.87, supporting relatively equal intensity of heavy to light-labeled peptides (90% of RH/L values in a range of 0.6–1.4) (Figure 4C–D and Supplementary Figure 4). In contrast, the median RH/L values in E2 is 5.15 (Figure 4C–D), supporting a higher level of glutathionylation in the presence of palmitate (heavy) versus its absence (light). The coefficient of variation (CV) in E1 showed the median value of 14.6% (80% of CV in a range of 1–43%), supporting the relatively consistent RH/L values among triplicate experiments. In the E2 experiment, the median value of CV is 50.2% (80% of CV in a range of 17–88%). The relatively higher CV values for E2 versus E1 mostly resulted from low signals of glutathionylation detected in light-labeled peptides. Examination of RH/L values and MS peaks for individual peptides in the E2 experiment showed a broad range of RH/L values upon addition of palmitate (Figure 4D). For example, PYC C55 and ALDOA C338 have relatively low RH/L values, 0.83 and 2.25, respectively (Figure 4D). On the other hand, BAG3 C184, CTTN C111, and CTTN C245 have high RH/L values, 9.66, 11.0, and 18.9, respectively (Figure 4D), suggesting their potentially differential susceptibility to glutathionylation in response to palmitate availability.
Bioinformatic analysis identifies glutathionylated cysteines associated with sarcomere, mitochondria, and cardiomyopathy
All identified proteins (n = 248) were examined for their localizations in DAVID gene ontology (GO) analysis, finding that a high number of proteins are involved in the cytoplasm (62.5%) in addition to the membrane (51.6%), nucleus (43.5%), mitochondria (22.2%), and endoplasmic reticulum (9.6%) (Supplementary Figure 5). Next, we sought to examine the major clusters of protein networks impacted by glutathionylation. The STRING and clustering analysis44 found a large cluster of proteins in cytoskeleton organization & remodeling with their median RH/L values of 6.0 (Figure 5A, left). Glutathionylated proteins in this cluster (listed with RH/L in E2) include GTPase for actin filament remodeling [RAC1 C178 (RH/L 5.33), RHOA C16 (RH/L 5.13)], G protein subunits [GNB2 C182 (RH/L 9.18) and GNB1 C204 (RH/L 5.50)], myosin motor proteins [MYH9 C988 (RH/L 9.62) and MYL6 C2 (RH/L 9.05)], actin-binding and structural constituent [CAPZB C206 (RH/L 7.17), SPTAN1 C315, C1622 (RH/L 4.96, 5.14), TLN1 C1087, C1939 (RH/L 1.55 and 5.36)], suggesting that glutathionylation may alter dynamics of cytoskeletal structural organization (via modulation of GTPase activity or modification of structural proteins) that is essentially integrated with myofibrillar and sarcomeric contractility in muscle.45 In addition, a large cluster of proteins are implicated in translation with their median RH/L value of 6, including translation initiation and elongation factor [EIF3g C139 (RH/L 13.7), EEF2 C693 (RH/L 8.44)] and ribosomal proteins (Figure 5A, middle). An additional large cluster of proteins was found in carbohydrate metabolism with their median RH/L value of 6, as seen with glycolytic proteins [LDHA C163 (RH/L 9.67), PGK1 C50, C316 (RH/L 3.71, 7.52) and GAPDH C245, C22 (RH/L 6.14, 7.45)] (Figure 5A, right).
Figure 5. Bioinformatic analysis of glutathionylated proteins upon addition of palmitate.
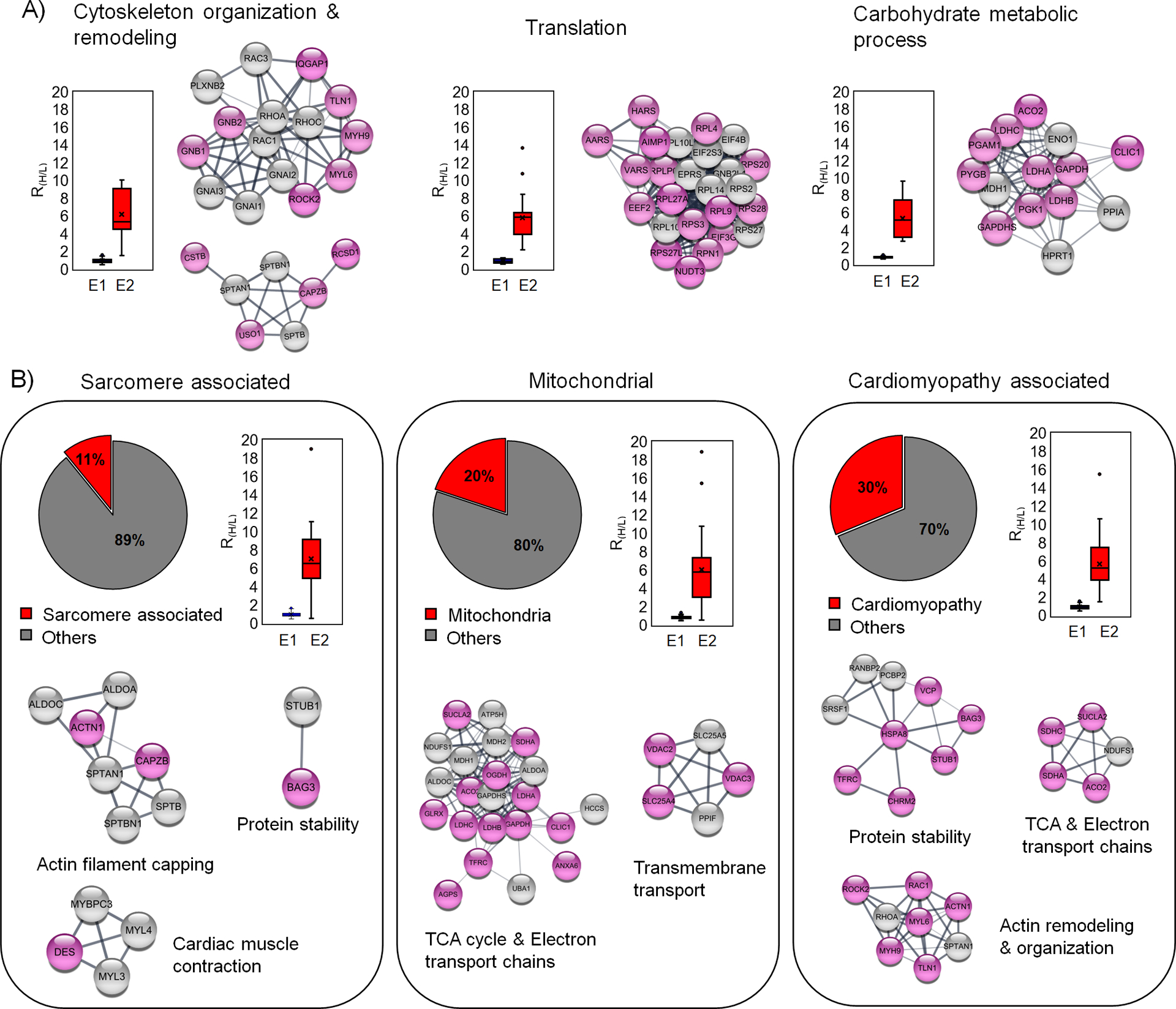
(A) The major biological process associated with the identified glutathionylated proteins. Each biological process is shown with a plot of RH/L values and selected clusters in STRING analysis. (B) Analysis of glutathionylated proteins in association with sarcomere, mitochondria, and cardiomyopathy. Among identified proteins, sarcomere-associated proteins (left) and mitochondrial proteins (middle) were identified from the DAVID GO database. Glutathionylated proteins associated with cardiomyopathy (right) were identified from cardiomyopathy disease query in STRING analysis. The identified proteins are shown after STRING and cluster analysis with plots of RH/L values. The names of clusters were assigned based on proteins in individual clusters after DAVID GO analysis. Glutathionylated proteins with low RH/L values (below 50% in the group) and high RH/L values [over 50% in the group and NL (no light but heavy label found)] are shown with white and pink colors in circles, respectively.
Next, we analyzed a group of sarcomeric or myofibrillar proteins and mitochondrial metabolic proteins because their oxidations under ischemic reperfusion have been found impactful to cardiac contractility.18, 28 We found 24 glutathionylated sarcomeric proteins (DAVID GO annotation, 11% among 221 proteins with RH/L value > 2, Supplementary Table 1) with their median RH/L values of 6.8 (Figure 5B, left). The STRING and clustering analysis found a few groups of clusters centered on contractile proteins [MYBPC3 C715 (RH/L 4.24), MYL3 C74 (RH/L 3.96)], sarcomeric structural proteins that interact with actin filaments [FLNC C1349, C713 (RH/L 6.52, 8.92), CAPZB C206 (RH/L 7.17), ACTN1 C370 (RH/L 9.17), SPTAN1 C315, 1622 (RH/L 4.92, 5.14)], chaperon and E3 ligase that regulate protein stability [BAG3 C185 (RH/L 9.66), STUB1 C200 (RH/L 5.66)] (Figure 5B, left, and Supplementary Figure 6A). Therefore, identification of these proteins with their RH/L values suggests the potential impact of glutathionylation on contractility or structural integrity of myofibrils upon ischemic stress.
In addition, 44 mitochondrial proteins were found (DAVID GO annotation, 20% among 221 proteins with RH/L value > 2, Supplementary Table 1) with their median RH/L values of 6.0 (Figure 5B, middle). The STRING and clustering analysis of mitochondrial proteins found a large group clustered with the electron transport chain (ETC) subunits and ATP synthase [SDHC C107 (RH/L 15.47), NDUFS1 C727 (RH/L 4.94), ATP5H C101 (RH/L 2.88)] and the tricarboxylic acid cycle (TCA) metabolic enzyme [OGDH C395 (RH/L 8.11), MDH2 C275 (RH/L 3.62)], PCCA C107 (RH/L 18.9)]. An additional cluster was found with ATP transport and ion channel [SLC25A4 C160 (RH/L 6.69), SLC25A5 C160 (RH/L 5.73), VDAC3 C65 (RH/L 7.13), VDAC2 C77, C48 (RH/L 6.10, 5.55)] (Figure 5B, middle, and Supplementary Figure 6B). Glutathionylation of these proteins may dampen ATP production and alter membrane permeability in mitochondria, which is implicated in the impairment of mitochondrial function after ischemic stress.46
Lastly, we compared our list of glutathionylated proteins (221 proteins with RH/L value > 2, ‘glutathionylation network’) with all proteins whose dysregulations are associated with cardiomyopathy (2000 proteins, ‘cardiomyopathy’ network) available in the STRING disease query, which identified 66 glutathionylated proteins associated with cardiomyopathy (30% among 221 proteins with RH/L value > 2, Supplementary Table 1) with their median RH/L values of 5.2 (Figure 5B, right). The STRING and clustering analysis found several similar clusters to sarcomere and mitochondrial proteins, such as clusters involved in protein stability with chaperone and E3 ligase [BAG3 C185 (RH/L 9.66), STUB1 C200 (RH/L 5.66),], ATP production with TCA and ETC [SDHC C107 (RH/L 15.5), NDUFS1 C727 (RH/L 4.94), ATP5H C101 (RH/L 2.88)], and cytoskeleton remodeling and organization [RAC1 C178 (RH/L 5.33), RHOA C16 (RH/L 5.13), MYH9 C988 (RH/L 9.62) and MYL6 C2 (RH/L 9.05)] (Figure 5B, right, and Supplementary Figure 6C), suggesting the potential implication of their glutathionylation in the etiology of cardiomyopathy. However, to better predict functional implications of glutathionylation on identified proteins, we examined all glutathionylated cysteines (n = 249, from mouse) whether variants of the identified cysteines or their flanking residues (in human orthologs) are linked to any muscular disorders in the UniProt database.47 We found mutations of 18 cysteines or their nearby residues (± 1 positions) in association with muscle dystrophy or neuromuscular diseases (Table 1). Notably, mutations of 7 cysteines (listed with RH/L values in E2) are linked to amyotrophic lateral sclerosis 1 (ALS1) [SOD1 C147R (RH/L 1.37)], autosomal recessive axonal neuropathy with neuromyotonia (NMAN) [HINT1 C84R (RH/L 2.18)], dilated cardiomyopathy [LMNA C522R (RH/L 4.71)], early infantile epileptic encephalopathy [Sptan1 C315R (RH/L 4.93)], mitochondrial complex II deficiency [SDHA C654R/S/G (RH/L NL)], hypertrophic cardiomyopathy [MYBPC3 C715R (RH/L 4.24)], or phosphoglycerate kinase 1 deficiency associated with hemolytic anemia or myopathy [(PGK1 C316R (RH/L 7.52)] (Table 1). These analyses imply that their glutathionylations may yield adverse functional outcomes associated with ischemic reperfusion.
Table 1.
A list of glutathionylated cysteines with variants of the cysteines or their neighboring residues (±1) implicated in diseases.
| Gene Name | Protein Name | Peptidea | RH/L | Natural Variantb | Disease Relevancec | |||
|---|---|---|---|---|---|---|---|---|
| E1 | E2 | Xn-1 | Cn | Yn+1 | ||||
| SOD1 | Superoxide dismutase [Cu-Zn] | LAC147GVIGIAQ | 0.70 | 1.37 | A→T/D | C→R | G→D/R | Amyotrophic lateral sclerosis 1 (ALS1) [#105400] |
| HINT1 | Histidine triad nucleotide-binding protein 1 | C84AADLGLK | 0.69 | 2.18 | C→R | A→V* | Autosomal recessive axonal neuropathy with neuromyotonia (NMAN) [#137200] | |
| PLOD3 | Procollagen-lysine,2-oxoglutarate 5-dioxygenase 3 | GVDYEGGGC694R | 0.91 | 3.45 | C→X | R→C* | Bone fragility with contractures, arterial rupture, and deafness [#612394] | |
| BAG3 | BAG family molecular chaperone regulator 3 | SQSPAASDC185SSSSSSASLPSSGR | 0.83 | 9.66 | C→stop | Dilated cardiomyopathy [#613881], myofibrillar myopathy [#612954] | ||
| LMNA | Prelamin-A/C; Lamin-A/C | AQNTWGC522GSSLR | 0.88 | 4.71 | C→R | G→R | Charcot-Marie-Tooth disease [#605588]. dilated cardiomyopathy [#115200], congenital muscular dystrophy [#613205] | |
| SPTAN1 | Spectrin alpha chain, non-erythrocytic 1 | ALC315AEADR | 1.01 | 4.93 | C→R | Early infantile epileptic encephalopathy [#613477] | ||
| PLOD1 | Procollagen-lysine,2-oxoglutarate 5-dioxygenase 1 | VGEDYEGGGC681R | 0.73 | 3.56 | G→V | R→W | Ehlers-Danlos syndrome, hydroxylysine-deficient [#225400] | |
| SUN2 | SUN domain-containing protein 2 | C577SETYETK | 1.09 | NL | R→stop | Emery-Dreifuss muscular dystrophy [#310300] | ||
| MYH6 | Myosin-6 | LEDEC949SELK | 0.44 | 1.58 | E→K | C→Y* | Familial hypertrophic cardiomyopathy 14 [#613251] | |
| SDHA | Succinate dehydrogenase [ubiquinone] flavoprotein subunit | TLNEADC654ATVPPAIR | 0.67 | NL | C→R/S/G | Mitochondrial complex II deficiency [#252011] | ||
| MYBPC3 | Myosin-binding protein C, cardiac-type | LLC715ETEGR | 1.36 | 4.24 | L→P | C→R | Hypertrophic cardiomyopathy [#115197] | |
| HCCS | Cytochrome c-type heme lyase | AYDYVEC70PVTGAR | 0.57 | 3.10 | P→T | Linear skin defects with multiple congenital anomalies 1 [#309801] | ||
| DES | Desmin | HQIQSYTC332EIDALK | 0.83 | NL | T→I | C→R* | E→K | myofibrillar myopathy [#601419] |
| FLNC | Filamin-C | LYAQDADGC713PIDIK | 0.86 | 8.92 | G→S* | C→W* | Hypertrophic cardiomyopathy [#617047], myofibrillar myopathy [609524] | |
| PGK1 | Phosphoglycerate kinase 1 | TGQATVASGIPAGWMGLDC316GTESSK | 0.82 | 7.52 | D→N | C→R | Phosphoglycerate kinase 1 deficiency [#300653] | |
| PCCA | Propionyl-CoA carboxylase alpha chain, mitochondrial | MADEAVC107VGPAPTSK | 0.83 | 18.9 | V→L | C→S* | C→X | Propionic acidemia [#606054] |
| SLC39A13 | Zinc transporter ZIP13 | SLGAAAAC46R | 0.68 | 0.62 | C→Y* | R→C | Spondylocheirodysplasia, Ehlers-Danlos syndrome-like [#612350] | |
| CSTB | Cystatin-B | MMC3GAPSATMPATAETQEVADQVK | 1.46 | 7.09 | C→W* | G→W/R | Unverricht-Lundborg syndrome [#254800] | |
All cysteine numbers are from mouse.
Variants are found from the UniProt database and shown with the residues before and after mutation indicated by arrows.
Diseases associated with indicated variants were found from UniProt database47 and shown with phenotype-MIM numbers.63 The symbol (*) indicates variants annotated and predicted as ‘probably damaging’ in UniProt and PolyPhen-2 prediction tool.64 Stop, stop codon. X, frameshift. NL = no light detected and found heavy isotope only.
Desmin and BAG3 are glutathionylated upon addition of palmitate
Among identified cysteines, we sought to validate glutathionylation of cysteines in two proteins, desmin and Bcl2-related athanogene 3 (BAG3), in response to palmitate availability (i.e., OGD/OGF vs. OGD/OG). Desmin is a muscle-specific intermediate filament associated with Z-disks in sarcomeres.48 Desmin is essential for the integrity of myofibrils, and its mutations are linked to myofibrillar myopathy (desminopathy).49 Desmin has only one cysteine, C332, and we have previously validated its glutathionylation in response to hydrogen peroxide (H2O2).34 BAG3 is a multi-functional abundant co-chaperone forming a complex with Hsc/Hsp70,50 which plays an important role in protein quality control in muscle via its activity in autophagy, anti-apoptosis, and stabilization of myofibrillar integrity.50–51 Under stress, BAG3 shifts to Z-discs of the sarcomere, where it stabilizes actin capping protein (CapzB).51 BAG3 mutations with functional loss are linked to impaired Z-disc integrity,52 cardiomyopathy,52 and myofibrillar myopathy.53 BAG3 has four cysteines (C154, C185, C295, and C378 in mouse), among which we identified glutathionylation of C185 (C179 in human). To confirm glutathionylation of desmin and BAG3, HL-1 cells expressing GS M4 were subjected to OGD/OGF versus OGD/OG. After click reactions with biotin-alkyne and pull-downs of glutathionylated proteins, western blot analysis found that repletion of glucose and oxygen after OGD (i.e., OGD/OG) did not increase levels of glutathionylation of desmin (Figure 6A, lane 1 vs 2) and BAG3 (Figure 6B, lane 1 vs 2). However, addition of palmitate during repletion (i.e. OGD/OGF) induced high levels of glutathionylation of both desmin (Figure 6A, lane 1 vs. 3) and BAG3 (Figure 6B, lane 1 vs. 3), confirming that addition of palmitate is responsible for glutathionylation of both desmin and BAG3. To determine glutathionylation at the specific cysteines, cysteine mutants of mouse desmin (Des C332S) and human BAG3 (BAG3 C179S), along with their wild types (WT), were produced and expressed to HEK293/GS M4. Similarly, desmin WT (Figure 6A, lane 4 and 5 vs. 6) and BAG3 WT (Figure 6B, lane 4 and 5 vs. 6) showed their glutathionylations only upon addition of palmitate during repletion (OGD/OGF vs. OGD/OG). Importantly, both desmin C332S and BAG3 C179S showed no or low level of glutathionylation even in the presence of palmitate during repletion (OGD/OGF) as opposed to their WT (Figure 6A–B, lane 6 vs 9). These data confirm glutathionylation of their specific cysteines during OGD/R.
Figure 6. Desmin and BAG3 are glutathionylated upon addition of palmitate during OGD/R.
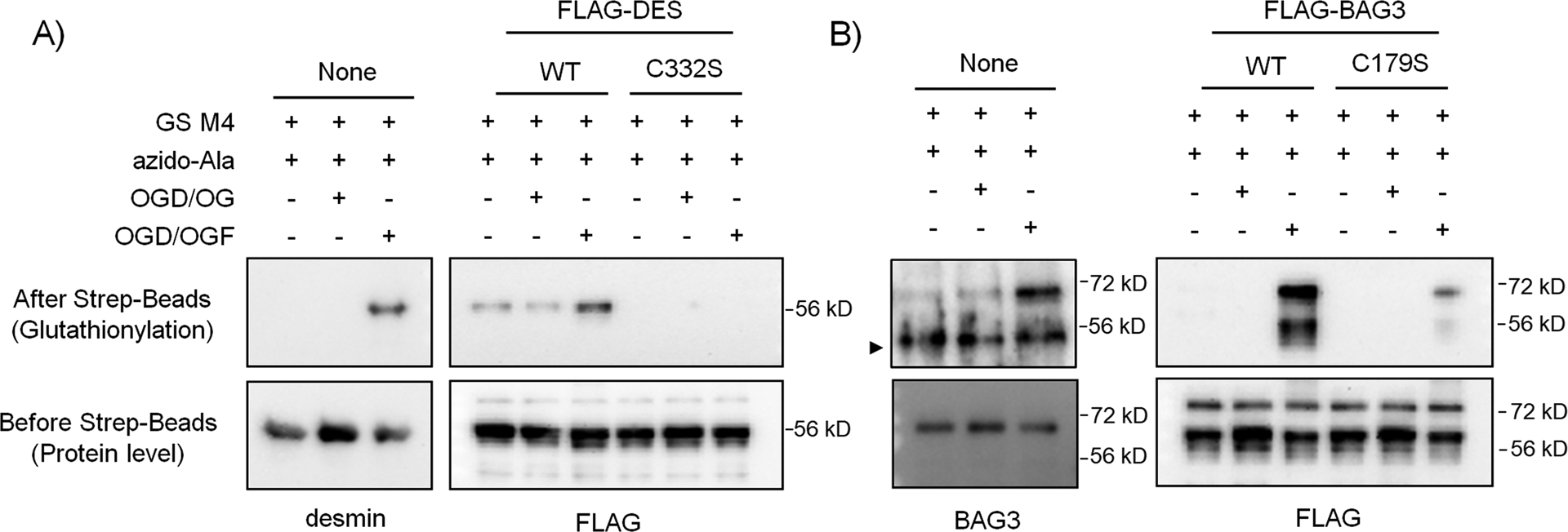
(A) Glutathionylation of desmin C332. (B) Glutathionylation of BAG3 C179. HL-1 cells expressing GS M4 were used without transfection. Alternatively, HEK293/GS M4 cells were transfected with wild-type or cysteine mutants of desmin or BAG3. Cells were then subjected to OGD for 8 h, followed by the addition of oxygen, glucose, and palmitate with BSA (OGD/OGF) or oxygen-glucose only (OGD/OG) for 2 h. Glutathionylated proteins were processed for click reaction with biotin-alkyne and pull-downs with streptavidin beads and analyzed by western blotting with individual antibodies for desmin, BAG3, and FLAG. Data are representative of 2 independent experiments. A symbol (►) indicates a non-specific band. Two bands in the FLAG-BAG3 blot appear to result from two BAG3 forms.62
Discussion
The mitochondrial ROS during ischemic reperfusion is well-known to cause muscle damages, presumably via oxidative modifications of many muscle proteins.18 In this report, we have monitored a global level of glutathionylation in response to metabolic alterations, such as depletion of glucose and oxygen, or upon reoxygenation with repletion of glucose and fatty acid (palmitate), which has been commonly used as a cellular model for ischemia and ischemic reperfusion injury.1 It is interesting to note that a global level of glutathionylation is apparent under glucose depletion in normoxia (ambient O2), whereas comparably low or absent in hypoxia (1% O2) and OGD (Figure 2). Overall, it suggests that despite the adverse roles of hypoxia and OGD in muscle, molecular mechanisms behind their pathological outcomes may not be significantly attributed to global induction of protein glutathionylation, at least under current experimental conditions. On the other hand, reoxygenation after OGD is a significant inducer for glutathionylation, but the level of glutathionylation decreases when glucose is provided (Figure 3A), supporting the important role of glucose in providing NADPH and restoring redox homeostasis. However, the addition of palmitate during reoxygenation, even in the presence of glucose, resulted in a high level of glutathionylation (Figure 3B). Additions of fatty acid oxidation inhibitors (Et, Ra, PA) decreased levels of glutathionylation (Figure 3C). This agrees with reports that enhancing glycolysis over mitochondrial beta-oxidation or blocking beta-oxidation relieves detrimental effects of reperfusion injury.14–16 Accordingly, a decreased level of viability was more apparent in the presence of palmitate than its absence. Our data are consistent with a previous report that repletion of palmitate to the ischemic heart aggravates the infarcted tissue, showing the significance of fatty acid (or palmitate) during ischemic reperfusion injury.13 However, it is important to note the limitations of the current observations. The current analyses have been made in an in-vitro model of OGD/R. While an OGD/R model has reproduced similar outcomes to the in vivo system,1 it uses artificial amounts of glucose (0–25 mM) and oxygen (1–21%) in a culture system to which HL-1 cells have adapted for growth and metabolic activity. For example, a high concentration of glucose (25 mM) used in this cell line may be an adapted metabolic condition to maintain its redox homeostasis. The exact oxygen concentration available to cells in in-vitro models varies depending on the culture system.54 Therefore, the amounts of nutrients or oxygen that elicit the observed effects in our in-vitro condition could differ from those in the in-vivo system.
Considering the significance of fatty acid and its plasma availability during reperfusion,9 we identified glutathionylated cysteines upon addition of palmitate versus its absence during repletion of oxygen and glucose (OGD/OGF vs. OGD/OG). Using our isotopically labeled clickable glutathione with mass spectrometric analysis, we identified 221 cysteines with RH/L values higher than 2. It is important to point out that this approach identifies peptides conjugated with glutathione derivative on it,33–34 thus unequivocally specifying the position of glutathionylated cysteines in the peptides. Among identified glutathionylated proteins, a large cluster of glutathionylated proteins was found involved in controlling dynamics of cytoskeletal structural organization (e.g., GTPase activity and actin filament remodeling), suggesting that the cellular structural integrity could be compromised, as seen in cardiomyocytes under ischemic reperfusion injury.5, 55–56 Because sarcomere-associated proteins are essential for contractile activity and their mutations contribute to many muscle diseases,57–58 we also analyzed glutathionylated proteins associated with sarcomere or cardiomyopathy. Selected clusters include contractile proteins, structural proteins, and proteostasis (chaperone and E3 ligase), many of which were identified in our previous proteomic analysis,33–34 albeit without quantification. The quantitative analyses with RH/L values would support a potential impact of glutathionylation on contractile activity or structural stability of sarcomeres and myofibrils. Lastly, it is important to note that 18 cysteines susceptible to glutathionylation are found to have variants on their own or neighboring residues (± 1) associated with muscular or other disorders (Table 1), thus suggesting potential functional alterations upon their glutathionylation. This also highlights the importance of site-specific identification as well as quantification, which enables to foresee potential functional outcomes even in the absence of structural information.
Finally, we have validated two proteins, desmin and BAG3, whose mutations and dysfunctions are strongly linked to cardiomyopathy and myofibrillar myopathy.49, 53 Our in vitro data validate glutathionylation of C332 (number in mouse) in desmin and C179 (number in human) in BAG3 under OGD/OGF. C332 in desmin is located in a coiled-coil domain that is involved in its oligomerizations of intermediate filaments.49 C332 is conserved in other intermediate filaments, including vimentin C328,59 which has been shown for its susceptibility to electrophile and interaction with zinc, resulting in a loss of intermediate filament integrity.60–61 Thus, it is expected that desmin C332 glutathionylation would result in a similar outcome in myocytes. Although structural information is not available, C179 in BAG3 is distant from BAG (420–499) and PXXP (302–412) domains that are essential for their chaperone and autophagy activities via binding to Hsp70 and dynein, respectively.50 However, C179 may be in a flexible and surface-exposed region, probably due to its proximity to poly-serine residues (180–188). Overall, it will be important to examine functional outcomes of glutathionylation on the identified cysteines, including ones found in desmin and BAG3, in myocytes in further analyses.
Supplementary Material
Figure S-1. Analysis of protein glutathionylation in HEK293 cell line during GD and hypoxia.
Figure S-2. Analysis of protein glutathionylation in HEK293 cell line during OGD/R
Figure S-3. Structures of isotopically labelled clickable glutathione.
Figure S-4. RH/L values of identified glutathionylated peptides at E1 experiment.
Figure S-5. Localization of glutathionylated proteins.
Figure S-6. Analysis of glutathionylated proteins in association with sarcomere, mitochondria, and cardiomyopathy
Figure S-7. Original western blots.
Table S-1: A list of glutathionylated proteins and peptides in the E1 and E2 experiments.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grant R01 HL131740 (Y.-H.A.) and the Wayne State competitive GRA award (Y. M). The Wayne State University Proteomics Core was supported through the NIH Center Grant P30 ES 020957, the NIH Cancer Center Support Grant P30 CA 022453, and the NIH Shared Instrumentation Grant S10 OD 010700. We thank Dr. Paul Stemmer and Dr. Nicholas Carruthers at WSU proteomic center for analysis. We thank the Ahn Lab members for their help and discussion.
1. The authors declare no competing financial interest.
2. The mass spectrometry data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE [1] partner repository with the dataset identifier PXD024766.
Supporting Information
The article contains Supplementary Figure S1–S7, and Supplementary Table S1.
References
- 1.Granger DN; Kvietys PR, Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol 2015, 6, 524–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kalogeris T; Baines CP; Krenz M; Korthuis RJ, Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol 2012, 298, 229–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giordano FJ, Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest 2005, 115 (3), 500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neubauer S, The failing heart--an engine out of fuel. N Engl J Med 2007, 356 (11), 1140–1151. [DOI] [PubMed] [Google Scholar]
- 5.Ruiz-Meana M; Garcia-Dorado D, Translational cardiovascular medicine (II). Pathophysiology of ischemia-reperfusion injury: new therapeutic options for acute myocardial infarction. Rev Esp Cardiol 2009, 62 (2), 199–209. [DOI] [PubMed] [Google Scholar]
- 6.Ruiz-Meana M; Garcia-Dorado D; Hofstaetter B; Piper HM; Soler-Soler J, Propagation of cardiomyocyte hypercontracture by passage of Na(+) through gap junctions. Circ Res 1999, 85 (3), 280–287. [DOI] [PubMed] [Google Scholar]
- 7.Depre C; Vatner SF, Cardioprotection in stunned and hibernating myocardium. Heart Fail Rev 2007, 12 (3–4), 307–317. [DOI] [PubMed] [Google Scholar]
- 8.Kolwicz SC Jr.; Purohit S; Tian R, Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res 2013, 113 (5), 603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lopaschuk GD; Collins-Nakai R; Olley PM; Montague TJ; McNeil G; Gayle M; Penkoske P; Finegan BA, Plasma fatty acid levels in infants and adults after myocardial ischemia. Am Heart J 1994, 128 (1), 61–67. [DOI] [PubMed] [Google Scholar]
- 10.De Leiris J; Opie LH; Lubbe WF, Effects of free fatty acid and enzyme release in experimental glucose on myocardial infarction. Nature 1975, 253 (5494), 746–747. [DOI] [PubMed] [Google Scholar]
- 11.Shipp JC; Opie LH; Challoner D, Fatty Acid and Glucose Metabolism in the Perfused Heart. Nature 1961, 189, 1018–1019. [Google Scholar]
- 12.Liu Q; Docherty JC; Rendell JC; Clanachan AS; Lopaschuk GD, High levels of fatty acids delay the recovery of intracellular pH and cardiac efficiency in post-ischemic hearts by inhibiting glucose oxidation. J Am Coll Cardiol 2002, 39 (4), 718–725. [DOI] [PubMed] [Google Scholar]
- 13.Tamm C; Benzi R; Papageorgiou I; Tardy I; Lerch R, Substrate competition in postischemic myocardium. Effect of substrate availability during reperfusion on metabolic and contractile recovery in isolated rat hearts. Circ Res 1994, 75 (6), 1103–1012. [DOI] [PubMed] [Google Scholar]
- 14.Ussher JR; Wang W; Gandhi M; Keung W; Samokhvalov V; Oka T; Wagg CS; Jaswal JS; Harris RA; Clanachan AS; Dyck JR; Lopaschuk GD, Stimulation of glucose oxidation protects against acute myocardial infarction and reperfusion injury. Cardiovasc Res 2012, 94 (2), 359–369. [DOI] [PubMed] [Google Scholar]
- 15.Dyck JR; Hopkins TA; Bonnet S; Michelakis ED; Young ME; Watanabe M; Kawase Y; Jishage K; Lopaschuk GD, Absence of malonyl coenzyme A decarboxylase in mice increases cardiac glucose oxidation and protects the heart from ischemic injury. Circulation 2006, 114 (16), 1721–1728. [DOI] [PubMed] [Google Scholar]
- 16.Lopaschuk GD; Barr R; Thomas PD; Dyck JR, Beneficial effects of trimetazidine in ex vivo working ischemic hearts are due to a stimulation of glucose oxidation secondary to inhibition of long-chain 3-ketoacyl coenzyme a thiolase. Circ Res 2003, 93 (3), e33–e37. [DOI] [PubMed] [Google Scholar]
- 17.Guarnieri C; Flamigni F; Caldarera CM, Role of oxygen in the cellular damage induced by re-oxygenation of hypoxic heart. J Mol Cell Cardiol 1980, 12 (8), 797–808. [DOI] [PubMed] [Google Scholar]
- 18.Beckendorf L; Linke WA, Emerging importance of oxidative stress in regulating striated muscle elasticity. J Muscle Res Cell M 2015, 36 (1), 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar V; Kleffmann T; Hampton MB; Cannell MB; Winterbourn CC, Redox proteomics of thiol proteins in mouse heart during ischemia/reperfusion using ICAT reagents and mass spectrometry. Free Radical Bio Med 2013, 58, 109–117. [DOI] [PubMed] [Google Scholar]
- 20.Canton M; Neverova I; Menabo R; Van Eyk J; Di Lisa F, Evidence of myofibrillar protein oxidation induced by postischemic reperfusion in isolated rat hearts. Am J Physiol- Heart C 2004, 286 (3), H870–H877. [DOI] [PubMed] [Google Scholar]
- 21.Eaton P; Byers HL; Leeds N; Ward MA; Shattock MJ, Detection, quantitation, purification, and identification of cardiac proteins S-thiolated during ischemia and reperfusion. J Biol Chem 2002, 277 (12), 9806–9811. [DOI] [PubMed] [Google Scholar]
- 22.Eaton P; Ward M; Byers H; Leung KY; Campbell J; Leeds N; Shattock MJ, Proteomic analysis identifies mitochondrial proteins which are S-thiolated during ischemia and reperfusion. Circulation 2002, 106 (19), 50–50.12093769 [Google Scholar]
- 23.Polewicz D; Cadete VJJ; Doroszko A; Hunter BE; Sawicka J; Szczesna-Cordary D; Light PE; Sawicki G, Ischemia induced peroxynitrite dependent modifications of cardiomyocyte MLC1 increases its degradation by MMP-2 leading to contractile dysfunction. J Cell Mol Med 2011, 15 (5), 1136–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kohr MJ; Sun J; Aponte A; Wang G; Gucek M; Murphy E; Steenbergen C, Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circ Res 2011, 108 (4), 418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J; Henderson GI; Freeman GL, Role of 4-hydroxynonenal in modification of cytochrome c oxidase in ischemia/reperfused rat heart. J Mol Cell Cardiol 2001, 33 (11), 1919–1927. [DOI] [PubMed] [Google Scholar]
- 26.Chen FC; Ogut O, Decline of contractility during ischemia-reperfusion injury: actin glutathionylation and its effect on allosteric interaction with tropomyosin. Am J Physiol- Cell Ph 2006, 290 (3), C719–C727. [DOI] [PubMed] [Google Scholar]
- 27.Mollica JP; Dutka TL; Merry TL; Lamboley CR; McConell GK; McKenna MJ; Murphy RM; Lamb GD, S-Glutathionylation of troponin I (fast) increases contractile apparatus Ca2+ sensitivity in fast-twitch muscle fibres of rats and humans. J Physiol- London 2012, 590 (6), 1443–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen YR; Chen CL; Pfeiffer DR; Zweier JL, Mitochondrial complex II in the post- ischemic heart - Oxidative injury and the role of protein S-glutathionylation. J Biol Chem 2007, 282 (45), 32640–32654. [DOI] [PubMed] [Google Scholar]
- 29.Eaton P; Wright N; Hearse DJ; Shattock MJ, Glyceraldehyde phosphate dehydrogenase oxidation during cardiac ischemia and reperfusion. J Mol Cell Cardiol 2002, 34 (11), 1549–1560. [DOI] [PubMed] [Google Scholar]
- 30.Mieyal JJ; Gallogly MM; Qanungo S; Sabens EA; Shelton MD, Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid Redox Sign 2008, 10 (11), 1941–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samarasinghe KTG; Godage DNPM; VanHecke GC; Ahn YH, Metabolic Synthesis of Clickable Glutathione for Chemoselective Detection of Glutathionylation. J Am Chem Soc 2014, 136 (33), 11566–11569. [DOI] [PubMed] [Google Scholar]
- 32.Samarasinghe KTG; Ahn Y-H, Synthesizing Clickable Glutathione by Glutathione Synthetase Mutant for Detecting Protein Glutathionylation. Synlett 2015, 26 (03), 285–293. [Google Scholar]
- 33.VanHecke GC; Abeywardana MY; Ahn YH, Proteomic Identification of Protein Glutathionylation in Cardiomyocytes. J Proteome Res 2019, 18 (4), 1806–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.VanHecke GC; Yapa Abeywardana M; Huang B; Ahn YH, Isotopically Labeled Clickable Glutathione to Quantify Protein S-Glutathionylation. Chembiochem 2020, 21 (6), 853–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samarasinghe KTG; Godage DNPM; Zhou YN; Ndombera FT; Weerapana E; Ahn YH, A clickable glutathione approach for identification of protein glutathionylation in response to glucose metabolism. Mol Biosyst 2016, 12 (8), 2471–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Claycomb WC; Lanson NA Jr.; Stallworth BS; Egeland DB; Delcarpio JB; Bahinski A; Izzo NJ Jr., HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A 1998, 95 (6), 2979–2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Munkanatta Godage DNP; VanHecke GC; Samarasinghe KTG; Feng HZ; Hiske M; Holcomb J; Yang Z; Jin JP; Chung CS; Ahn YH, SMYD2 glutathionylation contributes to degradation of sarcomeric proteins. Nat Commun 2018, 9 (1), 4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cherkas A; Holota S; Mdzinarashvili T; Gabbianelli R; Zarkovic N, Glucose as a Major Antioxidant: When, What for and Why It Fails? Antioxidants (Basel) 2020, 9 (2), 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lopaschuk GD; Wall SR; Olley PM; Davies NJ, Etomoxir, a carnitine palmitoyltransferase I inhibitor, protects hearts from fatty acid-induced ischemic injury independent of changes in long chain acylcarnitine. Circ Res 1988, 63 (6), 1036–1043. [DOI] [PubMed] [Google Scholar]
- 40.Shryock JC; Belardinelli L, Inhibition of late sodium current to reduce electrical and mechanical dysfunction of ischaemic myocardium. Br J Pharmacol 2008, 153 (6), 1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bhandari B; Subramanian L, Ranolazine, a partial fatty acid oxidation inhibitor, its potential benefit in angina and other cardiovascular disorders. Recent Pat Cardiovasc Drug Discov 2007, 2 (1), 35–39. [DOI] [PubMed] [Google Scholar]
- 42.Schulz H, Metabolism of 4-pentenoic acid and inhibition of thiolase by metabolites of 4- pentenoic acid. Biochemistry-Us 1983, 22 (8), 1827–1832. [DOI] [PubMed] [Google Scholar]
- 43.Szychowski J; Mahdavi A; Hodas JJ; Bagert JD; Ngo JT; Landgraf P; Dieterich DC; Schuman EM; Tirrell DA, Cleavable biotin probes for labeling of biomolecules via azide-alkyne cycloaddition. J Am Chem Soc 2010, 132 (51), 18351–18360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szklarczyk D; Morris JH; Cook H; Kuhn M; Wyder S; Simonovic M; Santos A; Doncheva NT; Roth A; Bork P; Jensen LJ; von Mering C, The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 2017, 45 (D1), D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henderson CA; Gomez CG; Novak SM; Mi-Mi L; Gregorio CC, Overview of the Muscle Cytoskeleton. Compr Physiol 2017, 7 (3), 891–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ong SB; Samangouei P; Kalkhoran SB; Hausenloy DJ, The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J Mol Cell Cardiol 2015, 78, 23–34. [DOI] [PubMed] [Google Scholar]
- 47.Famiglietti ML; Estreicher A; Gos A; Bolleman J; Gehant S; Breuza L; Bridge A; Poux S; Redaschi N; Bougueleret L; Xenarios I; UniProt C, Genetic variations and diseases in UniProtKB/Swiss-Prot: the ins and outs of expert manual curation. Hum Mutat 2014, 35 (8), 927–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shah SB; Davis J; Weisleder N; Kostavassili I; McCulloch AD; Ralston E; Capetanaki Y; Lieber RL, Structural and functional roles of desmin in mouse skeletal muscle during passive deformation. Biophys J 2004, 86 (5), 2993–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clemen CS; Herrmann H; Strelkov SV; Schroder R, Desminopathies: pathology and mechanisms. Acta Neuropathologica 2013, 125 (1), 47–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Behl C, Breaking BAG: The Co-Chaperone BAG3 in Health and Disease. Trends Pharmacol Sci 2016, 37 (8), 672–688. [DOI] [PubMed] [Google Scholar]
- 51.Hishiya A; Kitazawa T; Takayama S, BAG3 and Hsc70 interact with actin capping protein CapZ to maintain myofibrillar integrity under mechanical stress. Circ Res 2010, 107 (10), 1220–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arimura T; Ishikawa T; Nunoda S; Kawai S; Kimura A, Dilated cardiomyopathy- associated BAG3 mutations impair Z-disc assembly and enhance sensitivity to apoptosis in cardiomyocytes. Hum Mutat 2011, 32 (12), 1481–1491. [DOI] [PubMed] [Google Scholar]
- 53.Selcen D; Muntoni F; Burton BK; Pegoraro E; Sewry C; Bite AV; Engel AG, Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann Neurol 2009, 65 (1), 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wolff M; Fandrey J; Jelkmann W, Microelectrode measurements of pericellular PO2 in erythropoietin-producing human hepatoma cell cultures. Am J Physiol 1993, 265 (5 Pt 1), C1266–C1270. [DOI] [PubMed] [Google Scholar]
- 55.Steenbergen C; Hill ML; Jennings RB, Cytoskeletal Damage during Myocardial- Ischemia - Changes in Vinculin Immunofluorescence Staining during Total Invitro Ischemia in Canine Heart. Circ Res 1987, 60 (4), 478–486. [DOI] [PubMed] [Google Scholar]
- 56.Piper HM; Garcia-Dorado D; Ovize M, A fresh look at reperfusion injury. Cardiovasc Res 1998, 38 (2), 291–300. [DOI] [PubMed] [Google Scholar]
- 57.Seidman CE; Seidman JG, Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: a personal history. Circ Res 2011, 108 (6), 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zaunbrecher R; Regnier M, Connecting Sarcomere Protein Mutations to Pathogenesis in Cardiomyopathies: The Development of “Disease in a Dish” Models. Front Physiol 2016, 7, 566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Strelkov SV; Herrmann H; Geisler N; Wedig T; Zimbelmann R; Aebi U; Burkhard P, Conserved segments 1A and 2B of the intermediate filament dimer: their atomic structures and role in filament assembly. Embo J 2002, 21 (6), 1255–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perez-Sala D; Oeste CL; Martinez AE; Carrasco MJ; Garzon B; Canada FJ, Vimentin filament organization and stress sensing depend on its single cysteine residue and zinc binding. Nat Commun 2015, 6, 7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Monico A; Duarte S; Pajares MA; Perez-Sala D, Vimentin disruption by lipoxidation and electrophiles: Role of the cysteine residue and filament dynamics. Redox Biol 2019, 23, 101098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rosati A; Graziano V; De Laurenzi V; Pascale M; Turco MC, BAG3: a multifaceted protein that regulates major cell pathways. Cell Death Dis 2011, 2, e141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hamosh A; Scott AF; Amberger JS; Bocchini CA; McKusick VA, Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res 2005, 33 (Database issue), D514–D517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adzhubei IA; Schmidt S; Peshkin L; Ramensky VE; Gerasimova A; Bork P; Kondrashov AS; Sunyaev SR, A method and server for predicting damaging missense mutations. Nat Methods 2010, 7 (4), 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S-1. Analysis of protein glutathionylation in HEK293 cell line during GD and hypoxia.
Figure S-2. Analysis of protein glutathionylation in HEK293 cell line during OGD/R
Figure S-3. Structures of isotopically labelled clickable glutathione.
Figure S-4. RH/L values of identified glutathionylated peptides at E1 experiment.
Figure S-5. Localization of glutathionylated proteins.
Figure S-6. Analysis of glutathionylated proteins in association with sarcomere, mitochondria, and cardiomyopathy
Figure S-7. Original western blots.
Table S-1: A list of glutathionylated proteins and peptides in the E1 and E2 experiments.