

Abstract
Aducanumab is a human immunoglobulin G1 anti‐amyloid beta (Aβ) antibody currently being evaluated for potential treatment of patients with early Alzheimer’s disease. This paper describes the relationship between the population pharmacokinetics (PopPKs) and pharmacokinetics‐pharmacodynamics (PKs‐PDs) of aducanumab using data from phase I to III clinical studies, with standard uptake value ratio (SUVR) used as a PD marker. Across clinical studies, aducanumab was administered intravenously either as a single dose ranging from 0.3 to 60 mg/kg or as multiple doses of 1, 3, 6, or 10 mg/kg every 4 weeks. A titration regimen with maintenance doses of 3, 6, or 10 mg/kg was also evaluated. Aducanumab PK was characterized with a two‐compartment model with first‐order elimination. No nonlinearities in PKs were observed. The PopPK‐PD model was developed using a sequential estimation approach. The time course of amyloid plaques, as expressed by composite SUVR measured using positron emission tomography, was described using an indirect response model with drug effect stimulating the elimination of SUVR. None of the identified covariates on PK and the PopPK‐PD model were clinically relevant. The PopPK‐PD model showed that magnitude, duration, and consistency of dosing are important factors determining the degree of Aβ removal. The intrinsic pharmacology of aducanumab remained consistent across studies.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Two large, identical phase III trials of aducanumab, an IgG1 anti‐amyloid beta (Aβ) antibody, showed differential removal of plaque and efficacy over 18 months of treatment.
WHAT QUESTION DID THIS STUDY ADDRESS?
Pharmacokinetic (PK) characteristics and the relationship between exposure to aducanumab and changes in standard uptake value ratio (SUVR) in patients with early Alzheimer’s disease were explored.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
PKs of aducanumab was well‐behaved, with linear, time‐invariant kinetics, and low variability. Aβ removal, as measured by amyloid positron emission tomography, increased with increasing exposure. Dose titration to 10 mg/kg showed the greatest reduction in composite SUVR compared with titration to 3 or 6 mg/kg. Intrinsic pharmacology remained consistent across studies. Magnitude, duration, and consistency of dosing were identified as important factors determining the degree of Aβ removal.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
The population PK‐pharmacodynamic (PD) modeling framework can be leveraged to characterize the influence of changes in drug exposure on PD measures of drug action and inform optimal doses/dosing regimens for anti‐Aβ therapeutics.
INTRODUCTION
Alzheimer’s disease (AD) is the most common cause of dementia, accounting for 50% to 75% of all cases. Pathologically, AD is defined by the presence in the brain of extracellular neuritic plaques containing amyloid beta (Aβ) peptide and intraneuronal neurofibrillary tangles composed of hyperphosphorylated tau proteins. The pathogenesis of these plaques and tangles and how they contribute to the clinical syndrome remains to be fully elucidated. The amyloid hypothesis postulates that Aβ‐related toxicity is the primary cause of neurodegeneration underlying the progression characteristic of AD. An association between the presence of antibodies that recognize amyloid plaques and a slowing of cognitive decline in patients with early AD has been described. 1
Aducanumab is a human immunoglobulin G1 (IgG1) monoclonal anti‐Aβ antibody that selectively targets aggregated forms of Aβ, including soluble oligomers and insoluble fibrils. 2 In clinical trials for AD, aducanumab demonstrated concentration‐dependent reductions in composite standard uptake value ratio (SUVR), a sensitive pharmacodynamic (PD) marker of brain Aβ removal, and slowing of clinical decline with fixed and titration‐based dosing. 2 Aducanumab is currently being investigated as a disease‐modifying treatment for AD.
The pharmacokinetics (PKs) of aducanumab were dose proportional and exhibited time‐invariant kinetics. 3 After 18 months of treatment, fixed doses of 10 mg/kg administered every 4 weeks reduced SUVR from 1.44 to 1.10, 2 a value purported to be the quantitative cutoff point that discriminates between positive and negative scans. 4 An up titration regimen with steady‐state doses of 10 mg/kg delayed the attainment to 1.10 by ≈4 months. 5
The development of aducanumab was guided by PK‐PD modeling to quantify the effect of exposure on Aβ removal. 5 In this article, we present a comprehensive population PK (PopPK)‐PD analysis based on five clinical studies (including phase III) in patients with AD following treatment with aducanumab. The main objectives of this analysis were to (1) characterize aducanumab PK after intravenous administration, (2) describe the aducanumab PK‐PD relationship, and (3) quantify the impact of covariates that may contribute to differences in aducanumab PK and its PK‐PD relationship.
METHODS
Study design
The PopPK‐PD model was developed based on data from patients who participated in three phase I (221AD101, 221AD103 [PRIME, NCT01677572], and 221AD104 [PROPEL, NCT02434718]) and two phase III (221AD301 [ENGAGE, NCT02477800] and 221AD302 [EMERGE, NCT02484547]) studies. 2 , 3 , 5
More details on treatment regimens and study description across these studies are summarized in Table S1. All clinical studies were performed in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Clinical Practice.
Bioanalysis and SUVR measurements
The concentration of aducanumab in serum was determined using a validated sandwich enzyme‐linked immunosorbent assay. The value below the limit of quantitation (BLOQ) for the assay was 0.5 mg/L, with a coefficient of variation of 15%.
The effect of aducanumab on cerebral amyloid level was measured by amyloid positron emission tomography (PET) scanning with 18F‐florbetapir as a PET ligand. 2 The SUVR was calculated for a composite region of interest, with whole cerebellum as the reference region. The composite region of interest included parts of major cortical regions (frontal, parietal, lateral temporal, sensorimotor, and anterior and posterior cingulate) and served as a summary measure of global cerebral amyloid level. A negative change from baseline in composite region‐of‐interest SUVR indicated a reduction in amyloid level. The intra‐patient test‐retest reliability for cortical composite SUVR was 2.40% ± 1.41% for patients with AD. 4 , 6
PopPK‐PD model development
Model development was conducted in three stages: structural model selection, covariate analysis, and final model evaluation. The PopPK‐PD model was developed using a sequential estimation approach.
A linear two‐compartment model with first‐order elimination was selected as the structural model to characterize aducanumab PK (Figure 1). The model was parameterized in terms of clearance (CL), central volume of distribution (V1), distribution clearance (Q), and peripheral volume of distribution (V2). Because monoclonal antibodies have been shown to exhibit nonlinear kinetics, time‐varying clearance or Michaelis‐Menten models were also assessed. The effective half‐life of the drug was also reported using PK model parameters (Method discussed in Supplementary section).
FIGURE 1.
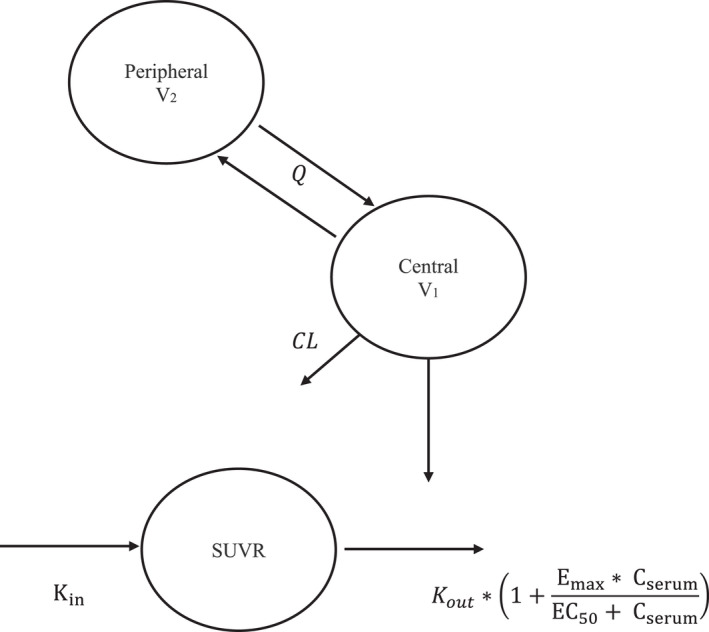
Structure of the serum population PK‐PD model for aducanumab. CL, systemic clearance; Cserum, model‐predicted aducanumab serum concentration; Emax, maximum fold change in the elimination of SUVR; EC50, aducanumab exposure that produces 50% of the maximum attainable stimulation; Kin, zero‐order rate for production of SUVR; K out, first‐order rate of elimination of SUVR; PopPK‐PD, population pharmacokinetic‐pharmacodynamic; Q, distribution clearance; SUVR, standard uptake value ratio; V1, central volume of distribution; V2, peripheral volume of distribution
Upon development of the PopPK model, individual PK parameter estimates were used to predict aducanumab exposures, which in turn were used in the PopPK‐PD model to predict the SUVR response. Based on the mechanism of action of aducanumab in removing Aβ, the relationship between aducanumab exposure and the SUVR reduction time course was best described by an indirect response model with a stimulatory effect on SUVR elimination rate (Figure 1). The functional form of the model is as provided in Equations (1), (2), (3):
| (1) |
| (2) |
| (3) |
where K in is the zero‐order rate for production of SUVR, K out is the first‐order rate of elimination of SUVR, SUVR t =0 is the SUVR at baseline, BaselineSUVR is the model‐predicted baseline SUVR prior to the first dose, Emax is the maximum fold change in the elimination of SUVR as a response to aducanumab exposure, C serum is the aducanumab serum concentration, and EC50 is the aducanumab exposure that produces 50% of the maximum attainable stimulation.
The PopPK‐PD analysis was initially conducted excluding all post‐baseline placebo observations, as these were not expected to impact drug effect parameters. Upon identification of the appropriate interindividual variability (IIV) and residual error structure, post‐baseline placebo measurements were included and the influence on the base model was evaluated. Following assessment of the stability and consistency of model parameters with and without post‐baseline placebo measurements, the covariate analysis was conducted.
The IIV in parameters was assumed to follow a log‐normal distribution in the following form (Equations 4 and 5):
| (4) |
| (5) |
where is the nth PK parameter for the patient i, θTV is the population typical value for the nth parameter (e.g., clearance), and ηi, n is the random interpatient effect on the nth parameter for patient i. The random effects (η1, …, η m ) are normally distributed with mean vector 0 and covariance matrix omega (Ω). Correlations between random effects were explored in the evaluation of the Ω structure.
Residual variability was evaluated using a combined proportional plus additive residual error model (Equations 6 and 7):
| (6) |
| (7) |
where Ci , j denotes the observed concentration for the ith individual at time tj , denotes the corresponding predicted concentration based on the PK model, ε ij denotes the intra‐individual (residual) random effect (zero mean and unit variance), and wi , j denotes the residual standard deviation with corresponding proportional and additive variance components, σ1 2 and σ2 2, respectively.
Furthermore, the prospect of having separate residual errors to account for differences between studies that collected intensive samples and those that collected sparse samples along with an interindividual error term on residual variability was assessed. 7
The first‐order conditional estimation with interaction (FOCE‐I) method was used for all runs. Examination of objective function using likelihood ratio tests, goodness‐of‐fit plots, visual inspection of fits, and scientific plausibility was used to discriminate models.
Covariate effects
Baseline covariates of interest, including weight, age, Mini‐Mental State Examination (MMSE), apolipoprotein E (ApoE) ε4 status, sex, and race, were tested on all PK or PK‐PD parameters with an IIV component. The incidence of anti‐drug antibodies in the PopPK‐PD data set was low (1%). Therefore, the effect of these antibodies as a covariate on PopPK or PopPK‐PD was not evaluated.
The covariate analysis was conducted using a full covariate modeling approach, testing all covariates for their influence. Subsequently, a stepwise backward elimination procedure was used to identify a parsimonious final model containing similar “information” content as the full model but with fewer covariates. At each step of the backward elimination procedure, the covariate‐parameter relationship that had the lowest change in objective function value (OFV) and met the exclusion criteria was eliminated, and the procedure was repeated until none of the remaining covariate parameters met the exclusion criteria (i.e., covariate was excluded if ΔOFV <10.83 [α = 0.001, 1 degree of freedom]).
The impact of significant covariates on PK parameters and PK‐SUVR was summarized using forest plots. Additionally, the influence of covariates on SUVR change from baseline (ΔSUVR) was also evaluated, as this would aid clinicians to better interpret the impact of covariates on amyloid removal. A detailed summary of the steps involved in the generation of the forest plots are provided in the Appendix S1.
PopPK‐PD model evaluation
Standard diagnostic methods for assessing the performance of the PopPK and PopPK‐PD models were applied. Successful minimization, diagnostic plots, plausibility and precision of parameter estimates, OFV comparisons, and shrinkage in the empirical Bayes estimates were used to guide model development and evaluation.
An internal assessment of the model was performed through a prediction‐corrected visual predictive check (pcVPC). VPCs allow visual comparison of the simulated and observed distributions of data. One thousand replicates of the data were simulated to build the VPC. The 95% (or 90%) prediction intervals for median and extreme quantiles in the simulated data were then plotted for each bin, and the median and extreme quantiles observed in the original data were overlaid onto the bands along with the full set of original observations. 8
Nonparametric bootstrapping (1000 replicates) was also performed to evaluate precision of the final PopPK and PK‐PD model parameter estimates and to generate 95% confidence intervals (CIs).
Simulations to understand variable outcomes in phase III studies
Modulation and in turn reduction in SUVR—the desired effect with respect to efficacy—lead to unfavorable safety events, such as amyloid‐related imaging abnormalities (ARIAs). The incidence of ARIAs appeared to be both dose and ApoE ε4 carrier dependent. 2
Following the initiation of phase III studies, two major protocol amendments were made. Patients who suspended dosing due to ARIAs could, after resolution of findings, resume dosing at the same dose and continue titration to the target dose (rather than resume at the next lower dose, with no further increases in dose permitted), allowing those randomized to the high‐dose ApoE ε4 carrier group to be titrated to 10 mg/kg instead of 6 mg/kg. 5
Upon futility assessment, both the 221AD301 and 221AD302 studies were halted. Investigations post futility demonstrated a statistically significant clinical effect and greater reduction in SUVR in the high‐dose aducanumab arms in study 302, but an insignificant clinical effect and numerically lesser reduction in SUVR in study 301. Although identical in design, 301 and 302 were executed differently due to protocol amendments, leading to differential dosing patterns and exposures. This differential dosing was more pronounced in the high‐dose groups, where the dose was increased from 6 to 10 mg/kg for ApoE ε4 carriers (approximately two‐thirds of the population) and there were more dose interruptions due to ARIAs. 5 Therefore, understanding and comparing exposure distribution differences following the 10‐mg/kg dose between studies 301 and 302 were particularly relevant. The final PopPK‐PD model was used to assist in the evaluation of these study discrepancies.
The exposure metric used to assess the above hypothesis was the uninterrupted time spent at steady‐state following the 10‐mg/kg titration regimens. Toward this goal, post hoc parameters from the final PopPK model were used to simulate exposures in patients from studies 301 and 302. Using these exposures, durations (expressed as number of aducanumab infusions) spent at steady‐state were calculated and compared. The duration was determined as the period when the time‐averaged exposure was greater than or equal to 0.9 times the time‐averaged exposure of the dose that each patient was expected to receive per the latest version of the protocol.
In the high‐dose group (10 mg/kg administered every 4 weeks), after titration for 24 weeks, the maximum number of 10‐mg/kg doses that a patient could potentially receive was 14. Because it takes ≈4 months for an individual to reach steady‐state, this suggests that a patient had an opportunity to spend 10 uninterrupted infusion cycles at steady‐state. Using the PopPK model, the number of individuals with greater than or equal to 10 uninterrupted infusions, spent at steady‐state, was determined and compared between studies 301 and 302.
The final PopPK‐PD model was also used to substantiate the presence of a dose exposure–SUVR relationship and understand the impact of exposures resulting from dose suspension due to ARIA on SUVR over time. For dose interruption, simulation of SUVR over time for a typical patient assumed an ARIA event at month 5, with an average dose suspension of 12 weeks and subsequent recovery doses at the patient’s dose level before the suspension.
Software for modeling
Models were developed using the nonlinear mixed‐effect modeling software (NONMEM version 7.4.3; ICON Development Solutions, Ellicott City, MD) running on a Linux cluster of multiprocessor computers. 9 FOCE‐I was used for all runs. Graphical visualization and model diagnostics were performed using R version 3.0.1. Covariate analysis and VPCs were performed using Perl‐speaks‐NONMEM version 4.9.0.
RESULTS
Analysis population
Aducanumab PK data from 2961 patients contributing to 50,306 individual concentration values were included in the PopPK analysis. For the PopPK‐PD analysis, data were pooled from 1125 patients and 3655 SUVR measurements. SUVR data were not available for the single‐dose and PROPEL studies (the observed PK, SUVR, and dose interruption characteristics are descriptively summarized and illustrated through Figures S1–S4 in the Supplementary section).
Approximately 3% of the observed aducanumab concentration records were BLOQ. Given this low percentage of BLOQ samples, all BLOQ samples were excluded from the modeling analysis, as it is not likely that ignoring the censoring from these BLOQ samples is likely to unduly bias the parameter estimates of the model.
Covariate characteristics at baseline for the PopPK and PK‐PD data sets are provided in Table S2. The patients were primarily White (79%) and ApoE ε4 carriers (69%), with a median age of 71.3 years. Overall, there were 52% female and 48% male subjects. Most patients (80%) were characterized with mild cognitive impairment due to AD.
PopPK model
A two‐compartment model with linear kinetics best characterized the concentration‐time profiles of aducanumab after intravenous administration (Figure 1, Table S3). The model was parameterized in terms of CL, V1, Q, and V2. The IIV was evaluated on CL, V1, and V2 with a block covariance matrix. Inclusion of IIV for Q resulted in an unstable model with high condition number and η on Q not centered around 0; hence, this random effect was not included in the final PK model.
The PKs of aducanumab was determined to be linear, as examination of nonlinearity in aducanumab elimination with time‐varying clearance or a Michaelis‐Menten model resulted in an insignificant change in OFV, indicating lack of concentration and time‐variant PKs (Table S3).
The residual variability was best explained by a combined residual error with an IIV component suggesting that the magnitude of residual variability varies between patients. The benefit of individual information content correction for the modeling of the residual error was reflected by a greater decrease in OFV and better distribution properties for conditional weighted residuals (symmetric and centered around 0; Table S3).
No obvious trends were observed in the standard goodness‐of‐fit plots for the base model (Figure S5).
A covariate search was run to assess the influence of pre‐specified covariates on the PK model parameters. Due to small sample size in the “other race” category, race as a covariate on PK was tested as White + Other versus Asians. The analysis identified weight and sex as covariates on CL and weight, sex, and race as covariates on V1 and V2, with V2 having an additional influence due to age and MMSE score at baseline (Table S4). Of the overall variability, inclusion of covariates explained ≈23% in CL and V2 and 32% in V1.
The significance of the included covariates was further supported by bootstrap analyses, as none of the 95% CIs for the covariate effects included zero. The magnitude of the covariate effects is shown in the forest plot in Figure 2a. Covariate effects were scaled to the magnitude of the parameter estimates for the reference patient (female White patient weighing 71.9 kg), as depicted by the vertical line at a reference value of 1.0. Note that the CIs for these covariate effects did not overlap this reference value, suggesting that these covariate effects are statistically significant. However, each of these CIs was contained within the bioequivalence region (the dotted line from 0.8 to 1.25), suggesting that none of these covariate effects are clinically relevant.
FIGURE 2.
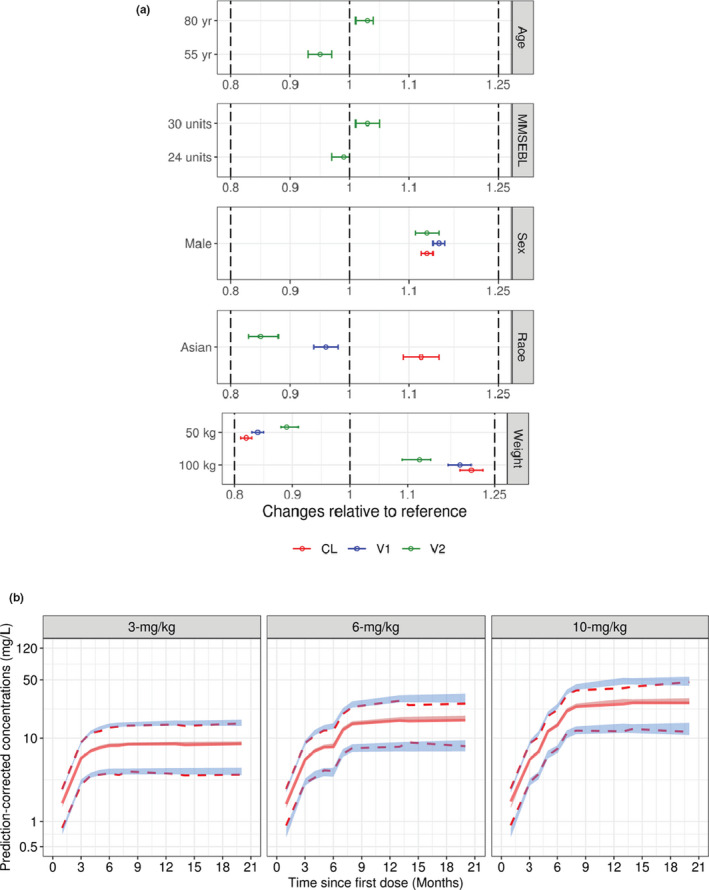
Aducanumab PopPK model evaluation. (a) Clinical relevance of statistically significant covariates; effects on PK parameters are expressed relative to a reference patient (female, White, with baseline body weight = 70.9 kg, MMSEBL = 26, and age = 71 years). Data are mean (90% CI). Dotted lines represent reference ±20%. (b) Prediction‐corrected visual predictive check for the final PopPK model of aducanumab. Solid lines represent the observed median, while dotted lines represent the observed prediction intervals. Shaded bands are simulation‐based 95% prediction intervals for the median, 5th, and 95th percentiles. CI, confidence interval; CL, systemic clearance; MMSEBL, Mini‐Mental State Examination score at baseline; PK, pharmacokinetics; PopPK, population pharmacokinetic; V1, central volume of distribution; V2, peripheral volume of distribution
The NONMEM parameter estimates for the final PopPK model and the bootstrap‐derived nonparametric 95% CIs are shown in Table 1. All fixed and random effect parameters were adequately estimated, with relative standard error less than 25% and low shrinkage (<20%). The diagnostic plots for the final PopPK model are presented in Figure S6. These plots suggest that the model provides an adequate fit to the data. The plots of observed concentrations versus population or individual predictions showed a homogeneous distribution of data points around the identity line without a notable lack of fit.
TABLE 1.
Parameter estimates for the final population PK model
| Parameter (unit) | NONMEM | Bootstrap | |||
|---|---|---|---|---|---|
| Estimate | 95% CI | Median | 95% CI a | ||
| Fixed effect | CL (L/h) | 0.0159 | 0.0157–0.0161 | 0.0159 | 0.0156, 0.0161 |
| V1 (L) | 3.59 | 3.55, 3.63 | 3.59 | 3.55, 3.62 | |
| V2 (L) | 6.04 | 5.94, 6.14 | 6.05 | 5.93, 6.17 | |
| Q (L/h) | 0.0194 | 0.0189, 0.0199 | 0.0195 | 0.0187, 0.0202 | |
| Covariate effect | |||||
| Effect of weight | WT on CL | 0.561 | 0.512, 0.610 | 0.561 | 0.512, 0.613 |
| WT on V1 | 0.506 | 0.467, 0.545 | 0.505 | 0.465, 0.544 | |
| WT on V2 | 0.320 | 0.269, 0.371 | 0.319 | 0.263, 0.374 | |
| Effect of age | Age on V2 | 0.207 | 0.139, 0.275 | 0.207 | 0.136, 0.276 |
| Effect of race, Asian | CL | 0.125 | 0.0903, 0.160 | 0.125 | 0.0917, 0.161 |
| V1 | −0.044 | −0.067, −0.020 | −0.044 | −0.067, −0.022 | |
| V2 | −0.148 | −0.176, −0.120 | −0.149 | −0.180, −0.116 | |
| Effect of sex, male | CL | 0.134 | 0.112, 0.156 | 0.135 | 0.111, 0.156 |
| V1 | 0.146 | 0.128, 0.164 | 0.146 | 0.127, 0.164 | |
| V2 | 0.129 | 0.106, 0.152 | 0.129 | 0.106, 0.156 | |
| Effect of MMSEBL | MMSEBL on V2 | 0.182 | 0.0963, 0.268 | 0.183 | 0.0929, 0.279 |
| IIV b | CL, % CV | 21.6 | 21.0, 22.2 | 21.6 | 20.9, 22.2 |
| V1, % CV | 14.8 | 14.2, 15.4 | 14.8 | 14.2, 15.5 | |
| V2, % CV | 17.0 | 16.2, 17.8 | 17.0 | 16.0, 17.9 | |
| Weighting on residual error, % CV | 34.9 | 33.6, 36.2 | 34.6 | 31.5, 37.7 | |
| ρ (CL, V1) | 0.378 | 0.352, 0.400 | 0.378 | 0.352, 0.402 | |
| ρ (CL, V2) | −0.407 | −0.500, −0.327 | −0.408 | −0.517, −0.317 | |
| ρ (V1, V2) | 0.392 | 0.360, 0.408 | 0.390 | 0.360, 0.403 | |
| Residual error | Proportional error (%) | 14.8 | 14.6, 15.0 | 14.8 | 14.5, 15.0 |
| Additive error (mg/L) | 0.202 | 0.189, 0.215 | 0.200 | 0.175, 0.227 | |
Abbreviations: CI, confidence interval; CL, clearance; CV, coefficient of variation; IIV, interindividual variability; MMSEBL, Mini‐Mental State Examination score at baseline; NONMEM, nonlinear mixed‐effect modeling software; ρ, correlation; PK, pharmacokinetic; Q, intercompartmental clearance; V1, central volume; V2, peripheral volume; WT, weight.
Twenty‐seven runs were skipped while calculating bootstrap summaries.
%CV = .
The qualification of the final aducanumab PopPK model was evaluated by pcVPC. Results for the final PopPK model, stratified by dosing regimen, are presented in Figure 2b (placebo‐controlled [PC] period of the data) and Figure S7 (PC + long term extension (LTE) data). Overall, the 5th, 50th, and 95th percentiles of observed concentrations were included in the respective 95% prediction intervals for these percentiles, suggesting that the model accurately predicted aducanumab concentrations in patients for a wide range of doses and times.
PopPK‐PD model
The time course of SUVR following administration of aducanumab was described by an indirect response model linking aducanumab concentrations with SUVR via a stimulatory effect, as shown in Figure 1. Inclusion of the Hill coefficient on drug effect function resulted in a model that failed to converge; therefore, it was excluded.
The structural model was then extended by incorporating IIV on all the PopPK‐PD model parameters with a full block covariance matrix. However, this resulted in an unstable model with a large condition number (>1 million) and a near‐perfect negative correlation between K out and Emax (ρ = −1). Upon further investigation, it was found that three of the six correlations of random effects were small, suggesting that a banded covariance structure with covariances for K out and Emax, Emax and baseline, and baseline and EC50 was parsimonious, improving the stability of the model while still allowing for the estimation of IIV on all PopPK‐PD model parameters. Residual variability was characterized using a combined proportional and additive error.
Adding post‐baseline placebo measurements resulted in minimal changes (<1% difference) to the fixed and random effect parameter estimates. The only finding was that the additive error estimate was very small (1E‐05) and hence was dropped from the model (Table S5). The base model diagnostic plots are provided in Figure S8.
The strategy for conducting a covariate analysis was to develop a full model and then perform a backward elimination procedure. However, the full model had issues with convergence (failure to iterate). Therefore, stepwise covariate modeling was performed in Perl‐speaks‐NONMEM by using the forward selection (α = 0.05) and backward elimination (α = 0.01) methods. After covariate selection, the final PopPK‐PD model included three covariates on baseline (age, ApoE ε4 status, and MMSE baseline), an age effect on Emax, and a weight effect on K out (Table S6 and Table S7).
The final PopPK‐PD model parameter estimates translated into a 70.2% maximum induction in SUVR elimination, with an EC50 of 46.4 mg/L. All the fixed, random, and covariate effect parameters were well‐estimated (CV <25%) except for ApoE effect (characterized as a trivariate covariate; 1‐copy of ε4 allele vs. 2‐copies of ε4 allele vs. noncarriers) on SUVR baseline. Although a statistically significant drop in OFV was observed, the ApoE ε4 carrier homozygote effect (2‐copies of ApoE ε4 allele) on baseline SUVR was identified to be weak (95% CI of the covariate coefficient includes 0). This could be attributed to fewer number of ApoE ε4 carrier homozygote subjects relative to ApoE ε4 carrier heterozygotes (1‐copy of ApoE ε4 allele) and noncarriers within the population. Based on literature evidence, physiological relevance, along with the significant improvement in OFV, both homozygote and heterozygote carriers (as well as noncarriers) were included as covariates, despite the large CI for carrier homozygote. 10 , 11 , 12
The relevance of these associated covariates is shown in Figure 3a and Figure 3b. Overall, it can be inferred that, except for age, all other identified covariates had a minimal effect on PK‐PD parameters (Baseline, Emax, and K out) or as measured using ∆SUVR at steady‐state. Additionally, post hoc model parameters were comparable between studies 301 and 302 (Figure S9).
FIGURE 3.
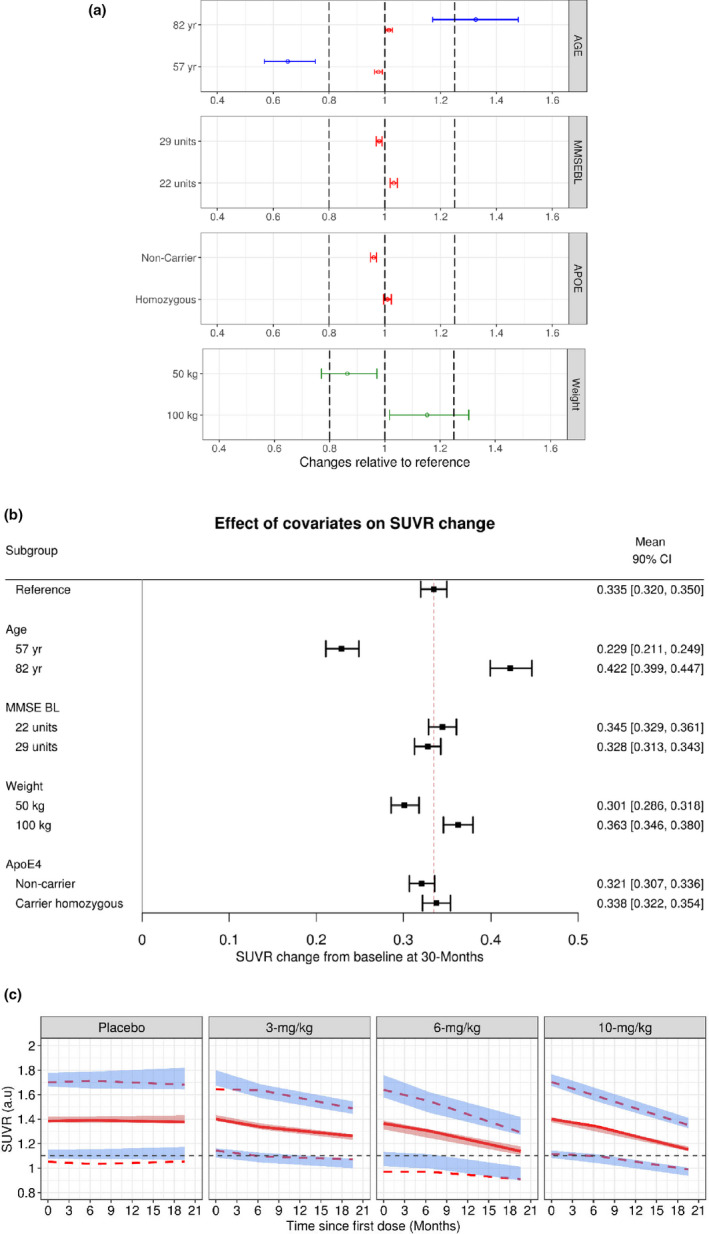
Aducanumab population PopPK‐PD model evaluation. (a) Clinical relevance of statistically significant covariates; effects on PK‐PD parameters are expressed relative to a reference patient (ApoE4 heterozygous, MMSEBL = 26, age = 71 years, and baseline body weight = 70.9 kg). Data are mean (90% CI). Dotted lines represent bioequivalence limits of 0.8 and 1.25. The blue lines represent Emax, red lines represent baseline, and the green lines represent K out parameters, respectively. (b) Impact of covariates on SUVR change; effects are expressed as ΔSUVR at 30 months relative to a reference patient (ApoE4 heterozygous, MMSEBL = 26, age = 71 years, and baseline body weight = 70.9 kg). Data are mean (90% CI). (c) Prediction‐corrected visual predictive check for the final PopPK‐PD model of aducanumab. Solid lines represent the observed median, while dotted lines represent the observed prediction intervals. Shaded bands are simulation‐based 95% prediction intervals for the median, 5th, and 95th percentiles. ApoE, apolipoprotein E; Emax, maximum fold change in the elimination of SUVR; K out, first‐order rate of elimination of SUVR; MMSEBL, Mini‐Mental State Examination score at baseline; PK‐PD, pharmacokinetic‐pharmacodynamic; SUVR, standard uptake value ratio
The proportional residual error was low (4%) and of the same magnitude as the expected accuracy of the SUVR assay 5 (Table 2). Diagnostic plots, including goodness‐of‐fit (Figure S10) and pcVPC by a phase I or phase III study or PC, PC + LTE period (Figure 3c, Figure S11, and Figure S12), indicated that the final PopPK‐PD model described the distribution and central tendency of the SUVR data–time profile adequately.
TABLE 2.
Parameter estimates for the final population PK‐PD model
| Parameter (unit) | NONMEM | Bootstrap | |||
|---|---|---|---|---|---|
| Estimate | 95% CI | Median | 95% CI a | ||
| Fixed effect | BL | 1.40 | 1.39, 1.41 | 1.40 | 1.39, 1.41 |
| K out (1/h) | 8.51 E−05 | 7.59E−5, 9.42E−5 | 8.40E−05 | 7.55E−5, 9.35E−5 | |
| Emax | 0.702 | 0.606, 0.797 | 0.697 | 0.616, 0.820 | |
| EC50 (mg/L) | 46.4 | 33.8, 59.0 | 45.2 | 35.2, 61.0 | |
| Covariate effect | |||||
| Effect of weight | WT on K out | 0.414 | 0.155, 0.673 | 0.403 | 0.132, 0.645 |
| Effect of age | BL | 0.101 | 0.0326, 0.169 | 0.0954 | 0.0282, 0.177 |
| Effect of ApoE ε4 | Noncarrier on BL | −0.0404 | −0.0547, −0.0260 | −0.0402 | −0.0571, −0.0246 |
| Carrier (2 copies) on BL | 0.00896 | −0.0096, 0.0275 | 0.00923 | −0.00987, −0.0270 | |
| Effect of MMSEBL | MMSEBL on BL | −0.186 | −0.256, −0.116 | −0.187 | −0.263, −0.116 |
| IIV b | K out, % CV | 44.2 | 29.8, 54.9 | 43.1 | 31.3, 55.8 |
| Emax, % CV | 25.3 | 15.8, 32.0 | 24.0 | 13.1, 32.6 | |
| BL, % CV | 12.8 | 12.2, 13.3 | 12.8 | 12.1, 13.5 | |
| EC50, % CV | 84.1 | 72.6, 94.2 | 84.9 | 69.9, 100 | |
| ρ (K out, Emax) | −0.872 | −3.17, −0.245 | −0.863 | −3.71, −0.232 | |
| ρ (Emax, BL) | 0.439 | 0.157, 0.593 | 0.378 | 0.107, 0.601 | |
| ρ (BL, EC50) | −0.406 | −0.772, −0.151 | −0.457 | −0.854, −0.143 | |
| Residual error | Proportional error (%) | 4.04 | 3.89, 4.18 | 4.03 | 3.86, 4.20 |
ApoE, apolipoprotein E; BL, baseline SUVR; CV, coefficient of variation; EC50, aducanumab exposure that produces 50% of the maximum attainable stimulation; Emax, maximum fold change in the elimination of SUVR as a response to aducanumab exposure; K in, zero‐order rate for production of SUVR; K out, first‐order rate of elimination of SUVR; MMSEBL, Mini‐Mental State Examination score at baseline; NONMEM, nonlinear mixed‐effect modeling software; ρ, correlation; PD, pharmacodynamic; PK, pharmacokinetic; WT, weight.
One hundred twenty‐two runs were skipped while calculating bootstrap summaries.
%CV = .
DISCUSSION
The PKs of aducanumab in patients with early AD was best described by a two‐compartment model with first‐order elimination. Individual concentration‐time profiles for the clinically relevant dose‐titration regimens were well‐described by the current model. Model diagnostics and VPCs demonstrated robustness and predictive performance of the model.
The estimated half‐life of aducanumab was ≈24.8 days for the reference patient (individual estimates ranged from 14.8 to 37.9 days [5th and 95th percentiles, respectively]), which is typical of human IgG1 (25 days) and monoclonal antibodies in the IgG1 subclass. 13
The strongest identified covariate on CL, V1, and V2 was body weight, of which ≈15% to 30% of the overall variability was explained. The exponent of body‐weight effect on CL, V1, and V2 was estimated to be 0.561, 0.506, and 0.320, respectively. Using the estimated exponents on body weight, it can be calculated that a 10% increase in body weight was associated with a 3% to 5% increase in CL, V1, and V2, respectively.
The residual error values for both the base and final models were very low. The additive error was less than half of the lower limit of quantitation. The proportional error was of the same magnitude as the precision of the serum aducanumab concentration assay (coefficient of variation <10%). This indicated that the final PopPK model almost completely extracted the information contained in the data; the unexplained variance was almost exclusively due to assay measurement errors.
Using the PopPK model, the number of patients with greater than or equal to 10 uninterrupted infusions spent at steady‐state was determined and compared between studies 301 and 302. Approximately 30% of patients across both studies never had any 10‐mg/kg exposure at steady state (Figure 4). Additionally, a higher percentage of patients in study 302 had greater than or equal to 10 uninterrupted infusions spent at steady‐state compared with study 301 (111/547 [20%] vs. 81/553 [15%]).
FIGURE 4.
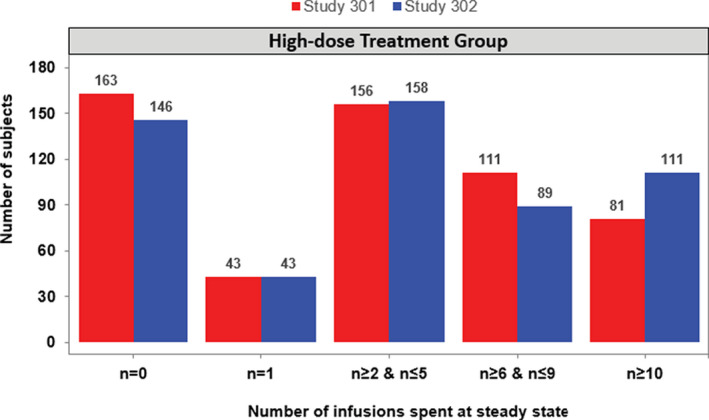
Bar plot of number of uninterrupted infusions spent at steady‐state in the high‐dose 10‐mg/kg treatment arms across studies 301 and 302
Although no difference in PK characteristics was detected between studies 301 and 302 when comparing post hoc PK parameters (Figure S13), changes to dose and dosing interruptions impacted the duration a patient spent at steady‐state, thereby potentially affecting pharmacology.
The time course of SUVR following aducanumab administration was well‐described, with indirect‐response PopPK‐PD models linking concentrations of aducanumab with SUVR via a stimulatory effect on the elimination rate of SUVR. The model herein was consistent with the mechanism of action of aducanumab in terms of Aβ removal.
In the model, SUVR was assumed to remain constant over time for placebo patients. This was because aducanumab evaluated in clinical studies primarily enrolled patients with early AD where amyloid levels are reported to be plateaued with minimal changes in time. 14 In this context, the placebo subjects are expected to have minimal drift over time and this hypothesis is also supported by the observed data (Figure S14). This is further supported by VPCs stratified by treatment group including placebo (Figure 3c) where an SUVR model with no placebo‐drift provides adequate predictive performance for both central tendency and the extremes (e.g., 10th and 90th percentiles) for the placebo‐time course. Therefore, it can be concluded that a placebo model with no SUVR changes in time is reasonable from the data collected in aducanumab clinical studies.
None of the identified covariates except for age had an appreciable impact on ∆SUVR over time. The ∆SUVR varied from 0.23 for a 57‐year‐old patient to 0.42 for an 82‐year‐old patient (Figure 3b), which was symmetrically different (about ±0.1) from the population mean reference value (∆SUVR = 0.335). This finding was consistent with observations from the phase III studies. Although no direct evidence exists for this observed effect, it has been reported that older patients tend to have a leaky blood‐brain barrier, which could lead to greater penetration of aducanumab into the brain and thereby a larger SUVR effect. 15
The exposure–Aβ removal relationship and population mean (typical individual: ApoE4 heterozygous, Mini‐Mental State Examination score at baseline [MMSEBL] = 26, age = 71 years, and baseline body weight = 70.9 kgs) SUVR‐time profiles for the 3‐, 6‐, and 10‐mg/kg doses without any dose interruption are presented in Figure 5a. The model indicated a marked difference in the SUVR profiles for the three dose groups. Additionally, it took ≈2.25 years with dosing at 10 mg/kg to reach an average brain Aβ PET composite SUVR of 1.102 compared with greater than 4 years with 6‐mg/kg dosing; the 3‐mg/kg group never achieved this threshold. These results indicate that both magnitude and duration of exposure are important factors in determining the onset and degree of Aβ removal.
FIGURE 5.
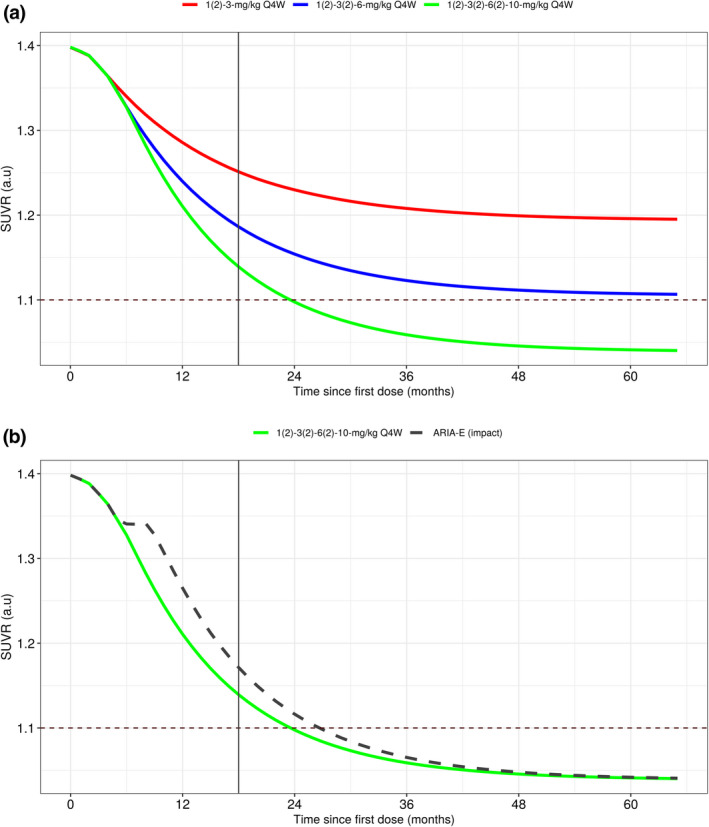
Population mean simulations of nominal SUVR‐time profiles. (a) Exposure‐SUVR simulations illustrating dose response using the standard titration regimens evaluated in phase III studies. (b) Effect of dose interruption due to ARIA on the time profile of SUVR. ARIA, amyloid‐related imaging abnormalities; Q4W, every 4 weeks; SUVR, standard uptake value ratio
It should be noted that predictions displayed in Figure 5a appear to contrast the SUVR changes observed in phase III studies (Figure 3c), however, they answer different questions and do not contradict the data. The pcVPC plot illustrated in Figure 3c explains what happened in the actual phase III trials and how well the model describes the same. The pcVPC are simulations evaluating model performance conditional on actual dosing histories observed in the phase III trials. From these simulations, we can infer the model characterizes the data adequately. Conversely, Figure 5a tries to depict typical individual dose exposure response predictions when the dosing is maintained without any interruption, thereby providing an understanding of benefit conferred by the 10‐mg/kg dose.
The delay between aducanumab exposure and Aβ removal has another important implication, as illustrated in Figure 5b, which shows predicted brain Aβ PET composite SUVR profiles for patients titrated to 10 mg/kg with monthly dosing over 5 years without dose interruptions and for patients with a 12‐week dose interruption at month 5 (3 missed doses). Patients with ARIA had a median of three missed doses. Hence, although this dosing pattern was representative, many variations in dosing interruptions and suspensions were observed.
The interruption in dosing had a prolonged influence on brain Aβ PET composite SUVR reduction at the week 78 primary end point, but this influence diminished with time beyond week 78 with treatment. Therefore, in addition to magnitude and duration, the consistency of dosing is an important factor in the degree of Aβ removal.
Handling ARIAs is an emerging field, with understanding of dosing through mild and moderate forms of ARIAs evolving with time. Hence, in the future, we except more subjects to be dosed through mild and moderate forms of ARIA, leading to fewer dose interruptions. Consequently, the predictions presented in this manuscript could still be considered relevant from the standpoint of discerning dose‐response and benefit of regular dosing.
In conclusion, results from the modeling herein show that the PK and SUVR response of aducanumab are well‐understood and that the relationship between them is adequately characterized by the models. The intrinsic pharmacology of the drug remains consistent across the evaluated studies and provides key insights on the importance of magnitude, duration, and consistency of dosing and its critical role in pharmacology outcomes. These models can be extended and used as a framework to understand the relationship between exposure to aducanumab and clinical response and contrasting efficacy outcomes.
CONFLICTS OF INTEREST
K.K.M., X.T., R.R., S.B.H., L.L., and I.N. are employees of and hold stock/stock options in Biogen. K.G.K. was a paid consultant for Biogen.
AUTHOR CONTRIBUTIONS
K.K.M., X.T., I.N., and K.G.K, R.R., L.L., S.B.H, and I.N. designed the research. K.K.M wrote the manuscript. K.G.K, R.R., L.L., S.B.H, and I.N. performed the research and analyzed the data.
Supporting information
Supplementary Material
Supplementary Material
ACKNOWLEDGEMENTS
Editorial support, under direction of the authors, was provided by Nucleus Global and was funded by Biogen. The authors also thank the entire Aducanumab Clinical and Biostatistics team for their support on this work.
Kandadi Muralidharan K, Tong X, Kowalski KG, et al. Population pharmacokinetics and standard uptake value ratio of aducanumab, an amyloid plaque–removing agent, in patients with Alzheimer’s disease. CPT Pharmacometrics Syst Pharmacol. 2022;11:7–19. doi: 10.1002/psp4.12728
Kumar Kandadi Muralidharan and Xiao Tong are co‐lead authors.
Funding information
This study was funded by Biogen. Biogen designed the study and collected, analyzed, and interpreted the data.
REFERENCES
- 1. Hock C, Konietzko U, Streffer JR, et al. Antibodies against beta‐amyloid slow cognitive decline in Alzheimer’s disease. Neuron. 2003;38(4):547‐554. [DOI] [PubMed] [Google Scholar]
- 2. Sevigny J, Chiao P, Bussière T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537(7618):50‐56. Update. In: Nature. 2017 June 21;546(7659):564.27582220 [Google Scholar]
- 3. Ferrero J, Williams L, Stella H, et al. First‐in‐human, double‐blind, placebo‐controlled, single‐dose escalation study of aducanumab (BIIB037) in mild‐to‐moderate Alzheimer’s disease. Alzheimer’s Dement (NY). 2016;2(3):169‐176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Joshi AD, Pontecorvo MJ, Clark CM, et al. 18 Study Investigators. Performance characteristics of amyloid PET with florbetapir F 18 in patients with Alzheimer’s disease and cognitively normal patients. J Nucl Med. 2012;53(3):378‐384. [DOI] [PubMed] [Google Scholar]
- 5. Budd Haeberlein S. EMERGE and ENGAGE topline results: Phase 3 studies of aducanumab in early Alzheimer's disease. Alzheimer's Dement. 2020;16(suppl 9):e047259. [Google Scholar]
- 6. Landau SM, Breault C, Joshi AD, et al. Amyloid‐β imaging with Pittsburgh compound B and florbetapir: comparing radiotracers and quantification methods. J Nucl Med. 2013;54:70‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Karlsson MO, Jonsson EN, Wiltse CG, Wade JR. Assumption testing in population pharmacokinetic models: illustrated with an analysis of moxonidine data from congestive heart failure patients. J Pharmacokinet Biopharm. 1998;26(2):207‐246. [DOI] [PubMed] [Google Scholar]
- 8. Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 2011;13(2):143‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Beal SL, Sheiner LB, Boeckmann A, Bauer RJ. NONMEM User’s Guides. Icon Development Solutions; 2009. [Google Scholar]
- 10. Baek MS, Cho H, Lee HS, et al. Effect of APOE ε4 genotype on amyloid‐β and tau accumulation in Alzheimer’s disease. Alzheimer Res Ther. 2020;12:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kanekiyo T, Xu H, Bu G. ApoE and Aβ in Alzheimer’s disease: accidental encounters or partners? Neuron. 2014;81(4):740‐754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Therriault J, Benedet AL, Pascoal TA, et al. APOEε4 potentiates the relationship between amyloid‐β and tau pathologies [published online ahead of print March 11, 2020]. Mol Psychiatry. https://doi.org/ 10.1038/s41380-020-0688-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ryman JT, Meibohm B. Pharmacokinetics of monoclonal antibodies. CPT Pharmacometrics Syst Pharmacol. 2017;6(9):576‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8(6):595‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rosenberg GA. Blood‐brain barrier permeability in aging and Alzheimer’s Disease. J Prev Alzheimers Dis. 2014;1(3):138‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material