

Abstract
Ablation of protein acyltransferase DHHC3 selectively enhanced the anti-cancer cell activities of several chemotherapeutic agents, but not kinase inhibitors. To understand why this occurs, we used comparative mass spectrometry-based palmitoyl-proteomic analysis of breast and prostate cancer cell lines, ± DHHC3 ablation, to obtain the first comprehensive lists of candidate protein substrates palmitoylated by DHHC3. Putative substrates included 22–28 antioxidant/redox-regulatory proteins, thus predicting that DHHC3 should have antioxidant functions. Consistent with this, DHHC3 ablation elevated oxidative stress. Furthermore, DHHC3 ablation, together with chemotherapeutic drug treatment, (a) elevated oxidative stress, with a greater than additive effect, and (b) enhanced the anti-growth effects of the chemotherapeutic agents. These results suggest that DHHC3 ablation enhances chemotherapeutic drug potency by disabling the antioxidant protections that contribute to drug resistance. Affirming this concept, DHHC3 ablation synergized with another anti-cancer drug, PARP inhibitor PJ-34, to decrease cell proliferation and increase oxidative stress. Hence, DHHC3 targeting can be a useful strategy for selectively enhancing potency of oxidative stress-inducing anti-cancer drugs. Also, comprehensive identification of DHHC3 substrates provides insight into other DHHC3 functions, relevant to in vivo tumor growth modulation.
Electronic supplementary material
The online version of this article (10.1007/s00018-020-03635-3) contains supplementary material, which is available to authorized users.
Keywords: Chemotherapeutic agents, DHHC3, Oxidative stress, PARP inhibitor, Protein palmitoylation, Protein acyl transferases
Introduction
The DHHC (Asp-His-His-Cys) enzyme family, which includes at least 23 members in mammals, is responsible for most protein S-palmitoylation (hereafter termed palmitoylation) in the animal kingdom [1]. DHHC enzymes can have oncogenic, tumor suppressor and prognostic roles in cancer [2, 3]. The DHHC3 (GODZ) enzyme is elevated in human breast cancer and elevated gene expression correlates with reduced survival in patients with breast cancer and six other types of cancer [4]. Upon DHHC3 ablation, in vivo tumor xenograft growth was markedly diminished, in parallel with enhanced recruitment of innate immune cell types typically associated with clearance of senescent tumor cells [4].
Although DHHC3 ablation diminished tumor growth in vivo, it did not diminish primary tumor xenograft angiogenesis, proliferation, or apoptosis. Also, DHHC3 ablation did not affect in vitro cell proliferation or 3D soft agar growth [4]. Nonetheless, we tested whether DHHC3 ablation may affect tumor cell sensitivity to a variety of anti-cancer drugs. Indeed, cell growth inhibitory effects of a subset of anti-cancer agents were markedly enhanced in DHHC3-ablated cells. To understand why DHHC3 ablation may enhance the potency of some but not other anti-cancer drugs, we used a previously developed global palmitoyl-proteomic method called PalmPISC (Palmitoyl protein identification and site characterization) for palmitoyl-protein enrichment and mass spectrometric identification [5]. PalmPISC was coupled with triplex stable isotope labeling by amino acids in cell culture (SILAC) [6] to yield unbiased identification of DHHC3 substrates from both breast and prostate cancer cell lines. Results point to a major role for DHHC3 in palmitoylating proteins that have antioxidant functions and explain why DHHC3 ablation selectively enhances functions of anti-cancer drugs that elevate oxidative stress.
Material and methods
Antibodies, chemicals and other reagents
Antibodies to NPC1 (#ab134113), TMEM192 (#ab186737) and PRDX4 (#ab16943) were from Abcam. Others were to CMTM6 (# HPA026980; Sigma LifeSciences), cleaved caspase-3 (#9664S; Cell Signaling Co.) and HSP70 (#sc-298; Santa Cruz Biotechnologies. Gefitinib (#13166) and 5-fluorouracil (#14416) were from Cayman Chemicals (Ann Arbor, MI, USA). Iodoacetamide was from GE Healthcare. Camptothecin (#C9911), oxaliplatin (#O9512) drugs and PARP inhibitor (PJ34) (#P-4365), N-ethylmaleimide (NEM) were from Sigma-Aldrich. Lapatinib was from LC Laboratories, Woburn, MA and nifuroxazide was from Dr. David Frank, Dana-Farber Cancer Institute. Cell-ROX labeling (#C10422), CellTrace CFSE cell proliferation kit (#C34554) and FxCycle PI/RNase cell cycle staining solution (#F10797) were from Invitrogen. Tris(2-carboxyethyl)phosphine (TCEP), N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)propionamide (biotin-HPDP), high-capacity streptavidin agarose beads, and Micro BCA protein assay were from Pierce. Mass spec-grade trypsin was from Promega. Control and DHHC3 shRNA and siRNA sequences and DHHC3 wild type and active site mutant reconstitution vectors were described [4]. Knockdown and reconstitution was achieved using a single vector, containing either wild type or mutant DHHC3 cDNA’s, downstream of knockdown DHHC3 shRNA [4].
Mass spectrometry analyses of DHHC3 substrates
DHHC3 substrate identification was as described [5]. Briefly, control and DHHC3-ablated MDA-MB-231 and PC3 cells were labelled with normal media (Lys0, Arg0) or media containing medium (Lys4, Arg6) and heavy isotopes (Lys8, Arg10) for three passages (~ 8 doublings) to achieve complete labeling. Subsequently, membrane fractions were sonicated, purified, and solubilized in 4% SDS buffer. Proteins were reduced (TCEP 10 µM), alkylated (NEM 50 mM), then chloroform/methanol precipitated 5 times. Samples were then divided and treated with hydroxylamine (HA+) buffer or Tris buffer (HA−). Exposed SH moieties were then biotinylated using biotin-HPDP. Following three sequential methanol chloroform precipitations, proteins were dissolved in 2% SDS buffer and precipitated using streptavidin agarose beads for 1 h at room temperature. Finally, after washing beads 6 times with equilibration buffer, proteins were eluted using 20 mM TCEP, further chloroform/methanol precipitated, then dissolved in 15 µl reducing sample buffer. Proteins separated by SDS-PAGE were in-gel digested into tryptic peptides for MS analysis, using a C18 nanoflow reversed-phase HPLC (Eksigent nanoLC•2D™) connected to an LTQ Orbitrap mass spectrometer (Thermo Scientific). Tryptic peptides were loaded to an in-house packed (inner diameter 100 μm) 15-cm C18 column (Magic C18, 5 μm, 200 Å, Michrom Bioresources Inc.), followed by separation at 200 nl/min with 80-min linear gradient from 5 to 35% acetonitrile in 0.4% formic acid. Survey spectra were acquired in the Orbitrap with resolution of 30,000. Up to five most intense ions per cycle were fragmented and analyzed in the linear ion trap. The RAW files were analyzed by MaxQuant (v1.3.0.5) in combination with Andromeda search engine [7, 8], essentially as described [9, 10]. The parameters were as follows: Oxidation (M), acetyl (protein N-term), NEM (C), and carbamidomethyl (C) were set as variable modifications; the IPI_human database (v3.36; 69,012 sequences) was used; mass tolerance was set as ± 20 ppm for MS spectra and ± 0.7 Da for MS/MS spectra. The maximum false discovery rates for peptide and protein identifications were set as 0.01.
Immunofluorescence
MDA-MB-231 cells were fixed (4% paraformaldehyde), permeabilized (0.1% Triton X-100), preincubated (5% horse serum), then incubated with specific antibodies (NPC1, TMEM192, PRDX4) at 4 °C overnight. After PBS washing (3×), secondary antibody conjugated with fluorophore (Alexa Fluor 594) was added (1 h, room temperature). After washing (5×, with PBS), slides were mounted with Prolong Gold antifade mounting media containing DAPI (Invitrogen) and photographed (after 24 h) using Leica SP5X laser scanning microscope. Staining intensity was quantified using Scion Image software (Scion Corp., Frederick MD). CMTM6 immunostaining was performed as described [11].
Oxidative stress estimation
MDA-MB231, PC3 and BT-549 cells (± DHHC3 ablation) were treated (DMSO control, camptothecin, PJ-34, 5-Fu, oxaliplatin) for 24 h in complete media containing 5% serum. Oxidative stress was assessed using CellROX fluorescent dye as described [4]. Mean fluorescent intensities (MFI) were compared using FlowJo software.
Cell proliferation and cell cycle analysis
Control and DHHC3-ablated MDA-MB-231 cells were trypsinized and loaded with CFSE (5 mM) per manufacturer. After plating (6-well plates, 24 h), cells were treated with PJ-34 (10 µM) for another 24 h, washed, and CFSE staining was analyzed by flow cytometry and data finalized using FlowJo software. Alternatively, after PJ-34 treatment, cells were labeled with FxCycle PI/RNase staining solution per manufacturer, then staining was assessed by flow cytometry assisted by FlowJo “cell cycle analysis” function.
Results
DHHC3 ablation enhances chemotherapeutic drug sensitivity
Following shRNA knockdown or siRNA knockdown in MDA-MB-231 and PC3 cells, DHHC3 was undetectable (Fig. 1a) or minimally detectable (Fig. 1b). Next, we tested whether DHHC3 ablation might affect anti-cancer drug sensitivity. For MDA-MB-231 cells, DHHC3 knockdown alone (Fig. 1c, lane 2) minimally stimulated apoptosis as indicated by caspase 3 cleavage. However, DHHC3 ablation enhanced the effects of treatment with camptothecin (Fig. 1c, lanes 3, 4) or 5-Fu (Fig. 1c, lanes 7, 8). By contrast, treatment of MDA-MB-231 cells with gefitinib, an EGFR inhibitor, did not show enhanced effectiveness in DHHC3-ablated cells (Fig. 1c, lanes 5, 6), thus serving as a negative control. In addition, apoptosis induction by active doses of Jak2/Tyk2 kinase inhibitor nifuroxazide (Fig. 1d, lanes 5,6) and EGFR inhibitor lapatinib (Fig. 1d, lanes 7,8) were not enhanced by DHHC3 shRNA ablation, while in the same experiment, effects of camptothecin were again enhanced (Fig. 1d, lanes 3, 4). DHHC3 knockdown enhanced camptothecin effects on cleaved caspase-3 by a mean of 2.1-fold (± 0.8 SD, N = 4) and 5-FU effects by 4.9-fold (± 1.2 SD, N = 3). The three different kinase inhibitors (GEF, NEF, LAPT) yielded a mean enhancement of only 0.58-fold (± 0.04 SD, N = 3).
Fig. 1.
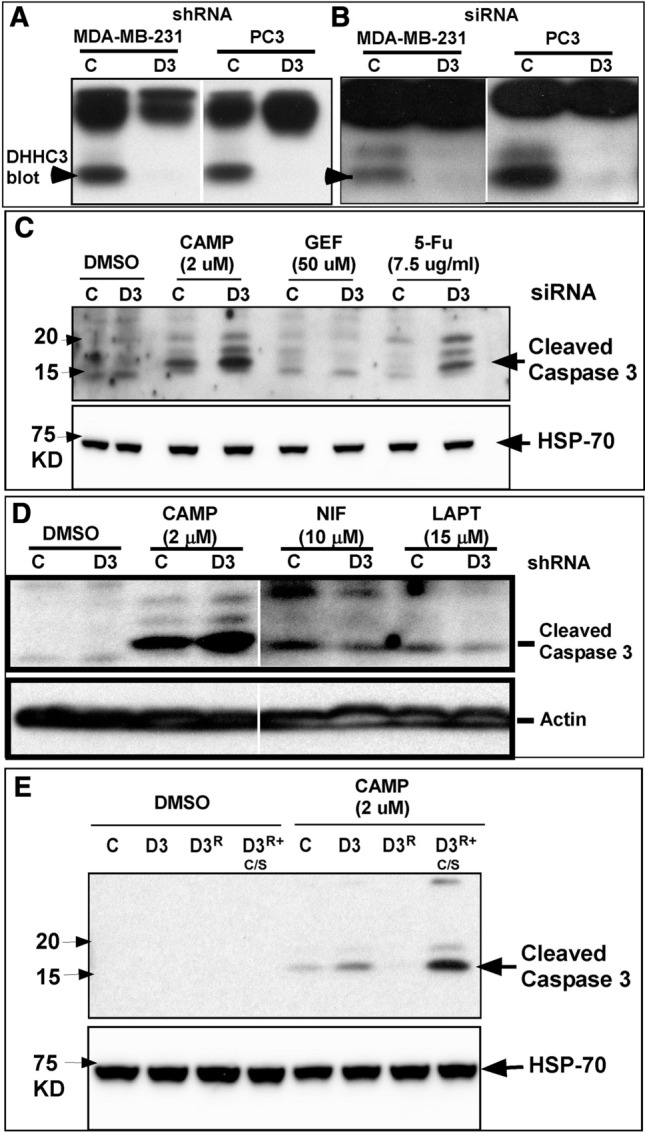
DHHC3 ablation enhances chemotherapeutic drug effects. DHHC3 expression is shown for MDA-MB-231 and PC3 cells a stably ablated for DHHC3, or b transiently ablated for DHHC3 using control (C) or DHHC3 (D) shRNA or siRNA. c Cleaved caspase 3 expression (indicator of apoptosis) is shown for MDA-MB-231 cells transiently transfected with control (C), or DHHC3 siRNA (D3), and treated with DMSO, camptothecin, gefitinib and 5-Fu for 24 h at indicated concentrations in 5% complete media. HSP-70 expression is used as a loading control (bottom panel). d MDA-MB-231 cells (control, C; DHHC3 ablated, D3) were treated with DMSO, camptothecin, nifuroxazide, or lapatinib at the indicated concentrations, for 24 h. Actin expression is used as a loading control (bottom panel). e Cleaved caspase-3 expression is shown for MDA-MB-231 cells, either control ablated (C) or stably DHHC3-ablated (D3) and reconstituted with wild-type DHHC3 (D3R) or reconstituted with palmitoylation-deficient mutant DHHC3 (D3R+C/S). All cells were treated with DMSO alone, or camptothecin (in DMSO) for 24 h in 5% complete media. HSP-70 expression is used as a loading control (bottom panel)
Whereas DHHC3 ablation rendered MDA-MB-231 cells more susceptible to camptothecin-triggered apoptosis (Fig. 1e, lanes 5, 6), this effect was reversed by reconstitution with wild type DHHC3 (Fig. 1e, lane 7), but not mutant DHHC3 (C → S point mutant; lacking palmitoylation activity; lane 8). Cleaved caspase 3 was diminished in DHHC3-reconstituted cells (lane 7) to a level even lower than in control cells (expressing endogenous DHHC3, lane 5), perhaps because levels of reconstituted DHHC3 are 1.5–2-fold greater than endogenous DHHC3 (not shown). The substantially elevated level of cleaved caspase 3 seen in lane 8 suggests that palmitoylation-deficient DHHC3 may exert a dominant negative effect. In control experiments, DHHC3 ablation, reconstitution and mutation did not trigger apoptosis in cells not treated with camptothecin (Fig. 1e, lanes 1–4).
Identification of DHHC3 substrates in breast and prostate cancer cells
To understand why DHHC3 ablation might enhance activities of select anti-cancer drugs, we needed to identify substrate proteins palmitoylated by DHHC3. For multiple reasons, breast and prostate cancer cell lines MDA-MB-231 and PC3 were chosen for this purpose. First, DHHC3 expression is elevated in malignant and metastatic breast cancer [4], and in malignant and metastatic prostate cancer (Suppl. Figure 1A). Second, zDHHC3 upregulation correlates with diminished patient survival in breast cancer [4], and in prostate cancer (Suppl. Figure 1B). Third, DHHC3 ablation markedly diminishes in vivo xenograft growth of tumors arising from both MDA-MB-231 [4] and PC3 cells (Suppl. Figure 1C).
Both breast (MDA-MB-231) and prostate (PC3) cell lines were treated with control shRNA or DHHC3-targeting shRNA. DHHC3 substrates were identified by quantitative palmitoyl-proteomic comparison of cells with, versus without, DHHC3 ablation, via integrating triplex SILAC with PalmPISC (Suppl. Figure 2). Totals of 1,097 and 1,137 candidate proteins were identified from MDA-MB-231 and PC3 cells, respectively, with false discovery rate of ≤ 1% (Raw data in Suppl. Tables 1, 2). Focusing on palmitoylated proteins, only proteins with + HA/− HA ratio of ≥ 1.5 were considered further. In DHHC3-ablated cells, compared to control cells, putative DHHC3 substrates have significantly lower palmitoylation levels. Potential DHHC3 substrates include 85 proteins from MDA-MB-231 cells (ranked in Table 1), 47 proteins from PC3 cells (Table 2), and 26 proteins common to both cell types (Table 3).
Table 1.
DHHC3 substrates identified from MDA-MB-231 cells
| Protein name | Ratio (+ D3/ − D3) | # of peptides | |
|---|---|---|---|
| 1 | SUMF2* | 13.2 | 2 |
| 2 | NOL6 | 3.3 | 2 |
| 3 | LAPTM4A | 3.0 | 3 |
| 4 | CMTM3 | 2.9 | 2 |
| 5 | ITGA6(20526329) | 2.7 | 2 |
| 6 | TMEM192 | 2.6 | 2 |
| 7 | FAM108A1 | 2.5 | 2 |
| 8 | TMEM97(31109310) | 2.5 | 6 |
| 9 | NPC1 (22216111) | 2.4 | 10 |
| 10 | HIST1H1C | 2.4 | 3 |
| 11 | TMEM179B | 2.1 | 2 |
| 12 | OSTC | 2.0 | 2 |
| 13 | TMED1 | 1.9 | 5 |
| 14 | GPX8(28751022) | 1.9 | 6 |
| 15 | M6PR(23984879) | 1.9 | 9 |
| 16 | AGPAT1 | 1.9 | 9 |
| 17 | METTL7B | 1.9 | 2 |
| 18 | SLC4A7 | 1.8 | 6 |
| 19 | BET1 | 1.8 | 2 |
| 20 | DERL2* | 1.8 | 5 |
| 21 | PTRH2 | 1.8 | 18 |
| 22 | CDKAL1 | 1.8 | 4 |
| 23 | SPRY2(20489163) | 1.8 | 2 |
| 24 | AUP1 | 1.8 | 10 |
| 25 | CKAP4 | 1.8 | 79 |
| 26 | SAR1A(31409740) | 1.8 | 2 |
| 27 | PRDX4(24098506) | 1.7 | 3 |
| 28 | SOAT1 | 1.7 | 3 |
| 29 | SCARB1(29976771) | 1.7 | 3 |
| 30 | SPINT2* | 1.7 | 2 |
| 31 | LMF2 | 1.7 | 15 |
| 32 | PCBP2(26907686) | 1.7 | 4 |
| 33 | SCAP*(29902864) | 1.7 | 2 |
| 34 | ZDHHC6 | 1.7 | 2 |
| 35 | ERGIC3(27588471) | 1.7 | 8 |
| 36 | SPRY4 | 1.7 | 24 |
| 37 | TFRC (21447374) | 1.6 | 63 |
| 38 | SRPRB | 1.6 | 2 |
| 39 | MDC1(26870895) | 1.6 | 3 |
| 40 | PTTG1IP | 1.6 | 9 |
| 41 | RCE1 | 1.6 | 3 |
| 42 | CYB5B | 1.6 | 17 |
| 43 | PRDX1(27653015) | 1.6 | 3 |
| 44 | COPA(25030084) | 1.6 | 3 |
| 45 | TSPAN6 | 1.6 | 4 |
| 46 | TIMP2*(15351863) | 1.6 | 2 |
| 47 | TUBB2C | 1.6 | 2 |
| 48 | FAR1(26714049) | 1.5 | 3 |
| 49 | GANAB(25658244) | 1.5 | 2 |
| 50 | TUBA1B | 1.5 | 37 |
| 51 | CD58 | 1.5 | 4 |
| 52 | CLPTM1 | 1.5 | 4 |
| 53 | RPL36 | 1.5 | 2 |
| 54 | MRPS18B | 1.5 | 2 |
| 55 | RHBDL7 | 1.5 | 2 |
| 56 | EBP | 1.5 | 2 |
| 57 | ARL6IP6 | 1.5 | 7 |
| 58 | TM4SF18* | 1.5 | 3 |
| 59 | FADS2(30732887) | 1.5 | 7 |
| 60 | NUP155 | 1.5 | 3 |
| 61 | AP2M1 | 1.5 | 3 |
| 62 | NHP2L1 | 1.5 | 6 |
| 63 | RER1 | 1.5 | 4 |
| 64 | NRSN2 | 1.5 | 2 |
| 65 | GLCE | 1.5 | 2 |
| 66 | TMEM55B | 1.5 | 3 |
| 67 | TOP2A(24995306) | 1.5 | 45 |
| 68 | SLC30A7 | 1.5 | 3 |
| 69 | MGST1(21428695) | 1.5 | 5 |
| 70 | RRP9 | 1.5 | 4 |
| 71 | SHISA3* | 1.5 | 2 |
| 72 | ITM2C | 1.5 | 3 |
| 73 | CD63 | 1.5 | 26 |
| 74 | SCAMP1 | 1.5 | 4 |
| 75 | SERPINH1(24412200) | 1.5 | 6 |
| 76 | CYP51A1(28658622) | 1.5 | 9 |
| 77 | TBL2 | 1.5 | 4 |
| 78 | NOMO2 | 1.5 | 12 |
| 79 | TMEM222 | 1.5 | 2 |
| 80 | RAB7A(27383256) | 1.5 | 17 |
| 81 | MAGT1(31559137) | 1.5 | 6 |
| 82 | NDUFS1(27799543) | 1.5 | 5 |
| 83 | MBOAT7 | 1.5 | 9 |
| 84 | TMX3(31304984) | 1.5 | 3 |
| 85 | PTDSS1 | 1.5 | 5 |
Criteria for inclusion in table: ≥ 2 peptides; + D3/-D3 ratio ≥ 1.5. Proteins that are bolded and italicized, with superscript PMID#s, are functionally linked to oxidative stress, reactive oxygen species and/or endoplasmic reticulum stress
*Proteins that have not been cross-referenced in the swisspalm.org database but contain likely cytoplasmic cysteines as potential sites for palmitoylation
Table 2.
DHHC3 substrates identified from PC3 cells
| Protein name | Ratio (+ D3/ − D3) | # of peptides | |
|---|---|---|---|
| 1 | ZDHHC3(29055014) | 4.1 | 2 |
| 2 | CBX5 | 3.0 | 2 |
| 3 | ANO1* | 2.5 | 3 |
| 4 | ITGA6(20526329) | 2.5 | 12 |
| 5 | TMEM192 | 2.2 | 4 |
| 6 | TMED1 | 2.2 | 8 |
| 7 | CD58 | 2.1 | 3 |
| 8 | CKAP4 | 2.0 | 80 |
| 9 | CMTM6 | 2.0 | 2 |
| 10 | B4GALT1(30551084) | 1.9 | 5 |
| 11 | TMEM97(31109310) | 1.8 | 4 |
| 12 | DHCR24(24489783) | 1.8 | 4 |
| 13 | TMX3(31304984) | 1.7 | 13 |
| 14 | CPD(23656787) | 1.7 | 2 |
| 15 | ITGB4(17604276) | 1.7 | 50 |
| 16 | SURF4 | 1.7 | 7 |
| 17 | CD44(27815445) | 1.7 | 60 |
| 18 | FAS(21711425) | 1.6 | 2 |
| 19 | GNB2L1 | 1.6 | 12 |
| 20 | ERGIC2(24303950) | 1.6 | 2 |
| 21 | M6PR(23984879) | 1.6 | 11 |
| 22 | ASAH1(30413652) | 1.5 | 10 |
| 23 | ADAM17 | 1.5 | 3 |
| 24 | JAG1 | 1.5 | 3 |
| 25 | FAF2 | 1.5 | 2 |
| 26 | BRI3BP | 1.4 | 3 |
| 27 | TSPAN13 | 1.4 | 4 |
| 28 | ERGIC1 | 1.4 | 5 |
| 29 | ERGIC3(27588471) | 1.4 | 11 |
| 30 | GAPD | 1.4 | 7 |
| 31 | GPX8(28751022) | 1.4 | 5 |
| 32 | PRDX5(17937766) | 1.4 | 2 |
| 33 | IGFR2 | 1.3 | 15 |
| 34 | PSEN1(10097174) | 1.3 | 6 |
| 35 | LOC3927(30401882) | 1.3 | 5 |
| 36 | BAT1 | 1.3 | 3 |
| 37 | NPC1(22216111) | 1.3 | 11 |
| 38 | AFG3L2(20700718) | 1.3 | 4 |
| 39 | TMEM55B | 1.3 | 6 |
| 40 | TRMT61A* | 1.3 | 2 |
| 41 | VKORC1L1(21367861) | 1.3 | 2 |
| 42 | KRAS | 1.3 | 2 |
| 43 | MREG | 1.3 | 9 |
| 44 | DHCR7 | 1.3 | 21 |
| 45 | NDUFA11(30531981) | 1.3 | 2 |
| 46 | C8orf55 | 1.3 | 14 |
| 47 | RER1 | 1.3 | 8 |
Criteria for inclusion in table: ≥ 2 peptides; + D3/-D3 ratio ≥ 1.3. Proteins that are bolded and italicized, with superscript PMID#s, are functionally linked to oxidative stress, reactive oxygen species and/or endoplasmic reticulum stress
*Proteins that have not been cross-referenced in the swisspalm.org database but contain likely cytoplasmic cysteines as potential sites for palmitoylation
Table 3.
DHHC3 substrates shared between MDA-MB-231 and PC3 cellsa
| Protein name | Ratio in MDA-MB-231 cells | Ratio in PC3 cells | # of peptides | Link with redox regulation (PMID #) | |
|---|---|---|---|---|---|
| 1 | ITGA6 | 2.7 | 2.5 | 2, 12 | OS (20526329) |
| 2 | TMEM192 | 2.6 | 2.2 | 2, 4 | |
| 3 | TMEM97 | 2.5 | 1.8 | 6, 4 | ES (30042674) |
| 4 | NPC1 | 2.4 | 1.3 | 10, 11 | OS (19458211) |
| 5 | TMED1 | 1.9 | 2.2 | 5, 8 | |
| 6 | GPX8 | 1.9 | 1.4 | 6, 5 | OS (24566470) |
| 7 | M6PR | 1.9 | 1.6 | 9, 11 | |
| 8 | CKAP4 | 1.8 | 2.0 | 79, 80 | |
| 9 | ERGIC3 | 1.7 | 1.4 | 8, 11 | ES (27588471) |
| 10 | CD58 | 1.5 | 2.1 | 4, 3 | |
| 11 | RER1 | 1.5 | 1.3 | 4, 8 | |
| 12 | TMEM55B | 1.5 | 1.3 | 3, 6 | |
| 13 | CMTM6 | 2.1 | 2.0 | 1, 2 | |
| 14 | DHCR24 | 1.3 | 1.8 | 7, 4 | OS (24489783) |
| 15 | TMX3 | 1.5 | 1.7 | 3, 13 | OS (31304984) |
| 16 | ITGB4 | 1.2 | 1.7 | 28, 50 | OS (20364299) |
| 17 | SURF4 | 1.4 | 1.7 | 5, 7 | |
| 18 | ASAH1 | 2.0 | 1.5 | 1, 10 | OS (30413652) |
| 19 | ADAM17 | 1.2 | 1.5 | 7, 3 | |
| 20 | BRI3BP | 2.0 | 1.4 | 1, 3 | |
| 21 | ERGIC1 | 1.2 | 1.4 | 2, 5 | |
| 22 | GAPD | 1.5 | 1.4 | 4, 7 | |
| 23 | IGF2R | 1.2 | 1.3 | 18, 15 | OS (24667322) |
| 24 | PSEN1 | 1.3 | 1.3 | 5, 6 | OS (9501245) |
| 25 | MREG | 2.6 | 1.3 | 1, 9 | |
| 26 | C8orf55 | 1.20 | 1.3 | 12, 14 |
Ingenuity Pathway Analysis (IPA) software was used for unbiased determination of direct and indirect relationships connecting candidate DHHC3 substrates with various biological and/or disease functions. MDA-MB-231 substrates yielded IPA-derived “Top Tox Lists”, in which 4 of the top 5 categories (including “Oxidative stress” and “NRF2-mediated oxidative stress response”) emphasize oxidative stress-related themes (Fig. 2a). Similarly, putative DHHC3 substrates from PC3 cells yielded a “Top Tox List” with top categories of “Mitochondrial Dysfunction” and “Increase Damage of Mitochondria” (Fig. 2b). PC3 cell substrates were further analyzed to yield an IPA-derived list of “Molecular and Cellular Functions”, which included “Cellular Compromise”, “Free Radical Scavenging”, and “Lipid Metabolism” as top categories (Fig. 2c). Further illustrating connections between candidate DHHC3 substrates and oxidative stress regulation, IPA-created networks show that 8 proteins from MDA-MB-231 cells (Suppl. Figure 3A), 11 from PC3 cells (Suppl. Figure 3B), and 5/26 of the shared proteins (Suppl. Figure 3C) are directly linked to “Metabolism of Reactive Oxygen Species”. Literature searches using key words “oxidative stress”, “reactive oxygen species” or “ER stress” (which is closely linked to oxidative stress) show additional candidate substrate proteins functionally linked to oxidative stress as annotated (Table 1, 28/85; Table 2, 22/47; Table 3, 11/26).
Fig. 2.

DHHC3 substrates linked to functional categories, using ingenuity pathway analysis. a “Top Toxicity” functions associated with 85 DHHC3 substrates from MDA-MB-231 breast cancer cells; b “Top Toxicity” functions associated with 47 DHHC3 substrates from PC3 prostate cancer cells; c top “Molecular and Cellular Functions” associated with 47 DHHC3 substrates from PC3 prostate cancer cells
Validation of candidate DHHC3 substrates
We independently assessed whether DHHC3 ablation indeed affected palmitoylation and subcellular distribution of representative potential substrates appearing in the top half of the shared list (Table 3), including redox regulatory protein NPC1 [12], TMEM192, which has been associated with oxidative stress sensitivity [13], and CMTM6. From lysates of DHHC3-ablated MDA-MB-231 cells, recoveries of biotinylated (originally palmitoylated) NPC1, TMEM192, and CMTM6 were reduced by 93% (Fig. 3a, lanes 3, 4), 46% (Fig. 3c, lanes 3,4) and 97% (Suppl. Figure 4A, lanes 3,4) respectively, relative to control-ablated cells. In additional control experiments, protein abundance of NPC1, TMEM192 and CMTM6 in whole cell lysates was minimally affected by DHHC3 ablation (decreases of 11%, 3%, 11%, respectively; Fig. 3a, c; Suppl. Figure 4A; lanes 1, 2 in each). In concert with altered palmitoylation, NPC1, TMEM192 and CMTM6 also showed altered subcellular localization in DHHC3-ablated MDA-MB-231 cells. In control cells, NPC1 showed relatively uniform cytoplasmic expression visualized by immunofluorescence staining (Fig. 3b, left panels). By contrast, DHHC3-ablated cells showed NPC1 more abundantly concentrated around the nucleus (Fig. 3b, right panels). In control-ablated MDA-MB-231 cells, TMEM192 appeared mostly in single large perinuclear clusters (Fig. 3d, left panels), consistent with expected lysosomal localization [14]. By contrast, TMEM192 in DHHC3-ablated cells appeared in several smaller puncta, with broader perinuclear distribution (Fig. 3d, right panels). Also, control ablated MDA-MB-231 cells showed more uniform CMTM6 distribution on the cell surface and in the cytoplasm, whereas DHHC3-ablated cells displayed a more intense signal proximal to the nucleus and reduced signal on cell surface and cytoplasm (Suppl. Figure 4B). Altered distributions of NPC1, TMEM192 and CMTM6 were also evident from individual cell ImageJ color intensity profiles (Fig. 3b, d; Suppl. 4B, bottom panels). Peroxiredoxin (PRDX4), another key redox regulator [15], is a potential DHHC3 substrate in MDA-MB-231 cells (protein #27; Table 1). In control-ablated MDA-MB-231 cells, PRDX4 shows concentrated peri-nuclear staining. In DHHC3-ablated cells, it is more uniformly distributed around the nucleus (Suppl. Figure 4C). ImageJ color intensity profiles are consistent with altered PRDX4 distribution (Suppl. Figure 4C, bottom panels). Validation of additional substrates (e.g. GPX8, TMED1) was unfeasible due to unavailability of suitable antibodies. Many palmitoylated proteins are unaffected by DHHC3 knockdown, thus serving as negative controls. For example, Suppl. Figure 4A shows an unknown background control protein (lanes 3, 4, bottom), which appears upon HA treatment (and thus is palmitoylated) but is not diminished when DHHC3 is knocked down. Also, many other known protein palmitoylations are unaffected by DHHC3. In this regard, the set of proteins least affected by DHHC3 knockdown (± 1.1 fold change in peptide recovery; within Suppl. Tables 1 and 2) includes many highly palmitoylated proteins [e.g. VAMP3, CLDN3, CAV1, CAV2, CD9, CD151, ZDHHC13 from MDA-MB-231 cells (within Suppl. Table 1); e.g. CMTM7, FLOT1, SCAMP2, SCAMP3, SCAMP5, SNAP23, VAMP7, TMX4 from PC3 cells (within Suppl. Table 2)]. Assessment of palmitoylation is based on enhanced recovery due to HA treatment (data in Suppl. Tables 1 and 2), which is consistent with information in the SwissPalm database.
Fig. 3.

Substrate validation for NPC1 and TMTM192. a Total cell lysates from control (C) or DHHC3-ablated (D3) MDA-MB-231 cells were blotted for major oxidative stress regulator NPC1 (lanes 1, 2). Lysates from MDA-MB-231 cells were reduced, alkylated, treated with hydroxylamine (HA, to remove palmitate), and then exposed cysteines were biotinylated. After immunoprecipitation with neutrAvidin agarose beads, protein complexes were resolved and then blotted for NPC1 (lanes 3, 4). Numbers at the bottom indicate NPC1 recovery ratios − D3/ + D3. b NPC1 cell distribution in control and D3 ablated cells was analyzed by immunofluorescence using anti-NPC1 antibody (red). Blue color depicts nuclei staining (DAPI). Line graphs (bottom panels) indicate representative pixel density profiles of NPC1 staining in control and D3 ablated cells, obtained using Image J software. c As in a, total cell lysates and neutravidin immunoprecipitated proteins were prepared from MDA-MB-231 cells [control (C) and DHHC3 (D3)-ablated] and then blotted for TMEM192. TMEM192 recovery ratios, − D3/ + D3, are indicated at the bottom. d TMEM192 cell distribution was analyzed by immunofluorescence using anti-TMEM192 antibody (red). Blue color depicts nuclei staining (DAPI). Line graphs (bottom panels) indicate representative pixel density profiles of TMEM192 staining in control and D3 ablated cells, obtained using Image J software
Elevated oxidative stress
Consistent with disruption of antioxidant protections, DHHC3 ablation alone was sufficient to elevate oxidative stress, as seen by CellRox labeling in three different cell lines (Fig. 4a–c; gray curves). Treatment with chemotherapeutic agents camptothecin (Fig. 4a), oxaliplatin (Fig. 4b) and 5-fluorouracil (Fig. 4c) also increased oxidative stress (red curves). However, combined effects of DHHC3 ablation and camptothecin treatment were greater than sums of individual effects, by factors of 1.3, 2.1 and 1.2 respectively (Fig. 4a, top, middle, bottom dark blue curves). Similarly, in Fig. 4b, the sum of individual effects (DHHC3 ablation, ∆ 51 MFI; oxaliplatin treatment, ∆ 15 MFI) on BT-549 cells was exceeded by combined effects (∆ 119 MFI). Likewise, DHHC3 ablation plus 5-Fu treatment increased oxidative stress in PC3 cells (∆ 110 MFI) more than either condition alone (∆ 60 MFI; ∆ 40 MFI) (Fig. 4c).
Fig. 4.

DHHC3 ablation and chemotherapeutic drug effects on oxidative stress. a CellRox fluorescence is used to assess oxidative stress in cells (MDA-MB-231, top panel; BT-549, middle panel; PC3, bottom panel) that were DHHC3-ablated (D3 KD) or not ablated (Cont KD) and treated with either DMSO or camptothecin (CAMP) for 24 h in complete media with 5% serum. b Cells were treated as in panel A, except that oxaliplatin at a concentration of 10 µg/ml for 24 h in media with 5% serum was used. c Cells were treated as in a, except that 5-fluorouracil at a concentration of 7.5 µg/ml for 24 h in complete media with 5% serum was used
Another example of oxidative stress and drug synergy: DHHC3 ablation and a PARP inhibitor
Results above establish a link between DHHC3 ablation and oxidative stress and point to antioxidant functions of DHHC3 in suppressing chemotherapeutic drug effects on cancer cells. To test this further, we analyzed another anti-cancer drug, PARP inhibitor PJ-34, whose functions (inhibition of cell growth, triggering of oxidative stress) are reversed upon addition of an antioxidant [16]. Again, we observed that presence of DHHC3 exerted drug suppression and antioxidant effects. Consistent with other studies of PARP inhibitors [16, 17], we observed reduced proliferation (Fig. 5a) and elevated S-phase cell cycle arrest (Fig. 5b) in PJ-34-treated MDA-MB-231 cells. The magnitude of decreased CFSE staining (Fig. 5a) was most apparent for PJ-34 treatment of DHHC3-ablated MDA-MB-231 cells (26% decreased proliferation, bottom panel) compared to PJ-34 treatment of control KD cells (13% decreased proliferation, top panel). DHHC3 ablation alone did not diminish proliferation (Fig. 5a). Similarly, S phase accumulation (44.4%) was most obvious when cells were both ablated for DHHC3 and treated with PJ-34, compared to either alone (Fig. 5b). Addition of PJ-34 did not trigger cleaved caspase 3-mediated apoptosis in the cell types analyzed (not shown).
Fig. 5.

DHHC3 ablation enhances PARP inhibitor (PJ-34) effects. a The extent of CFSE staining decrease is used to assess proliferation of MDA-MB-231 cells that were DHHC3-ablated (D3 KD; bottom panel) or not ablated (Cont KD; top panel) using specific siRNA. Cells were CFSE loaded (for 36 h) and then either not further incubated (0 h; black, blue curves) or treated with DMSO (gray, red curves) or 10 µM PJ-34 (yellow, green curves) for an additional 48 h. b Propidium iodide fluorescence is used for cell cycle analysis of MDA-MB-231 cells transfected with control and DHHC3 siRNA, and treated with DMSO or PJ-34 (20 µM) for 24 h. c CellRox fluorescence is used to assess oxidative stress in cells (MDA-MB-231, top panel; BT-549, middle panel; PC3, bottom panel) that were DHHC3-ablated (D3 KD) or not ablated (Cont KD) using siRNA, and treated with either DMSO or PJ-34 for 24 h in complete media with 5% serum
DHHC3 ablation (Fig. 5c, gray curves) or PJ-34 treatment (olive curves) alone caused relatively small increases in oxidative stress in MDA-MB-231, BT-549 and PC3 cells, as seen by CellRox fluorescence. However, both ablation and drug treatment together caused shifts that were substantially greater (by 2.7-, 4.6-, 1.8-fold; dark blue curves) than sums of individual treatments of MDA-MB-231, BT-549 and PC3 cells respectively.
Discussion
Increased anti-cancer drug potency
DHHC3 ablation by itself had minimal effect on tumor cell apoptosis. However, when combined with chemotherapeutic drug treatment, drug-induced apoptosis was markedly enhanced. Results were confirmed using multiple chemotherapeutic agents, and multiple tumor cell lines. Protection from drug-induced oxidative stress and apoptosis was restored upon reconstitution with functional DHHC3, but not palmitoylation-deficient (active site mutant) DHHC3, thus ruling out off-target effects of DHHC3 ablation. Expression of inactivated DHHC3 enabled drug-induced cell death even more than did DHHC3 ablation, suggesting a possible dominant negative effect. Inactive DHHC3 could perhaps block key substrates from being palmitoylated by compensatory DHHC enzymes, such as the closely related DHHC7 [18].
DHHC3 substrates establish a mechanistic link to oxidative stress regulation
To gain insight into why DHHC3 ablation might enhance chemotherapeutic drug activity, we needed information about its protein substrates. A few DHHC3 protein substrates were previously identified, including integrin α6 and β4 subunits [19], G protein α subunit [20], GABA(A) receptor [21], regulator of G protein signaling 4 (RGS4) [22] and phosphatidylinositol 4-kinase IIα [23]. However, unbiased and comprehensive substrate analysis had not been reported. To achieve this, we used quantitative palmitoyl-proteomics [5] to identify candidate DHHC3 substrates (85 from MDA-MB-231 cells; 47 from PC3 cells) with 26 proteins appearing in both cell type lists. Selection criteria were that (a) proteins must be palmitoylated (i.e. HA sensitive) and (b) preferentially recovered when DHHC3 is present vs absent. It seems unlikely, but perhaps not impossible, that expression of a palmitoylated protein would diminish, due to DHHC3 ablation, for some reason other than loss of palmitoylation.
The DHHC3 substrate list is validated by presence of previously identified proteins (e.g. ITGA6 [19], ITGB4 [19], ERGIC3 [4]) plus DHHC3 itself, which undergoes autopalmitoylation [24]. DHHC3-dependent palmitoylation was independently verified for representative substrates (NPC1, TMEM192 and CMTM6) newly identified from both MDA-MB-231 and PC3 cells. Effects of DHHC3 ablation on substrate palmitoylation were similar in magnitude whether detected by mass spectrometry or Western blotting. Subcellular distributions of these proteins, as well as PRDX4, were markedly altered in DHHC3-ablated cells, as typically observed when protein palmitoylation is impaired [25, 26]. Among DHHC3 substrate cysteines known (within integrin α6, integrin β4) [19], or predicted using CSS Palmitoylation Prediction program (e.g. in ERGIC3, CMTM6, TMEM192, NPC1) there is no apparent flanking sequence similarity. Thus, DHHC3 substrate specificity must be determined elsewhere in substrate proteins.
Putative substrates identified were broadly involved in redox regulation, including 28/85 from MDA-MB-231 cells, 22/47 from PC3 cells, and 11/26 from the shared substrate list. Unbiased Ingenuity Pathway Analysis (IPA) assessments link both sets of candidate substrates to oxidative stress, free radical scavenging, and mitochondrial dysfunction, which is closely linked to oxidative stress [27]. Furthermore, IPA-assembled networks directly link 8 substrates from MDA-MB-231 cells, 11 from PC3 cells, and 5 from both lists to “Metabolism of reactive oxygen species”. Hence, simultaneous disruption of palmitoylation and function of many redox regulatory proteins likely explains elevated oxidative stress in DHHC3-ablated cells, such as first noticed in breast cancer cell lines [4] and confirmed here in additional cell lines and extended to prostate cancer cell line PC3.
Oxidative stress plays a key role during enhanced drug activity
Potency of chemotherapeutic agents is typically attenuated by antioxidants, which help to protect cells by preventing the triggering of excess oxidative stress [28, 29]. Our results are consistent with DHHC3 also exerting a protective antioxidant function that diminishes chemotherapeutic drug potency (summarized in Suppl. Figure 5). Indeed, when DHHC3 was present, chemotherapeutic drug-induced apoptosis was diminished, in concert with reduced oxidative stress. It is notable that DHHC3 ablation did not enhance the activities of EGFR inhibitors gefitinib and lapatinib or Jak2/Tyk2 inhibitor nifuroxazide, which do not trigger oxidative stress.
As a test of our hypothesis that DHHC3 antioxidant functions can protect cancer cells from oxidative stress-inducing anti-cancer drugs, we studied PARP inhibitor PJ-34. In concert with diminished cell growth, PARP inhibitors, such as PJ-34, can induce oxidative stress in cancer cells as seen previously [16] and confirmed here in multiple cell lines. Furthermore, inhibitory functions of PJ-34 were shown to be attenuated in the presence of an antioxidant [16]. Presence or absence of DHHC3 ablation alone had little effect on cell proliferation or S-phase arrest. However, ablation of DHHC3 markedly enhanced anti-proliferative effects of PJ-34, as seen in tumor cell proliferation and cell cycle arrest assays. The absence of DHHC3 also synergistically enhanced the triggering of oxidative stress by PJ-34. These results are again consistent with DHHC3 having an antioxidant function that blunts the effectiveness of the oxidative stress-inducing drug.
Other implications regarding DHHC3 substrates
Antioxidant protein TMX3 and related protein TMX1 support melanoma progression by a mechanism involving suppression of oxidative stress [30]. To function properly, TMX proteins require palmitoylation [31]. TMX3 is a substrate for DHHC3 in both MDA-MB-231 and PC3 cells. It remains to be seen whether loss of TMX3 palmitoylation may affect tumor cell growth in DHHC3-ablated cells.
It was previously suggested that DHHC3 ablation diminishes breast tumor xenograft growth by a mechanism involving oxidative stress-triggered senescence-associated secretory phenotype (SASP), resulting in tumor clearance by innate immune cells [4]. We now understand that diminished palmitoylation of several antioxidant-type proteins provides a key link between DHHC3 ablation and elevated oxidative stress.
We show here that DHHC3-ablated prostate carcinoma PC3 cells, as seen previously for breast cancer cells, showed (a) altered redox regulation, (b) elevated oxidative stress, and (c) reduced xenograft growth. These results help explain correlations between elevated ZDHHC3 expression and diminished survival in patients with prostate cancer (shown here), and other cancers [4].
Identification of CMTM6 as a substrate suggests that DHHC3 ablation could at least partly diminish tumor growth by a mechanism involving enhanced adaptive immunity. Removal of CMTM6 markedly decreases PD-L1 levels [11, 32], and DHHC3 ablation may enhance adaptive immunity [33]. We show that DHHC3 ablation alters CMTM6 palmitoylation and distribution, which could lead to decreased PD-L1 levels. Effects of diminished CMTM6 palmitoylation on PD-L1 expression and adaptive immunity are currently under investigation. Notably, our substrate lists did not include PD-L1. Also, extensive cell labeling with [H3]-palmitate did not yield detectable PD-L1 palmitoylation (not shown). These results contrast with reports that PD-L1 itself may be palmitoylated [33, 34].
While our current studies are focused on pre-existing cellular drug resistance mechanisms, we suggest that DHHC3 ablation and/or inhibition should also help to overcome longer term acquired resistance to chemotherapeutic agents, since that also involves antioxidant upregulation [28, 35].
Conclusions
First, DHHC3 ablation did not affect activities of multiple anti-cancer kinase inhibitors but did increase the potency of multiple anti-cancer drugs (i.e. chemotherapeutic agents, PARP inhibitor) that trigger elevated oxidative stress. Second, the first comprehensive identification of candidate DHHC3 substrates now explains how DHHC3 ablation can elevate oxidative stress by simultaneously disrupting the antioxidant functions of many proteins. Third, DHHC3 ablation and chemotherapeutic drug treatment synergistically enhance both drug function and oxidative stress. This reinforces the idea that antioxidant protection mechanisms that blunt effects of the anti-cancer drugs, are being disabled. Fourth, DHHC3 utility as potential drug target and prognostic marker now expands beyond breast cancer [4], to include prostate cancer. Fifth, lists of newly identified putative DHHC3 substrates provide clues regarding additional mechanisms by which DHHC3 may affect tumor behavior. In summary, DHHC3 targeting, either alone or combined with other oxidative stress-inducing agents, should be a useful anti-cancer strategy.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Abbreviations
- DHHC3
Enzyme #3 in a family containing Asp–His–His–Cys motif
- NPC1
NPC intracellular cholesterol transporter 1
- PalmPISC
Palmitoyl protein identification and site characterization
- PARP
Poly (ADP-ribose) polymerase
- PRDX4
Peroxiredoxin 4
- SILAC
Stable isotope labeling by amino acids in cell culture
- TMEM192
Transmembrane protein 192
- TMX1 and TMX3
Thioredoxin related transmembrane proteins 1 and 3
Author contributions
WY and HS carried out mass spectrometry experiments, assisted by CS. CS carried out all other experiments. MF helped to direct the project. CS prepared figures and tables and wrote the manuscript, with assistance from MEH.
Funding
This study was supported by NIH grant CA42368 to M Hemler and C Sharma, DoD grant W81XWH-11-1-0112 to M Hemler, and DoD grant W81XWH-11-1-0113 to M Freeman. The funding body played no role in the design of the study, collection, analysis, and interpretation of data, or in writing the manuscript.
Availability of data and materials
Datasets generated and analyzed during the current study will be available as supplemental tables, and data with additional details will be available in the appropriate repository.
Compliance with ethical standards
Conflict of interest
There are no financial or non-financial competing or conflicting interests to be declared.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Chandan Sharma, Email: chandan_sharma@dfci.harvard.edu.
Martin E. Hemler, Email: martin_hemler@dfci.harvard.edu
References
- 1.Mitchell DA, Vasudevan A, Linder ME, Deschenes RJ. Protein palmitoylation by a family of DHHC protein S-acyltransferases. J Lipid Res. 2006;47:1118–1127. doi: 10.1194/jlr.R600007-JLR200. [DOI] [PubMed] [Google Scholar]
- 2.Yeste-Velasco M, Linder ME, Lu YJ. Protein S-palmitoylation and cancer. Biochim Biophys Acta. 2015;1856:107–120. doi: 10.1016/j.bbcan.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Ko PJ, Dixon SJ. Protein palmitoylation and cancer. EMBO Rep. 2018;19:e46666. doi: 10.15252/embr.201846666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma C, Wang HX, Li Q, Knoblich K, Reisenbichler ES, Richardson A, Hemler ME. Protein acyltransferase DHHC3 regulates breast tumor growth, oxidative stress and senescence. Can Res. 2017;77:6880–6890. doi: 10.1158/0008-5472.CAN-17-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang W, Di Vizio D, Kirchner M, Steen H, Freeman MR. Proteome scale characterization of human S-acylated proteins in lipid raft-enriched and non-raft membranes. Mol Cell Proteomics. 2010;9:54–70. doi: 10.1074/mcp.M800448-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blagoev B, Kratchmarova I, Ong SE, Nielsen M, Foster LJ, Mann M. A proteomics strategy to elucidate functional protein-protein interactions applied to EGF signaling. Nat Biotechnol. 2003;21:315–318. doi: 10.1038/nbt790. [DOI] [PubMed] [Google Scholar]
- 7.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 8.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 9.Xu Q, Biener-Ramanujan E, Yang W, Ramanujan VK. Targeting metabolic plasticity in breast cancer cells via mitochondrial complex I modulation. Breast Cancer Res Treat. 2015;150:43–56. doi: 10.1007/s10549-015-3304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Minciacchi VR, You S, Spinelli C, Morley S, Zandian M, Aspuria PJ, Cavallini L, Ciardiello C, Reis SM, Morello M, Kharmate G, Jang SC, Kim DK, Hosseini-Beheshti E, Tomlinson GE, Gleave M, Gho YS, Mathivanan S, Yang W, Freeman MR, Di VD. Large oncosomes contain distinct protein cargo and represent a separate functional class of tumor-derived extracellular vesicles. Oncotarget. 2015;6:11327–11341. doi: 10.18632/oncotarget.3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burr ML, Sparbier CE, Chan YC, Williamson JC, Woods K, Beavis PA, Lam EYN, Henderson MA, Bell CC, Stolzenburg S, Gilan O, Bloor S, Noori T, Morgens DW, Bassik MC, Neeson PJ, Behren A, Darcy PK, Dawson SJ, Voskoboinik I, Trapani JA, Cebon J, Lehner PJ, Dawson MA. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature. 2017;549:101–105. doi: 10.1038/nature23643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu R, Yanjanin NM, Bianconi S, Pavan WJ, Porter FD. Oxidative stress in Niemann-Pick disease, type C. Mol Genet Metab. 2010;101:214–218. doi: 10.1016/j.ymgme.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chandiramani N, Wang X, Margeta M. Molecular basis for vulnerability to mitochondrial and oxidative stress in a neuroendocrine CRI-G1 cell line. PLoS ONE. 2011;6:e14485. doi: 10.1371/journal.pone.0014485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Z, Lv YJ, Song YP, Li XH, Du YN, Wang CH, Hu LK. Lysosomal membrane protein TMEM192 deficiency triggers crosstalk between autophagy and apoptosis in HepG2 hepatoma cells. Oncol Rep. 2012;28:985–991. doi: 10.3892/or.2012.1881. [DOI] [PubMed] [Google Scholar]
- 15.Nystrom T, Yang J, Molin M. Peroxiredoxins, gerontogenes linking aging to genome instability and cancer. Genes Dev. 2012;26:2001–2008. doi: 10.1101/gad.200006.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hou D, Liu Z, Xu X, Liu Q, Zhang X, Kong B, Wei JJ, Gong Y, Shao C. Increased oxidative stress mediates the antitumor effect of PARP inhibition in ovarian cancer. Redox Biol. 2018;17:99–111. doi: 10.1016/j.redox.2018.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Slade D. Mitotic functions of poly(ADP-ribose) polymerases. Biochem Pharmacol. 2019;167:33–43. doi: 10.1016/j.bcp.2019.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kilpatrick CL, Murakami S, Feng M, Wu X, Lal R, Chen G, Du K, Luscher B. Dissociation of Golgi-associated DHHC-type zinc finger protein (GODZ)- and Sertoli cell gene with a zinc finger domain-beta (SERZ-beta)-mediated palmitoylation by loss of function analyses in knock-out mice. J Biol Chem. 2016;291:27371–27386. doi: 10.1074/jbc.M116.732768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma C, Rabinovitz I, Hemler ME. Palmitoylation by DHHC3 is critical for the function, expression and stability of integrin a6b4. Cell Mol Life Sci. 2012;69:2233–2244. doi: 10.1007/s00018-012-0924-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsutsumi R, Fukata Y, Noritake J, Iwanaga T, Perez F, Fukata M. Identification of G protein alpha subunit-palmitoylating enzyme. Mol Cell Biol. 2009;29:435–447. doi: 10.1128/MCB.01144-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keller CA, Yuan X, Panzanelli P, Martin ML, Alldred M, Sassoe-Pognetto M, Luscher B. The gamma2 subunit of GABA(A) receptors is a substrate for palmitoylation by GODZ. J Neurosci. 2004;24:5881–5891. doi: 10.1523/JNEUROSCI.1037-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J, Xie Y, Wolff DW, Abel PW, Tu Y. DHHC protein-dependent palmitoylation protects regulator of G-protein signaling 4 from proteasome degradation. FEBS Lett. 2010;584:4570–4574. doi: 10.1016/j.febslet.2010.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lu D, Sun HQ, Wang H, Barylko B, Fukata Y, Fukata M, Albanesi JP, Yin HL. Phosphatidylinositol 4-kinase IIalpha is palmitoylated by Golgi-localized palmitoyl transferases in cholesterol-dependent manner. J Biol Chem. 2012;287:21856–21865. doi: 10.1074/jbc.M112.348094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jennings BC, Linder ME. DHHC protein S-acyltransferases use similar ping-pong kinetic mechanisms but display different acyl-CoA specificities. J Biol Chem. 2012;287:7236–7245. doi: 10.1074/jbc.M111.337246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Linder ME, Deschenes RJ. Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol. 2007;8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- 26.Aicart-Ramos C, Valero RA, Rodriguez-Crespo I. Protein palmitoylation and subcellular trafficking. Biochim Biophys Acta. 2011;1808:2981–2994. doi: 10.1016/j.bbamem.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 27.Handy DE, Loscalzo J. Redox regulation of mitochondrial function. Antioxid Redox Signal. 2012;16:1323–1367. doi: 10.1089/ars.2011.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 29.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X, Gibhardt CS, Will T, Stanisz H, Korbel C, Mitkovski M, Stejerean I, Cappello S, Pacheu-Grau D, Dudek J, Tahbaz N, Mina L, Simmen T, Laschke MW, Menger MD, Schon MP, Helms V, Niemeyer BA, Rehling P, Vultur A, Bogeski I. Redox signals at the ER-mitochondria interface control melanoma progression. EMBO J. 2019;38:e100871. doi: 10.15252/embj.2018100871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raturi A, Gutierrez T, Ortiz-Sandoval C, Ruangkittisakul A, Herrera-Cruz MS, Rockley JP, Gesson K, Ourdev D, Lou PH, Lucchinetti E, Tahbaz N, Zaugg M, Baksh S, Ballanyi K, Simmen T. TMX1 determines cancer cell metabolism as a thiol-based modulator of ER-mitochondria Ca2+ flux. J Cell Biol. 2016;214:433–444. doi: 10.1083/jcb.201512077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries, Wu W, Logtenberg MEW, Slagter M, Rozeman EA, Hofland I, Broeks A, Horlings HM, Wessels LFA, Blank CU, Xiao Y, Heck AJR, Borst J, Brummelkamp TR, Schumacher TNM (2017)Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 549:106–110 [DOI] [PMC free article] [PubMed]
- 33.Yao H, Lan J, Li C, Shi H, Brosseau JP, Wang H, Lu H, Fang C, Zhang Y, Liang L, Zhou X, Wang C, Xue Y, Cui Y, Xu J. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat Biomed Eng. 2019;3:306–317. doi: 10.1038/s41551-019-0375-6. [DOI] [PubMed] [Google Scholar]
- 34.Yang Y, Hsu JM, Sun L, Chan LC, Li CW, Hsu JL, Wei Y, Xia W, Hou J, Qiu Y, Hung MC. Palmitoylation stabilizes PD-L1 to promote breast tumor growth. Cell Res. 2019;29:83–86. doi: 10.1038/s41422-018-0124-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Landriscina M, Maddalena F, Laudiero G, Esposito F. Adaptation to oxidative stress, chemoresistance, and cell survival. Antioxid Redox Signal. 2009;11:2701–2716. doi: 10.1089/ars.2009.2692. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Datasets generated and analyzed during the current study will be available as supplemental tables, and data with additional details will be available in the appropriate repository.