

Abstract
Intravenous (i.v.) morphine is a safe, robust, and recommended treatment for severe pain using the titration principle. Despite its high efficacy, it is impacted by organizational constraints related to venous access. Nebulized (NEB) morphine may represent an alternative for titration but pharmacokinetic (PK) properties of short nebulization using routine devices need evaluation. Twenty‐seven healthy volunteers were included to receive NEB or i.v. morphine administration using increasing amounts according to Dixon’s reference method. Plasma morphine, morphine‐3‐glucuronide (M3G), and morphine‐6‐glucuronide (M6G) were quantified. PK modeling and simulations were performed using Monolix. Dixon’s method exhibited a significantly higher morphine dose regimen in the NEB group versus the i.v. group (6.2 [5.3–7.1] vs. 3.0 [2.0–4.0] mg, p < 0.001). Morphine, M3G, and M6G dose‐normalized exposure were significantly lower in the NEB group versus the i.v. group: morphine (19 [13–23] vs. 1044 [702–1266] µg min/L, p < 0.001), M3G (245 [162–287] vs. 3752 [2487–5165] µg min/L, p < 0.001) and M6G (28 [21–43] vs. 466 [370–723] µg min/L, p < 0.001). The model that best fitted the data consisted in a transit compartment for morphine absorption, three compartments for morphine distribution followed by multiple transit compartments (8.2 and 57.5‐min transit time for M3G and M6G, respectively) and a first order elimination for M3G and M6G. Morphine bioavailability in the NEB group was 3.5% using the i.v. group as reference. Administration route and sex significantly influenced morphine and metabolite PKs. This work aimed to evaluate the PKs of NEB morphine compared with the i.v. route. Despite a bioavailability to improve, NEB morphine administration using a routine device is suitable to plan morphine titration.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Intravenous morphine titration is the gold standard of pain management but suffers from organizational constraints: nebulized morphine could be an alternative.
WHAT QUESTION DID THIS STUDY ADDRESS?
The aim of this study was to compare the population pharmacokinetics (PopPKs) of intravenous (i.v.) versus nebulized (NEB) morphine in healthy phase I/II trial participants in order to assess the feasibility and robustness of NEB morphine using an inhalator device suitable in emergency departments and postanesthesia care units.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Despite the low bioavailability of NEB morphine linked mainly to leakage from the mask, it was possible using the developed PopPK model to predict confidently the PKs and variability of NEB morphine.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
NEB morphine could be used in acute pain situations with predictable morphine concentrations. Sex and route of administration were found to have potentially clinically relevant effects on morphine and metabolite PK in healthy participants.
INTRODUCTION
Intravenous (i.v.) morphine titration is an efficient and safe way to induce analgesia in emergency departments (EDs) and postanesthesia care units (PACUs) 1 , 2 , 3 and to fight against the oligo‐analgesia problematic, which is linked to opioid undertreatment. 2 However, this technique requires venous access and is nurse time‐consuming. As a consequence, it is associated with a 30% deviation from standard protocol in EDs. 4 Thus, morphine titration must be modernized to meet the new challenges of noninvasive practices while keeping an unchanged risk‐benefit ratio compared with the i.v. route.
Nebulized (NEB) morphine may represent an interesting alternative, improving morphine accessibility in patients with poor vein access (sickle cell anemia, children, elderly, etc.). This easy‐to‐use route could also be more robust in crowding situations and increase morphine tolerance. 5
The lack of homogenous aerosol techniques (doses, administration routes, and duration time), evaluation goals, and available data in the literature make it difficult to assess the effectiveness and appropriateness of NEB administration. 6 , 7 , 8 Moreover, pharmacological studies conducted in healthy volunteers lack relevance for clinical objectives and there is currently no gold‐standard pain model. The need for stronger fundamentals, especially regarding routine devices, is essential to improve our knowledge and challenge i.v. morphine titration at the bedside. 6 , 8 , 9 , 10 , 11 , 12
Morphine is the major alkaloid constituent of opium and belongs to the large family of opioids. It provides central analgesia by agonist action on µ‐opioid receptor, a G protein‐coupled receptor, which leads to inhibition of the nociceptive transmission in afferent fibers. 13 , 14 After administration, morphine is extensively metabolized by glucuronidation catalyzed by UDP‐glucuronosyltransferase enzymes with UGT2B7 known to be the major isoform responsible for 3‐ and 6‐glucuronidation of morphine in humans. 15 The main morphine metabolite, morphine‐3‐glucuronide (M3G), represents nearly 90% of morphine metabolism in comparison with ~ 10% for morphine‐6‐glucuronide (M6G). 16 Toxicity and analgesia related to morphine are also linked to M3G and M6G, respectively. 17
A better characterization of the pharmacokinetic (PK) profile of morphine and metabolites using short NEB administration would help to estimate the most appropriate dose of morphine to use as an alternative to i.v. titrations in EDs and PACUs.
Population PK (PopPK) modeling using compartmental analysis is a widely used approach for NEB drugs in clinical practice because it provides the ability to use transit compartments to explain delayed appearance 18 and drug concentration prediction, which is particularly useful in the case of repeated administration over a short period of time, as in the case of morphine titration.
In this context, the aim of the present study was to assess in healthy volunteers the PK profile of morphine, M3G, and M6G in an experimental model of short NEB morphine administration with a routine ED device for inhaled therapeutics, in comparison with i.v. administration, using a compartmental PopPK parent‐metabolite model.
MATERIALS AND METHODS
Consent and ethics
This study was approved by the French National Agency for Medicines and Health Products Safety (reference 130976A‐32), the local ethics committee of Rouen Normandy University Hospital (reference 02/17/2013), and was registered on https://clinicaltrials.gov/ on October 29, 2013 (reference NCT01975753, principal investigator: Dr V. Lvovschi). Written informed consent was obtained from healthy volunteers at the inclusion visit.
Population
Healthy volunteers eligible for this study had to be aged between 18 and 60 years old with a body mass index (BMI) ranging from 19 to 29 kg/m² with effective contraception for women of childbearing age. Noninclusion criteria were chronic antalgic and/or psychoactive medication, healthy volunteers with chronic pain, use of narcotics, active smoking, chronic neuropsychiatric conditions that may alter the pain threshold, chronic obstructive or restrictive respiratory disease, sleep apnea syndrome, risk factors for chronic kidney disease, known progressive disease, all chronic medications except oral contraception, impaired judgment, poor understanding of the French language, pregnancy or breastfeeding, poor venous capital, history of abnormal reaction to local anesthesia or known hypersensitivity to opioids, heart rate lower than 50 beats per minute, hypotension with systolic blood pressure (SBP) below 100 mmHg, respiratory rate lower than 12, atrioventricular block with a PR interval higher than 200 ms, a period of exclusion from other biomedical research, and volunteers placed under judicial protection, guardianship, or curatorship.
Study design
Either i.v. or NEB morphine was administered using the Dixon’s up‐and‐down method, classically used in this research area, for the determination of the median effective dose (ED50). 19 A standard RIII reflex model (sural nerve) 20 was adapted to this therapeutic purpose. One of the main judgment criteria was the estimation of a coefficient between the ED50 observed in both groups. The study is a parallel group randomized trial with patients receiving either a single dose of i.v. morphine and NEB sodium chloride or a single dose of NEB morphine and i.v. sodium chloride. The inhaler device consisted in a transparent polyvinyl chloride (PVC) nebulizer mask (CE0120 – Class IIa; Hudson RCI). A 2.1‐m transparent PVC tube (Doran International) and a 10 ml transparent reservoir suitable for emergency use (Figure S1). This device allows drug nebulization with an average mass diameter of 3.6 microns, at an average speed of 0.8 ml/min, allowing a greater potential to lung rather than mouth‐throat deposition. 21 A 5‐min inhalation was assessed diluting 10 mg/ml morphine chlorhydrate in sodium chloride to a final volume of 3 ml. At a constant airflow rate of 10 L/min, all drug particles were nebulized at the end of the procedure. Morphine doses ranged from 1 to 5 mg and 3 to 8 mg for i.v. and NEB route, respectively. A 0.3 mg increase or decrease in dose was performed according to the previous patient pain visual analogic scale (PVAS). During opiate test session, heart rate, respiratory rate, SBP, diastolic blood pressure, and PVAS values were recorded. Electrostimulations were carried out every minute for 5 min and 2, 10, 15, and 30 min after the end of the inhalation. Blood sampling was performed on 4‐ml lithium heparin tubes (BD Vacutainer) just before each electrostimulation with additional blood samples drawn at 60, 120, 180, 240, and 300 min after the nebulization was stopped. After centrifugation at 3000 g for 10 min at +5°C, the plasma was stored at −80°C until morphine, M3G, and M6G quantification.
Quantification of morphine, M3G, and M6G
Morphine, M3G, and M6G were quantified with a validated method for clinical practice by on‐line solid‐phase extraction followed by high performance liquid chromatography coupled with tandem mass spectrometry using a Prominence Shimadzu UFLC system (Shimadzu, Prominence, Kyoto, Japan) in combination with a 4500QTRAP equipped with an electrospray ionization source operating in positive ion mode (Sciex, Toronto, Canada). Briefly, 100 µl of plasma were mixed with 200 µl of methanol (MeOH) spiked with internal standards (2 µg/L of morphine‐d3, 20 µg/L of M3G‐d3, and 20 µg/L of M6G‐d3; Euromedex, France) for protein precipitation. After centrifugation at 13,200 g for 5 min, 200 µl of supernatant were collected and mixed with 200 µl of 0.2% ammonium hydroxide before injection. Quantitation was performed using the following precursor ion to product ion transition (quantifier/qualifier): morphine m/z 286.0 greater than 152.0/286.0 greater than 201.0, M3G and M6G m/z 462.0 greater than 285.9/462.0 greater than 152.0. Separation of M3G and M6G was based on retention time difference between the two metabolites. Chromatographic separation was performed on a Raptor Biphenyl column (2.7 μm particle size, 50 mm length ×3 mm inner diameter; Restek, France) after flush/flush online extraction using an Oasis HLB on‐line analytical column (25‐μm particle size, 20 mm length ×2.1 mm inner diameter; Waters SAS, France) using 0.2% of NH4OH in pure water. The auto‐sampler temperature was set at 8°C, the column oven at 30°C, the injected volume was 50 μl, and the flow rate was 700 μl/min. The mobile phase consisted in 0.2% formic acid in pure MeOH (solvent A) and 2 mM ammonium formate with 0.2% formic acid in water (solvent B). The following elution gradient was performed: 80% B (0–0.2 min), 80–40% B (0.2–1 min), 40–0% B (1–1.2 min), 0% B (1.2–2.2 min), 0–80% B (2.2–2.3 min), and 80% B (2.3–4 min). Calibration curves ranged from 0.1 to 10 µg/L for morphine and 1–100 µg/L for M3G and M6G. Samples above the upper limit of quantification for morphine, M3G, or M6G were 50‐fold diluted in morphine, M3G, and M6G serum‐free.
Pharmacokinetic modeling
PK analysis was performed using MonolixSuite version 2020R1 (Lixoft, Antony, France). 22 Morphine, M3G, and M6G concentrations were converted from µg/L to nM for the parent‐metabolite model to take into account the difference between morphine and glucuronides molecular weights (285.34 and 461.46 µg/µmol, respectively, molar ratio of 1.62). All data below the lower limit of quantification (LLOQ) were censored and imputed in an interval between 0 and the LLOQ not to lose information. Structural models were designed using a user‐defined ordinary differential equation written in Mlxtran language. PK parameters were considered as lognormal (solutions ∈ > 0); except bioavailability, which was considered as logit‐normal (solutions ∈]0;1[). For the development of the statistical models (covariates and residual error model), continuous covariates (COVs) age, weight, and BMI were centered to the mean and log‐transformed. Categorical covariates (CAVs) were coded as zero for women and the i.v. route (reference) and one for men and the NEB route. The impact of COV and CAV were tested as follows: log(θ) = log(θpop) + βCOV × COV and log(θ) = log(θpop) + βCAV, respectively. Both structural and statistical model selection and diagnosis were evaluated. A drop in corrected Bayesian Information Criterion (BICc) derives from the objective function value 23 was considered as a significant improvement of the model. 24
Model building was carried out in the following way: (1°) the i.v. group was used to determine the best PK model for morphine alone. Initial population parameters were estimated using the Stochastic Approximation Expectation‐Maximization algorithm starting with large omega population parameters and simulated annealing. (2°) The optimal parent‐metabolite model for the i.v. group was built to assess M3G and M6G production and elimination. (3°) The final parent‐metabolite model of the i.v. group was used in the whole dataset (both the NEB and i.v. groups) to determine initial absorption parameters for the NEB group. (4°) The determination of covariate effects was carried out disabling the simulated annealing allowing to remove parameter random effects when the variability tends toward zero. Then, covariates were added or removed based on correlation tests. (5°) The Fisher Information Matrix calculated by stochastic approximation was used to fix parameters fixed effect determined by the Monolix estimation for relative standard error higher than 50%. (6°) A “relative” sensitivity analysis was performed to study the impact of a 20% increase of each fixed parameter fixed effect on morphine, M3G, and M6G PK profiles.
Simulation
The final PopPK model was used to simulate concentration–time curves with Simulx version 2020R1. For the simulation, patients received either three NEB (one 5‐min NEB every 10 min) or three i.v. injections (one bolus every 5 min) to mimic morphine titration to treat severe pain in the EDs. 4 Doses were 15 mg and 3 mg for NEB and i.v. administrations, respectively. NEB and i.v. administrations were simulated for 1000 patients using the population parameters (both fixed and random effects including covariates) estimated by Monolix for each of the following conditions: NEB Male, NEB Female, i.v. Male, and i.v. Female. Morphine as well as M3G and M6G concentrations were plotted versus time.
Statistical analysis
For patient characteristics, data were expressed as median interquartile range or n (%). The p values comparing healthy volunteers between the i.v. and NEB routes were computed using χ² test or Mann‐Whitney U test when appropriate using R software version 4.0.0. 25
RESULTS
Population
Twenty‐seven healthy volunteers were included in the study, 13 in the NEB group and 14 in the i.v. group. In the NEB group, 161 of 178 (90%) morphine, 94 of 178 (53%) M3G, and 17 of 178 (10%) M6G were included for PK analysis. In the i.v. group, 174 of 188 (93%) morphine, 154 of 188 M3G (82%), and 98 of 188 (52%) M6G plasma samples were used for PK analysis. In the i.v. group, all samples collected at t = 1 min (n = 14) were excluded due to value higher than the upper limit of quantification, whereas other missing data were due to values below the LLOQ (Supplementary Material S2 – Dataset.csv). No significant differences were found for age, sex, BMI, nor hemodynamics between the NEB and i.v. groups. The number of samples per patient for M3G and M6G was significantly lower for the NEB group compared with the i.v. group (7 [7–8] vs. 11 [11–12], p < 0.001 and 2 [0–2] vs. 7 [7–8], p < 0.001, respectively). Dixon’s up‐and‐down method exhibited a significantly higher morphine dose regimen in the NEB group compared with the i.v. group (6.2 [5.3–7.1] vs. 3.0 [2.0–4.0] mg, p < 0.001; Table 1, Supplementary Material S1 – Table S1).
TABLE 1.
Summary of demographics, biological, and clinical characteristics
| Parameters |
Intravenous route (N = 14) |
Nebulization route (N = 13) |
p value |
|---|---|---|---|
| Age, years | 25 [24–34] | 27 [25–50] | 0.239 |
| Male, n (%) | 7 (50%) | 7 (54%) | 0.842 |
| Weight, kg | 71 [62–76] | 68 [63–75] | 0.715 |
| Height, cm | 174 [166–177] | 178 [164–178] | 0.789 |
| Body mass index, kg/m² | 24 [23–25] | 23 [21–25] | 0.308 |
| No. of samples per patient | |||
| Morphine | 13 [13–13] | 13 [12–13] | 0.809 |
| M3G | 11 [11–12] | 7 [7–8] | <0.001 |
| M6G | 7 [7–8] | 2 [0–2] | <0.001 |
| Morphine dose regimen, mg | 3.0 [2.0–4.0] | 6.2 [5.3–7.1] | <0.001 |
Demographics of the 27 healthy volunteers included. Data are expressed as median (IQR), n (%). The p values comparing intravenous versus nebulization route were computed using χ² test and Mann‐Whitney U test.
P‐value below 0.05 are in bold.
Abbreviations: M3G, morphine‐3‐glucuronide; M6G, morphine‐6‐glucuronide.
Quantification of morphine, M3G, and M6G
We observed significantly lower concentrations for the NEB group compared with the i.v. group for all samples at 5, 7, 15, 35, 65, 125, 245, and 305 min (p < 0.001) except for M6G at 5 and 7 min. The dose‐normalized area under the curve (AUC) were significantly lower in the NEB group compared with the i.v. group for morphine (19 [13–23] vs. 1044 [702–1266] µg min/L, p < 0.001), M3G (245 [162–287] vs. 3752 [2487–5165] µg min/L, p < 0.001), and M6G (28 [21–43] vs. 466 [370–723] µg min/L, p < 0.001). Finally, M3G‐to‐morphine and M6G‐to‐morphine AUC ratios were significantly higher in the NEB group compared with the i.v. group (12.47 [11.70–12.81] vs. 4.46 [2.84–5.27], p < 0.001 and 1.54 [1.36–1.67] vs. 0.53 [0.42–0.74], p < 0.001, respectively; Table 2).
TABLE 2.
Summary of morphine, M3G, and M6G plasma concentrations and dose‐normalized concentrations
| Parameters | Intravenous route | Nebulization route(N = 13) | p value |
|---|---|---|---|
| Morphine/morphine (dose‐normalized), µg/L | |||
| T5 | 33.0 [24.1–53.4]/12.6 [8.3–21.1] | 1.6 [1.1–3.1]/0.27 [0.22–0.43] | <0.001 |
| T7 | 29.7 [12.3–33.4]/8.2 [4.6–9.9] | 1.7 [1.1–2.8]/0.29 [0.22–0.39] | <0.001 |
| T15 | 16.7 [7.2–21.6]/4.6 [3.0–7.7] | 1.1 [0.5–1.5]/0.17 [0.13–0.22] | <0.001 |
| T35 | 5.5 [3.2–11.4]/2.3 [1.7–2.8] | 0.9 [0.4–1.0]/0.12 [0.08–0.14] | <0.001 |
| T65 | 4.4 [2.2–8.6]/1.5 [1.2–1.8] | 0.4 [0.3–0.5]/0.07 [0.06–0.07] | <0.001 |
| T125 | 2.7 [1.2–5.1]/0.9 [0.7–1.2] | 0.3 [0.2–0.4]/0.05 [0.04–0.06] | <0.001 |
| T185 | 1.9 [1.0–3.4]/0.7 [0.5–0.8] | 0.2 [0.2–0.2]/0.03 [0.03–0.05] | <0.001 |
| T245 | 1.4 [0.7–2.1]/0.5 [0.4–0.5] | 0.2 [0.1–0.2]/0.03 [0.02–0.04] | <0.001 |
| T305 | 1.1 [0.5–1.5]/0.3 [0.2–0.4] | 0.1 [0.1–0.1]/0.02 [0.02–0.02] | <0.001 |
| M3G/M3G (dose‐normalized), µg/L | |||
| T5 | 8.0 [2.9–13.7]/2.0 [1.3–4.7] | 0.0 [0.0–0.0]/0.0 [0.0–0.0] | <0.001 |
| T7 | 15.5 [5.6–22.1]/4.2 [3.0–6.2] | 0.0 [0.0–0.0]/0.0 [0.0–0.0] | <0.001 |
| T15 | 43.4 [21.1–60.0]/12.9 [8.4–17.5] | 2.8 [1.8–3.6]/0.42 [0.32–0.52] | <0.001 |
| T35 | 48.9 [26.6–81.3]/17.4 [10.7–20.4] | 5.6 [3.2–7.3]/0.81 [0.58–1.03] | <0.001 |
| T65 | 45.5 [25.0–80.0]/16.6 [9.9–18.7] | 4.5 [3.2–8.4]/1.12 [0.61–1.23] | <0.001 |
| T125 | 39.0 [18.7–68.6]/13.4 [7.7–18.6] | 6.8 [3.9–9.5]/0.95 [0.79–1.28] | <0.001 |
| T185 | 30.6 [13.9–55.8]/11.0 [6.2–15.6] | 5.9 [3.6–7.5]/0.89 [0.65–0.97] | <0.001 |
| T245 | 24.0 [11.0–42.2]/8.6 [4.7–12.3] | 4.3 [2.7–6.2]/0.70 [0.50–0.89] | <0.001 |
| T305 | 27.0 [11.0–37.0]/7.0 [4.8–10.1] | 4.0 [2.8–5.0]/0.59 [0.49–0.78] | <0.001 |
| M6G/M6G (dose‐normalized), µg/L | |||
| T5 | 0.0 [0.0–0.0]/0.0 [0.0–0.0] | 0.0 [0.0–0.0]/0.00 [0.00–0.00] | 0.356 |
| T7 | 0.0 [0.0–0.0]/0.0 [0.0–0.0] | 0.0 [0.0–0.0]/0.00 [0.00–0.00] | 0.166 |
| T15 | 2.7 [1.9–3.6]/0.8 [0.5–1.2] | 0.0 [0.0–0.0]/0.00 [0.00–0.00] | <0.001 |
| T35 | 7.0 [3.7–9.1]/2.1 [1.5–2.9] | 0.0 [0.0–0.0]/0.00 [0.00–0.00] | <0.001 |
| T65 | 7.2 [4.0–10.3]/2.2 [1.7–3.2] | 0.0 [0.0–1.2]/0.00 [0.00–0.19] | <0.001 |
| T125 | 5.6 [2.6–8.9]/1.8 [1.2–2.4] | 1.0 [0.0–1.5]/0.15 [0.00–0.19] | <0.001 |
| T185 | 4.0 [1.9–6.3]/1.3 [1.0–1.7] | 0.0 [0.0–0.0]/0.00 [0.00–0.00] | <0.001 |
| T245 | 3.1 [1.4–4.2]/1.0 [0.7–1.2] | 0.0 [0.0–0.0]/0.00 [0.00–0.00] | <0.001 |
| T305 | 2.5 [1.6–4.4]/0.8 [0.7–1.0] | 0.0 [0.0–0.0]/0.00 [0.00–0.00] | <0.001 |
| AUC (dose‐normalized), (µg min/L) | |||
| Morphine | 1044 [702–1266] | 19 [13–23] | <0.001 |
| M3G | 3752 [2487–5165] | 245 [162–287] | <0.001 |
| M6G | 466 [370–723] | 28 [21–43] | <0.001 |
| M3G‐to‐morphine AUC ratio | 4.46 [2.84–5.27] | 12.47 [11.70–12.81] | <0.001 |
| M6G‐to‐morphine AUC ratio | 0.53 [0.42–0.74] | 1.54 [1.36–1.67] | <0.001 |
Data are expressed as median [IQR]. The p values comparing intravenous versus nebulization route were computed using Mann‐Whitney U test.
Abbreviations: AUC, area under curves; M3G, morphine‐3‐glucuronide; M6G, morphine‐6‐glucuronide.
P‐value below 0.05 are in bold.
Pharmacokinetic modeling
The best model for morphine consisted in a three‐compartment model. The use of transit compartments with logarithm of factorial function (factln) for the estimation of the number of compartments for M3G and M6G production with a mean transit time (MTT) of 8.2 min and 57.5 min, respectively, strongly decreased BICc. The NEB absorption process was also best described using transit compartments with an MTT of 2.35 min and an absolute bioavailability of 3.5%. Finally, M3G and M6G elimination was modeled using first order elimination (Figure 1). Statistical model analysis revealed a significantly higher transfer rate from the central compartment (Vc) to the peripheral compartment 2 (Vp2) when the NEB route was used compared with the i.v. route (1.30 ± 0.47 vs. 0.35 ± 0.14) as well as a significantly higher M3G elimination (0.0038 ± 0.0007 vs. 0.0010 ± 0.0002). Furthermore, men exhibited a significantly lower elimination rate of M6G compared with women (0.005 vs. 0.008; Table 3). Residual variability was best described using proportional error models and a correlation between k13 and km3 (corr_km3_k13) improved the PK model. Bioavailability, ka, Ktr, k21, k31, Mtt1, ktr2, km6, and kam6 random effects were fixed due to values below 0.05. Bioavailability, ka, Ktr, k21, k13, k31, Mtt1, ktr2, and kam6 fixed effects were fixed according to the Fisher Information Matrix (Supplementary File S1 – Table S2). Population parameter estimates including the influence of the administration route and sex are summarized in Table 3. Captions and BICc of tested models and Mlxtran script of the final model are provided in the Supplementary data (Supplementary File S1 – Supplementary Material S3 and S4). Individual predictions are depicted for morphine (upper panels, Figure 2a,b), M3G (middle panels, Figure 2c,d), and M6G (lower panels, Figure 2e,f) following i.v. (left panels, Figure 2a,c,e) and NEB (right panels, Figure 2b,d,f) administration. Goodness‐of‐fit plots for the population as individual predictions and individual weighted residual, based on conditional distribution and normalized prediction distribution error diagnostic plots, are shown for morphine (Figure 3a–d, respectively), M3G (Figure 3e–h, respectively), and M6G (Figure 3i–l, respectively). Sensitivity analysis performed on fixed effect revealed that a 20% increase in F, ka, Mtt1, and ktr2 and to a lesser extent k13 may influence the PK profiles of morphine, M3G and M6G (Supplementary File S1 – Supplementary Material S5). R code for Figure 2, Table 2, and Supplementary Material S5 can be found in Supplementary File S1 – Supplementary Material S6.
FIGURE 1.
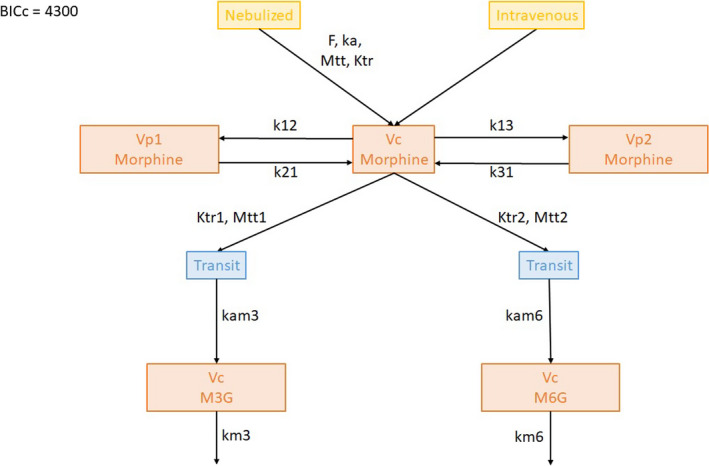
Schematic of the best pharmacokinetic model developed to describe plasma concentration of morphine, morphine‐3‐glucuronide (M3G), and morphine‐6‐glucuronide (M6G) following nebulization (NEB) or intravenous (i.v.) administration. BICc, corrected Bayesian information criterion; F, bioavailability; k12, transfer rate from Vc to Vp1; k13, transfer rate from Vc to Vp2; k21, transfer rate from Vp1 to Vc; k31, transfer rate from Vp2 to Vc; ka, absorption rate; kam3, transfer rate from the last transit compartment to M3G compartment Vc; kam6, transfer rate from the last transit compartment to M6G compartment Vc; km3, elimination rate of M3G; km6, elimination rate of M6G; ktr, transfer rate of morphine absorption transit compartments; ktr1, transfer rate of M3G transit compartments; ktr2, transfer rate of M6G transit compartments; MTT, mean transit time for absorption process of NEB morphine; MTT 1, mean transit time for delayed M3G appearance; MTT 2, mean transit time for delayed M6G appearance; Vc, morphine central compartment; Vp1, morphine peripheral compartment 1; Vp2, morphine peripheral compartment 2; a, coefficient of the proportional residual error model
TABLE 3.
Population pharmacokinetic parameter estimates
| Parameters | Estimate (RSE%) | BSV (RSE%) | Median [IQR] |
|---|---|---|---|
| F | 0.035 (−) | [−] | 0.035 [0.035–0.035] |
| ka [min−1] | 0.046 (−) | [−] | 0.046 [0.046–0.046] |
| ktr [min−1] | 1.23 (−) | [−] | 1.23 [1.23–1.23] |
| Mean transit time (MTT) [min] | 2.35 (6.83) | 0.21 (27.5) | 2.41 [2.31–2.46] |
| Vc [L] | 1.75 (10.6) | 0.49 (15.8) | 1.79 [1.25–2.11] |
| k12 [min−1] | 0.188 (19.2) | 0.61 (32.9) | 0.209 [0.160–0.249] |
| k21 [min−1] | 0.143 (−) | [−] | 0.143 [0.143–0.143] |
|
k13 [min−1] β_km3_Route (=NEB) |
0.306 (−) 1.39 (−) |
0.403 (−) | 0.564 [0.351–1.28] |
| k31 [min−1] | 0.010 (−) | [−] | 0.010 [0.010–0.010] |
| ktr1 [min−1] | 0.642 (3.77) | 0.114 (20.3) | 0.654 [0.611–0.670] |
| ktr2 [min−1] | 0.040 (−) | [−] | 0.040 [0.040–0.040] |
| MTT 1 (M3G production) | 8.16 (−) | [−] | 8.16 [8.16–8.16] |
| MTT 2 (M6G production) | 57.5 (2.72) | 0.090 (22.5) | 57.5 [55.9–60.3] |
| kam3 [min−1] | 0.172 (19.7) | 0.942 (16) | 0.131 [0.101–0.390] |
| kam6 [min−1] | 0.186 (−) | [−] | 0.186 [0.186–0.186] |
|
km3 [min−1] β_km3_Route (=NEB) |
0.0038 (1.78) −1.33 (−) |
0.18 (8.35) |
0.0029 [0.0010–0.0036] |
|
km6 [min−1] β_km6_Sex (=Male) |
0.0081 (1.79) −0.483 (−) |
[−] |
0.0050 [0.0050–0.0081] |
| corr_km3_k13 | −1 (0.81) | ||
| Residual error for morphine a | 0.29 (5.01) | ||
| Residual error for M3G a | 0.18 (6.86) | ||
| Residual error for M6G a | 0.27 (9.07) |
Parameter estimates and between subject variability (BSV) were computed using the Stochastic Approximation Expectation‐Maximization (SAEM) algorithm. Median and interquartile range (IQR) were derived from conditional distribution using Monlix default MCMC convergence assessment. Relative standard error (RSE) was obtained from the Fisher Information Matrix. The metabolite volumes of distribution for M3G and M6G were parametrized as being equal to Vc. Due to the absence of crossover study, F BSV was imputed to Vc which became an apparent volume of distribution equal to V/F. Estimates of BSV are shown as approximated coefficients of variation. [−], (−) and – symbols indicate fixed values.
Abbreviations: F, bioavailability; k12, transfer rate from Vc to Vp1; k13, transfer rate from Vc to Vp2; k21, transfer rate from Vp1 to Vc; k31, transfer rate from Vp2 to Vc; ka, absorption rate; kam3, transfer rate from the last transit compartment to M3G compartment Vc; kam6, transfer rate from the last transit compartment to M6G compartment Vc; km3, elimination rate of M3G; km6, elimination rate of M6G; ktr, transfer rate of morphine absorption transit compartments; ktr1, transfer rate of M3G transit compartments; ktr2, transfer rate of M6G transit compartments; M3G, morphine‐3‐glucuronide; M6G, morphine‐6‐glucuronide; MTT, mean transit time for absorption process of NEB morphine; MTT 1, mean transit time for delayed M3G appearance; MTT 2, mean transit time for delayed M6G appearance; Vc, morphine central compartment; Vp1, morphine peripheral compartment 1; Vp2, morphine peripheral compartment 2.
Coefficient of the proportional residual error model.
FIGURE 2.
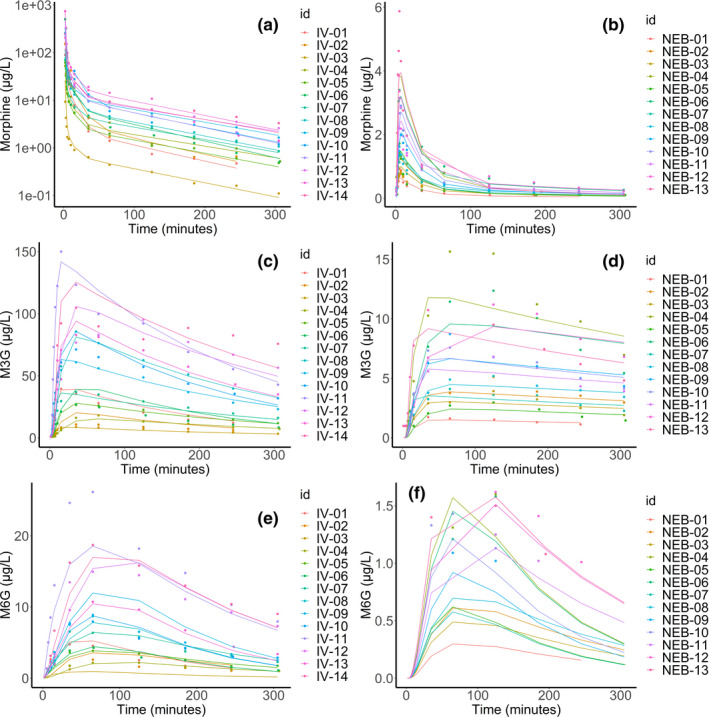
Individual predictions of morphine (upper panels, a, b), morphine‐3‐glucuronide (M3G; middle panels, c, d) and morphine‐6‐glucuronide (M6G; lower panels, e, f) following intravenous (left panels, a, c, e) and nebulized (right panels, b, d, f) administration. IV, intravenous; NEB, nebulized
FIGURE 3.
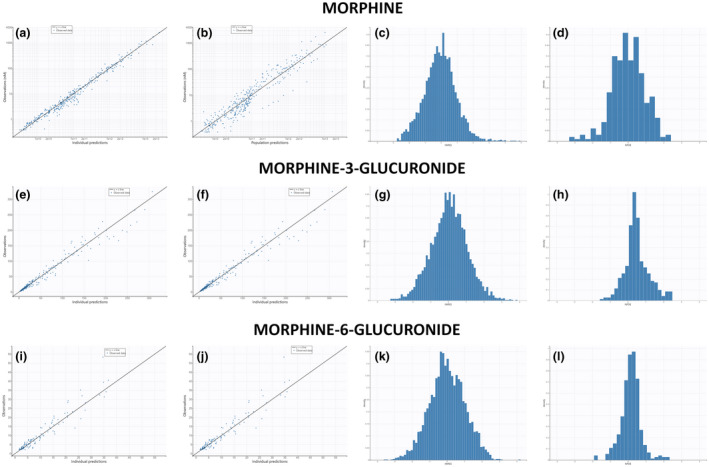
Population and individual predictions, individual weighted residual (IWRES) based on conditional distribution and normalized prediction distribution error (NPDE) of morphine (a–d), M3G (e–h) and M6G (i–l). M3G, morphine‐3‐glucuronide; M6G, morphine‐6‐glucuronide
Simulations
Based on significant predictors of the statistical model, simulations were performed with marked covariate differences (route of administration and sex). M3G and M6G maximal concentrations were ~ 50‐ and fivefold higher with i.v. administration compared with NEB. We observed furthermore that men exhibited lower M6G elimination compared with women resulting in a 2.5‐fold higher concentration at 300 min. Morphine, M3G, and M6G profiles according to covariates were plotted in Figure 4a–c, respectively.
FIGURE 4.
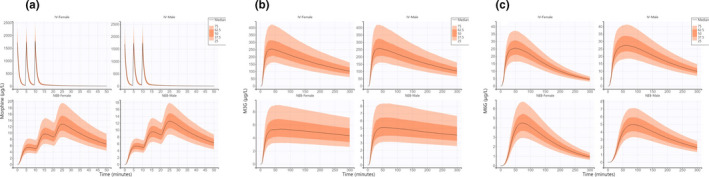
Predicted morphine (a), M3G (b), and M6G (c) concentrations over time based on 1000 simulations of the following conditions: IV Female, IV Male, NEB Female, and NEB Male using population parameters (both fixed and random effects including covariates) estimated by Monolix. IV, intravenous; M3G, morphine‐3‐glucuronide; M6G, morphine‐6‐glucuronide; NEB, nebulized
DISCUSSION
In this phase I trial, the design aimed to evaluate a simple and accessible procedure for short NEB morphine administration for subsequent titration. This PK analysis has brought new insights giving the opportunity to use morphine nebulization in clinical practice. Interestingly, a previous work suggested a two‐compartment absorption model with a prototype of pulmonary drug delivery system. 26 Due to our limited sample size and different administration settings, a transit compartment model was used to describe the absorption as a multiple step process. We also observed lower absolute bioavailability (3.5%) that could be mainly explained by drug leakage from the mask and to a lesser extent by local pulmonary UGT‐induced metabolism. 27 Low bioavailability is already known and has been observed for NEB analgesics, such as fentanyl, and could be improved using a more airtight mask limiting drug leakage. 28 , 29 NEB administration exhibited satisfying PK profiles with an increase exposure in a dose‐dependent manner, which is of particular importance for clinical purposes. The i.v. morphine titration is known to be a robust and safe way to respond to the efficacy/safety problematic at bedside, as in research protocols. Our results support the hypothesis of NEB as a substitute to i.v. morphine titration, expecting feasibility and reliability at bedside. The hypothesis that analgesia from NEB opioids may be different from that after other routes of administration has to be considered. 29 Compared with the i.v. route, NEB administration could represent an advantage regarding the absorption phase, which exhibits a progressive increase in morphine concentrations during drug inhalation. This could lead to improve morphine tolerance during analgesia and to counteract the “shooting effects” that could be induced by i.v. morphine titration. 5 The use of transit compartments is relevant to explain the inherent complex absorption during NEB administration where several sites are involved (lung, ear, nose, and throat). 30 At this point, NEB duration is a critical parameter because morphine titration aims to deliver multiple subtherapeutic doses until pain relief.
Concerning morphine, PK modeling often used one 31 or two 32 peripheral compartments to describe drug extravasation from blood and its distribution to tissues.
Importantly, morphine metabolism into M3G and M6G is delayed, which can be challenging to model. Hence again, the use of transit compartments provides a convenient solution. The use of N‐transit compartments for delayed appearance has been widely used since Savic et al. 33 described an analytical solution for the computation of factorial N (N!). In our model, the use of the mathematical operator factln() provided the best BICc for transit compartments modeling. Interestingly, transit compartments for M3G and M6G formation have already been implemented 34 by testing a varying number of compartments suggesting manual addition of compartments. The automatic determination of the number of transit compartments is a useful tool to determine the optimal MTT. M3G and M6G PK profiles showed a log‐linear relationship of metabolite concentration over time even if peripheral compartments are sometimes modeled. 32 Interestingly, M3G‐to‐morphine and M6G‐to‐morphine AUC ratios were a bit less than threefold higher for the NEB group compared with the i.v. group so the hypothesis of hepatic first‐pass cannot be excluded. However, M3G and M6G production were delayed in the NEB compared with the i.v. group, suggesting that metabolism saturation and concentration‐dependent cell permeability 35 of morphine in the i.v. group could also be an explanation. In our model, NEB administration resulted in a higher transfer rate from Vc to Vp1 suggesting the possibility of low expressed high affinity receptors. This finding can lead to the hypothesis that morphine distribution is a nonlinear process due to possible saturation of influx and efflux transporters, such as OCT1, OATP2, P‐gp, MRP2, and MRP3. 35 , 36 , 37 , 38 Unfortunately, the use of Michaelis‐Menten or Hill derived equations did not improve the model so the route of administration was considered as a covariate. Particular attention must be paid to the fixed parameters because they could be associated with unidentifiability. The sensitivity analysis allowed us to evaluate the impact of a 20% increase of these parameters. Of note, the bioavailability F has been fixed so variability could be attributed to the apparent volume of the central compartment but an increase in Mtt1 or ktr2 could also lead to important changes in concentration over time for M3G and M6G and need to be further investigated. Consequently, one way to avoid unidentifiability could be to calculate expected standard errors given the model and design with software like PFIM. 39 However, it is difficult to transpose the syntax of the MLXTRAN model into PFIM especially for complex models. An interesting way to solve this problem might be the direct importation of a MLXTRAN model into PFIM software or calculating expected SE in Monolix. In this study, the choice has been made to fix values that come from monolix estimation rather than following the literature. Indeed, most of the published morphine parent‐metabolite models used a specific volume of distribution for metabolites, 34 do not use transit compartments for metabolite production, 40 , 41 or were built for specific populations, such as morbid obesity in adults, 34 patients with cancer, 41 or neonates. 40
Although analgesically inactive, M3G has been reported to antagonize morphine and to produce stimulatory effects responsible for side effects, such as myoclonus, seizure, and allodynia. 42 On the other hand, M6G is a much more potent analgesic than morphine but the penetration rate of the blood‐brain‐barrier is lower than morphine itself. 43 In case of NEB titration, time between administrations must take into account M3G and M6G delayed formation because it can be involved in morphine safety and efficacy. Similarly, because M3G production is delayed with NEB, the duration of monitoring following morphine titration has to be determined and compared with the standard 2 h for the i.v. route.
Based on simulated data, even if sex improved the overall goodness‐of‐fit of the model, they only reflect minor changes in the morphine and metabolite concentrations over time. In agreement, the literature provided contradictory results about sex 44 , 45 , 46 and weight and/or BMI may only be relevant in extreme cases, such as morbid obesity. 34 Finally, because NEB administration relies, at least partially, on the cardiorespiratory system, special attention must be paid in clinical practice to respiratory and cardiac rates because moderate pain can induce tachypnea and cardiovascular disorders (tachycardia, bradycardia, and hypotension) that could affect bioavailability and absorption processes.
In this pain‐induced model, small boluses of morphine were tested, especially because induced pain was moderate according to both scientific and ethical considerations. Most of boluses were expected to be subtherapeutic according to Dixon’s method but priority was given to minimize the risk of toxicity, in line with phase I trial objectives. Maximum doses for both the NEB and i.v. groups were finally reached before ED50 could be observed. Thus, pharmacodynamics and its relationship with PKs could not be investigated.
Finally, because this model was built based on healthy volunteers, included covariates need to be confirmed in severe acute pain conditions with patients exhibiting extreme clinico‐biological characteristics, such as morbid obesity, severe malnutrition, or inflammation. Moreover, our results cannot solve the question of the balance between the accessibility of routine devices and their pharmacological relevance. Nebulization must remain readily available without increased cost or risk for caregivers in the treatment room.
To conclude, taken together, these results suggest that short NEB morphine could be suitable for pain management by titration approach in the EDs or in PACUs but determination of the optimal dose required for a NEB bolus remains unclear. Based on the present results, simulation could be carried out to predict the most efficient dose for pain relief using an NEB device suitable for clinical practice in various care units.
Comparison of pain relief between short NEB and i.v. morphine by titration is currently under evaluation in the ongoing CLIN‐AEROMORPH trial (NCT03257319), a multicenter clinical study on a large scale with a noninferiority design.
CONFLICTS OF INTEREST
The authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
T.D., V.E.L., and F.A. wrote the manuscript. V.E.L. and R.J. designed the research. V.E.L., T.D., M.P.T., T.P., and F.B. performed the research. T.D. and V.E.L. analyzed the data. T.P. and F.B. contributed new reagents/analytical tools.
Supporting information
Supplementary Material S1
Supplementary Material S2
ACKNOWLEDGEMENTS
The authors would like to thank Armelle Guidotti of the Department of Clinical Research and Innovation for her help in promoting the study and to all technicians and nurses of the Clinical Investigation Center (CIC), for logistical support at bedside. The authors are also grateful to Patricia Compagnon, Benny Charbit, and Bruno Riou for major contributions to the study design, Jessica Bortzmeyer for the demonstration of the nebulization device, and Nikki Sabourin‐Gibbs for editorial assistance and English writing support. The authors also would like to thank Dr. Ayral for R script provided in the Webinar: QSP modeling with MonolixSuite – Session 1 for the sensitivity analysis.
Duflot T, Pereira T, Tavolacci M‐P, et al. Pharmacokinetic modeling of morphine and its glucuronides: Comparison of nebulization versus intravenous route in healthy volunteers. CPT Pharmacometrics Syst Pharmacol. 2022;11:82–93. doi: 10.1002/psp4.12735
Funding information
The study was supported by a grant from the French Ministry of Health (“Programme Hospitalier de Recherche Clinique 2012” of the French Ministry of Health). The study was sponsored by Rouen University Hospital and monitored by the Clinical Investigation Center (CIC) of Rouen University Hospital. The funder has no role in the study design, collection, management, analysis or interpretation of data, or in the writing of the report and decision to submit. The investigators of this experiment have no competing interest with this study. The study has received neither industry funding nor financial/non‐financial support by any organization that may have an interest in its results
REFERENCES
- 1. Aubrun F, Monsel S, Langeron O, Coriat P, Riou B. Postoperative titration of intravenous morphine. Eur J Anaesthesiol. 2001;18(3):159‐165. [DOI] [PubMed] [Google Scholar]
- 2. Aubrun F, Langeron O, Quesnel C, Coriat P, Riou B. Relationships between measurement of pain using visual analog score and morphine requirements during postoperative intravenous morphine titration. Anesthesiology. 2003;98(6):1415‐1421. [DOI] [PubMed] [Google Scholar]
- 3. Aubrun F, Amour J, Rosenthal D, Coriat P, Riou B. Effects of a loading dose of morphine before i.v. morphine titration for postoperative pain relief: a randomized, double‐blind, placebo‐control study. Br J Anaesth. 2007;98(1):124‐130. [DOI] [PubMed] [Google Scholar]
- 4. Lvovschi V, Aubrun F, Bonnet P, et al. Intravenous morphine titration to treat severe pain in the ED. Am J Emerg Med. 2008;26(6):676‐682. [DOI] [PubMed] [Google Scholar]
- 5. Grissa MH, Boubaker H, Zorgati A, et al. Efficacy and safety of nebulized morphine given at 2 different doses compared to IV titrated morphine in trauma pain. Am J Emerg Med. 2015;33(11):1557‐1561. [DOI] [PubMed] [Google Scholar]
- 6. Bruera E, Sala R, Spruyt O, Palmer JL, Zhang T, Willey J. Nebulized versus subcutaneous morphine for patients with cancer dyspnea: a preliminary study. J Pain Symptom Manage. 2005;29(6):613‐618. [DOI] [PubMed] [Google Scholar]
- 7. Bounes V, Ducassé JL, Bona AM, Battefort F, Houze‐Cerfon CH, Lauque D. Nebulized morphine for analgesia in an emergency setting. J Opioid Manag. 2009;5(1):23‐26. [DOI] [PubMed] [Google Scholar]
- 8. Lasheen W, Panneerselvam A, Davis MP. Can we really say that nebulized morphine works? J Pain Symptom Manage. 2006;32(2):101‐103. [DOI] [PubMed] [Google Scholar]
- 9. Stephany T. Nebulized morphine: another point of view. Am J Hosp Palliat Care. 1995;12(5):7. [DOI] [PubMed] [Google Scholar]
- 10. Baydur A. Nebulized morphine: a convenient and safe alternative to dyspnea relief? Chest. 2004;125(2):363‐365. [DOI] [PubMed] [Google Scholar]
- 11. Fulda GJ, Giberson F, Fagraeus L. A prospective randomized trial of nebulized morphine compared with patient‐controlled analgesia morphine in the management of acute thoracic pain. J Trauma. 2005;59(2):383‐390. [DOI] [PubMed] [Google Scholar]
- 12. Masood AR, Thomas SH. Systemic absorption of nebulized morphine compared with oral morphine in healthy subjects. Br J Clin Pharmacol. 1996;41(3):250‐252. [DOI] [PubMed] [Google Scholar]
- 13. Pasternak GW, Pan YX. Mu opioids and their receptors: evolution of a concept. Pharmacol Rev. 2013;65(4):1257‐1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McDonald J, Lambert DG. Opioid receptors. Continu Educat Anaesthesia Crit Care Pain. 2005;5(1):22‐25. [Google Scholar]
- 15. De Gregori S, De Gregori M, Ranzani GN, Allegri M, Minella C, Regazzi M. Morphine metabolism, transport and brain disposition. Metab Brain Dis. 2012;27(1):1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smith HS. Opioid metabolism. Mayo Clin Proc. 2009;84(7):613‐624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smith TW, Binning AR, Dahan A. Efficacy and safety of morphine‐6‐glucuronide (M6G) for postoperative pain relief: a randomized, double‐blind study. Eur J Pain. 2009;13(3):293‐299. [DOI] [PubMed] [Google Scholar]
- 18. Borghardt JM, Weber B, Staab A, Kloft C. Pharmacometric models for characterizing the pharmacokinetics of orally inhaled drugs. AAPS J. 2015;17(4):853‐870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dixon WJ. Staircase bioassay: the up‐and‐down method. Neurosci Biobehav Rev. 1991;15(1):47‐50. [DOI] [PubMed] [Google Scholar]
- 20. Willer JC. Comparative study of perceived pain and nociceptive flexion reflex in man. Pain. 1977;3(1):69‐80. [DOI] [PubMed] [Google Scholar]
- 21. Borghardt JM, Kloft C, Sharma A. Inhaled therapy in respiratory disease: the complex interplay of pulmonary kinetic processes. Can Respir J. 2018;2018:2732017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Traynard P, Ayral G, Twarogowska M, Chauvin J. Efficient pharmacokinetic modeling workflow with the monolixsuite: a case study of remifentanil. CPT Pharmacometrics Syst Pharmacol. 2020;9(4):198‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Delattre M, Lavielle M, Poursat MA. A note on BIC in mixed‐effects models. Electron J Statist. 2014;1:456‐475. [Google Scholar]
- 24. Mould DR, Upton RN. Basic concepts in population modeling, simulation, and model‐based drug development‐part 2: introduction to pharmacokinetic modeling methods. CPT Pharmacometrics Syst Pharmacol. 2013;2(4):e38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. R Core Team . R: A language and environment for statistical computing. R Foundation for Statistical Computing; 2020. https://www.R‐project.org/. [Google Scholar]
- 26. Dershwitz M, Walsh JL, Morishige RJ, et al. Pharmacokinetics and pharmacodynamics of inhaled versus intravenous morphine in healthy volunteers. Anesthesiology. 2000;93(3):619‐628. [DOI] [PubMed] [Google Scholar]
- 27. Yang Z‐Z, Li LI, Wang LU, et al. The regioselective glucuronidation of morphine by dimerized human UGT2B7, 1A1, 1A9 and their allelic variants. Acta Pharmacol Sin. 2017;38(8):1184‐1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Higgins MJ, Asbury AJ, Brodie MJ. Inhaled nebulised fentanyl for postoperative analgesia. Anaesthesia. 1991;46(11):973‐976. [DOI] [PubMed] [Google Scholar]
- 29. Worsley MH, MacLeod AD, Brodie MJ, Asbury AJ, Clark C. Inhaled fentanyl as a method of analgesia. Anaesthesia. 1990;45(6):449‐451. [DOI] [PubMed] [Google Scholar]
- 30. Zebraski SE, Kochenash SM, Raffa RB. Lung opioid receptors: pharmacology and possible target for nebulized morphine in dyspnea. Life Sci. 2000;66(23):2221‐2231. [DOI] [PubMed] [Google Scholar]
- 31. Staahl C, Upton R, Foster DJ, et al. Pharmacokinetic‐pharmacodynamic modeling of morphine and oxycodone concentrations and analgesic effect in a multimodal experimental pain model. J Clin Pharmacol. 2008;48(5):619‐631. [DOI] [PubMed] [Google Scholar]
- 32. Lötsch J, Skarke C, Schmidt H, Liefhold J, Geisslinger G. Pharmacokinetic modeling to predict morphine and morphine‐6‐glucuronide plasma concentrations in healthy young volunteers. Clin Pharmacol Ther. 2002;72(2):151‐162. [DOI] [PubMed] [Google Scholar]
- 33. Savic RM, Jonker DM, Kerbusch T, Karlsson MO. Implementation of a transit compartment model for describing drug absorption in pharmacokinetic studies. J Pharmacokinet Pharmacodyn. 2007;34(5):711‐726. [DOI] [PubMed] [Google Scholar]
- 34. de Hoogd S, Välitalo PAJ, Dahan A, et al. Influence of Morbid Obesity on the Pharmacokinetics of Morphine, Morphine‐3‐Glucuronide, and Morphine‐6‐Glucuronide. Clin Pharmacokinet. 2017;56(12):1577‐1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mashayekhi SO, Sattari MR, Routledge PA. Evidence of active transport involvement in morphine transport via MDCKII and MDCK‐PGP cell lines. Res Pharm Sci. 2010;5(2):99‐106. [PMC free article] [PubMed] [Google Scholar]
- 36. Groenendaal D, Freijer J, de Mik D, Bouw MR, Danhof M, de Lange EC. Population pharmacokinetic modeling of non‐linear brain distribution of morphine: influence of active saturable influx and P‐glycoprotein mediated efflux. Br J Pharmacol. 2007;151(5):701‐712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zelcer N, van de Wetering K, Hillebrand M, et al. Mice lacking multidrug resistance protein 3 show altered morphine pharmacokinetics and morphine‐6‐glucuronide antinociception. Proc Natl Acad Sci USA. 2005;102(20):7274‐7279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Venkatasubramanian R, Fukuda T, Niu J, et al. ABCC3 and OCT1 genotypes influence pharmacokinetics of morphine in children. Pharmacogenomics. 2014;15(10):1297‐1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bazzoli C, Retout S, Mentré F. Design evaluation and optimisation in multiple response nonlinear mixed effect models: PFIM 3.0. Comput Methods Programs Biomed. 2010;98:55‐65. [DOI] [PubMed] [Google Scholar]
- 40. Knøsgaard KR, Foster DJR, Kreilgaard M, et al. Pharmacokinetic models of morphine and its metabolites in neonates: systematic comparisons of models from the literature, and development of a new meta‐model. Eur J Pharm Sci. 2016;92:117‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Oosten AW, Abrantes JA, Jönsson S, et al. A prospective population pharmacokinetic study on morphine metabolism in cancer patients. Clin Pharmacokinet. 2017;56:733‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Smith MT. Neuroexcitatory effects of morphine and hydromorphone: evidence implicating the 3‐glucuronide metabolites. Clin Exp Pharmacol Physiol. 2000;27(7):524‐528. [DOI] [PubMed] [Google Scholar]
- 43. Yoshimura H, Îda S, Oguri K, Tsukamoto H. Biochemical basis for analgesic activity of morphine‐6‐glucuronide. I. Penetration of morphine‐6‐glucuronide in the brain of rats. Biochem Pharmacol. 1973;22(12):1423‐1430. [DOI] [PubMed] [Google Scholar]
- 44. Ionescu TI, Drost RH, Roelofs JMM, et al. The pharmacokinetics of intradural morphine in major abdominal surgery. Clin Pharmakinet. 1988;14:178‐186. [DOI] [PubMed] [Google Scholar]
- 45. Sarton E, Olofsen E, Romberg R, et al. Sex differences in morphine analgesia. Anesthesiology. 2000;93:1245‐1254. [DOI] [PubMed] [Google Scholar]
- 46. Murthy BP, Pollack GM, Brouwer KLR. Contribution of morphine‐6‐glucuronide to antinociception following intravenous administration of morphine to healthy volunteers. J Clin Pharmacol. 2002;42:569‐576. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material S1
Supplementary Material S2