

Abstract
Background
Inflammation in epicardial adipose tissue (EAT) may contribute to coronary atherosclerosis. Myocardial bridge is a congenital anomaly in which the left anterior descending coronary artery takes a “tunneled” course under a bridge of myocardium: while atherosclerosis develops in the proximal left anterior descending coronary artery, the bridged portion is spared, highlighting the possibility that geographic separation from inflamed EAT is protective. We tested the hypothesis that inflammation in EAT was related to atherosclerosis by comparing EAT from proximal and bridge depots in individuals with myocardial bridge and varying degrees of atherosclerotic plaque.
Methods and Results
Maximal plaque burden was quantified by intravascular ultrasound, and inflammation was quantified by pericoronary EAT signal attenuation (pericoronary adipose tissue attenuation) from cardiac computed tomography scans. EAT overlying the proximal left anterior descending coronary artery and myocardial bridge was harvested for measurement of mRNA and microRNA (miRNA) using custom chips by Nanostring; inflammatory cytokines were measured in tissue culture supernatants. Pericoronary adipose tissue attenuation was increased, indicating inflammation, in proximal versus bridge EAT, in proportion to atherosclerotic plaque. Individuals with moderate‐high versus low plaque burden exhibited greater expression of inflammation and hypoxia genes, and lower expression of adipogenesis genes. Comparison of gene expression in proximal versus bridge depots revealed differences only in participants with moderate‐high plaque: inflammation was higher in proximal and adipogenesis lower in bridge EAT. Secreted inflammatory cytokines tended to be higher in proximal EAT. Hypoxia‐inducible factor 1a was highly associated with inflammatory gene expression. Seven miRNAs were differentially expressed by depot: 3192‐5P, 518D‐3P, and 532‐5P were upregulated in proximal EAT, whereas miR 630, 575, 16‐5P, and 320E were upregulated in bridge EAT. miR 630 correlated directly with plaque burden and inversely with adipogenesis genes. miR 3192‐5P, 518D‐3P, and 532‐5P correlated inversely with hypoxia/oxidative stress, peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha (PCG1a), adipogenesis, and angiogenesis genes.
Conclusions
Inflammation is specifically elevated in EAT overlying atherosclerotic plaque, suggesting that EAT inflammation is caused by atherogenic molecular signals, including hypoxia‐inducible factor 1a and/or miRNAs in an “inside‐to‐out” relationship. Adipogenesis was suppressed in the bridge EAT, but only in the presence of atherosclerotic plaque, supporting cross talk between the vasculature and EAT. miR 630 in EAT, expressed differentially according to burden of atherosclerotic plaque, and 3 other miRNAs appear to inhibit key genes related to adipogenesis, angiogenesis, hypoxia/oxidative stress, and thermogenesis in EAT, highlighting a role for miRNA in mediating cross talk between the coronary vasculature and EAT.
Keywords: atherogenesis, atherosclerosis, epicardial fat, inflammation, myocardial bridging
Subject Categories: Coronary Artery Disease, Vascular Disease
Nonstandard Abbreviations and Acronyms
- CCTA
cardiac computed tomography angiography
- EAT
epicardial adipose tissue
- FFA
free fatty acid
- HIF
hypoxia‐inducible factor
- IVUS
intravascular ultrasound
- MB
myocardial bridge
- MCP‐1
monocyte chemoattractant protein 1
- MPB
maximal plaque burden
- PCAT
pericoronary adipose tissue attenuation
Clinical Perspective
What Is New?
In patients with myocardial bridge, the left anterior descending coronary artery tunnels through the myocardium and remains free of atherosclerotic plaque, whereas the left anterior descending coronary artery proximal to the myocardial bridge develops atherosclerotic plaque: we noted increased inflammation in the epicardial adipose tissue (EAT) proximal to the myocardial bridge but not over the myocardial bridge: inflammation was proportional to degree of atherosclerotic plaque, suggesting that geographic location per se was not the basis for increased inflammation, and that atherogenic molecular signals from the intravascular plaque may have contributed to EAT inflammation in an “inside‐to‐out” direction, particularly given the absence of physical barrier between the coronary artery and the EAT.
We identified 4 novel EAT microRNAs (3192‐5P, 518D‐3P, 532‐5P, and 630) that inhibit adipogenesis, based on strong inverse relationships with mRNA expression for adipose differentiation genes in the same tissue sample: of these 4, miR630 correlated with maximal plaque burden, pointing to a novel role for microRNAs in mediating cross talk between coronary atherosclerosis and adipose cell function in human EAT.
What Are the Clinical Implications?
In clinical and epidemiological studies, an increase in the volume or thickness of human EAT is associated with increased coronary atheroma burden and the occurrence of major adverse coronary events.
Our data support EAT inflammation as a risk marker for atherosclerotic coronary artery disease; if inflammatory molecules produced in EAT also diffuse inside the coronary to contribute to atherogenesis, EAT inflammation might be considered a coronary artery disease risk factor.
Select EAT microRNAs represent potential therapeutic targets to prevent the adverse effects of atherosclerotic plaque on EAT, and merit investigation as inhibitors of adipogenesis in other fat depots.
In clinical and epidemiological studies, an increase in the volume or thickness of human epicardial adipose tissue (EAT) is associated with increased coronary atheroma burden and the occurrence of major adverse coronary events. 1 , 2 , 3 EAT adjacent to severe atherosclerotic coronary artery disease (CAD) in patients undergoing coronary artery bypass grafting, compared with EAT abutting nonatherosclerotic coronaries in patients undergoing aortic or mitral valve replacement, shows increased mRNA expression of inflammatory cytokine, vasoconstrictor, prothrombotic, angiogenic, and pro‐oxidant and antioxidant enzyme genes that are known to play a role in inflammation‐mediated atherogenesis, 4 , 5 , 6 , 7 as well as increased density of T‐lymphocytes and proinflammatory M1 macrophages, as quantified by immunohistochemistry. 8 More important, hematoxylin and eosin histological features of human coronary arteries embedded in EAT show no fibrous layer next to the coronary adventitia that would act as a physical barrier to the passage of molecules between EAT and the coronary intima and media; furthermore, there is evidence that microcirculation connects epicardial fat and coronary wall via vasa vasora. 9 Together, these physical characteristics make it highly plausible that there is direct cross talk between the epicardial fat and coronary arteries. This is the basis for the hypothesis that EAT contributes to underlying CAD for which a putative mechanism involves bidirectional molecular and cellular cross talk across the coronary wall, beginning as signaling from intimal plaque to EAT inside‐out followed by outside‐in, as originally suggested by Mazurek et al 10 and recently supported experimentally by Antonopoulos et al. 11 Despite this intriguing hypothesis and the well‐established correlations between EAT mass and inflammation with CAD, there is no direct evidence in humans that EAT plays a causal role in the pathogenesis of CAD, and recently, some authors report that coronary disease is not associated with robust alterations in inflammatory gene expression in human epicardial fat. 12 That said, surgical removal of EAT overlying a segment of the left anterior descending branch of the left main coronary artery (left anterior descending coronary artery [LAD]) in an Ossabaw pig model of early‐stage CAD inhibits plaque progression. 13 Confirmatory experiments have not been reported in other porcine species or other animal models with CAD.
The myocardial bridge (MB) is a congenital anomaly in which a segment of the epicardial coronary artery, almost always the LAD, takes a “tunneled” intramuscular course under a bridge of overlying myocardium. 14 This causes vessel compression during systole, impacting vessel size in early‐mid diastole, resulting in hemodynamic changes in coronary blood flow that may be associated with angina pectoris, myocardial ischemia, acute coronary syndrome, left ventricular dysfunction, arrhythmias, and sudden cardiac death. 15 , 16 Atherosclerosis typically develops in the epicardial LAD proximal to the bridge, with maximum plaque burden usually found about 20 mm proximal to bridge entrance into the myocardium. 17 The tunneled coronary segment is spared. 18 Although this interesting phenomenon may result from turbulence and interrupted laminar flow in the proximal segment of the vessel as the myocardium compresses the MB segment of the vessel during systole, 19 , 20 an alternative explanation is the direct contact between the proximal segment and EAT, in contrast to the geographic separation of the tunneled segment from the EAT, thus preventing the inflammatory cascade that results from free fatty acid (FFA) stimulation of Toll‐like receptors and resultant proinflammatory cytokine secretion, macrophage activation, and endothelial dysfunction. 21 Alternatively, endothelial dysfunction and/or atheroma in the proximal LAD could potentiate adipocyte dysfunction, because exposure to inflammatory cytokines has been linked to impaired adipocyte differentiation/fat storage, decreased adiponectin secretion, and proinflammatory phenotype, 22 , 23 , 24 , 25 which, in turn, would further potentiate development of atherosclerotic plaque.
The MB could thus be a useful experiment of nature by which to investigate whether atheroma in the coronary arteries is related to inflammation or other biological changes in the adjacent EAT. We hypothesized that EAT overlying an atheromatous vessel proximal to the MB would be inflamed compared with EAT overlying the MB, wherein a nonatheromatous portion of the same vessel tunnels through the myocardium. We further investigated whether inflammation in the EAT overlying the proximal vessel differed according to the degree of atheroma present in the underlying vessel, and examined differential expression of microRNAs (miRNAs), which have been shown to regulate differentiation and function of both adipocytes and macrophages. 26 , 27 , 28 , 29 , 30
Methods
The data that support the findings of this study are available from the corresponding author on reasonable request.
Participants
Participants were recruited from the Stanford Myocardial Bridge clinic during a preoperative clinic visit. Eligible participants included individuals aged >18 years who were scheduled for elective unroofing surgery of an LAD MB via a sternotomy. 31 The study protocol was approved by the Stanford Internal Review Board, and written informed consent was obtained by the study coordinator.
Imaging Studies
Presurgical evaluation for all participants included an invasive coronary angiogram with intravascular ultrasound (IVUS) and a coronary computed tomography angiogram.
Intravascular Ultrasound
IVUS measurements were performed, as previously described, 32 with a 40‐MHz mechanical IVUS catheter (Atlantis SR Pro2; Boston Scientific, Marlborough, MA), placed as far distally in the LAD as safely possible. Recordings were obtained during an automated pullback and at a stationary wire position. Two experienced invasive cardiologists from an independent core laboratory (Cardiovascular Core Analysis Laboratory at Stanford University) reviewed all IVUS recordings. MB was defined as an echo‐lucent half‐moon (“halo”) and/or systolic luminal area compression >10%. The systolic compression of the bridged segment obtained from the stationary wire position was calculated by the following equation: 100× (vessel area in diastole−vessel area in systole)/vessel area in diastole. The length and location of the MB were assessed by the presence of a halo. The MB halo thickness was measured in mm. The maximal plaque burden (MPB) was calculated as follows: (vessel area−lumen area)/vessel area (%) at the location of the largest plaque.
Cardiac Computed Tomography Angiography
All cardiac computed tomography angiography (CCTA) scans were performed as retrospectively ECG‐gated scans of the heart, with slice thickness between 0.6 and 1 mm, and range from the tracheal bifurcation to the diaphragm. The reconstructed field of view was individually adjusted to encompass the heart. The CCTA images were originally acquired on the Siemens Somatom Definition, Siemens Somatom Definition Flash (Siemens, Forchheim, Germany), the GE Lightspeed VCT, and the GE Discovery HD750 (General Electric, Milwaukee, WI). All CCTA image data were reevaluated for research purposes as part of this analysis on an external workstation (SyngoVia, VA30A; Siemens Medical Solutions, Forchheim, Germany). The image quality of all computed tomography scans was adequate in all cases. Multiplanar and curved reformations were used for the assessment of MBs in long‐ and short‐axis planes. The thickness of the epicardial fat was measured above the MB segment as well as above and below the coronary artery proximal and distal to MB.
Pericoronary Adipose Tissue Attenuation as a Measure of Inflammation
Pericoronary adipose tissue (PCAT) attenuation (in Hounsfield units) was measured systematically from CCTA, in coronary arteries with distal diameter ≥2.0 mm, as outlined in Goeller et al, 33 , 34 by blinded expert readers, using semiautomated software (Autoplaque 2.5; Cedars‐Sinai Medical Center, Los Angeles, CA). PCAT measurements were automatic following delineation of the coronary vessel wall. In this study, for the LAD, plaque features and PCAT attenuation were specifically measured proximal to the MB, over the bridge length, and distal to the bridge. PCAT attenuation over 3 mm radius around the vessel wall was used for analysis, as in previous studies. 33 , 35 PCAT was expressed as mean Hounsfield units, in which pure fat values are −1000 Hounsfield units, and attenuated, or less negative values, indicate the presence of other soft tissue components, including collagen and immune cells. The PCAT has been validated as a measure of inflammation in humans with and without coronary artery disease. 36
Classification of Atherosclerotic Burden
The MPB in the proximal LAD located 0.8 to 5.6 cm from the entry of the LAD into the myocardium was determined by IVUS in all participants. The MPB ranged from a minimum 15.8% to a maximum of 61.8%, with a mean of 32.0%. MPB tended to cluster into 2 clear groups above and below 30%. Patients with MPB >30.0% were thus classified as having moderate‐to‐high atherosclerotic plaque burden, and those with MPB <30.0% were classified as having low atherosclerotic plaque. Subgroups were referred to as “moderate‐high” and “low” atherosclerotic plaque subgroups.
Tissue Harvesting
For consented participants, EAT was harvested intraoperatively via sharp dissection during the MB unroofing procedure. Three EAT samples (50–200 mg) were harvested: (1) proximal to where the coronary artery dipped into the myocardium (proximal EAT); (2) above the area where the coronary artery traversed the myocardium (bridge EAT); and (3) distal to the MB (distal EAT). Adipose tissue samples were cleaned of blood in the operating suite and then flash frozen and stored at −80°C for later RNA preparation.
RNA Isolation and Purification
Total RNA was isolated using RNeasy Mini Kit (QIAgen), according to the manufacturer's instructions. Briefly, 30 mg of fat tissue from each depot was minced and homogenized. RNA was extracted and column purification was performed according to the manufacturer's protocol. RNA was eluted, and concentrations and purities (measured by 280/260 and 260/230 ratios) were recorded by nanodrop. Purity requirement was a 280/260 absorbance ratio ≥1.9.
Gene Expression Measured by NanoString nCounter Array
We preselected 25 genes, including 2 housekeeping genes, GADPH and cyclophilin A, which have been validated as control genes for epicardial fat, 37 related to adipocyte differentiation, thermogenesis, inflammation, hypoxia, and oxidative stress, to measure using a custom‐Codeset by Nanostring, Inc (Seattle, WA) specifically for this study. A total of 150 mg of total RNA from each qualifying sample was applied to the cartridge to assay the gene expression. Normalization of nCounter results was performed using nSolver Analysis Software Version 2.5 (NanoString Technologies), according to the manufacturer's guidelines. RNA counts were normalized using the expression of the 2 housekeeping genes, as previously described. 38
miRNA Expression
miRNA was prepared on total RNA (without amplification or reverse transcription) using the nCounter miRNA Sample Preparation Kit, according to the manufacturer's instructions. Data analysis was performed by Canopy Biosciences with the nSolver software from NanoString. Advanced analysis was done with the Rosalind platform from OnRamp Bioinformatics (https://www.onramp.bio/). Log2‐fold changes of miRNAs were calculated against the common reference sample. Comparisons between EAT depots were expressed as fold‐change depots.
Cytokine Secretion
Supernatants from proximal and bridge epicardial fat samples were assayed by Luminex using the Human Adipocyte Panel (catalog No. HADCYMAG‐61K; EMD Millipore Corp, Burlington, MA) and performed by the Human Immune Monitoring Center at Stanford University. After 48‐hour incubation, assays were performed according to the manufacturer's recommendations. Custom Assay Chex control beads were purchased from Radix Biosolutions (Georgetown, TX), and added to all wells. Cytokines were expressed as mean fluorescence intensity and adjusted for total protein concentration.
Statistical Analysis
Comparison of participant demographic variables, adipose tissue variables, and PCAT, according to presence/absence of significant atherosclerotic plaque, used unpaired Student t tests, adjusted for age, which was the only variable that differed between groups. Comparison of adipose tissue variables in different EAT depots (proximal versus bridge) used paired Student t tests. Correlational analyses used general linear regression with formal tests for interaction between variables of interest and maximum plaque burden (moderate‐high versus low plaque group). Variables that were not normally distributed (fibroblast growth factor 21, interleukin 6, and MCP‐1 [monocyte chemoattractant protein 1]) were log transformed for both t tests and correlational analyses. Results are presented as mean±SD. P<0.05 was considered statistically significant.
Results
Twenty‐five participants completed the study. Demographic and clinical characteristics are shown in Table 1. Mean age was 46 years, sex was balanced equally, and mean body mass index was 28.9 kg/m2. One participant had type 1 diabetes, and all others were nondiabetic. Twenty‐two of the participants had good quality RNA from the 2 epicardial depots of interest: proximal to the MB (proximal EAT) and over the MB (bridge EAT). Fat harvested from the depot distal to the bridge was small and RNA of inconsistent quality, and therefore was excluded from gene expression analyses. All participants underwent IVUS to quantify atherosclerotic plaque. MPB ranged from 15% to 62%, with 11 participants qualifying as having moderate‐high atherosclerotic plaque burden based on prespecified criteria (see Methods). PCAT attenuation was calculated from CCTA on 18 participants: 6 scans were not available for analysis, and 1 was excluded because of angiographic dye artifact. By design, the average MPB was significantly greater in the moderate‐high atherosclerotic plaque group versus the low plaque group (n=14) (48.2±8.0 versus 20.4±3.5; P<0.001). Participants classified as having moderate‐high plaque burden were older than those with low plaque burden (58±8 versus 42±12; P=0.017); body mass index and sex did not differ significantly between groups (28.9±5.6 versus 28.9±6.2 kg/m2, and 36% versus 57% female, respectively), nor did race. A total of 10 and 12 from each group, respectively, had good quality RNA for comparisons.
Table 1.
Demographic and Clinical Characteristics of 25 Patients With MB and Quantification of Atherosclerotic Plaque in LAD, as Measured by IVUS
| Characteristic | Mean±SD | Range |
|---|---|---|
| Age, y | 46±11 | 22–63 |
| Sex, women/men | 12/13 | … |
| Race/ethnicity, n (%) | ||
| White | 20 (80) | … |
| Hispanic | 4 (16) | … |
| Black | 1 (4) | … |
| Weight, kg | 86.4±20.7 | 42.7–130.4 |
| Height, cm | 172.4±10.1 | 146–188 |
| BMI, kg/m2 | 28.9±5.8 | 19.3–38.6 |
| Fasting blood glucose, mg/dL | 97±14 | 74–142 |
| HbA1c, % | 5.4±0.7 | 4.8–8.0 |
| Total cholesterol, mg/dL | 159.0±36.7 | 92–234 |
| LDL cholesterol, mg/dL | 81.7±27.3 | 39–146 |
| HDL cholesterol, mg/dL | 64.8±20.6 | 37–109 |
| Triglyceride, mg/dL | 91.0±122.3 | 23–603 |
| hsCRP, mg/L | 1.2±1.0 | 0.2–3.2 |
| Statin therapy, n | 15 | … |
| ACEI/ARB, n | 2 | … |
| Aspirin, n | 15 | … |
| Current smoker, n | 1 | … |
| Previous smoker >1 y, n | 11 | … |
| MPB, % | 32.3±15.3 | 15–62 |
| Location of MPB from myocardial bridge entrance, mm | 25.1±12.3 | 8.0–56.0 |
| Location of myocardial bridge entrance from LAD/circumflex bifurcation, mm* | 40.2±10.8 | 24–71 |
| MB length, mm* | 26.5±15.5 | 3.3–65.0 |
| MB thickness, mm* | 0.62±0.27 | 0.3–1.23 |
ACEI indicates angiotensin‐converting enzyme inhibitor; ARB, angiotensin II receptor blocker; BMI, body mass index; HbA1c, hemoglobin A1c; HDL, high‐density lipoprotein; hsCRP, high‐sensitivity C‐reactive protein; IVUS, intravascular ultrasound; LAD, left anterior descending coronary artery; LDL, low‐density lipoprotein; MB, myocardial bridge; and MPB, maximal plaque burden.
Includes measures of multiple MBs from 4 participants with 2 anatomical MBs and 1 participant with 3 anatomical MBs.
PCAT Attenuation by Depot and Plaque Subgroup
PCAT attenuation was higher (less negative), consistent with inflammation, in the proximal versus the bridge depots (Figure 1A). Furthermore, PCAT in the proximal EAT depot was significantly higher in the subgroup with moderate‐high plaque than the subgroup with low plaque (−71.3±6.7 versus −79.6±8.4 HU; P=0.037), and correlated significantly with MPB (Figure 1B; r=0.60, P=0.009). There was also a trend toward correlation between PCAT over the bridge and MPB (r=0.42, P=0.089), as well as a significant correlation between PCAT in the proximal and bridge EAT depots (r=0.65, P=0.004).
Figure 1. (A) Pericoronary adipose tissue (PCAT) attenuation is reduced in proximal epicardial adipose tissue versus bridge adipose tissue in patients with myocardial bridge; (B) PCAT attenuation is reduced in proportion to maximal plaque burden.
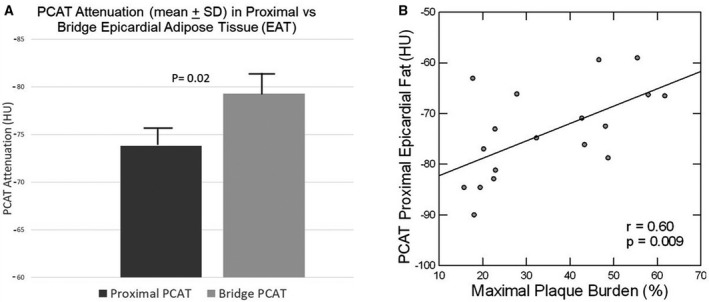
HU indicates Hounsfield unit.
Gene Expression According to Plaque Burden and Relationship to Hypoxia‐Inducible Factor 1a
For each fat depot, gene expression was compared between those with moderate‐high and low atherosclerotic plaque burden, as quantified by IVUS. As shown in Table 2, age‐adjusted expression of interleukin 6 and MCP‐1 in proximal EAT was 3‐ to 5‐fold higher (P<0.005) in the group with moderate‐high plaque compared with the group with low plaque, with significant correlations to MPB (interleukin 6: r=0.53, P=0.012; and MCP‐1: r=0.77, P<0.001). Other inflammatory genes showed a similar trend but did not reach statistical significance. Hypoxia‐inducible factor (HIF) 1a, a marker of hypoxia, was significantly upregulated in proximal EAT of the moderate‐high plaque compared with the low‐plaque group (P=0.017), and correlated significantly with MPB (r=0.53, P=0.025). Angiogenic gene expression did not differ in the proximal EAT between groups, nor did adipogenic, thermogenic, or oxidative stress genes. In the proximal EAT for the total cohort, HIF1a expression correlated highly with MCP‐1 (age‐adjusted r=0.88, P<0.001) and interleukin 6 expression (age‐adjusted r=0.68, P<0.001), as well as hypoxia‐regulated angiogenic genes, such as vascular endothelial growth factor (r=0.68, P<0.001) and platelet‐derived growth factor (r=0.87, P<0.001) (Figure 2). Furthermore, expression of interleukin 6 and MCP‐1 in proximal EAT was inversely correlated with adipogenic gene expression in bridge EAT (age‐adjusted r=−0.44 to −0.55 with P<0.05 for adiponectin, adipose tissue triglyceride lipase, perilipin 1 (PLIN1), perilipin 5 (PLIN5), and lipoprotein lipase).
Table 2.
Gene Expression (relative to housekeeping genes) in Epicardial Fat in Participants With Moderate‐High Versus Low Atherosclerotic Plaque Burden
| Gene | Participants with moderate‐high plaque (n=10) | Participants with low plaque (n=12) | P value* |
|---|---|---|---|
| Inflammation proximal fat | |||
| Interleukin 6 † | 1420±1054 | 402±428 | 0.001 |
| CCL2–MCP‐1 † | 1703±1188 | 347±331 | 0.004 |
| CD68 | 237±152 | 195±157 | 0.26 |
| CD44 | 366±194 | 258±138 | 0.11 |
| Hypoxia proximal fat | |||
| HIF1a | 812±471 | 506±296 | 0.017 |
| P22PHOX | 459±226 | 372±226 | 0.20 |
| SOD2 | 2352±194 | 1772±952 | 0.09 |
| Adipogenesis bridge fat | |||
| PPARG | 138±58.6 | 216±129 | 0.107 |
| Adiponectin | 1918±851 | 4095±2313 | 0.009 |
| GLUT4 | 83±35.9 | 181±146 | 0.065 |
| CD36‐AP2 | 3028±1490 | 5123±2750 | 0.053 |
| ATGL | 624±247 | 1086±637 | 0.036 |
| FABP4 | 13 845±7360 | 26 418±15 929 | 0.041 |
| Perilipin 1 | 1560±1006 | 3422±2341 | 0.038 |
| Perilipin 5 | 58±24 | 133±101 | 0.046 |
| LPL | 1246±686 | 2532±1680 | 0.044 |
| Inflammation bridge fat | |||
| Interleukin 6 † | 1212±1329 | 529±1077 | 0.06 |
| CCL2–MCP‐1 † | 682±643 | 253±158 | 0.15 |
| CD68 | 209±182 | 167±99.2 | 0.51 |
| CD44 | 348±338 | 245±264 | 0.58 |
Data are given as mean±SD. Depot‐specific functional gene categories with no significant differences between plaque subgroups are omitted from the table. AP2 indicates adipocyte Protein 2; ATGL, adipose tissue triglyceride lipase; CCL2‐MCP‐1: monocyte chemoattractant protein‐1; CD, cluster of differentiation; FABP4, fatty acid binding protein 4; GLUT4, glucose transporter 4; HIF, hypoxia‐inducible factor; LPL, lipoprotein lipase; MCP‐1, monocyte chemoattractant protein 1; P22PHOX, NADH oxidase subunit, phox22; PPARG, peroxisome proliferator‐activated receptor γ; and SOD, superoxide dismutase.
ANCOVA with adjustment for age.
Test for significance done on log value.
Figure 2. Age‐adjusted correlations between hypoxia‐inducible factor (HIF) 1a and inflammatory and angiogenic gene expression in bridge and proximal epicardial adipose tissue.
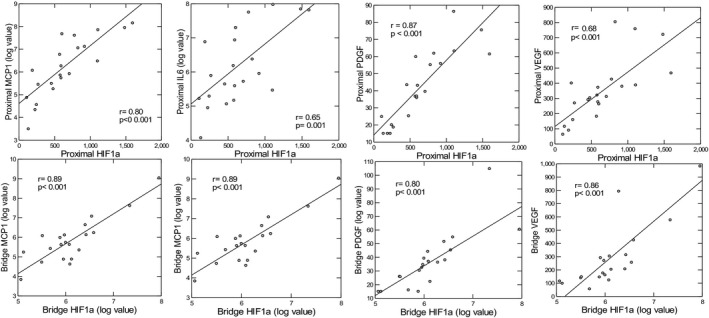
IL6 indicates interleukin‐6; MCP1, monocyte chemoattractant protein 1; PDGF, platelet‐derived growth factor; and VEGF, vascular endothelial growth factor.
In bridge EAT, individuals with moderate‐high plaque exhibited significant or near‐significant downregulation of virtually all adipogenic genes compared with those with low plaque (Table 2). Inflammatory gene expression in bridge EAT was higher in the moderate‐high plaque versus the low plaque group, and although between‐group comparisons did not reach statistical significance, the age‐adjusted correlation between MPB and interleukin 6 was statistically significant (r=0.51, P=0.17). Other gene groups did not differ according to plaque group (and were therefore omitted from Table 2). As in proximal EAT, HIF1a in bridge EAT was highly correlated with inflammatory and angiogenic genes, as shown in Figure 2. Interleukin 6 but not MCP‐1 expression in bridge EAT was modestly inversely correlated with bridge peroxisome proliferator‐activated receptor γ (r=−0.44, P=0.045); P values were borderline for the remainder of adipogenic genes (0.05–0.09).
Comparison of Gene Expression by Depot
Comparing different depots in the same individuals allows for investigation of the regional impact of geographical location (depot comparison) and atherosclerotic plaque (depot comparison in subgroups with high versus moderate‐low plaque burden), independent of confounders that limit results from comparing 2 separate patient groups (eg, those with CAD versus those without). In the group as a whole, gene expression in proximal compared with bridge EAT did not differ significantly. Among those with moderate‐high plaque burden, however, gene expression in multiple functional groups differed significantly between depots. Results are shown in Table 3. Among the participants with moderate‐high atherosclerotic plaque burden, inflammatory genes MCP‐1 and interleukin 6 were upregulated in the proximal versus the bridge depot. Adipogenesis genes were also significantly downregulated in the bridge compared with the proximal depot, whereas fibroblast growth factor 21, a marker of thermogenesis/beige adipose cells, was significantly downregulated in the proximal versus the bridge depot. Unlike the participants with moderate‐high atherosclerotic plaque, the 12 participants with low atherosclerotic plaque demonstrated no significant depot‐related differences in adipogenic/lipogenic or inflammatory genes. There were no significant between‐depot differences in expression of other thermogenic genes (UCP1 [uncoupling protein 1] or peroxisome proliferator‐activated receptor γ coactivator 1‐α), or genes related to oxidative stress or angiogenesis.
Table 3.
Expression (relative to housekeeping genes) of Genes by Depot in Epicardial Fat in Individuals With Moderate‐High Plaque Versus Low Atherosclerotic Plaque Burden
| Gene group | Participants with moderate‐high plaque (n=11) | Participants with low plaque (n=14) | ||||
|---|---|---|---|---|---|---|
| Proximal EAT | Bridge EAT | P value * | Proximal EAT | Bridge EAT | P value * | |
| Inflammation | ||||||
| Interleukin 6 † | 1420±1054 | 1212±1329 | 0.03 | 402±428 | 529±1077 | 0.17 |
| CCL2–MCP‐1 † | 1703±1189 | 682±643 | <0.03 | 347±331 | 253±158 | 0.27 |
| CD68 | 237±152 | 209±182 | 0.66 | 195±157 | 167±99.2 | 0.55 |
| CD44 | 366±194 | 348±338 | 0.99 | 258±138 | 245±264 | 0.88 |
| Adipogenesis | ||||||
| PPARG | 258±137 | 138±58.6 | 0.029 | 254±175 | 216±129 | 0.49 |
| Adiponectin | 4372±2402 | 1918±851 | 0.014 | 5227±4518 | 4095±2313 | 0.38 |
| GLUT4 | 161±97.3 | 83±35.9 | 0.005 | 206±185 | 181±146 | 0.70 |
| CD36‐AP2 | 4850±2837 | 3028±1490 | 0.04 | 5679±4215 | 5123±2750 | 0.63 |
| ATGL | 1322±626 | 624±247 | 0.003 | 1631±1291 | 1086±637 | 0.17 |
| FABP4 | 25 578±15 851 | 13 845±7360 | 0.049 | 32 917±29 223 | 26 418±15 929 | 0.35 |
| Perilipin 1 | 3908±2275 | 1560±1006 | 0.011 | 4737±3761 | 3422±2341 | 0.28 |
| Perilipin 5 | 162±80.9 | 58±24 | 0.006 | 222±235 | 133±101 | 0.17 |
| LPL | 2756±1808 | 1246±686 | 0.039 | 2590±1944 | 2532±1680 | 0.93 |
| Thermogenesis | ||||||
| UCP1 | 30.5±14.3 | 28.3±11.9 | 0.65 | 28.6±9.9 | 35.0±28.3 | 0.44 |
| FGF21 † | 15.8±1.6 | 24.9±10.1 | 0.03 | 19±9.8 | 22.7±11.4 | 0.10 |
| PGC1a | 37±16.2 | 39.5±14.5 | 0.73 | 41.9±19.6 | 56.9±62.5 | 0.48 |
| Hypoxia/oxidative stress | ||||||
| HIF1a | 812±471 | 805±868 | 0.92 | 506±296 | 628±718 | 0.57 |
| P22PHOX | 459±226 | 360±246 | 0.50 | 372±226 | 329±182 | 0.55 |
| P47PHOX | 155±114 | 124±66.4 | 0.68 | 98.8±63.6 | 90.7±35.0 | 0.42 |
| SOD2 | 2352±1326 | 2711±3883 | 0.77 | 1773±952 | 2402±3378 | 0.51 |
| Angiogenesis | ||||||
| PDGF | 46.1±19.6 | 42.7±28.1 | 0.58 | 39.2±21.6 | 35.1±13.9 | 0.56 |
| VEGF | 346±175 | 297±296 | 0.61 | 347±226 | 341±275 | 0.96 |
Data are given as mean±SD. AP2 indicates adipocyte Protein 2; ATGL, adipose tissue triglyceride lipase; CCL2‐MCP‐1, monocyte chemoattractant protein‐1; CD, cluster of differentiation; EAT, epicardial adipose tissue; FABP4, fatty acid binding protein 4; FGF21, fibroblast growth factor 21; GLUT4, glucose transporter 4; HIF, hypoxia‐inducible factor; LPL, lipoprotein lipase; MCP‐1, monocyte chemoattractant protein 1; P22PHOX, NADH oxidase subunit, phox22; P47PHOX, neutrophil cytosol factor 1; PDGF, platelet‐derived growth factor; PGC1a, peroxisome proliferator‐activated receptor γ coactivator 1‐α; PPARG, peroxisome proliferator‐activated receptor γ; SOD, superoxide dismutase; UCP1, uncoupling protein 1; and VEGF, vascular endothelial growth factor.
Paired Student t test.
Test for significance done on log‐transformed value.
Cytokine Secretion
Inflammatory cytokine concentrations, adjusted for total protein, were 2‐ to 3‐fold higher in the supernatant from the proximal compared with the bridge depot. Only 7 participants had paired supernatants available, limiting ability to attain statistical significance or correlate with atherosclerotic plaque burden, but several cytokines reached near‐significant levels (interleukin 8: P=0.05; MCP‐1: P=0.06), supporting the mRNA findings (Table S1).
miRNA Expression by Depot
A total of 798 miRNAs were evaluated in proximal and bridge depots. With a filter of 1.25‐fold‐change difference and P<0.05 applied, 7 miRNAs showed significant upregulation or downregulation in proximal compared with bridge EAT in the cohort as a whole, as shown in Figure 3. miR 3192‐5P, 518D‐3P, and 532‐5P were upregulated in proximal compared with bridge EAT, whereas miR 630, 575, 16‐5P, and 320E were downregulated in proximal compared with bridge EAT. Of these miRNAs, only 630 in bridge EAT were directly correlated with MPB (age‐adjusted r=0.55, P=0.014). As shown in Figure 4, miR 630 in bridge EAT was highly significantly inversely correlated with expression of all proximal EAT adipogenesis genes (age‐adjusted r values, −0.63 to −0.68; P<0.004). miR 630 in proximal EAT was also inversely correlated with all proximal EAT adipogenesis genes (age‐adjusted r values, −0.51 to −0.57; P<0.02). The other 3 miRNAs that were upregulated in bridge EAT did not correlate with any functional mRNA groups in bridge or proximal EAT. Similar to miR 630, the 3 miRNAs that were overexpressed in proximal EAT, miR 3192‐5P, 518D‐3P, and 532‐5P, demonstrated significant age‐adjusted inverse correlations with all 9 adipogenic genes in proximal EAT, with r values of −0.50 to −0.67, and P≤0.01, as well as with angiogenesis genes (vascular endothelial growth factor: r=−0.51 to −0.76, P<0.001 to 0.01; platelet‐derived growth factor: r=−0.48 to −0.71, P<0.001 to 0.027), hypoxia/oxidative stress genes (HIF1a: r=−0.49 to −0.53, P=0.01 to 0.026; superoxide dismutase 2: r=−0.45, P=0.04; NADH oxidase subunit, phox22: r=−0.44 to −0.58, P=0.001 to 0.045), and a single thermogenesis gene, PCG1a (r=−0.59 to −0.67, P=0.001 to 0.009) (Figure 5). miR 3192‐5P and 532P were also inversely correlated with HIF1a (r=−0.49 to −0.53, P=0.01 to 0.026). In bridge EAT, these 3 miRNAs were not significantly correlated with any functional gene groups. All 4 miRNAs that correlated inversely with adipogenic gene expression in proximal EAT (miR 630, 3192‐5P, 518D‐3P, and 532‐5P) were highly intercorrelated, with r values from 0.70 to 0.89, and P<0.001.
Figure 3. Volcano plot demonstrating 7 differentially expressed microRNAs (miRNAs) in proximal vs bridge epicardial adipose tissue.
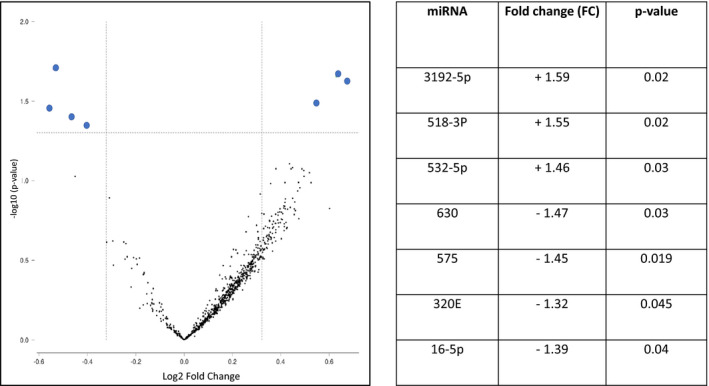
Table on right shows fold change in proximal relative to bridge depot.
Figure 4. Age‐adjusted correlations between miR 630 in bridge and proximal epicardial adipose tissue and adipogenic gene expression in proximal epicardial adipose tissue.
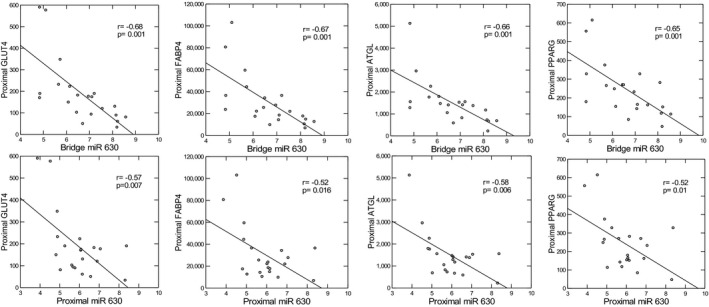
ATGL indicates adipose tissue triglyceride lipase; FABP4, fatty acid binding protein 4; GLUT4, glucose transporter 4; and PPARG, peroxisome proliferator‐activated receptor γ.
Figure 5. MicroRNA (miRNA) expression in proximal and bridge epicardial fat and associations with mRNA gene expression.
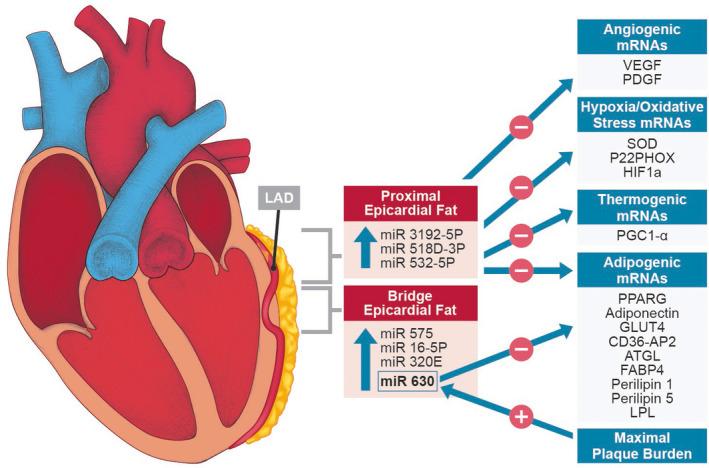
miRNAs upregulated in proximal vs bridge epicardial fat (epicardial adipose tissue [EAT]) include 3192‐5P, 518D‐3P, and 532‐5P, which correlated inversely with mRNA expression for adipogenesis, angiogenesis, hypoxia/oxidative stress, and thermogenesis in proximal EAT. miRNAs upregulated in bridge EAT included 630, which correlated directly with plaque burden and inversely with adipogenic gene expression in both proximal and bridge EAT. AP2 indicates adipocyte Protein 2; ATGL, adipose tissue triglyceride lipase; CD36, cluster of differentiation 36; FABP4, fatty acid binding protein 4; GLUT4, glucose transporter 4; LAD, left anterior descending coronary artery; LPL, lipoprotein lipase; P22PHOX, NADH oxidase subunit, phox22; PDGF, platelet‐derived growth factor; PGC1‐α, peroxisome proliferator‐activated receptor γ coactivator 1‐α; PPARG, peroxisome proliferator‐activated receptor γ; SOD, superoxide dismutase; and VEGF, vascular endothelial growth factor.
Discussion
To our knowledge, this is the first study to compare physical and molecular differences in the epicardial fat from different depots in the same patients with MB, in which the proximal LAD is prone to developing atherosclerotic plaque, whereas the bridged segment of the LAD, which tunnels through the myocardium and is therefore not in direct contact with the epicardial fat, remains plaque free. We hypothesized that the EAT overlying the proximal coronary artery would exhibit signs of inflammation, oxidative stress, and impaired adipocyte function, in contrast to the EAT overlying the bridge, in which there was no direct contact between coronary artery and epicardial fat.
The results of the current study clearly demonstrate that in individuals with MB, inflammation is increased in proximal EAT compared with bridge EAT but only among those with high atherosclerotic plaque burden. Inflammation was demonstrated by both physical characteristics (computed tomography attenuation of the pericoronary fat) and molecular characteristics (mRNA expression and secreted cytokines). More important, computed tomography attenuation of the pericoronary fat, indicating inflammation, bore a strong relationship to the degree of plaque burden, consistent with the hypothesis that inflammation in epicardial fat is related to the atherosclerotic process in the adjacent vessel. mRNA analysis of epicardial fat also demonstrated significant upregulation of key inflammatory genes in relationship to degree of plaque burden. Secreted inflammatory cytokines were also higher in the supernatants from proximal compared with bridge fat, although the number with available supernatant was too small to correlate with degree of plaque burden. The fact that inflammation was increased in proximal versus bridge fat only in the setting of atherosclerotic plaque indicates that rather than geographical location per se, 39 inflammation is related to signals derived from the process affecting the atherosclerotic vessel: endothelium‐derived secreted factors, effects of activated immune cells, or hypoxia related to impaired blood flow. Furthermore, obtaining tissues from the same individuals as their own control eliminates potential confounding from interindividual factors, such as metabolic syndrome or systemic inflammation, that might account for observed differences in the epicardial fat between patients with or without atherosclerotic cardiovascular disease, and makes a strong case for regional variation in EAT inflammation as a function of proximity to atherosclerotic plaque. Thus, the current results extend prior studies demonstrating inflammation in epicardial fat compared with subcutaneous fat of individuals undergoing coronary artery bypass grafting 4 , 5 , 10 and higher inflammation in epicardial fat obtained from individuals with versus without CAD. 6 , 36 Whether inflammation in the epicardial fat overlying a vessel with plaque is cause or consequence cannot be determined from this study. However, if inflammation in EAT was a primary driver of atherosclerosis, it would not likely be restricted to the subset of individuals who demonstrated higher plaque burden. Thus, our findings are consistent with the inside‐to‐out hypothesis in which changes in the atherosclerotic artery lead to inflammation in adjacent EAT, which, in turn, may further contribute the pathogenesis of plaque and/or plaque destabilization.
The significantly higher expression of HIF1a, the driver of hypoxia‐mediated cellular responses, in the proximal EAT of those with higher plaque burden, and the robust association between HIF1a and inflammatory cytokine expression, points to regional hypoxia as a potential mechanism linking atherosclerosis to inflammation within the epicardial fat. Hypoxia has been clearly related to regional inflammation in adipose tissue in animals, 40 and has been demonstrated, along with decreased capillary density and increased cluster of differentiation 68 expression, in the subcutaneous adipose tissue of obese versus lean individuals, 41 leading some to hypothesize that in obesity, adipose mass exceeds available capillary blood supply, leading to hypoxia and inflammation. In the current study, the observation that HIF1a was selectively upregulated in relation to plaque burden points to regional ischemia, endothelial dysfunction, inflammation, and/or other paracrine or mechanical factors related to the atherosclerotic process, rather than obesity and adipocyte hypertrophy per se. Furthermore, HIF1a was robustly associated not only with inflammatory but also with angiogenic gene expression, pointing to stimulation of neoangiogenesis that extends to the vasa vasorum of the coronary arteries, facilitating cross talk between the EAT and vasculature via transport of adipose tissue macrophages and secreted factors that promote plaque progression. 42 , 43 Indeed, plaque neoangiogenesis is associated with plaque progression and intraplaque hemorrhage, which is thought to arise from the vasa vasorum. 43 Thus, although the current data point to an inside‐out relationship between atherosclerotic plaque and EAT inflammation, it is plausible that the relationship is bidirectional.
Contrary to our hypothesis that adipocyte function would be impaired in the proximal fat depot as a result of higher inflammation, we found a striking downregulation of all adipogenic genes in the fat overlying the bridge compared with the proximal EAT depot: this downregulation, however, was restricted to the subgroup with moderate‐high atherosclerotic plaque. The other 3 subgroups (proximal EAT with/without plaque and bridge EAT without plaque) demonstrated similar gene expression, pointing to specific downregulation in bridge EAT related to the high plaque burden. Expression of adipogenic genes in bridge EAT was inversely correlated with inflammatory gene expression in proximal, and to a lesser extent, bridge EAT, pointing to a paracrine or downstream effect of secreted factors from inflamed proximal EAT. These findings extend abundant prior literature demonstrating that inflammation can induce adipocyte dysfunction 39 , 40 , 41 , 42 and are in keeping with recent studies on human epicardial fat by Antonopoulos et al. 36 Decreased expression of genes related to adipocyte differentiation and FFA uptake/storage also indicates that release of FFA is higher, which may lead to cardiac (myocardial) steatosis, or alternatively, may increase FFA supply in response to increased energy demands in the setting of ischemia. The notion that adipose tissue remodeling, including inflammation, angiogenesis, and altered adipocyte function, is an adaptive response to adipose tissue stress has gained traction in recent years. 44 , 45 , 46 , 47 Adipogenesis is tightly coupled to angiogenesis, 41 and while in vitro, inflammatory cytokines impair adipose differentiation and fat storage, in vivo, inflammation has also been shown to be essential and adaptive for adipose tissue remodeling and metabolic health. 40 Signals determining these coordinated responses to adipose tissue stressors, such as hypoxia and/or lipotoxicity, have not been fully elucidated but likely include transcription regulators, such as HIF1a and peroxisome proliferator‐activated receptor γ. Thus, it is important to consider that the current findings pointing to inflammation, angiogenesis, and impaired adipogenic gene expression in the setting of atherosclerotic plaque may point not only to a pathologic process in EAT, but also may reflect an adaptive process.
In addition to transcription factors, such as HIF1a and peroxisome proliferator‐activated receptor γ, that may coordinate multicellular responses to regional adipose tissue stressors, recent interest has focused on miRNAs, small noncoding RNAs that regulate translation of mRNA to >60% of all proteins. These molecules may serve as a signal between atheroma and adipose tissue. 48 Abundant literature supports a role for miRNA in regulating adipogenesis 28 , 49 as well as cross talk between immune and adipose cells. 50 miRNA analysis in this study uniquely compared expression in the 2 epicardial fat depots that differed not only in geographic location, but also with respect to proximity to atherosclerotic plaque. We identified 7 miRNAs that were differentially expressed between depots. One miRNA was significantly associated with plaque burden: miR 630. This miRNA, which was highly associated with 3 other miRNAs that all inhibited adipogenesis, is known to be upregulated by hypoxia, 51 and has been noted in the literature to inhibit endothelial‐to‐mesenchymal transition in which sheer stress or hypoxia in a vessel leads perivascular endothelial cells to dedifferentiate to mesenchymal cells, which in turn can differentiate into fibroblasts, myofibroblasts, mural cells, osteoblasts, chondrocytes, and adipocytes, 52 , 53 likely contributing to tissue remodeling and adaptation. The novel finding presented in the current study, that atherosclerotic plaque is positively associated with miR 630, which, in turn, appears to broadly inhibit adipogenesis in EAT, points to miR 630 as a novel molecule to explore as a determinant of adipose tissue remodeling in the presence of hypoxia and inflammation. Three other miRNAs, 3192‐5P, 518D, and 532‐5p, which were highly intercorrelated with each other and with miR 630, were upregulated in proximal relative to bridge EAT, and like miR 630, were significantly inversely associated with virtually all adipogenic genes in the proximal depot, as well as with PCG1a, a cofactor for peroxisome proliferator‐activated receptor γ stimulation and a key player in lipid metabolism, HIF1a, and genes related to oxidative stress and angiogenesis. In light of prior reports that consistently reveal miRNA to play a role in regulating adipogenesis, 28 , 48 , 49 the finding that adipogenic genes were downregulated as a group in relationship to miRNA expression in adipose tissue is not unexpected. The miRNAs shown to downregulate adipogenesis in the current study, however, have not previously been linked to adipocyte differentiation or function, and prior studies have been done under experimental conditions with preadipocytes in culture. Thus, the human data presented herein reveal a novel and potentially important role for miR 3192‐5P, 518D, 532‐5p, and 630 in regulating adipocyte differentiation in vivo, and point to their potential as key mediators between atherosclerosis, hypoxia, inflammation, and adipocyte dysfunction, as illustrated in Figure 5. It is necessary to point out that inhibition of genes related to adipocyte differentiation and FFA uptake and storage may not necessarily be maladaptive, as in the setting of hypoxia, release of FFA from adipose tissue may provide needed fuel to minimize ischemic damage to the myocardium. Further research is warranted to delineate the role of these miRNAs in human cells and their potential as novel therapeutic targets for metabolic and cardiovascular disease.
The current study has several strengths and limitations. A major strength is the examination of mRNA and miRNA from 2 depots of EAT from the same individual that uniquely includes one depot that is adjacent to the coronary artery and another depot that is not. Furthermore, this is the first study with the ability to discriminate between geographic location per se and the presence of atherosclerotic plaque in relationship to the inflammation and gene expression in EAT, compared with studies comparing EAT in 2 groups of patients according to presence/absence of atherosclerotic cardiovascular disease. Having target mRNA quantified in the same samples from which miRNA was prepared allows for real measurement of target gene regulation by miRNAs compared with estimates from databases. The results presented cannot necessarily be extrapolated to atherosclerotic heart disease in general, as all of our participants had MB. Future studies should examine these relationships in other patient groups, such as those with obesity, diabetes, and traditional atherosclerotic cardiovascular disease. We identified several miRNAs as potential regulators of adipogenesis, as well as thermogenesis and hypoxia/oxidative stress, that differed by either geographical location or plaque burden. The results presented are correlational, however, and cannot prove causality, so additional experimental studies (eg, RNA silencing) should be done to confirm miRNA regulation of target genes related to adipocyte function.
In summary, the forgoing data suggest that plaque burden rather than geographical location per se is a determinant of the adjacent EAT structural and molecular characteristics, including hypoxia, inflammation, and impaired adipogenesis. These results are consistent with the well‐established response to lipoprotein retention hypothesis that atheroma starts in the intima and then media, not in the fat around the adventitia. 54 Atheroma‐related hypoxia in the adjacent epicardial fat attributable to changes in the coronary microcirculation supplying the epicardial fat may incite inflammation, which, in turn, impairs adipocyte function, and promote angiogenesis, both of which may contribute to plaque progression in a bidirectional manner via increased circulation of inflammatory immune cells and cytokines from EAT back to the large vessels. miRNA signals, most notably miR 630, are another likely mediator between atheromatous plaque, regional hypoxia, and adipocyte function. Although the current results cannot confirm causality, they reveal new findings about the physiological processes that occur in the epicardial fat in association with atherosclerotic plaque, and highlight several molecules that may mediate these relationships.
Sources of Funding
This work was supported by a grant from the Cardiometabolic Disease Research Foundation, Los Angeles, California. Cytokine work was supported in part by the Diabetes Immune Monitoring Core of the Stanford Diabetes Research Center (P30DK116074). Dr Dey was supported by National Institutes of Health/National Heart, Lung, and Blood Institute grant 1R01HL148787‐01A1.
Disclosures
Dr Nieman reports unrestricted, institutional research support from Siemens Healthineers, Bayer, and HeartFlow Inc outside of the current work. The remaining authors have no disclosures to report.
Supporting information
Table S1
Acknowledgments
We would like to thank the team at the Stanford Cardiothoracic Surgery Clinic for supporting our recruitment efforts.
For Sources of Funding and Disclosures, see page 13.
References
- 1. Nerlekar N, Brown AJ, Muthalaly RG, Talman A, Hettige T, Cameron JD, Wong DTL. Association of epicardial adipose tissue and high‐risk plaque characteristics: a systematic review and meta‐analysis. J Am Heart Assoc. 2017;6:e006379. doi: 10.1161/JAHA.117.006379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mahabadi AA, Berg MH, Lehmann N, Kälsch H, Bauer M, Kara K, Dragano N, Moebus S, Jöckel K‐H, Erbel R, et al. Association of epicardial fat with cardiovascular risk factors and incident myocardial infarction in the general population: the Heinz Nixdorf Recall Study. J Am Coll Cardiol. 2013;61:1388–1395. doi: 10.1016/J.JACC.2012.11.062 [DOI] [PubMed] [Google Scholar]
- 3. Alexopoulos N, Raggi P. Epicardial adipose tissue: another tassel in the complex fabric of atherosclerosis. Cardiovasc Hematol Disord Drug Targets. 2018;18:17–26. doi: 10.2174/1871529X17666170125103555 [DOI] [PubMed] [Google Scholar]
- 4. Cheng KH, Chu CS, Lee KT, Lin TH, Hsieh CC, Chiu CC, Voon WC, Sheu SH, Lai WT. Adipocytokines and proinflammatory mediators from abdominal and epicardial adipose tissue in patients with coronary artery disease. Int J Obes. 2007;32:268–274. doi: 10.1038/SJ.IJO.0803726 [DOI] [PubMed] [Google Scholar]
- 5. Eiras SE, Teijeira‐Fernández E, Shamagian LG, Fernandez AL, Vazquez‐Boquete A, Gonzalez‐Juanateya JR. Extension of coronary artery disease is associated with increased IL‐6 and decreased adiponectin gene expression in epicardial adipose tissue. Cytokine. 2008;43:174–180. doi: 10.1016/J.CYTO.2008.05.006 [DOI] [PubMed] [Google Scholar]
- 6. Sacks HS, Fain JN, Cheema P, Bahouth SW, Garrett E, Wolf RY, Wolford D, Samaha J. Depot‐specific overexpression of proinflammatory, redox, endothelial cell, and angiogenic genes in epicardial fat adjacent to severe stable coronary atherosclerosis. Metab Syndr Relat Disord. 2011;9:433–439. doi: 10.1089/MET.2011.0024 [DOI] [PubMed] [Google Scholar]
- 7. Karastergiou K, Evans I, Ogston N, Miheisi N, Nair D, Kaski JC, Jahangiri M, Mohamed‐Ali V. Epicardial adipokines in obesity and coronary artery disease induce atherogenic changes in monocytes and endothelial cells. Arterioscler Thromb Vasc Biol. 2010;30:1340–1346. doi: 10.1161/ATVBAHA.110.204719 [DOI] [PubMed] [Google Scholar]
- 8. Hirata Y, Tabata M, Kurobe H, Motoki T, Akaike M, Nishio C, Higashida M, Mikasa H, Nakaya Y, Takanashi S, et al. Coronary atherosclerosis is associated with macrophage polarization in epicardial adipose tissue. J Am Coll Cardiol. 2011;58:248–255. doi: 10.1016/J.JACC.2011.01.048 [DOI] [PubMed] [Google Scholar]
- 9. Iacobellis G. Local and systemic effects of the multifaceted epicardial adipose tissue depot. Nat Clin Pract Cardiovasc Med. 2005;2:536–543. doi: 10.1038/nrendo.2015.58 [DOI] [PubMed] [Google Scholar]
- 10. Mazurek T, Zhang L, Zalewski A, Mannion JD, Diehl JT, Arafat H, Sarov‐Blat L, O’Brien S, Keiper EA, Johnson AG, et al. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation. 2003;108:2460–2466. doi: 10.1161/01.CIR.0000099542.57313.C5 [DOI] [PubMed] [Google Scholar]
- 11. Antonopoulos AS, Margaritis M, Coutinho P, Shirodaria C, Psarros C, Herdman L, Sanna F, De Silva R, Petrou M, Sayeed R, et al. Adiponectin as a link between type 2 diabetes and vascular NADPH oxidase activity in the human arterial wall: the regulatory role of perivascular adipose tissue. Diabetes. 2015;64:2207–2219. doi: 10.2337/db14-1011 [DOI] [PubMed] [Google Scholar]
- 12. Fitzgibbons TP, Lee N, Tran K‐V, Nicoloro S, Kelly M, Tam SKC, Czech MP. Coronary disease is not associated with robust alterations in inflammatory gene expression in human epicardial fat. JCI Insight. 2019;4:e124859. doi: 10.1172/JCI.Insight.124859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McKenney‐Drake ML, Rodenbeck SD, Bruning RS, Kole A, Yancey KW, Alloosh M, Sacks HS, Sturek M. Epicardial adipose tissue removal potentiates outward remodeling and arrests coronary atherogenesis. Ann Thorac Surg. 2017;103:1622–1630. doi: 10.1016/J.ATHORACSUR.2016.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ge J, Jeremias A, Rupp A, Abels M, Baumgart D, Liu F, Haude M, Gorge G, Von Birgelen C, Sack S, et al. New signs characteristic of myocardial bridging demonstrated by intracoronary ultrasound and Doppler. Eur Heart J. 1999;20:1707–1716. doi: 10.1053/EUHJ.1999.1661 [DOI] [PubMed] [Google Scholar]
- 15. Corban MT, Hung OY, Eshtehardi P, Rasoul‐Arzrumly E, McDaniel M, Mekonnen G, Timmins LH, Lutz J, Guyton RA, Samady H. Myocardial bridging: contemporary understanding of pathophysiology with implications for diagnostic and therapeutic strategies. J Am Coll Cardiol. 2014;63:2346–2355. doi: 10.1016/J.JACC.2014.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rogers IS, Tremmel JA, Schnittger I. Myocardial bridges: overview of diagnosis and management. Congenit Heart Dis. 2017;12:619–623. doi: 10.1111/CHD.12499 [DOI] [PubMed] [Google Scholar]
- 17. Ishikawa Y, Akasaka Y, Akishima‐Fukasawa Y, Iuchi A, Suzuki K, Uno M, Abe E, Yang Y, Li C‐P, Mukai K, et al. Histopathologic profiles of coronary atherosclerosis by myocardial bridge underlying myocardial infarction. Atherosclerosis. 2013;226:118–123. doi: 10.1016/J.ATHEROSCLEROSIS.2012.10.037 [DOI] [PubMed] [Google Scholar]
- 18. Ishii T, Asuwa N, Masuda S, Ishikawa Y, Kiguchi H, Shimada K. Atherosclerosis suppression in the left anterior descending coronary artery by the presence of a myocardial bridge: an ultrastructural study. Mod Pathol. 1991;4:424–431. [PubMed] [Google Scholar]
- 19. Cheng C, Tempel D, van Haperen R, van der Baan A, Grosveld F, Daemen MJ, Krams R, de Crom R. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation. 2006;113:2744–2753. doi: 10.1161/CIRCULATIONAHA.105.590018 [DOI] [PubMed] [Google Scholar]
- 20. Heo KS, Fujiwara K, Abe J. Shear stress and atherosclerosis. Mol Cells. 2014;37:435–440. doi: 10.14348/molcells.2014.0078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Iacobellis G, Barbaro G. Epicardial adipose tissue feeding and overfeeding the heart. Nutrition. 2019;59:1–6. doi: 10.1016/J.NUT.2018.07.002 [DOI] [PubMed] [Google Scholar]
- 22. Gustafson B, Smith U. Cytokines promote Wnt signaling and inflammation and impair the normal differentiation and lipid accumulation in 3T3‐L1 preadipocytes. J Biol Chem. 2006;281:9507–9516. doi: 10.1074/jbc.M512077200 [DOI] [PubMed] [Google Scholar]
- 23. Isakson P, Hammarstedt A, Gustafson B, Smith U. Impaired preadipocyte differentiation in human abdominal obesity: role of Wnt, tumor necrosis factor‐alpha, and inflammation. Diabetes. 2009;58:1550–1557. doi: 10.2337/db08-1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu LF, Craig CM, Tolentino LL, Choi O, Morton J, Rivas H, Cushman SW, Engleman EG, McLaughlin T. Adipose tissue macrophages impair preadipocyte differentiation in humans. PLoS One. 2017;12:e0170728. doi: 10.1371/journal.pone.0170728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: role of free fatty acids and tumor necrosis factor alpha. Arterioscler Thromb Vasc Biol. 2005;25:2062–2068. doi: 10.1161/01.ATV.0000183883.72263.13 [DOI] [PubMed] [Google Scholar]
- 26. Kloting N, Berthold S, Kovacs P, Schon MR, Fasshauer M, Ruschke K, Stumvoll M, Gluher M. MicroRNA expression in human omental and subcutaneous adipose tissue. PLoS One. 2009;4:e4699. doi: 10.1371/JOURLAN.PONE.0004699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xie H, Sun L, Lodish HF. Targeting microRNAs in obesity. Expert Opin Ther Targets. 2009;13:1227–1238. doi: 10.1517/14728220903190707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ortega FJ, Moreno‐Navarrete JM, Pardo G, Sabater M, Hummel M, Ferrer A, Rodriguez‐Hermosa JI, Ruiz B, Ricart W, Peral B, et al. MiRNA expression profile of human subcutaneous adipose and during adipocyte differentiation. PLoS One. 2010;5:e9022. doi: 10.1371/JOURNAL.PONE.0009022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miranda K, Yang X, Bam M, Murphy EA, Nagarkatti PS, Nagarkatti M. MicroRNA‐30 modulates metabolic inflammation by regulating Notch signaling in adipose tissue macrophages. Int J Obes. 2018;42:1140–1150. doi: 10.1038/s4166-018-0114-01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Esau C, Kang X, Peralta E, Hanson E, Marcusson EG, Ravichandran LV, Sun Y, Koo S, Perera RJ, Jain R, et al. Micro‐RNA 143 regulates adipocyte differentiation. J Biol Chem. 2004;279:52361–52365. doi: 10.1074/jbc.C400438200 [DOI] [PubMed] [Google Scholar]
- 31. Boyd JH, Pargaonkar VS, Scoville DH, Rogers IS, Kimura T, Tanaka S, Yamada R, Fischbein MP, Tremmel JA, Mitchell RS, et al. Surgical unroofing of hemodynamically significant left anterior descending myocardial bridges. Ann Thorac Surg. 2017;10:1443–1450. doi: 10.1016/j.athoracsur.2016.08.035 [DOI] [PubMed] [Google Scholar]
- 32. Yamada R, Jennifer A, Tremmel JA, Tanaka S, Lin S, Kobayashi Y, Hollak MB, Yock PG, Fitzgerald PJ, Schnittger I, et al. Functional versus anatomic assessment of myocardial bridging by intravascular ultrasound: impact of arterial compression on proximal atherosclerotic plaque. J Am Heart Assoc. 2016;5:e0017. doi: 10.1161/JAHA.114.001735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Goeller M, Achenbach S, Cadet S, Kwan AC, Commandeur F, Slomka PJ, Gransar H, Albrecht MH, Tamarappoo BK, Berman DS, et al. Pericoronary adipose tissue computed tomography attenuation and high‐risk plaque characteristics in acute coronary syndrome compared with stable coronary artery disease. JAMA Cardiol. 2018;3:858–863. doi: 10.1001/JAMACARDIO.2018.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goeller M, Tamarappoo BK, Kwan AC, Cadet S, Commandeur F, Razipour A, Slomka PJ, Gransar H, Chen XI, Otaki Y, et al. Relationship between changes in pericoronary adipose tissue attenuation and coronary plaque burden quantified from coronary computed tomography angiography. Eur Heart J Cardiovasc Imaging. 2019;20:636–643. doi: 10.1093/EHJCI/JEZ013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kwiecinski J, Dey D, Cadet S, Lee SE, Otaki Y, Huynh PT, Doris MK, Eisenberg E, Yun M, Jansen MA, et al. Peri‐coronary adipose tissue density is associated with 18F‐sodium fluoride coronary uptake in stable patients with high‐risk plaques. JACC Cardiovasc Imaging. 2019;12:2000–2010. doi: 10.1016/j.JCMG.2018.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Antonopoulos AS, Sanna F, Sabharwal N, Thomas S, Oikonomou EK, Herdman L, Margaritis M, Shirodaria C, Kampoli A‐M, Akoumianakis I, et al. Detecting human coronary inflammation by imaging perivascular fat. Sci Transl Med. 2017;9:eaal2658. doi: 10.1126/SCITRANSLMED.AAL2658 [DOI] [PubMed] [Google Scholar]
- 37. Chechi K, Gelinas Y, Mathieu P, Deshaies Y, Richard D. Validation of reference genes for the relative quantification of gene expression in human epicardial adipose tissue. PLoS One. 2012;7:e32265. doi: 10.1371/JOURNAL.PONE.0032265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Davé V, Yousefi P, Huen K, Volberg V, Holland N. Relationship between expression and methylation of obesity‐related genes in children. Mutagenesis. 2015;30:411–420. doi: 10.1093/MUTAGE/GEU089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gaborit B, Venteclef N, Ancel P, Pelloux V, Gariboldi V, Leprince P, Amour J, Hatem SN, Jouve E, Dutour A, et al. Human epicardial adipose tissue has a specific transcriptomic signature depending on its anatomical peri‐atrial, peri‐ventricular, or peri‐coronary location. Cardiovasc Res. 2015;108:62–73. doi: 10.1093/CVR/CVV208 [DOI] [PubMed] [Google Scholar]
- 40. Rausch ME, Weisberg S, Vardhana P, Tortoriello DV. Obesity in C57BL/6J mice is characterized by adipose tissue hypoxia and cytotoxic T‐cell infiltration. Int J Obes. 2008;32:451–463. doi: 10.1038/sj.ijo.0803744 [DOI] [PubMed] [Google Scholar]
- 41. Pasarica M, Sereda OR, Redman LM, Albarado DC, Hymel DT, Roan LE, Rood JC, Burk DH, Smith SR. Reduced adipose tissue oxygenation in human obesity: evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes. 2009;58:718–725. doi: 10.2337/db08-1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Akoumianakis I, Antoniades C. The interplay between adipose tissue and the cardiovascular system: is fat always bad? Cardiovasc Res. 2017;113:999–1008. doi: 10.1093/cvr/cvx111 [DOI] [PubMed] [Google Scholar]
- 43. Iozzo P. Myocardial, perivascular, and epicardial fat. Diabetes Care. 2011;34:S371–S379. doi: 10.2337/dc11-s250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Man K, Kutyavin VI, Chawla A. Tissue immunometabolism: development, physiology, and pathobiology. Cell Metab. 2017;25:11–26. doi: 10.1016/j.cmet.2016.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee YH, Petkova AP, Granneman JG. Identification of an adipogenic niche for adipose tissue remodeling and restoration. Cell Metab. 2013;18:355–367. doi: 10.1016/j.cmet.2013.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW II, DeFuria J, Jick Z, Greenberg AS, Obin MS. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes. 2007;56:2910–2918. doi: 10.2337/db07-0767 [DOI] [PubMed] [Google Scholar]
- 47. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vacca M, Di Eusanio M, Cariello M, Graziano G, D'Amore S, Petridis FD, D'orazio A, Salvatore L, Tamburro A, Folesani G, et al. Integrative miRNA and whole‐genome analyses of epicardial adipose tissue in patients with coronary atherosclerosis. Cardiovasc Res. 2016;109:228–239. doi: 10.1093/cvr/cvv266 [DOI] [PubMed] [Google Scholar]
- 49. Esau C, Kang X, Peralta E, Hanson E, Marcusson EG, Ravichandran LV, Sun Y, Koo S, Perera RJ, Jain R, et al. MicroRNA‐143 regulates adipocyte differentiation. J Biol Chem. 2004;279:52361–52365. doi: 10.1074/jbc.C400438200 [DOI] [PubMed] [Google Scholar]
- 50. Hulsmans M, De Keyzer D, Holvoet P. MicroRNAs regulating oxidative stress and inflammation in relation to obesity and atherosclerosis. FASEB J. 2011;25:2515–2527. doi: 10.1096/fj.11-181149 [DOI] [PubMed] [Google Scholar]
- 51. Rupaimoole R, Ivan C, Yang D, Gharpure KM, Wu SY, Pecot CV, Previs RA, Nagaraja AS, Armaiz‐Pena GN, McGuire M, et al. Hypoxia‐upregulated microRNA‐630 targets Dicer, leading to increased tumor progression. Oncogene. 2016;35:4312–4320. doi: 10.1038/onc.2015.492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sun Y, Cai J, Yu S, Chen S, Li F, Fan C. MiR‐630 inhibits endothelial‐mesenchymal transition by targeting slug in traumatic heterotopic ossification. Sci Rep. 2016;6:22729. doi: 10.1038/srep22729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Medici D, Kalluri R. Endothelial‐mesenchymal transition and its contribution to the emergence of stem cell phenotype. Semin Cancer Biol. 2012;225:379–384. doi: 10.1016/j.semcancer.2012.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tabas I, Williams KJ, Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832–1844: doi: 10.1161/CIRCULATIONAHA.106.676890 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1