

Abstract
Background
Recent data suggest that systemic inflammation can negatively affect atrioventricular conduction, regardless of acute cardiac injury. Indeed, gap‐junctions containing connexin43 coupling cardiomyocytes and inflammation‐related cells (macrophages) are increasingly recognized as important factors regulating the conduction in the atrioventricular node. The aim of this study was to evaluate the acute impact of systemic inflammatory activation on atrioventricular conduction, and elucidate underlying mechanisms.
Methods and Results
We analyzed: (1) the PR‐interval in patients with inflammatory diseases of different origins during active phase and recovery, and its association with inflammatory markers; (2) the existing correlation between connexin43 expression in the cardiac tissue and peripheral blood mononuclear cells (PBMC), and the changes occurring in patients with inflammatory diseases over time; (3) the acute effects of interleukin(IL)‐6 on atrioventricular conduction in an in vivo animal model, and on connexin43 expression in vitro. In patients with elevated C‐reactive protein levels, atrioventricular conduction indices are increased, but promptly normalized in association with inflammatory markers reduction, particularly IL‐6. In these subjects, connexin43 expression in PBMC, which is correlative of that measured in the cardiac tissue, inversely associated with IL‐6 changes. Moreover, direct IL‐6 administration increased atrioventricular conduction indices in vivo in a guinea pig model, and IL‐6 incubation in both cardiomyocytes and macrophages in culture, significantly reduced connexin43 proteins expression.
Conclusions
The data evidence that systemic inflammation can acutely worsen atrioventricular conduction, and that IL‐6‐induced down‐regulation of cardiac connexin43 is a mechanistic pathway putatively involved in the process. Though reversible, these alterations could significantly increase the risk of severe atrioventricular blocks during active inflammatory processes.
Keywords: atrioventricular blocks, atrioventricular conduction, cardiac electrical remodelling, connexin43, interleukin‐6, PR‐interval, systemic inflammation
Subject Categories: Arrhythmias, Growth Factors/Cytokines, Ion Channels/Membrane Transport
Nonstandard Abbreviations and Acronyms
- AVB
atrioventricular block
- DPBS
Dulbecco’s Phosphate Buffered Saline
- HR
heart rate
- IL‐1
interleukin‐1
- IL‐10
interleukin‐10
- IL‐6
interleukin‐6
- IL‐6R
IL‐6‐receptor
- PBMC
peripheral blood mononuclear cells
- PRc‐interval
heart rate‐corrected PR‐interval
- PRc‐segment
heart rate‐corrected PR‐segment
- qRT‐PCR
quantitative real time‐polymerase chain reaction
- TNFα
tumor necrosis factor‐α
Clinical Perspective
What Is New?
In patients with elevated C‐reactive protein levels, regardless of the specific underlying inflammatory condition, electrocardiographic indices of atrioventricular conduction are increased, but promptly normalized when inflammatory markers were reduced, particularly interleukin‐6 (IL‐6).
In these subjects, connexin43 expression in peripheral blood mononuclear cells, which correlates with that measured in the cardiac tissue, inversely associated with IL‐6 changes.
IL‐6 direct administration in a guinea pig model induced a significant increase of atrioventricular conduction indices; in in vitro experiments, IL‐6 incubation significantly reduced connexin43 protein expression in both cardiomyocytes and macrophages in culture.
What Are the Clinical Implications?
The study substantiates the recommendation to always evaluate the possible impact of a systemic inflammatory state on atrioventricular conduction, particularly when it is of high grade and occurs in elderly patients with history of cardiac disease and/or taking drugs negatively affecting theatrioventricular node function.
In subjects with severe atrioventricular blocks occurring/worsening during active systemic inflammation, a prompt and specific management of the inflammatory process may be an important, but often disregarded therapeutic measure for the conduction disturbance.
Anti‐cytokine therapies, specifically targeting the IL‐6, may be an innovative anti‐arrhythmic short‐term approach in these patients, possibly sparing or delaying pacemaker implantation.
The PR‐interval is routinely evaluated on the surface ECG as a measure of the delay between atrial and ventricular activation. A prolongation of the PR‐interval>200 ms, ie, first‐degree atrioventricular block (I°AVB), is traditionally considered a benign abnormality. 1 , 2
However, large population studies found that PR‐interval prolongation is an independent risk factor for cardiac events, including severe AVBs requiring pacemaker implantation, heart failure, atrial and ventricular tachyarrhythmias, and cardiac death. 3 , 4 , 5 , 6 , 7 In addition, recent data point to both gap‐junctions and the immune‐inflammatory system as important factors regulating atrioventricular conduction potentially involved in the pathogenesis of AVBs. 8 , 9 , 10 , 11 , 12
Physiologically, the electrical coupling between 2 adjacent cardiomyocytes is mediated by intercellular channels named gap‐junctions, resulting from 2 hemichannels or connexons, in turn constituted by 6 transmembrane ion‐channel proteins or connexins. 13 , 14 Among the different connexins present in the cardiac tissue, all characterized by a rapid turnover (half‐life 1–5 hours), 13 connexin43 is the most abundant, and expressed in atria, ventricles, and the conduction system, including the lower atrioventricular node. 14 Notably, gap‐junctions containing connexin43 also electrically couple cardiomyocytes with non‐cardiomyocyte cells, playing key modulatory roles in cardiac electrophysiology. 12 Hulsmans et al 10 recently demonstrated that cardiac macrophages, whose density is particularly high in the distal part of the AV node, facilitate electrical conduction through this region by coupling to cardiomyocytes via connexin43. Such a mechanism has a high physiological relevance, as either conditional deletion of connexin43 in macrophages or congenital absence of macrophages can critically delay atrioventricular conduction. 10
Accumulating data indicate that inflammation plays an important role for cardiac electric remodeling, via direct effects of cytokines on ion channels, including connexins (inflammatory cardiac channelopathies). 15 , 16 , 17 , 18 , 19 , 20 Specifically, several in vitro and ex vivo studies provided evidence that tumour necrosis factor‐α (TNFα), interleukin‐6 (IL‐6), and interleukin‐1 (IL‐1) can decrease connexin43 expression in cardiomyocytes, 21 , 22 , 23 , 24 , 25 an effect which arises rapidly (within hours‐days). 26 It is very likely, although not proven, that such changes might also occur in cardiac macrophages, given the central role of these cells in the innate immune‐inflammatory response. Thus, it is reasonable to hypothesize that systemic inflammatory processes can negatively affect atrioventricular conduction via cytokine‐mediated inhibitory effects on connexin43 expression in cardiac myocytes and macrophages, particularly those localized in the atrioventricular node. The potential clinical relevance of these mechanisms has been recently emphasized by findings from the COVID‐19, frequently characterized by an exaggerated immune‐inflammatory response with high levels of circulating cytokines, particularly IL‐6. 27 , 28 Large studies found that arrhythmic events are relatively common in COVID‐19, including bradyarrhythmias in ≈20% of the cases. 29 , 30 Specifically, several authors reported new‐onset AVBs in COVID‐19, mostly associated with elevated inflammation markers and high short‐term mortality rates, despite the absence of acute cardiac injury signs. 31 , 32 , 33 Moreover, Pavri et al 34 found that PR‐interval was significantly prolonged in 75 patients with COVID‐19 when compared with their pre‐COVID‐19 ECGs, and the presence of PR‐interval prolongation (or absence of shortening with increasing heart rate) was associated with a 4‐fold risk of death. Notably, patients who died not only very frequently showed PR‐interval prolongation (≈80%), but also showed higher peak values of CRP (C‐reactive protein) than survivors. 34 Consistent findings were also provided by Moey et al, 35 who reported significant PR‐interval prolongation in 107 patients with COVID‐19, regardless of medication status and troponin levels. In this cohort I°AVB was present in 18.7% of cases, and 1 patient developed transient Mobitz 2 II°AVB. 35
The aim of this study was to evaluate the acute impact of systemic inflammatory activation on atrioventricular conduction, regardless of specific cause, and to elucidate the underlying mechanisms. We therefore analyzed: (1) the behavior of PR‐interval in patients with inflammatory diseases from different origins during active phase and recovery, and its association with inflammatory markers; (2) the correlation existing between connexin43 expression in the cardiac tissue and circulating blood cells, and the changes occurring in patients with inflammatory diseases over time; (3) the acute effects of IL‐6 on atrioventricular conduction in an in vivo animal model, and on connexin43 expression in vitro.
Methods
The data underlying this article are available in the article and in its online supplementary material.
Local Ethical Committee approved the study, and patients from all groups gave their oral and written informed consent in accordance with the Principles of the Declaration of Helsinki. All experiments involving animals were approved by the local Animal‐Care and Use‐Committee.
Study Populations
The impact of systemic inflammation on atrioventricular conduction was evaluated by 46 prospectively enrolled patients with elevated CRP levels due to different inflammatory diseases, including acute inflammatory processes, septic or aseptic, or chronic immuno‐mediated diseases during flares. ECG recordings and blood sample withdrawals were performed in these subjects during active disease (PRE), and after different therapeutic interventions resulting in CRP decrease >75% compared with the baseline (POST). None of these patients was under treatment with drugs potentially affecting atrioventricular conduction, specifically antiarrhythmics (beta‐blockers, calcium channel blockers, class I or III antiarrhythmics), phenytoin, or lithium. Demographic and clinical characteristics of this population are detailed in Table S1.
A second group of 30 subjects comparable for age and sex, without any history of systemic inflammation or cardiac disease, was enrolled as controls (C) for ECG and laboratory parameters (Table S2).
Finally, a third additional sample of 14 patients undergoing cardiac surgery, different from the 2 above cohorts, was specifically recruited to study the possible correlation of connexins43 expression between peripheral blood mononuclear cells (PBMC) and cardiac tissue. Details on this population are provided below and in Table S3.
ECG Recordings
PR‐interval, representing the sum of P wave duration and PR segment, is a well‐established tool to estimate the atrioventricular conduction on the surface ECG. While P wave duration reflects the electrical signals that propagate through the atria, PR‐segment corresponds to the period during which electrical signals are delayed at the atrioventricular node, before they enter the ventricular branches. Thus, PR‐segment represents a more accurate ECG parameter to specifically evaluate atrioventricular nodal conduction. In all patients and controls, simultaneous twelve‐lead ECG (25 mm/s and 10 mV/cm; sampling rate 1kHz) was recorded (Cardioline ECT WS 2000, Remco Italia, Vignate‐Milano, Italy) in supine position, and during spontaneous breathing, in the morning hours (between 08:00 am and 12:00 noon). Paper‐printed ECGs were scanned and digitized in order to achieve greater precision in detecting and measuring PR‐intervals/segments. PR‐interval and PR‐segment were measured from 3 non‐consecutive beats (mean value) by a single investigator (M.A.) blinded to the subject clinical status. PR‐interval duration was measured from the beginning of the P‐wave deflection to the beginning of the QRS complex. PR‐interval duration <200 ms was considered normal. Thus, according to current American College of Cardiology Foundation/American Heart Association/Heart Rhythm Society guidelines, I°AVB is defined as abnormal prolongation of the PR interval (>200 ms). 36 To better evaluate the severity of the atrioventricular delay, an additional, more stringent cut‐off (>220 ms) was also considered. 2 , 4 It is well documented that most of the physiological PR‐interval beat to beat or day to day variability for a given patient is dependent on heart rate (HR) changes, 37 , 38 and that adjustment for the HR results in a high intra‐subject stability of this parameter throughout the time. 37 Accordingly, PR‐interval was then adjusted for the HR by using the Soliman‐Rautaharju’s formula (PR‐interval + 0.26 (HR—70) for age group younger than 60 years, or PR‐interval+0.42 (HR—70) for age group 60 years or older) to obtain the heart corrected PR‐interval (PRc‐interval). 39 In this case, PRc‐interval>205 ms (95th percentile) was considered as the limit for I°AVB. 39 PR‐segment duration was measured from the end of the P‐wave deflection to the beginning of the QRS complex. According to Soliman et al, 40 a PR‐segment duration <91 ms (95th percentile) was considered normal. Finally, given that no formulas to correct the PR‐segment for the HR presently exist and that, differently to PR‐interval, P‐wave duration is not significantly affected by heart rate changes 41 (thereby implying that PR‐segment is the specific component of the PR‐interval which actually require HR‐adjustment), the Soliman‐Rautaharju’s formula 39 was also applied to the PR‐segment to estimate, for research purposes, the heart corrected PR‐segment (PRc‐segment).
Laboratory Analysis
Blood samples were centrifuged at 2000g and serum samples were stored at −80 °C. CRP was assayed by a particle‐enhanced turbidimetric method (COBAS‐6000 platform, Roche Diagnostics GmbH; Mannheim, Germany) and the values were expressed as mg/dL (normal values <0.5). A multiplex assay for cytokine quantification (Bioplex, Bio‐Rad, Hercules, CA) was used to measure circulating levels of inflammatory cytokines IL‐6, TNFα, and IL‐1, and the anti‐inflammatory cytokine IL‐10. Cytokine concentrations were calculated using a standard curve established from serial dilutions of each cytokine standard as described in the manufacturer’s protocol and expressed as pg/mL. Since no established reference values for cytokine levels are currently available, an internal reference control group of 10 healthy subjects (mean age 55.5±4.3 years) without clinical signs of ongoing acute infections was used.
Correlation Study of Cardiac Tissue and Circulating Levels of Connexin43 in Patients Undergoing Cardiac Surgery
Previous studies already have demonstrated that a strong correlation exists between mRNA expression of several cardiac ion channels in PBMC and the cardiac tissue. 19 , 24 , 42 Moreover, the presence of a strict relationship between mRNA and protein levels of connexin43 has been demonstrated by several authors either in cardiac 43 , 44 , 45 or in PBMC. 46 Based on this evidence, in order to specifically evaluate whether connexin43 expression in PBMC may be a reflection of cardiac connexin43 expression, PBMC and cardiac tissue specimens from atria and/or ventricles were obtained from 14 consecutive patients undergoing cardiac surgery, including valve surgery (n=9), heart transplantation (n=3), septal myomectomy (n=1), and interatrial communication (n=1), for a total of 19 myocardial samples collected (right atrium n=7, left atrium n=5, left ventricle n=4, right ventricle n=3; Table S3). Blood samples were withdrawn 1–12 hours before surgical procedures, and cardiac tissues were obtained prior to the establishment of extracorporeal circulation. Tissue specimens were collected in a Dulbecco’s Phosphate Buffered Saline (DPBS) 1% penicillin‐streptomycin solution and immediately homogenized and stored at −80 °C for further RNA extraction. Heparinized peripheral blood samples were diluted 1:1 with DPBS and PBMCs collected after a “Lympholyte density gradient separation.” PBMCs were washed twice in DPBS and the pellet obtained was immediately lysed, frozen and stored at −80°C for further RNA extraction. For each of these patients, connexin43 mRNA levels were measured in both cardiac tissue and PBMCs.
Circulating Connexin43 Levels in Patients With Inflammatory Diseases
Peripheral blood samples were withdrawn in PRE and POST conditions in 13/46 consecutive, unselected patients with inflammatory diseases, as well as in 25 controls, to isolate PBMCs and analyze connexin43 mRNA expression in these cells.
Analysis of Connexin43 mRNA by qRT‐PCR
Total RNA was isolated from the heart tissue (30μg) and PBMCs (10×106) using the RNeasy Lipid Tissue Mini Kit and miRNeasy Mini Kit respectively, following the datasheet provided by the manufacturer. For cDNA synthesis, RT2 First Strand Kit was used, according to manufacturer’s instructions. qRT‐PCR was performed using customized RT2 PCR arrays with primers for connexin43 (GJA1). β‐actin and HPRT were selected and used as reference genes to normalize mRNA expression data obtained with qRT‐PCR experiments. 42 Primers for the genes of interest were synthesized by the manufacturer and provided already dispensed in customized RT2 PCR arrays. All kits and reagents were purchased at Qiagen S.p.A, Milano, Italy.
Guinea Pig Study
Dunkin‐Hartley guinea pigs (350–450 g) were anesthetized with 2%–4% isoflurane using a Tec‐3 precision vaporizer (Patterson Veterinary, Greeley, CO). Animal was placed on a pad heated with recirculating water (Patterson Veterinary, Prostration Rodent Workstation; T/Pump, Stryker, Portage, MI). After acclimation to the heating pad and anesthesia, basal ECG recordings in lead‐I were obtained followed by IL‐6 (200ng/mL) and IL‐6‐receptor (IL‐6R, 200 ng/mL) (Sigma Aldrich, St. Louis, MO) administered intraperitoneally. IL‐6R was co‐administered with IL‐6 to mimic the existing high levels of the soluble form of IL‐6R during inflammatory processes. ECG recordings were obtained every 30 minutes for 2 hours (sampling rate 1kHz). Acquisition hardware included an Animal Bio Amp (ML136), Dual Bio Amp (ML135), and PowerLab 8/30 (ML870) all from ADInstruments (Colorado Springs, CO). Software acquisition and analysis performed using LabChart6 (ADInstruments).
Protein Extraction and Western Blotting in Mouse HL‐1 Cardiomyocytes and Raw264.7 Macrophages in Cultures
HL‐1 mouse cardiomyocytesare one of the most currently used models for adult cardiac cells, 47 particularly to examine Cx43 expression. 48 , 49 , 50 HL‐1 cells cardiomyocytes were maintained in gelatin‐fibronectin‐coated flasks in Claycomb medium (Sigma‐Aldrich, Boston, MA) supplemented with 10% FBS (Sigma‐Aldrich, Boston, MA), 0.1 mmol/L norepinephrine (Sigma‐Aldrich, Boston, MA), 100 U/mL penicillin, 100 mg/mL streptomycin (Gibco, Invitrogen, Grand Island, NY), and 2 mmol/L L‐glutamine (Gibco, Invitrogen, Grand Island, NY) and kept semi‐confluent at all times.
Raw264.7 is a cell line of mouse blood monocytes/macrophages very similar to macrophages derived from circulating PBMCs. Physiologically, adult cardiac resident macrophages are distinct cells originated from embryo precursors that migrate to the heart from yolk sac and fetal liver prior to birth and maintain themselves by in situ renewal, mostly in the absence of the adult blood mononuclear cell pool. 51 Nevertheless, when self‐renewal declines with age or after cardiac insult, cardiac tissue can be repopulated by monocyte‐derived macrophages. 52 In this regard, it has been demonstrated that Raw264.7 (similar to circulating PBMC) can be recruited by expression of the chemokine (C–C motif) receptor‐2 and are capable of cell‐cell interaction. 53 , 54 Raw264.7 macrophages, kindly provided by C.T. Baldari, University of Siena, were grown in DMEM High‐Glucose (Sigma‐Aldrich, Milano, Italy) supplemented with 10% FBS (Euroclone, Milano, Italy).
Both cell lines were untreated or treated with IL‐6 (100 pg/mL, Sigma‐Aldrich) for 24 hours. Cell pellets were lysed in RIPA buffer with protease inhibitors (25 mmol/L Tris‐HCl, pH 7.6 containing 150 mmol/L NaCl, 1% NP‐40, 1% sodium deoxycholate, 0.1% SDS) for 30 minutes at 4 °C and centrifuged at 10 000g for 15 minutes. The supernatant after centrifugation was resolved by SDS‐PAGE on a 4%–15% Tris‐HCl gel (Bio‐Rad) and transferred on to a PVDF membrane (Bio‐Rad). Blots were blocked with 5% milk for an hour and probed with polyclonal rabbit anti‐connexin 43 antibody (Sigma‐Aldrich, 1:200) and, as loading controls, anti GAPDH (Sigma‐Aldrich, 1:5000) or mouse anti‑β actin (Merck‐Millipore 1:1000) antibodies, respectively, for HL‐1 or Raw264.7, overnight at 4 °C. Further it was probed with anti‐rabbit IgG HRP (Santa Cruz) at a 1:5000 or and anti‐mouse IgG HRP‐linked (1:10000, Invitrogen) dilution at room temperature for 1 hour. The signal was detected with Clarity ECL substrate (Bio‐Rad) and blots were scanned in a C‐Digit blot scanner (LI‐COR) at high sensitivity to obtain the image.
Statistical Analysis
Descriptive statistics is reported as frequency count and percentage for qualitative data and mean±SD or median and range of variation for quantitative data.
The following parametric or non‐parametric statistical analyses were carried out: the two‐tail Student’s paired “t” test, or the two‐tail Wilcoxon matched‐pairs test, or the two‐tail Student’s unpaired “t” test, or the two‐tail Mann‐Whitney test to evaluate differences in quantitative variables between 2 groups of data paired (changes in PR‐intervals/segments, CRP, cytokines, PBMC connexin43 expression in inflammatory patients, PRE versus POST; IL‐6‐dependent changes in connexin43 expression in cardiomyocytes or macrophages versus baseline) or unpaired (comparisons of age, PR‐intervals/segments, CRP, cytokines, PBMC connexin43 expression in inflammatory patients versus controls or in inflammatory patients with PRc>200 ms versus <200 ms), respectively; the Spearman rank correlation‐test to verify possible statistical association between quantitative variables in patients with inflammatory diseases (PR‐intervals/segments versus CRP, cytokines, or PBMC connexin43 expression; CRP or cytokines versus PBMC connexin43 expression) and in patients undergoing cardiac surgery (cardiac versus PBMC connexin43 expression); the two‐sided McNemar test, or the two‐sided Fisher’s exact test were performed to evaluate statistical association between categorical variables in patients with inflammatory diseases and controls (PR‐intervals/segments, PRE versus POST, PRE/POST versus controls, PRc>200 ms versus PRc<200 ms). The Dunnett’s test was applied when patients with inflammatory diseases in PRE/POST conditions were compared to controls.
We also performed a sample size and power analysis to define the number of subjects to include in both groups of patients and controls. For the study, an effect size of about 0.8, a first type error of 0.05 and a power of 90% were considered, obtaining 44 study cases and 28 controls, using Mann‐Whitney test. Nevertheless, considering 46 study patients for the PRE‐POST comparison, the effect size decreases to about 0.5, still maintaining a power of 90% based on Wilcoxon‐test. The sample size was estimated with the GPower software.
P values ≤0.05 were considered significant (GraphPad‐InStat, version 3.06 for Windows 2000).
Results
PR‐interval in Patients With Inflammatory Diseases and its Relationship With Inflammatory Markers
Patients with active inflammatory diseases showed PR‐interval prolongation in 15%–17% of cases (depending on the cutoff used: >200 ms, 8/46; >220 ms 7/46), a percentage greater than that observed in controls (>200 ms: 1/30 [3%], P=0.08; >220 ms: 0/30 [0%], P=0.038; two‐sided Fisher’s exact test). A similar trend was also observed for median PR‐interval values, which were 168.4 ms in inflammatory patients versus 156.7 ms in controls (P=0.10, two‐tails Mann‐Whitney test). However, in the inflammatory group the median HR was significantly higher than in control subjects (80.0 versus 66.5 bpm; P<0.0001, two‐tails Mann‐Whitney test), a factor which might have underestimated the differences. In fact, it is well known that HR and PR‐interval have inverse relationship, with PR‐interval becoming shorter with increasing HR and vice‐versa. 39 Accordingly, after correcting the PR‐interval values for the heart rate by using the Soliman‐Rautaharju’s formula, 39 the resulting PRc‐interval became significantly different in inflammatory versus control subjects, both in terms of median values (173.1 versus 156.9 ms; P=0.012, two‐tails Mann‐Whitney test) and prolongation percentages (>200 ms: 11/46 [24%] versus 0/30 [0%], P=0.0026; >205 ms: 9/46 [20%] versus 0/30 [0%], P=0.0097; >220 ms: 7/46 [15%] versus 0/30 [0%], P=0.038; two‐sided Fisher’s exact test; Table 1; Figure 1; Table S2).
Table 1.
Changes in Clinical, Electrocardiographic, Laboratory and Echocardiography Parameters in Patients With Inflammatory Diseases (n=46), During Active Disease (PRE) and After Therapeutic Interventions Resulting in a CRP decrease >75% When Compared With the Baseline (POST)
| PRE | POST | P value | |
|---|---|---|---|
| CRP, mg/dL (r.v.<0.5) | 12.5 (15.5) | 1.6 (1.9) | <0.0001* |
| IL‐6, pg/mL (r.v.0.49–1.25) | 14.0 (22.4) | 3.0 (4.2) | <0.0001* |
| IL‐1, pg/mL (r.v.0.08–0.29) | 0.37 (0.39) | 0.27 (0.21) | 0.045* |
| TNFα, pg/mL (r.v.0.60–3.24) | 0.75 (0.10) | 0.75 (0.15) | 0.57 |
| IL‐10, pg/mL (r.v.0–3.60) | 0.56 (0.52) | 0.55 (0.27) | 0.45 |
| Heart rate, bpm | 80.0 (14.0) | 74.0 (17.5) | 0.0007* |
| PR‐interval, ms | 168.4 (45.6) | 163.0 (28.6) | 0.19 |
| Patients with PR‐interval>200 ms, n | 8 (17%) | 5 (11%) | 0.37 |
| Patients with PR‐interval>220 ms, n | 7 (15%) | 3 (7%) | 0.13 |
| PRc‐interval, ms | 173.1 (39.4) | 162.5 (30.1) | 0.0091* |
| Patients with PRc‐interval>200 ms, n | 11 (24%) | 5 (11%) | 0.041* |
| Patients with PRc‐interval>205 ms, n | 9 (20%) | 5 (11%) | 0.07 |
| Patients with PRc‐interval>220 ms, n | 7 (15%) | 3 (7%) | 0.13 |
| PR‐segment, ms | 59.7 (36.2) | 57.3 (26.9) | 0.09 |
| Patients with PR‐segment>91 ms, n | 8 (17%) | 5 (11%) | 0.37 |
| PRc‐segment, ms | 63.7 (33.4) | 59.8 (23.4) | 0.0021* |
| Patients with PRc‐segment>91 ms, n | 9 (20%) | 5 (11%) | 0.13 |
| Potassium, mEq/L (r.v.3.5–5.5) | 4.0±0.5 | 4.2±0.6 | 0.33 |
| Calcium, mg/dL (r.v.8–11) | 8.5±0.8 | 8.6±0.6 | 0.92 |
| Magnesium, mg/dL (r.v.1.5–2.5) | 1.9±0.3 | 1.8±0.4 | 0.19 |
| Creatinine, mg/dL (r.v.0.7–1.2) | 1.1±0.5 | 0.9±0.2 | 0.10 |
| pO2, mm Hg (r.v.70–100) | 70.1±13.4 | 72.8±10.2 | 0.27 |
| pH (r.v.7.35–7.45) | 7.44±0.05 | 7.44±0.04 | 0.96 |
| Left atrial diameter, mm (r.v.<40) | 39.9±2.9 | 39.8±3.0 | 0.99 |
| Ejection fraction, % (r.v.>50) | 57.0±3.1 | 57.9±3.1 | 0.20 |
| Left ventricular internal dimension, mm (r.v.<56) | 47.8±4.5 | 47.7±4.9 | 0.19 |
| Estimated pulmonary artery pressure, mm Hg (r.v.<30) | 33.0±6.6 | 29.8±5.6 | 0.10 |
Cytokine level range measured in an internal reference group of healthy controls.
Values are expressed as median (interquartile range), or mean±SD, or frequency count and percentages.
Differences were evaluated by the two‐tail Student’s paired “t” test, or the two‐tail Wilcoxon matched pairs test. Difference in categorical variables were evaluated by McNemar test.
CRP indicates C‐reactive protein; IL‐1, interleukin‐1; IL‐10, interleukin‐10;,IL‐6, interleukin‐6; PRc interval, heart rate‐corrected PR interval; PRc segment, heart rate‐corrected PR segment; r.v., reference values; and TNFα, tumor necrosis factor alpha.
statistically significant difference.
Figure 1. PR‐interval changes and their correlation with C‐reactive protein (CRP) in patients with inflammatory diseases, during active disease (PRE) and after therapeutic interventions resulting in CRP decrease >75% when compared with the baseline (POST), and controls (C).
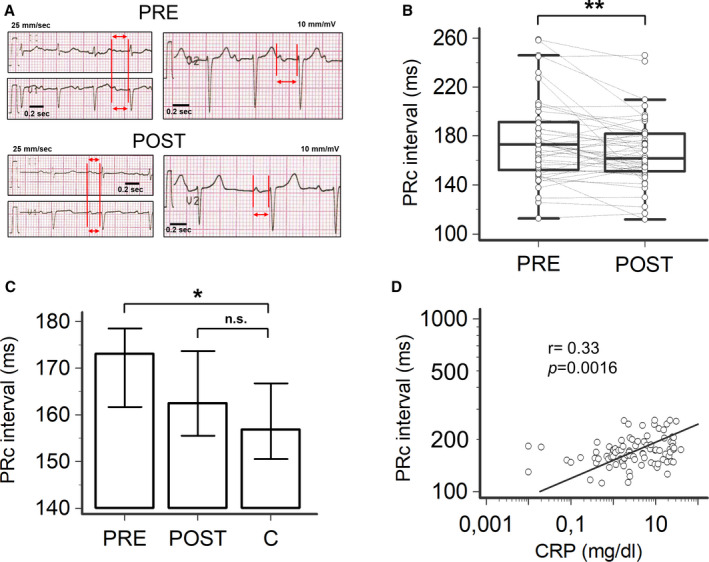
A, Representative ECG strips of an 82‐year‐old patient with polymyalgia rheumatica, during active disease (PRE: heart rate‐corrected PR[PRc]‐interval 253 ms) and after a 11‐day treatment with prednisone (POST: PRc‐interval 194 ms). Red vertical lines and horizontal arrows in leads II, V1 and V2, indicate PR‐interval. B, Changes in PRc‐interval observed in PRE and POST conditions in the entire population of patients with inflammatory diseases; two‐tail Wilcoxon matched‐pairs test, **P<0.01. C, Comparison of PRc‐interval in patients with inflammatory diseases, in PRE and POST conditions, and controls; two‐tail Dunnett’s test, *P<0.05, n.s. not significant. D, Relationship between PRc‐interval and CRP levels; Spearman test. Patients, n=46; controls, n=30.
Based on the underlying inflammatory disease, a specific treatment in each patient was initiated, including antibiotics, anti‐inflammatory drugs or protease inhibitors (Table S1). Such interventions lead to a rapid (mean follow‐up time 19.9±24.4 days, median 10.0 days) and significant decrease in CRP levels (median −87.2%) as well as PRc‐interval duration (median ΔPRc=−10.6 ms, from 173.1 to 162.5 ms) (Table 1; Figure 1A through 1C). Notably, PRc‐interval decreased reaching median values not significantly different from controls (Figure 1C). Hence, the percentage of patients showing PRc‐interval prolongation decreased (>200 ms: 5/46 [11%]; >205 ms: 5/46 [11%]; >220 ms: 3/46 [7%]). Notably, about a half of the subjects who presented I°AVB in PRE conditions showed a normal atrioventricular conduction when re‐evaluated in POST conditions (Table 1). Moreover, the prevalence of PRc‐interval prolongation found in the patients after treatment was no longer different to that observed in controls (>200 ms: 5/46 [11%] versus 0/30 [0%], P=0.15; >205 ms: 5/46 [11%] versus 0/30 [0%], P=0.15; >220 ms: 3/46 [7%] versus 0/30 [0%], P=0.27; two‐sided Fisher’s exact test).
Changes in CRP levels and PRc‐interval significantly correlated throughout the study period (r=0.33, P<0.01; Figure 1D). On the contrary, no appreciable variations in other laboratory (electrolytes, arterial blood gases, pH, renal function) or echocardiography findings was demonstrated when PRE and POST conditions were compared (Table 1).
Circulating IL‐6 levels showed a marked elevation during the active inflammatory phase, but rapidly reduced to near‐normal values when CRP decreased (Table 1; Figure 2A). Notably, IL‐6 levels significantly associated with PRc‐interval values throughout the study time, even more than that observed for CRP (r=0.38, P<0.001; Table 1; Figure 2B). A smaller but significant increase of IL‐1 concentration was also demonstrated at baseline which decreased after therapeutic interventions and correlated with PRc‐interval changes (r=0.27, P<0.05; Table 1; Figure 2C and 2D). On the contrary, TNFα and IL‐10 levels were not different when compared to controls and did not show any substantial change following therapy (Table 1).
Figure 2. Changes in cytokines and their correlation with heart rate‐corrected PR‐interval (PRc‐interval) in patients with inflammatory diseases, during active disease (PRE) and after therapeutic interventions resulting in CRP decrease >75% when compared with the baseline (POST), and controls (C).
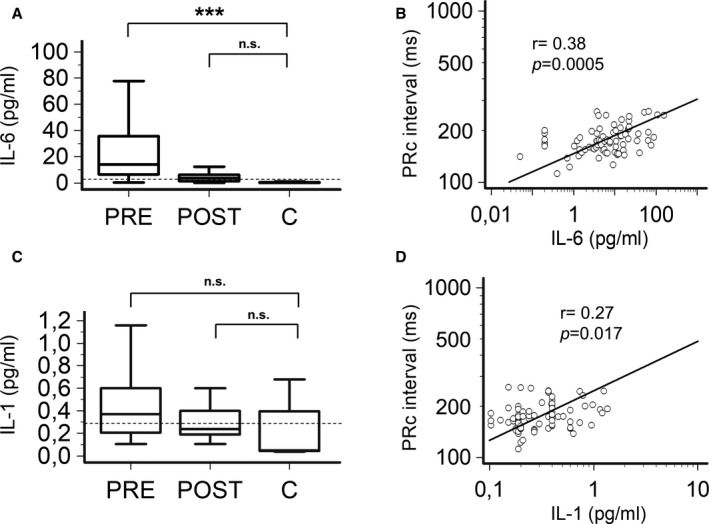
A, Comparison of interleukin‐6 (IL‐6) levels in patients with inflammatory diseases, in PRE and POST conditions, and controls; two‐tail Dunnett’s test, ***P<0.001, n.s. not significant. B, Relationship between PRc‐interval and IL‐6 levels; Spearman test; C, Comparison of interleukin‐1 (IL‐1) levels in patients with inflammatory diseases, in PRE and POST conditions, and controls; two‐tail Wilcoxon matched‐pairs test, *P<0.05; two‐tail Dunnett’s test, n.s. not significant. D, Relationship between PRc‐interval and IL‐1 levels; Spearman test. Patients, n=46; controls, n=30. Horizontal dotted line indicates the upper limit values in a reference healthy population, ie, 1.25 pg/mL for IL‐6, and 0.29 pg/mL for IL‐1.
PR‐Segment in Patients With Inflammatory Diseases and Association With Cytokine Levels
A previous study demonstrated that systemic inflammation, via IL‐6 increase, can affect intra‐atrial conduction. 24 Given that the PR‐interval reflects the summation of electric signal propagation in atria and atrioventricular node, we also evaluated the impact of inflammatory activation on the PR‐segment, as it corresponds to the period that the electric signals are delayed at the atrioventricular node. 55 By comparing patients with inflammation to controls, median PR‐segment values tended to be higher, but not significantly (59.7 versus 53.3 ms; P=0.13, two‐tails Mann‐Whitney test). However, the percentages of inflamed patients with a prolonged PR‐segments (>91 ms) was significantly higher than in controls (>91 ms: 8/46 [17%] versus 0/30 [0%], P=0.022; two‐sided Fisher’s exact test). Again, these differences became more evident when the PRc‐segment was used, both in term of median values (63.7 versus 55.6 ms; P=0.0073, two‐tails Mann‐Whitney test), and prolongation percentages (>91 ms: 9/46 [20%] versus 0/30 [0%], P=0.0097; two‐sided Fisher’s exact test; Table 1; Figure 3; Table S2).
Figure 3. PR‐segment changes and their correlation with interleukin(IL)‐6 levels in patients with inflammatory diseases, during active disease (PRE) and after therapeutic interventions resulting in CRP decrease >75% when compared with the baseline (POST), and controls (C).
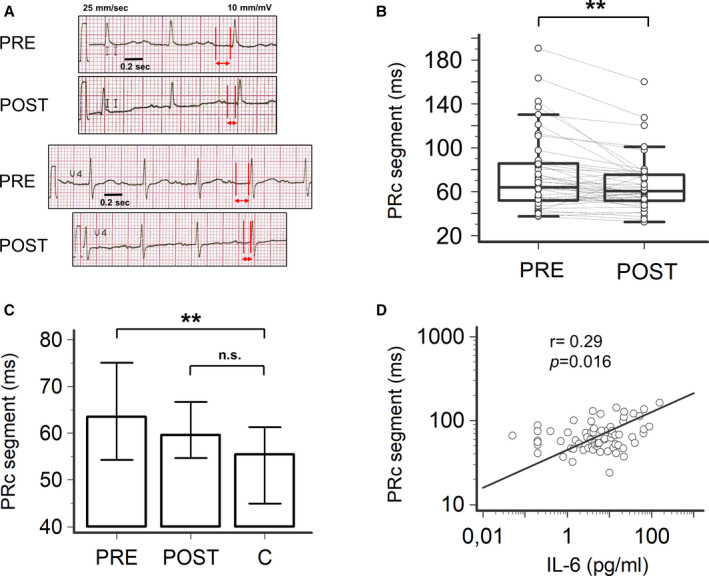
A, Representative ECG strips of a 74‐year‐old patient with acute cholangitis, during active disease (PRE: heart rate‐corrected PR[PRc]‐segment 178 ms) and after a 16‐day treatment with piperacillin/tazobactam (POST: PRc‐segment 105 ms). Red vertical lines and horizontal arrows in leads II and V4, indicate PR‐segment. B, Changes in PRc‐segment observed in PRE and POST conditions in the entire population of patients with inflammatory diseases; two‐tail Wilcoxon matched‐pairs test, **P<0.01. C, Comparison of PRc‐segment in patients with inflammatory diseases, in PRE and POST conditions, and controls; two‐tail Dunnett’s test, **P<0.01, n.s. not significant. D, Relationship between PRc‐segment and IL‐6 levels; Spearman test. Patients, n=46; controls, n=30.
In patients with inflammation, the therapeutic intervention resulted in a significant PRc‐segment shortening (median ΔPRc= −3.9 ms, from 63.7 to 59.8 ms; Table 1; Figure 3A through 3C), until reaching median values not significantly different from controls (Figure 3C). Moreover, while the prevalence of patients showing PRc‐segment prolongation after treatment was reduced, but not significantly when compared to PRE conditions (Table 1), such a percentage was no longer different to that observed in controls (5/46 [11%] versus 0/30 [0%], P=0.15; two‐sided Fisher’s exact test). Notably, a significant association with IL‐6 levels was also confirmed for PRc‐segment changes observed over time (r=0.29, P=0.016; Figure 3D). Altogether, these data suggest that inflammation can delay atrioventricular conduction via IL‐6‐mediated specific effects on the atrioventricular node.
Determinants of Different Magnitude of PR‐interval/PR‐Segment Changes in Patients With Inflammatory Diseases
As observed in Figures 1 and 3, the magnitude of PR‐interval/PR‐segment changes markedly differed throughout the patients with inflammatory diseases, the most evident variations occurring in those who presented I°AVB during active disease (≈ a quarter of the entire population). In order to better characterize this phenomenon and underlying causes, we conducted a subanalysis by dividing the patients in 2 groups based on PRc‐interval values in PRE conditions, ie >200 ms (11/46, 24%) versus <200 ms (35/46, 76%). We found that in the PRc>200 group, PRE/POST changes were noticeably more evident than those observed in the PRc<200 group, both in terms of PRc‐interval (PRc>200 group: median ΔPRc=−36.2 ms, from 231.6 to 195.4 ms; PRc<200 group: median ΔPRc= −7.7 ms, from 164.6 to 156.9 ms) and for PRc‐segment (PRc>200 group: median ΔPRc= −32.5 ms, from 112.7 to 80.2 ms; PRc>200 group: median ΔPRc= −4.1 ms, from 61.8 to 57.7 ms).
Comparisons of demographic, clinical, electrocardiographic and laboratory findings (Table S4) showed that patients in the PRc>200 group were older and reached significantly higher median IL‐6 levels during active disease (≈2‐fold) with respect to those in the PRc<200 group. Moreover, in the PRc>200 group both the median PRc‐interval and PRc‐segment durations, although markedly shortened after therapy, remained significantly longer than in the PR<200 group also when the active inflammatory phase was resolved (POST conditions). Conversely, no differences in terms of heart disease, hypertension or diabetes prevalence were observed between the 2 groups.
Overall, these findings suggest that patients with the higher risk of developing severe atrioventricular delays during inflammation are those in whom IL‐6 levels are substantially increased (>20 pg/mL), and a preexisting conduction reserve deficit is present, more commonly age‐related. Accordingly, in this group of subjects the correlation existing over time between conduction parameters and circulating IL‐6 was markedly stronger than that observed in the whole population (PRc‐interval/IL‐6: r=0.54 [versus 0.38]; PRc‐segment/IL‐6: r=0.48 [versus 0.29]; Figure S1).
Correlation Between Cardiac and Circulating Levels of Connexin43 in Patients Undergoing Cardiac Surgery
Connexin43 is expressed in PBMC, and critically regulates leukocyte recruitment and development, as well as T‐cell activation by accumulating at the level of the so‐called immunological synapse. 56 , 57 Paired analysis of mRNA levels of connexin43 from cadiac (atrial and/or ventricular) tissue and PBMC demonstrated a strong center‐periphery correlation (r=0.58, P=0.0091; Figure 4A), thereby providing evidence that PBMC‐derived expression of connexin43 is correlative to the their expression levels in the myocardial tissue in the atria and ventricles.
Figure 4. Correlation between cardiac and connexin43 transcript expression in patients undergoing cardiac surgery, and changes of connexin43 transcript expression in patients with inflammatory diseases.
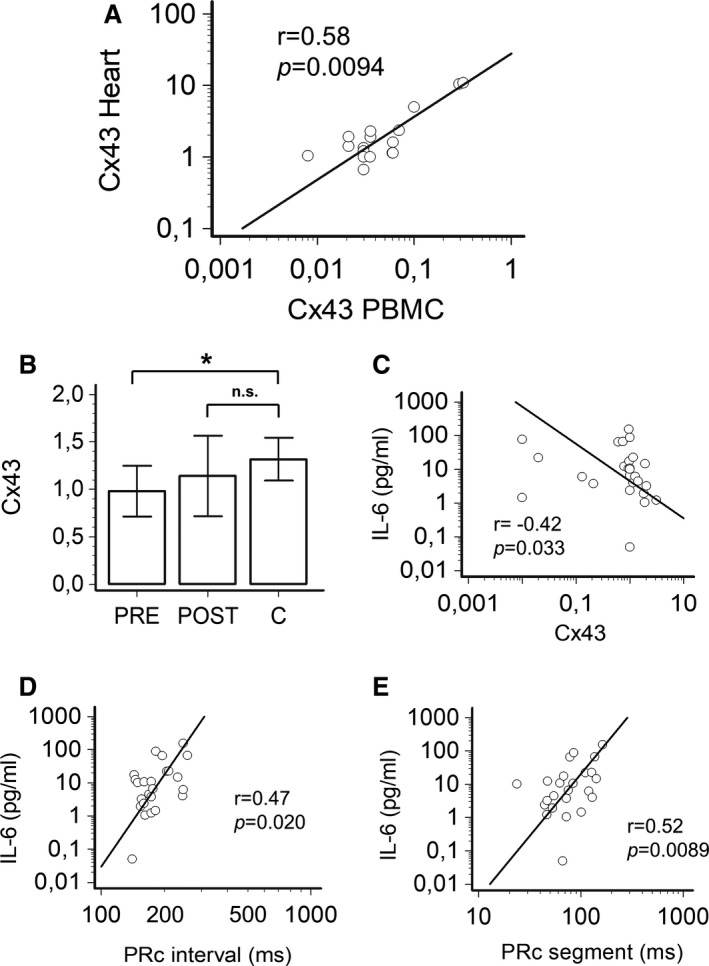
A, Relationship between connexin43 mRNA levels in atrial and/or ventricular tissue and peripheral blood mononuclear cells (PBMCs); Spearman test; patients, n=14; samples, n=19. B, Comparison of circulating levels of connexin43 mRNA levels in patients with inflammatory diseases, during active disease (PRE) and after therapeutic interventions resulting in CRP decrease >75% when compared with the baseline (POST), and controls; two‐tail Dunnett’s test, *P<0.05, n.s. not significant; patients, n=13; controls (C) n=25. C, Relationship over time between circulating connexin43 mRNA and IL‐6 levels in patients with inflammatory diseases; Spearman rank correlation. D, Relationship over time between PRc‐interval and IL‐6 levels in patients with inflammatory diseases; Spearman rank correlation; patients, n=13. E, Relationship over time between PRc‐segment and IL‐6 levels in patients with inflammatory diseases; Spearman rank correlation; patients, n=13.
Relationship Between Circulating Connexin43 levels, PRc‐interval/segment and Inflammatory Markers in Patients With Inflammatory Diseases
In 13 unselected, consecutive patients with inflammatory diseases, connexin43 expression analyses was performed in PBMCs, to compare PRE and POST conditions (mean follow‐up time 11.2±6.6 days, median 11.0 days). Values of PRc‐interval/segment and inflammatory markers of these subjects in PRE and POST conditions show that they were representative of the entire cohort (Table S5).
Although a rise in mRNA levels of connexin43 was observed in these patients after the CRP decrease of >75% when compared with baseline (1.01±0.44 [median 1.00] versus 1.24±0.87 [1.08]), such a difference did not reach the statistical significance (Table S5). Nevertheless, in these patients, connexin43 levels in PRE, but not in POST conditions, were significantly lower than those observed in controls (1.31±0.52 [1.10]; Figure 4B). Moreover, connexin43 expression significantly and inversely correlated with IL‐6 concentrations over time (r=−0.42, P=0.033; Figure 4C), which in turn were directly associated with both PRc‐interval (r=0.47, P=0.020) and PRc‐segment (r=0.52, P=0.0089) changes (Figure 4D and 4E).
Acute Effect of IL‐6 on Guinea Pig PR‐Interval
In order to further support the role of IL‐6 in the pathogenesis of inflammation‐induced atrioventricular conduction delay, IL‐6 was directly injected intraperitoneally into guinea pigs (n=5), a suitable animal model for PR‐interval studies because of the similarities of ECG features with humans. 58
As shown in Table 2 and Figure 5, IL‐6 intraperitoneal administration was associated with significant prolongation of both PR‐interval (mean ΔPR‐interval: +6.8 ms, P<0.01) and PR‐segment (mean ΔPR‐segment: +6.4 ms, P<0.05), despite no significant changes in heart rate or P wave duration. Figure 5A illustrates representative ECGs from a guinea pig before and after IL‐6 injection, respectively, with a ΔPR‐interval of +6 ms and a ΔPR‐segment of +6 ms.
Table 2.
Changes in Electrocardiographic Parameters of Guinea Pigs (n=5) Before and After Administration of Interleukin(IL)‐6
| BASELINE | IL‐6 | P value | |
|---|---|---|---|
| Heart rate, bpm | 260.1 (9.7) | 226.9 (11.7) | 0.13 |
| P‐wave, ms | 19.7 (1.0) | 20.1 (0.9) | 0.74 |
| PR‐interval, ms | 51.4 (2.4) | 58.2 (2.7) | 0.0068* |
| PR‐segment, ms | 31.7 (2.2) | 38.1 (2.7) | 0.042* |
Values are expressed as mean±SEM.
Differences were evaluated by the two‐tail Student’s paired “t” test, or the two‐tail Wilcoxon matched pairs test.
statistically significant difference.
Figure 5. Impact of interleukin(IL)‐6 on PR‐interval and PR‐segment in guinea pigs.
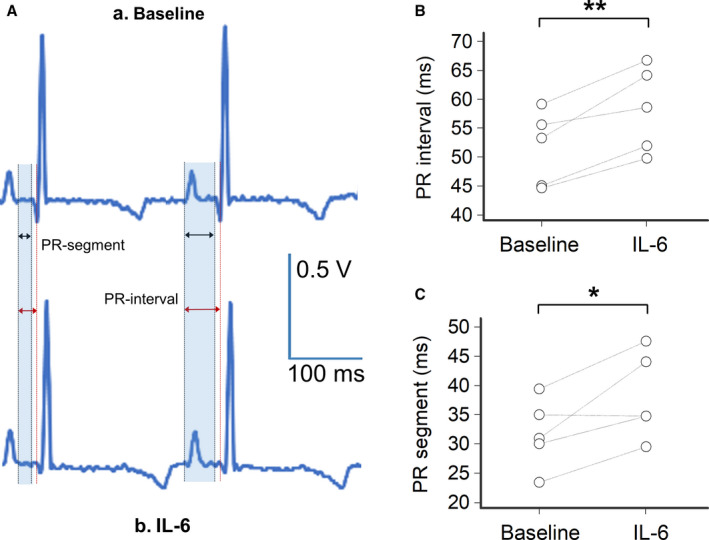
A, Representative lead‐I ECG from a female guinea pig during basal condition (A) with a normal sinus rhythm at 245 beat/min with a PR‐interval= 44 ms and a PR‐segment=23 ms, and (B) at 120 minutes after intraperitoneal injection with IL‐6/IL‐6‐receptor(R) (200 ng/mL each) in the same guinea pig, demonstrating a sinus rhythm at 219 beat/min, a PR‐interval=50 ms and a PR‐segment=29 ms; shaded areas in light blue indicate the PR‐segment and PR‐interval respectively. B, Changes in PR‐interval observed at baseline and 120 minutes after intraperitoneal injection with IL‐6/IL‐6R for each guinea pig (n=5); two‐tail paired T test, **P<0.01. C, Changes in PR‐segment observed at baseline and 120 minutes after intraperitoneal injection with IL‐6/IL‐6R for each guinea pig (n=5); two‐tail paired T test, *P<0.05.
Effect of IL‐6 on Connexin43 Protein Expression in HL‐1 Cardiomyocytes and Raw264.7 Macrophages
The direct effect of IL‐6 (100 pg/mL for 24 hours) on connexin43 protein expression was evaluated both in HL‐1 cardiomyocytes and in Raw264.7 macrophages by using western blots.
HL‐1 cell incubation with IL‐6 led to a significant decrease in connexin43 (−54%, P=0.007; Figure 6A and 6B). Although less marked, a significant IL‐6‐dependent decrease of connexin43 was also observed in cultured macrophages (−43%, P=0.008; Figure 6C and 6D).
Figure 6. Effects of IL‐6 on connexin43 protein expression in HL‐1 cardiomyocytes and Raw264.7 macrophages in culture.
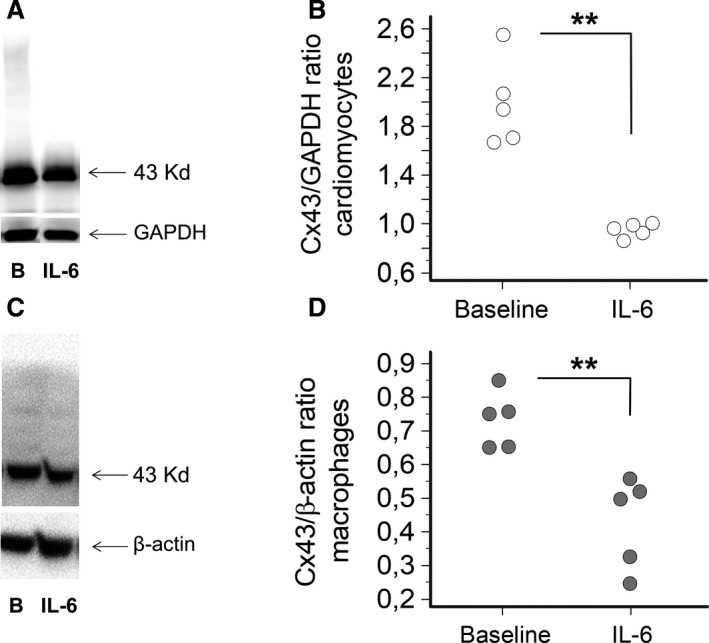
A, Western blot for connexin(Cx)43 in cardiomyocytes at baseline (B) and after treatment with IL‐6 (100 pg/mL) for 24 hours, and (B) the corresponding histograms for Cx43 normalized to GAPDH (n=5; mean percentage decrease=54%). Two‐tails Mann‐Whitney test: **P<0.01. C, Western blot for Cx43 in macrophages at baseline (B) and after treatment with IL‐6 (100 pg/mL) for 24 hours, and (D) the corresponding histograms for Cx43 normalized to β‐actin (n=5; mean percentage decrease= 43%). Two‐tails Unpaired “t” test: **P<0.01.
Discussion
The novel findings of the present study are: (1) in patients with elevated CRP levels, regardless of the specific underlying inflammatory condition, ECG indices of atrioventricular conduction are increased, but promptly normalized when inflammatory markers were reduced, particularly IL‐6; (2) in these subjects, connexin43 expression in PBMC, which correlates with that measured in the cardiac tissue, inversely associated with IL‐6 changes; (3) IL‐6 direct administration in a guinea pig model induced a significant increase of atrioventricular conduction indices; (4) in in vitro experiments, IL‐6 incubation significantly reduced connexin43 protein expression in both cardiomyocytes and macrophages in culture.
Accumulating data, mostly emerging from the recent COVID‐19 pandemic, suggest that systemic inflammation can negatively affect atrioventricular conduction, regardless of the cardiac injury, and that this alteration is associated with higher short‐term mortality rates. 29 , 31 , 32 , 33 , 34 , 35 Putative underlying mechanisms for death include the progression to severe AVBs, 3 , 6 atrioventricular dyssynchrony promoting heart failure, 4 , 5 , 6 and increased propensity for malignant arrhythmias. 7 Furthermore, previous studies identified PR‐interval prolongation as an independent predictor of cardiac mortality in large populations. 3 , 4 , 5 , 6 , 7
Although it is well known that long‐lasting inflammatory activation can promote coronary artery disease and heart failure, 59 , 60 , 61 representing 2 established causes of AVBs, 36 increasing evidence intriguingly suggests that inflammation could delay atrioventricular conduction also in the short‐term via direct cytokine‐mediated effects on the cardiac tissue. 16 , 17 , 18 By combining in vivo, ex vivo, and in vitro data from humans, animals and cell models, the present study, for the first time, demonstrated the existence of this second mechanism and its relevance in the clinical setting.
An important piece of evidence derives from our cohort of patients with elevated CRP levels due to different inflammatory processes, infective, immune‐mediated, or from other origins. In these subjects, both the PRc‐interval duration and the prevalence of PRc‐interval prolongation were found to be significantly increased when compared with controls, but rapidly normalize (median ≈10 days) after the treatment lowered CRP levels. Moreover, PRc‐interval length directly correlated over time with CRP and circulating cytokines, more strongly with IL‐6. Notably, similar changes and correlation with IL‐6 levels were also demonstrated when the PRc‐segment was specifically analyzed, thereby suggesting that inflammation‐mediated atrioventricular conduction delay is primarily due to direct effects of IL‐6 on the atrioventricular node. Importantly, the magnitude of PRc‐interval/segment prolongation was particularly relevant (>30 ms) in elderly subjects with a preexisting lower conduction reserve, in the presence of substantially high IL‐6 levels (>20 pg/mL). Based on these data, and on the evidence that PR‐interval in the general population progressively increases with age (median values from 140 ms [18–20 years] to almost 190 ms [over 80 years]), 62 it is anticipated that while usually not clinically relevant in the younger people, inflammation‐induced changes may conversely represent an important precipitant factor for higher degree AVBs in a number of elderly subjects.
In addition, the fact that such modifications are reversible in hours/days only, a timescale mismatched with structural changes due to collagen deposition and fibrosis, strongly points to functional mechanisms responsible for an electrical remodelling of the atrioventricular node. In this regard, the hypothesis that, in these patients, IL‐6 elevation can prolong the PR‐interval by down‐regulating connexin43 expression in atrioventricular node cells (myocytes and/or macrophages) is particularly attractive. Indeed, connexin43 is characterized by a very rapid physiological turnover, 13 and both in vitro and ex vivo studies demonstrated that inflammatory mediators significantly reduce its cardiac expression in the course of few hours/days. 24 , 26
In order to provide support to this pathogenic cascade, we performed ex‐vivo experiments to investigate changes of connexin43 expression in patients with inflammatory diseases. To address this point, we preliminarily evaluated a cohort of subjects that have undergone cardiac surgery and established that mRNA expression of connexin43 in PBMC and cardiac tissue (atrial and ventricular specimens, physiologically including both myocytes and macrophages) 12 are strongly correlated, thereby proving that circulating connexin43 levels are correlative of those expressed in the heart. Accordingly, we then analyzed PBMC connexin43 expression in patients with inflammatory diseases demonstrating that while it was significantly reduced in PRE conditions when compared with controls, such a difference was no longer detectable after therapeutic interventions (POST). Moreover, circulating connexin43 levels were inversely associated over time with IL‐6 concentrations, which in turn strictly correlated with both PRc‐interval and PRc‐segment duration. Overall, these data support the hypothesis that systemic inflammation can rapidly induce significant IL‐6‐mediated connexin43 remodelling throughout the heart, including in the atrioventricular node cells (myocytes and macrophages), and that these changes may contribute to PR‐interval prolongation and related clinical risks.
Further animal and in vitro experiments were performed in this study to verify the validity of our hypothesis, specifically to substantiate a role of key mediator for IL‐6 in acutely delaying atrioventricular conduction by decreasing connexin43 expression in cardiomyocytes and macrophages.
First, we established an animal model in which to investigate the direct impact of IL‐6 on PR‐interval and PR‐segment. For this purpose, we administered guinea pigs with IL‐6, along with IL‐6R to reproduce the conditions observed in vivo during inflammatory activation. In these animals, the treatment resulted in a significant, rapidly occurring (2 hours) prolongation of both PR‐interval and PR‐segment, without any appreciable change in P‐wave duration or heart rate.
Then, we investigated the in vitro effects of IL‐6 incubation on the connexin43 protein expression in cardiomyocytes and macrophages in culture. Our results showed a clear‐cut inhibitory activity of this cytokine in both the cell types, particularly in myocytes.
Altogether, these data provide additional mechanistic support to the view that IL‐6 can promote acute worsening of atrioventricular conduction at least in part by inducing remodelling of the gap‐junctions containing connexin43, which couple adjacent myocytes, as well as myocytes and macrophages, at the atrioventricular node (Figure 7).
Figure 7. IL‐6‐induced PR‐interval prolongation: proposed mechanism of action.

Systemic inflammatory activation, by increasing circulating IL‐6 levels could promote PR‐interval prolongation by affecting the electric properties of different cells in the atrioventricular (AV) node. The results of the present study suggest that these changes may consist, at least in part, in a down‐regulation of the gap‐junctions containing connexin43 (Cx43), which couple adjacent myocytes, as well as myocytes and macrophages, at the AV node.
The study has strengths and limitations. One important strength is the translational methodology used, by which human data were combined with basic/translational mechanistic experiments to dissect a specific clinical problem.
On the other hand, a possible limitation could be the lack of direct data from the atrioventricular node to confirm that the observed gap‐junction remodelling actually occurs in this specific part of the heart. In fact, while in the ex vivo study we established a correlation between connexin43 expression in atrial and ventricular specimens and PBMC, such an evaluation did not specifically include any sample from the atrioventricular node, as the collection of this tissue is not feasible in the clinical practice. Nevertheless, the evidence that such a correlation is present both above (atria) and below (ventricles) the atrioventricular node, ie heart regions in which likewise connexin43 is well‐expressed and both myocytes and macrophages are present, makes it highly probable that these findings can be extended to the atrioventricular node.
In addition, we did not explore the potential in vivo IL‐6‐dependent changes in connexin43 expression directly in the atrioventricular node by western blotting. However, it was not a possible approach because of anatomical considerations, specifically the need of significant amounts of atrioventricular node tissue to perform these experiments. Notably, while connexin43 plays a pivotal role in facilitating atrioventricular conduction, its expression is restricted to a specific part of the node only (the distal part), thereby amplifying the inherent technical challenge due to the small dimension of the guinea pig atrioventricular node.
Another possible limitation is that more biologically relevant cells could be used for the cellular studies, specifically human induced pluripotent stem cells (hiPSCs)‐derived cardiomyocytes differentiated into nodal cells. 63 However, their usability as a human adult cardiomyocyte model was limited by their functional immaturity, 64 as well as by the very small amount of nodal‐like cells among hiPSCs‐derived cardiomyocytes (less than 10%, with a shift into an atrial/ventricular‐like phenotype in the range of 7‐to‐95 days during development). 65 , 66 Moreover, there is no evidence to date that hiPSCs‐derived cardiomyocytes express larger amount of connexin43 than HL‐1 cells. 67
Finally, accumulating evidence in the recent years demonstrated that IL‐6 can induce significant cardiac electrical remodeling also independently of macrophages, by affecting trafficking and function of several ion channels in cardiac myocytes. For example, it has been demonstrated that IL‐6 can markedly blunt hERG channel protein levels and inhibit IKr current in guinea pig ventricular myocytes, 68 as well as significantly decrease connexin40 expression in mouse cardiomyocytes. 24 Given that both these channels are expressed in the atrioventricular node 69 , 70 and loss‐of‐function mutations in the corresponding encoding genes can cause AVB, 8 , 71 it is likely that IL‐6‐mediated delaying effects on atrioventricular conduction observed in vivo result from multiple and synergistically operating electrophysiological mechanisms, among which connexin43 downregulation in cardiomyocytes and macrophages is included (not necessarily it being the most important one). In this complex scenario, also connexin45 could be involved, given its important contributing role in atrioventricular node conduction. 9 , 72 Although this subject warrants further investigation, in this study we specifically focused on the role of connexin43 and for this reason the potential effects of IL‐6 on other ion channels and connexins expressed in the atrioventricular node was beyond the scope of our work.
In conclusion, this study for the first time provides evidence that systemic inflammation via IL‐6 elevation can acutely worsen atrioventricular conduction. Moreover, we demonstrate that these changes are associated with evident cardiac electrical remodeling, and that IL‐6‐induced down‐regulation of connexin43 is a mechanistic pathway putatively involved in the process. Though promptly reversible, these electrophysiological alterations may significantly increase the risk of severe AVBs and other adverse clinical events related to the prolongation of atrioventricular conduction times and, consequently, the risk of cardiac death in the presence of active inflammatory processes.
While our data can provide a possible explanatory mechanism contributing to COVID‐19‐associated AVBs, on the other hand, they more in general support the hypothesis that inflammatory activation is per se able to delay atrioventricular conduction, regardless the specific underlying disease involved. From a teleological point of view, it is intriguing to speculate that cytokine‐induced PR‐interval prolongation could be a protective mechanism, unless higher grade AVB develops. In fact, slowing atrioventricular nodal conduction in the face of cytokine‐induced tachycardia 16 , 19 , 73 could increase the diastolic interval and allow for maintenance of cardiac output.
The study also substantiates the recommendation for the clinician to always carefully evaluate the possible impact of a systemic inflammatory state on atrioventricular conduction and related complications, particularly when it is of high grade and occurs in elderly patients with history of cardiac disease and/or taking drugs negatively affecting the atrioventricular node function. In these subjects, a prompt management of intercurrent infections or other concomitant inflammatory processes may be an important therapeutic measure, although often disregarded. As such, it is an intriguing perspective that anti‐cytokine therapy, specifically targeting the IL‐6, may be an innovative anti‐arrhythmic short‐term approach for severe AVBs occurring/worsening during active inflammatory states, possibly sparing or delaying pacemaker implantation.
Sources of Funding
This work was supported by Ministero dell’Istruzione, dell’Università e della Ricerca (MIUR), Progetti di Rilevante Interesse Nazionale (PRIN), and Bando 2017, protocollo 2017XZMBYX, and by a Merit Review grant I01 BX002137 from Biomedical Laboratory Research & Development Service of Veterans Affairs Office of Research and Development (to M.B).
Disclosures
Dr Pietro Enea Lazzerini received a grant from Roche Italia S.p.A. outside the submitted work, in 2018. The remaining authors have no disclosures to report.
Supporting information
Tables S1–S5
Figure S1
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.121.022095
For Sources of Funding and Disclosures, see page 15.
References
- 1. Mymin D, Mathewson FA, Tate RB, Manfreda J. The natural history of primary first‐degree atrioventricular heart block. N Engl J Med. 1986;315:1183–1187. doi: 10.1056/NEJM198611063151902 [DOI] [PubMed] [Google Scholar]
- 2. Aro AL, Anttonen O, Kerola T, Junttila MJ, Tikkanen JT, Rissanen HA, Reunanen A, Huikuri HV. Prognostic significance of prolonged PR interval in the general population. Eur Heart J. 2014;35:123–129. doi: 10.1093/eurheartj/eht176 [DOI] [PubMed] [Google Scholar]
- 3. Cheng S, Keyes MJ, Larson MG, McCabe EL, Newton‐Cheh C, Levy D, Benjamin EJ, Vasan RS, Wang TJ. Long‐term outcomes in individuals with prolonged PR interval or first‐degree atrioventricular block. JAMA. 2009;301:2571–2577. doi: 10.1001/jama.2009.888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Crisel RK, Farzaneh‐Far R, Na B, Whooley MA. First‐degree atrioventricular block is associated with heart failure and death in persons with stable coronary artery disease: data from the heart and soul study. Eur Heart J. 2011;32:1875–1880. doi: 10.1093/eurheartj/ehr139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kwok CS, Rashid M, Beynon R, Barker D, Patwala A, Morley‐Davies A, Satchithananda D, Nolan J, Myint PK, Buchan I, et al. Prolonged PR interval, first‐degree heart block and adverse cardiovascular outcomes: a systematic review and meta‐analysis. Heart. 2016;102:672–680. doi: 10.1136/heartjnl-2015-308956. [DOI] [PubMed] [Google Scholar]
- 6. Rasmussen PV, Nielsen JB, Skov MW, Pietersen A, Graff C, Lind B, Struijk JJ, Olesen MS, Haunsø S, Køber L, et al. Electrocardiographic PR interval duration and cardiovascular risk: results from the Copenhagen ECG study. Can J Cardiol. 2017;33:674–681. doi: 10.1016/j.cjca.2017.02.015 [DOI] [PubMed] [Google Scholar]
- 7. Li YQ, Zhao S, Chen KP, Su YG, Hua W, Chen SL, Liang ZG, Xu W, Dai Y, Fan XH, et al. Heart rate‐adjusted PR as a prognostic marker of long‐term ventricular arrhythmias and cardiac death in ICD/CRT‐D recipients. J Geriatr Cardiol. 2019;16:259–264. doi: 10.11909/j.issn.1671-5411.2019.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Delmar M, Makita N. Cardiac connexins, mutations and arrhythmias. Curr Opin Cardiol. 2012;27:236–241. doi: 10.1097/HCO.0b013e328352220e [DOI] [PubMed] [Google Scholar]
- 9. Ellinor PT, Jameson HS. Connexin‐45 as a new gene underlying syndromic AV block. J Am Coll Cardiol. 2017;70:371–372. doi: 10.1016/j.jacc.2017.06.003 [DOI] [PubMed] [Google Scholar]
- 10. Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, Hucker WJ, Wülfers EM, Seemann G, Courties G, et al. Macrophages facilitate electrical conduction in the heart. Cell. 2017;169:510–522.e520. doi: 10.1016/j.cell.2017.03.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rosenthal N. A guardian of the heartbeat. N Engl J Med. 2017;377:84–86. doi: 10.1056/NEJMcibr1705327 [DOI] [PubMed] [Google Scholar]
- 12. Swirski FK, Nahrendorf M. Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat Rev Immunol. 2018;18:733–744. doi: 10.1038/s41577-018-0065-8 [DOI] [PubMed] [Google Scholar]
- 13. Saffitz JE, Laing JG, Yamada KA. Connexin expression and turnover: implications for cardiac excitability. Circ Res. 2000;86:723–728. doi: 10.1161/01.RES.86.7.723 [DOI] [PubMed] [Google Scholar]
- 14. Severs NJ, Bruce AF, Dupont E, Rothery S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res. 2008;80:9–19. doi: 10.1093/cvr/cvn133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu YF, Chen YJ, Lin YJ, Chen SA. Inflammation and the pathogenesis of atrial fibrillation. Nat Rev Cardiol. 2015;12:230–243. doi: 10.1038/nrcardio.2015.2 [DOI] [PubMed] [Google Scholar]
- 16. Lazzerini PE, Capecchi PL, Laghi‐Pasini F. Systemic inflammation and arrhythmic risk: lessons from rheumatoid arthritis. Eur Heart J. 2017;38:1717–1727. doi: 10.1093/eurheartj/ehw208 [DOI] [PubMed] [Google Scholar]
- 17. Lazzerini PE, Capecchi PL, El‐Sherif N, Laghi‐Pasini F, Boutjdir M. Emerging arrhythmic risk of autoimmune and inflammatory cardiac channelopathies. J Am Heart Assoc. 2018;7:e010595. doi: 10.1161/JAHA.118.010595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lazzerini PE, Laghi‐Pasini F, Boutjdir M, Capecchi PL. Cardioimmunology of arrhythmias: the role of autoimmune and inflammatory cardiac channelopathies. Nat Rev Immunol. 2019;19:63–64. doi: 10.1038/s41577-018-0098-z [DOI] [PubMed] [Google Scholar]
- 19. Lazzerini PE, Acampa M, Laghi‐Pasini F, Bertolozzi I, Finizola F, Vanni F, Natale M, Bisogno S, Cevenini G, Cartocci A, et al. Cardiac arrest risk during acute infections: systemic inflammation directly prolongs QTC interval via cytokine‐mediated effects on potassium channel expression. Circ Arrhythm Electrophysiol. 2020;13:e008627. doi: 10.1161/CIRCEP.120.008627 [DOI] [PubMed] [Google Scholar]
- 20. Lazzerini PE, Laghi‐Pasini F, Boutjdir M, Capecchi PL. Commentary: systemic effects of IL‐17 in inflammatory arthritis. Front Cardiovasc Med. 2019;6:183. doi: 10.3389/fcvm.2019.00183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sawaya SE, Rajawat YS, Rami TG, Szalai G, Price RL, Sivasubramanian N, Mann DL, Khoury DS. Downregulation of connexin40 and increased prevalence of atrial arrhythmias in transgenic mice with cardiac‐restricted overexpression of tumor necrosis factor. Am J Physiol Heart Circ Physiol. 2007;292:H1561–H1567. doi: 10.1152/ajpheart.00285.2006 [DOI] [PubMed] [Google Scholar]
- 22. Liew R, Khairunnisa K, Gu Y, Tee N, Yin NO, Naylynn TM, Moe KT. Role of tumor necrosis factor‐α in the pathogenesis of atrial fibrosis and development of an arrhythmogenic substrate. Circ J. 2013;77:1171–1179. doi: 10.1253/circj.CJ-12-1155 [DOI] [PubMed] [Google Scholar]
- 23. Sun Z, Zhou D, Xie X, Wang S, Wang Z, Zhao W, Xu H, Zheng L. Cross‐talk between macrophages and atrial myocytes in atrial fibrillation. Basic Res Cardiol. 2016;111:63. doi: 10.1007/s00395-016-0584-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lazzerini PE, Laghi‐Pasini F, Acampa M, Srivastava U, Bertolozzi I, Giabbani B, Finizola F, Vanni F, Dokollari A, Natale M, et al. Systemic inflammation rapidly induces reversible atrial electrical remodeling: the role of interleukin‐6‐mediated changes in connexin expression. J Am Heart Assoc. 2019;8:e011006. doi: 10.1161/JAHA.118.011006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Francis Stuart SD, De Jesus NM, Lindsey ML, Ripplinger CM. The crossroads of inflammation, fibrosis, and arrhythmia following myocardial infarction. J Mol Cell Cardiol. 2016;91:114–122. doi: 10.1016/j.yjmcc.2015.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fernandez‐Cobo M, Gingalewski C, Drujan D, De Maio A. Downregulation of connexin 43 gene expression in rat heart during inflammation. The role of tumour necrosis factor. Cytokine. 1999;11:216–224. doi: 10.1006/cyto.1998.0422 [DOI] [PubMed] [Google Scholar]
- 27. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, HLH Across Speciality Collaboration UK . Covid‐19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033–1034. doi: 10.1016/S0140-6736(20)30628-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lazzerini PE, Laghi‐Pasini F, Acampa M, Boutjdir M, Leopoldo CP. IL‐6 (interleukin 6) blockade and heart rate corrected QT interval prolongation in COVID‐19. Circ Arrhythm Electrophysiol. 2020;13:e008791. doi: 10.1161/CIRCEP.120.008791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bhatla A, Mayer MM, Adusumalli S, Hyman MC, Oh E, Tierney A, Moss J, Chahal AA, Anesi G, Denduluri S, et al. COVID‐19 and cardiac arrhythmias. Heart Rhythm. 2020;17:1439–1444. doi: 10.1016/j.hrthm.2020.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Coromilas EJ, Kochav S, Goldenthal I, Biviano A, Garan H, Goldbarg S, Kim JH, Yeo I, Tracy C, Ayanian S, et al. Worldwide survey of COVID‐19 associated arrhythmias. Circ Arrhythm Electrophysiol. 2021;14:e009458. doi: 10.1161/CIRCEP.120.009458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kochav SM, Coromilas E, Nalbandian A, Ranard LS, Gupta A, Chung MK, Gopinathannair R, Biviano AB, Garan H, Wan EY. Cardiac arrhythmias in COVID‐19 infection. Circ Arrhythm Electrophysiol. 2020;13:e008719. doi: 10.1161/CIRCEP.120.008719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gupta MD, Qamar A, Mp G, Safal S, Batra V, Basia D, Mandal SK, Yusuf J, Mukhopadhyay S, Bansal A. Bradyarrhythmias in patients with covid‐19: a case series. Indian Pacing Electrophysiol J. 2020;20:211–212. doi: 10.1016/j.ipej.2020.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chinitz JS, Goyal R, Harding M, Veseli G, Gruberg L, Jadonath R, Maccaro P, Gandotra P, Ong L, Epstein LM. Bradyarrhythmias in patients with COVID‐19: marker of poor prognosis? Pacing Clin Electrophysiol. 2020;43:1199–1204. doi: 10.1111/pace.14042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pavri BB, Kloo J, Farzad D, Riley JM. Behavior of the PR interval with increasing heart rate in patients with COVID‐19. Heart Rhythm. 2020;17:1434–1438. doi: 10.1016/j.hrthm.2020.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moey MYY, Sengodan PM, Shah N, McCallen JD, Eboh O, Nekkanti R, Carabello BA, Naniwadekar AR. Electrocardiographic changes and arrhythmias in hospitalized patients with COVID‐19. Circ Arrhythm Electrophysiol. 2020;13:e009023. doi: 10.1161/CIRCEP.120.009023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kusumoto FM, Schoenfeld MH, Barrett C, Edgerton JR, Ellenbogen KA, Gold MR, Goldschlager NF, Hamilton RM, Joglar JA, Kim RJ, et al. 2018 ACC/AHA/HRS guideline on the evaluation and management of patients with bradycardia and cardiac conduction delay: executive summary: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines, and the heart rhythm society. Circulation. 2019;140:e333–e381. doi: 10.1161/CIR.0000000000000627 [DOI] [PubMed] [Google Scholar]
- 37. Malik M, Hnatkova K, Sisakova M, Schmidt G. Subject‐specific heart rate dependency of electrocardiographic QT, PQ, and QRS intervals. J Electrocardiol. 2008;41:491–497. doi: 10.1016/j.jelectrocard.2008.06.022 [DOI] [PubMed] [Google Scholar]
- 38. Toman O, Hnatkova K, Smetana P, Huster KM, Šišáková M, Barthel P, Novotný T, Schmidt G, Malik M. Physiologic heart rate dependency of the PQ interval and its sex differences. Sci Rep. 2020;10:2551. doi: 10.1038/s41598-020-59480-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Soliman EZ, Rautaharju PM. Heart rate adjustment of PR interval in middle‐aged and older adults. J Electrocardiol. 2012;45:66–69. doi: 10.1016/j.jelectrocard.2011.06.003 [DOI] [PubMed] [Google Scholar]
- 40. Soliman EZ, Cammarata M, Li Y. Explaining the inconsistent associations of PR interval with mortality: the role of P‐duration contribution to the length of PR interval. Heart Rhythm. 2014;11:93–98. doi: 10.1016/j.hrthm.2013.10.003 [DOI] [PubMed] [Google Scholar]
- 41. Dilaveris PE, Färbom P, Batchvarov V, Ghuran A, Malik M. Circadian behavior of P‐wave duration, P ‐wave area, and PR interval in healthy subjects. Ann Noninvasive Electrocardiol. 2001;6:92–97. doi: 10.1111/j.1542-474X.2001.tb00092.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gao GE, Brahmanandam V, Raicu M, Gu L, Zhou LI, Kasturirangan S, Shah A, Negi SI, Wood MR, Desai AA, et al. Enhanced risk profiling of implanted defibrillator shocks with circulating SCN5A mRNA splicing variants: a pilot trial. J Am Coll Cardiol. 2014;63:2261–2269. doi: 10.1016/j.jacc.2014.02.588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Formigli L, Ibba‐Manneschi L, Perna AM, Pacini A, Polidori L, Nediani C, Modesti PA, Nosi D, Tani A, Celli A, et al. Altered Cx43 expression during myocardial adaptation to acute and chronic volume overloading. Histol Histopathol. 2003;18:359–369. [DOI] [PubMed] [Google Scholar]
- 44. Ai Z, Fischer A, Spray DC, Brown AM, Fishman GI. Wnt‐1 regulation of connexin43 in cardiac myocytes. J Clin Invest. 2000;105:161–171. doi: 10.1172/JCI7798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Salameh A, Krautblatter S, Karl S, Blanke K, Gomez DR, Dhein S, Pfeiffer D, Janousek J. The signal transduction cascade regulating the expression of the gap junction protein connexin43 by beta‐adrenoceptors. Br J Pharmacol. 2009;158:198–208. doi: 10.1111/j.1476-5381.2009.00344.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oviedo‐Orta E, Hoy T, Evans WH. Intercellular communication in the immune system: differential expression of connexin40 and 43, and perturbation of gap junction channel functions in peripheral blood and tonsil human lymphocyte subpopulations. Immunology. 2000;99:578–590. doi: 10.1046/j.1365-2567.2000.00991.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Claycomb WC, Lanson NA, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ. Hl‐1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA. 1998;95:2979–2984. doi: 10.1073/pnas.95.6.2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chen Y, Qiao X, Zhang L, Li X, Liu Q. Apelin‐13 regulates angiotensin II‐induced Cx43 downregulation and autophagy via the AMPK/mTOR signaling pathway in HL‐1 cells. Physiol Res. 2020;69:813–822. doi: 10.33549/physiolres.934488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dai W, Chao X, Li S, Zhou S, Zhong G, Jiang Z. Long noncoding RNA HOTAIR functions as a competitive endogenous RNA to regulate connexin43 remodeling in atrial fibrillation by sponging microRNA‐613. Cardiovasc Ther. 2020;2020:1–11. doi: 10.1155/2020/5925342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou E, Zhang T, Bi C, Wang C, Zhang Z. Vascular smooth muscle cell phenotypic transition regulates gap junctions of cardiomyocyte. Heart Vessels. 2020;35:1025–1035. doi: 10.1007/s00380-020-01602-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Epelman S, Lavine K, Beaudin A, Sojka D, Carrero J, Calderon B, Brija T, Gautier E, Ivanov S, Satpathy A, et al. Embryonic and adult‐derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40:91–104. doi: 10.1016/j.immuni.2013.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yap J, Cabrera‐Fuentes HA, Irei J, Hausenloy DJ, Boisvert WA. Role of macrophages in cardioprotection. Int J Mol Sci. 2019;20. doi: 10.3390/ijms20102474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ibi M, Horie S, Kyakumoto S, Chosa N, Yoshida M, Kamo M, Ohtsuka M, Ishisaki A. Cell‐cell interactions between monocytes/macrophages and synoviocyte‐like cells promote inflammatory cell infiltration mediated by augmentation of MCP‐1 production in temporomandibular joint. Biosci Rep. 2018;38. doi: 10.1042/BSR20171217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen Y, Fu WL, Gan XD, Xing WW, Xia WR, Zou MJ, Liu Q, Wang YY, Zhang C, Xu DG. SAK‐HV promotes RAW264.7 cells migration mediated by MCP‐1 via JNK and NF‐κB pathways. Int J Biol Sci. 2018;14:1993–2002. doi: 10.7150/ijbs.27459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Verweij N, Mateo Leach I, van den Boogaard M, van Veldhuisen DJ, Christoffels VM, Hillege HL, van Gilst WH, Barnett P, de Boer RA, van der Harst P, et al. Genetic determinants of P wave duration and PR segment. Circ Cardiovasc Genet. 2014;7:475–481. doi: 10.1161/CIRCGENETICS.113.000373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wong CW, Christen T, Kwak BR. Connexins in leukocytes: shuttling messages? Cardiovasc Res. 2004;62:357–367. doi: 10.1016/j.cardiores.2003.12.015 [DOI] [PubMed] [Google Scholar]
- 57. Mendoza‐Naranjo A, Bouma G, Pereda C, Ramírez M, Webb KF, Tittarelli A, López MN, Kalergis AM, Thrasher AJ, Becker DL, et al. Functional gap junctions accumulate at the immunological synapse and contribute to T cell activation. J Immunol. 2011;187:3121–3132. doi: 10.4049/jimmunol.1100378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hamlin RL, Kijtawornrat A, Keene BW, Hamlin DM. QT and RR intervals in conscious and anesthetized guinea pigs with highly varying RR intervals and given QTc‐lengthening test articles. Toxicol Sci. 2003;76:437–442. doi: 10.1093/toxsci/kfg254 [DOI] [PubMed] [Google Scholar]
- 59. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, Tokgözoğlu L, Lewis EF. Atherosclerosis. Nat Rev Dis Primers. 2019;5:56. doi: 10.1038/s41572-019-0106-z [DOI] [PubMed] [Google Scholar]
- 60. Briasoulis A, Androulakis E, Christophides T, Tousoulis D. The role of inflammation and cell death in the pathogenesis, progression and treatment of heart failure. Heart Fail Rev. 2016;21:169–176. doi: 10.1007/s10741-016-9533-z [DOI] [PubMed] [Google Scholar]
- 61. Lazzerini PE, Hamilton RM, Boutjdir M. Editorial: cardioimmunology: inflammation and immunity in cardiovascular disease. Front Cardiovasc Med. 2019;6:181. doi: 10.3389/fcvm.2019.00181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. van der Ende MY, Siland JE, Snieder H, van der Harst P, Rienstra M. Population‐based values and abnormalities of the electrocardiogram in the general Dutch population: the lifelines cohort study. Clin Cardiol. 2017;40:865–872. doi: 10.1002/clc.22737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Protze SI, Liu J, Nussinovitch U, Ohana L, Backx PH, Gepstein L, Keller GM. Sinoatrial node cardiomyocytes derived from human pluripotent cells function as a biological pacemaker. Nat Biotechnol. 2017;35:56–68. doi: 10.1038/nbt.3745 [DOI] [PubMed] [Google Scholar]
- 64. Koivumäki JT, Naumenko N, Tuomainen T, Takalo J, Oksanen M, Puttonen KA, Lehtonen Š, Kuusisto J, Laakso M, Koistinaho J, et al. Structural immaturity of human iPSC‐derived cardiomyocytes. Front Physiol. 2018;9:80. doi: 10.3389/fphys.2018.00080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ichimura H, Kadota S, Kashihara T, Yamada M, Ito K, Kobayashi H, Tanaka Y, Shiba N, Chuma S, Tohyama S, et al. Increased predominance of the matured ventricular subtype in embryonic stem cell‐derived cardiomyocytes in vivo. Sci Rep. 2020;10:11883. doi: 10.1038/s41598-020-68373-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ben‐Ari M, Naor S, Zeevi‐Levin N, Schick R, Ben Jehuda R, Reiter I, Raveh A, Grijnevitch I, Barak O, Rosen MR, et al. Developmental changes in electrophysiological characteristics of human‐induced pluripotent stem cell‐derived cardiomyocytes. Heart Rhythm. 2016;13:2379–2387. doi: 10.1016/j.hrthm.2016.08.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wells SP, Waddell HM, Sim CB, Lim SY, Bernasochi GB, Pavlovic D, Kirchhof P, Porrello ER, Delbridge LMD, Bell JR. Cardiomyocyte functional screening: interrogating comparative electrophysiology of high‐throughput model cell systems. Am J Physiol Cell Physiol. 2019;317:C1256–C1267. doi: 10.1152/ajpcell.00306.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Aromolaran AS, Srivastava U, Alí A, Chahine M, Lazaro D, El‐Sherif N, Capecchi PL, Laghi‐Pasini F, Lazzerini PE, Boutjdir M. Interleukin‐6 inhibition of hERG underlies risk for acquired long QT in cardiac and systemic inflammation. PLoS One. 2018;13:e0208321. doi: 10.1371/journal.pone.0208321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dobrzynski H, Anderson RH, Atkinson A, Borbas Z, D'Souza A, Fraser JF, Inada S, Logantha SJRJ, Monfredi O, Morris GM, et al. Structure, function and clinical relevance of the cardiac conduction system, including the atrioventricular ring and outflow tract tissues. Pharmacol Ther. 2013;139:260–288. doi: 10.1016/j.pharmthera.2013.04.010 [DOI] [PubMed] [Google Scholar]
- 70. Bartos DC, Grandi E, Ripplinger CM. Ion channels in the heart. Compr Physiol. 2015;5:1423–1464. doi: 10.1002/cphy.c140069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chevalier P, Bellocq C, Millat G, Piqueras E, Potet F, Schott JJ, Baró I, Lemarec H, Barhanin J, Rousson R, et al. Torsades de pointes complicating atrioventricular block: evidence for a genetic predisposition. Heart Rhythm. 2007;4:170–174. doi: 10.1016/j.hrthm.2006.10.004 [DOI] [PubMed] [Google Scholar]
- 72. Temple IP, Inada S, Dobrzynski H, Boyett MR. Connexins and the atrioventricular node. Heart Rhythm. 2013;10:297–304. doi: 10.1016/j.hrthm.2012.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lazzerini PE, Capecchi PL, Laghi‐Pasini F. Long QT syndrome: an emerging role for inflammation and immunity. Front Cardiovasc Med. 2015;2:26. doi: 10.3389/fcvm.2015.00026 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S5
Figure S1