

Abstract
Artemisinin (ART) is a bioactive molecule derived from the Chinese medicinal plant Artemisia annua (Asteraceae). ART and artemisinin derivatives (ARTs) have been effectively used for antimalaria treatment. The structure of ART is composed of a sesquiterpene lactone, including a peroxide internal bridge that is essential for its activity. In addition to their well-known antimalarial effects, ARTs have been shown recently to resist a wide range of tumors. The antineoplastic mechanisms of ART mainly include cell cycle inhibition, inhibition of tumor angiogenesis, DNA damage, and ferroptosis. In particular, ferroptosis is a novel nonapoptotic type of programmed cell death. However, the antitumor mechanisms of ARTs by regulating ferroptosis remain unclear. Through this review, we focus on the potential antitumor function of ARTs by acting on ferroptosis, including the regulation of iron metabolism, generation of reactive oxygen species (ROS), and activation of endoplasmic reticulum stress (ERS). This article systematically reviews the recent progress in ferroptosis research and provides a basis for ARTs as an anticancer drug in clinical practice.
1. Introduction
1.1. Artemisinin Derivatives
Qinghaosu, known as artemisinin (ART), is a bioactive molecule derived from Artemisia annua. In addition, the artemisinin derivatives comprise artemether (ARM), arteether (ARTE), dihydroartemisinin (DHA), and artesunate (ATS) (Figure 1). They are all sesquiterpene lactones containing peroxide internal bridges necessary for the maintenance of ART activity, collectively known as artemisinin and derivatives, whose pharmacophore is l,2,4-trioxane. ART can be divided into water-soluble derivatives (such as DHA and ATS) and oil-soluble derivatives (such as ARTE and ARM) [1, 2]. ART was first reported by the Chinese scientist Tu Youyou, who discovered its antimalarial effects and thus saved the lives of many malaria patients [3]. In addition, ART also has the effects of anti-inflammatory [4], antidiabetic [5], and antisystemic lupus erythematosus effects [6]. Because of the low cost and high safety of ART, many studies have shown its effects against human cancers. For example, ARTs have been proven to cause necrosis [7] and apoptosis [8] in multiple human cancer cells. ARTs also exhibit the following anticancer functions: cell cycle arrest [9], suppression of angiogenesis [10], inhibition of cell metastasis and invasion [11], DNA damage [12–14], and ferroptosis. ART-type drugs could be applied in combination with other therapeutic modalities in clinical oncology and can inhibit tumor growth in different ways, making them attractive chemotherapeutic agents for cancer treatment [15–17].
Figure 1.
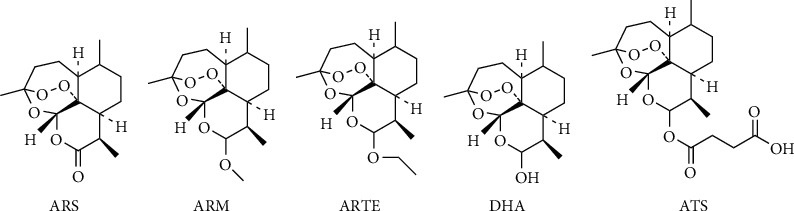
Artemisinin and its derivatives.
1.2. Ferroptosis
Ferroptosis is a new type of cell death defined by Dixon et al. in 2012 [18] and is characterized by the accumulation of lipid peroxidation and iron, the effects of which can be inhibited by iron chelators [19]. The cell death mode of ferroptosis is completely different from cell necrosis, autophagy, and apoptosis in terms of cell morphology, genetics, and biology [18]. In 2003, Dolma et al. found that a novel compound, erastin, could induce the death of RAS-mutated human foreskin fibroblasts (BJeLRs) in a nonapoptotic manner. In erastin-induced cell death, they found wrinkling of mitochondrial morphology and loss of mitochondrial cristae, while no nuclear sequestration or chromatin condensation was found [20]. Subsequently, in 2008, Stockwell et al. found that RSL3, RSL5, and erastin were similar in their ability to act with the accumulation of ROS and iron. Additionally, it could be inhibited by iron chelators and antioxidants [19, 21]. In 2012, this novel mode of cell death was officially named ferroptosis by Dixon et al. [18]. Since then, many studies have indicated that ferroptosis is associated with the development of several diseases, especially in cancer diagnosis and treatment. Due to the resistance of tumor cells to conventional chemotherapy and radiotherapy, ferroptosis has recently attracted extensive attention in the cancer research field because of its specific mechanism of action.
Glutathione peroxidase 4 (GPX4) is a GSH-dependent enzyme. It belongs to the glutathione peroxidase (GPXs) family, which plays a crucial regulatory role in ferroptosis [22]. GPX4 is considered to inhibit the accumulation of lipid peroxidation [23], as it degrades H2O2 and small or complex lipid peroxides with the degradation products being water or the corresponding alcohols [24]. For example, GPX4 can convert lipid hydroperoxides (LOOH) to lipid alcohols, and this process inhibits ferroptosis by reducing the production of lipid peroxidation. Inactivation of GPX4 can trigger ferroptosis through lipid peroxidation resulting in the accumulation of ROS [22, 25]. Common inducers of ferroptosis, such as erastin and RSL3, can induce cellular ferroptosis by reducing the expression of GPX4; however, overexpression of GPX4 can resist the effects caused by erastin and RSL3. Therefore, GPX4 is an important target of ferroptosis triggered by various ferroptosis inducers [22]. In vivo, knockout of GPX4 induced acute renal failure in mice, and this effect was inhibited by the ferroptosis inhibitors ferrostatin-l and necrostatin-1 [26]. Cellular ferroptosis and rapid death of mice can occur after knockdown of GPX4 in mouse neurons [27].
The cystine/glutamate transporter (system Xc-) is a membrane transporter that is composed of solute carrier family 3 member 2 (SLC3A2) and solute carrier family 7 member 11 (SLC7A11). The primary function of system Xc- is to mediate Na+-dependent cystine-glutamate exchange [28]. It transports intracellular glutamate (Glu) to the extracellular compartment and extracellular cystine (Cys2) to the intracellular compartment in the process of generating the antioxidant GSH [29] (Figure 2). GSH can inhibit the production of lipid peroxides and oxidative stress (OS) in cells. Erastin can inhibit the activity of GPX4 by regulating system Xc-, leading to ferroptosis. Reducing the expression of system Xc- by RNAi can increase the anticancer effects of erastin, and overexpression of SLC7A11 can inhibit ferroptosis [18, 30]. In HT-1080 cells, the oncogene p53 could cause cellular ferroptosis by suppressing the expression of SLC7A11 [31]. The anti-inflammatory drug sulfasalazine [32] and the tumor-targeting drug sorafenib [33] can inhibit the effect of system Xc-, thereby reducing intracellular Cys2 uptake and decreasing GSH synthesis, resulting in cell death.
Figure 2.

Possible mechanisms of ART against ferroptosis: ferroptosis is characterized by disruption of the homeostatic balance of iron and the accumulation of lipid peroxidation. (a) ART can degrade ferritin or directly increase the concentration of iron in the LIP, thereby interfering with iron metabolism. (b) GPX4 is an important negative regulator in the ferroptosis-inducing pathway, and ART can reduce the expression of GPX4 and increase lipid peroxidation, thereby promoting ferroptosis. (c) ART inhibits ferroptosis by the regulating Nrf2-ARE pathway. (d) ART acts on the PERK-ATF4-HSPA5 pathway and ATF4-CHOP-CHAC1 pathway in the endoplasmic reticulum stress pathway to regulate ferroptosis.
Voltage-dependent anion channels (VDACs) are widely distributed in the outer mitochondrial membrane. They have three isoforms, i.e., VDAC1, VDAC2, and VDAC3, which maintain the permeability of the mitochondrial outer membrane [34]. VDAC2 and VDAC3 are positive regulators of ferroptosis and can also be used as direct targets of erastin [18, 35]. In human hepatoma cells (HepG2), erastin can promote the accumulation of lipid ROS in mitochondria and limit aerobic glycolysis by damaging the interaction between microtubulin and VDAC, suggesting that energy metabolism and the cytoskeleton may play a potential role in the development of ferroptosis [36].
There are other important regulatory molecules of ferroptosis that can be divided into positive and negative regulatory molecules. The positive regulatory molecules include Ras, transferrin receptor 1 (TFR1), p53, and NADPH oxidase (NOX). Negative regulatory molecules include nuclear factor E2-related factor 2 (Nrf2) and heat shock protein beta-1 (HSPB1). Ras is a protooncogene, and erastin is more selective in killing Ras mutant cells [35]. ART can cause ferroptosis in Ras mutant pancreatic cancer cells [37]. In addition, it can also induce ferroptosis in leukemia cells in a non-Ras-dependent manner [38]. TFR1 is a cell membrane transferrin, a characteristic marker of ferroptosis, and its mechanism of action may be related to altered intracellular iron metabolism [39]. In BJeLR cells, low expression of TFR1 resists erastin-induced ferroptosis [19]. Gao et al. found that the knockdown of the TFR1 expression significantly inhibited ferroptosis in fibroblasts cells [40]. In addition, the iron can be transported to the labile iron pool (LIP) via divalent metal transporter 1 (DMT1). Ferroportin (FPN) is a transmembrane protein, which transports excess iron from the inside of a cell to the outside of it. The LIP, as a catalyst in the Fenton reaction, will lead to the accumulation of lipid peroxidation [41]. p53 can increase the levels of intracellular ROS and trigger the stress response induced by ROS, thereby promoting the susceptibility of tumor cells to ferroptosis [42, 43]. Another study has shown that p53 can suppress the uptake of Cys2 and sensitize cancer cells to ferroptosis by inhibiting the expression of SLC7A11 [43]. HSPB1 is a recently identified molecule that regulates ferroptosis. Knockdown of HSPB1 expression increases the expression of TFR and promotes ferroptosis induced by erastin. Additionally, overexpression of HSPB1 can inhibit ferroptosis induced by erastin. Protein kinase C- (PKC-) mediated phosphorylation of HSPB1 can reduce iron-mediated accumulation of lipid ROS, which in turn reduces ferroptosis in tumor cells. Moreover, suppression of HSPB1 phosphorylation and the heat shock factor-1- (HSF-1-) HSPB1 pathway can increase the anticancer effects of erastin in cancer cells [44]. Sun et al. found that Nrf2 inhibited ferroptosis in hepatocellular carcinoma cells, and knockdown of Nrf2 or inhibition of Nrf2-related gene expression accelerated erastin and sorafenib-induced ferroptosis [45]. More importantly, an accumulating body of research suggests that ART may induce ferroptosis in cancer cells by regulating the above molecules.
ARTs are a class of ferroptosis inducers and have been shown to inhibit the growth of various malignancies such as head and neck cancer [46], liver cancer [45], glioma [47], and bladder cancer [48]; however, the molecular mechanisms of how it affects ferroptosis remain unclear.
2. Mechanisms of ART-Induced Ferroptosis
2.1. The Potential Role of ART in Ferroptosis by Regulating Iron Metabolism
One of the characteristics of ferroptosis is Fe2+ accumulation and disturbance of iron metabolism. Many studies have shown that one of the mechanisms by which ART induces ferroptosis in tumor cells is the regulation of intracellular iron metabolism. Ooko et al. reported that the mRNA levels of twenty iron-related genes were associated with the activity of ART in a group of sixty cancer cell lines. In addition, several of the proteins related to the regulation of iron metabolism, including lactoferrin (LTF), transferrin receptors 1/2 (TFR1, TFR2), transferrin (TF), and ceruloplasmin (CP), have been identified as tumor markers [49].
Many studies have reported that the expression of TFR is very high in tumors [50–54]. Therefore, the regulation of iron metabolism has become an important target in the treatment and prevention of tumors. Additionally, ART can make better use of this mechanism to selectively kill tumor cells while preserving normal cells. DHA can cause intracellular iron depletion in a time- and dose-dependent manner, disrupting intracellular iron metabolism and leading to cell death [55]. Deferoxamine and ferrostatin-1, as iron scavengers, can inhibit ferroptosis by decreasing the levels of free iron [49, 56, 57]; however, ART antagonizes this effect, which indirectly demonstrates that ART can induce ferroptosis by acting on iron metabolism [46, 58–60]. In addition, an increasing body of evidence has discovered that tumor cells contain much more iron than normal cells, partially indicating that ART-type drugs prefer to kill cancer cells than normal cells [50, 52, 61].
Current studies have shown that the main mechanism of ART action is that ART can increase the degradation of ferritin by ferrous iron or transferrin, lysosomes, and autophagosomes, which contribute to the imbalance of intracellular iron metabolism and induce the accumulation of lipid peroxides and OS. DHA and erastin are both ferroptosis inducers. Erastin can lead to autophagic degradation of ferritin, which is called ferritinophagy [62]. Unlike erastin, DHA induces the accumulation of iron by inducing the degradation of ferritin by lysosomes. Chen et al. demonstrated that DHA affects iron homeostasis and increases the levels of cellular free iron, thereby inducing ferroptosis [63]. In Hela cells, ferritin binds with nuclear receptor coactivator 4 (NCOA4) in autophagosomes and is then transported to lysosomes, where ART can promote the degradation of ferritin. Then, it will produce free iron and activate the Fenton reaction, which eventually leads to the accumulation of lipid peroxides [64]. Yang et al. reported that ART can activate lysosomal function by promoting the degradation of ferritin, which will result in the release of iron. Finally, it leads to cell death [64]. Additionally, Eling et al. reported that administration of ART can increase lysosomal-free iron and promote ferroptosis in pancreatic ductal adenocarcinoma (PDAC) [37]. NCOA4 has been identified as a cargo receptor during ferritinophagy [65]. Recently, some studies have revealed that NCOA4 contributes to ferroptosis through ferritin degradation via autophagy in pancreatic cancer [65, 66]. Du et al. discovered that DHA can induce ferroptosis in the treatment of the HL-60 leukemic cell line. DHA-induced autophagy by activating the AMPK/mTOR/p70S6k pathway can increase the LIP and promote the accumulation of lipid peroxidation, thereby resulting in ferroptosis [67].
In conclusion, ART can promote the degradation of ferritin by lysosomes and autophagosomes (Figure 2), disruption of iron metabolism in tumor cells, development of the Fenton reaction, accumulation of lipid peroxides, and OS. Therefore, ART may have a potential role in tumor therapy by regulating intracellular iron metabolism and thus promoting ferroptosis.
2.2. The Potential Role of ART in Ferroptosis by Regulating Oxidative Stress
In normal organisms, there is a set of oxidative and antioxidant systems in equilibrium. The antioxidant system of the organism includes the glutathione reduction system, thioredoxin reduction system, detoxification enzymes (such as catalase, superoxide dismutase, and other modifying enzymes), and Nrf2 pathway. Once ROS accumulation significantly exceeds normal levels, it will lead to significant damage to DNA, lipids, and proteins [68]. ART was first known as an antimalarial drug. The interaction between heme-derived iron and ART will result in the production of ROS and thus the death of malaria parasites [69–72]. Some researchers have proposed a similar mechanism to explain the anticancer activity of ART [71, 73, 74]. ART may induce cancer cell death, similar to what happens in malaria parasites. Therefore, the induction of OS in tumor cells via the production of ROS is the key mechanism of ART against cancer [75–78]. ROS are highly reactive oxygen-containing molecules, including hydroxyl, superoxide, and hydrogen peroxide [79]. Excessive free iron can produce ROS through Fenton reactions [80]. Iron cycles between reduced forms and oxidized forms, resulting in the formation of free radicals [21]. The production of ROS can induce a large number of lipid peroxides and thus damage cells.
Through cooperation with the National Cancer Institute, researchers correlated the IC50 values of 55 tumor cell lines with their microarray-based transcriptome-wide expression for ART and found a statistically significant association between ART and oxidative stress-related gene expression. These genes were antioxidative protein 2, dihydrodiol dehydrogenase, catalase, diaphorase (NADH) (cytochrome b-5 reductase), glutaredoxin 2, γ-glutamylcysteine synthetase, glutathione S-transferases, glutathione peroxidases, reductase, and thioredoxin peroxidase [77, 81–83]. Researchers introduced antioxidant genes into tumor cells and found that the tumor cells were more resistant to ART [8, 84].
Many studies have discovered that ART or DHA can induce the generation of ROS in different kinds of tumor origins [7, 46, 58, 85–87]. Zhu et al. found that ART can promote ferritin degradation in lysosomes and regulate the system Xc-/GPX4 axis to induce ferroptosis [88]. Yi et al. found that the main mechanism by which DHA caused ferroptosis was the accumulation of lipid ROS and downregulation of GPX4. In addition, ferristatin-1, a specific ferroptosis inhibitor, can reverse all these changes in glioblastoma [89]. Induction of ROS overload by ARTs not only induces ferroptosis but also induces other cell death types, including apoptosis and necroptosis. High concentrations of ARTs have ROS- and iron-dependent cytotoxicity in ovarian cancer cells and can result in the death of HEY1 and HEY2 cells through the combined effects of cell cycle blockade leading to apoptosis and ferroptosis [58]. ART can induce the production of ROS, resulting in cell death mediated by apoptosis, ferroptosis, and necroptosis in adult T-cell leukemia/lymphoma [90].
In addition, Nrf2 is a major factor that regulates intracellular redox balance in the cell [91]. Nrf2 is also an oncogenic transcription factor that plays a key role against environmental or intracellular stress and controls the abundant cellular antioxidant systems responsible for GSH production in cancer cells [92]. The proteasomal activity of Keap1 (Kelch-like ECH-associated protein 1) binding to Nrf2 continuously degrades Nrf2. In addition, p62 is a negative regulator of Keap1. p62-Keap1 is related to Nrf2 expression in ferroptosis. The p62-Keap1-Nrf2 signaling pathway is an important negative regulator of ferroptosis in liver cancer [45]. The Nrf2-ARE pathway is an important antioxidant pathway in cancer cells. ART induces ferroptosis mainly through the accumulation of lipid peroxide and iron [93]. Roh et al. reported that ATS can selectively kill head and neck cancer (HNC) cells rather than normal tissue cells. In addition, they also found that suppression of Nrf2-ARE signaling promoted ATS sensitivity and reversed ferroptotic resistance in HNC cells [46]. Activation of the Nrf2-ARE signaling contributed to the ferroptotic resistance in cisplatin-resistant HNC cells [45]. Liu et al. discovered that ART covalently targets Keap1 at Cys151 to activate the Nrf2-dependent pathway [94, 95]. Inhibition of Nrf2 production can reduce resistance to ART in cisplatin-resistant HNCs [46]. The status of Nrf2 is critical in influencing the therapeutic efficacy of ferroptosis-treated hepatocellular carcinoma [45]. In addition, ART can selectively kill cisplatin-resistant HNC cells by inducing ferroptosis in a manner that leaves normal cells unharmed. Theoretically, the cytotoxicity of ART may be enhanced by suppressing the Nrf2-ARE pathway [46] (Figure 2). In other words, the combination of ART and Nrf2 inhibitors to promote ferroptosis may have more efficient anticancer effects without damaging normal cells. In summary, ART can stimulate the production of ROS, leading to the occurrence of ferroptosis as well as other modes of death (Figure 2). Therefore, ART may play a critical role in improving the efficacy of cancer therapy through the induction of ferroptosis and apoptosis. ART may function as a more effective anticancer drug by regulating oxidative stress.
2.3. The Potential Role of ART in Ferroptosis by Regulating Endoplasmic Reticulum Stress
The endoplasmic reticulum (ER) is a multifunctional organelle for protein modification, synthesis, transport, and processing. The accumulation of unfolded or misfolded proteins in the ER will result in ER stress (ERS). ERS and OS are highly correlated biological processes. It has been reported that alterations in redox homeostasis are sufficient to induce endoplasmic reticulum stress, which can induce ROS production in the ER [96]. Wang et al. found that upregulation of cation transport regulator like 1 (CHAC1) can result in the degradation of GSH. In addition, ART can activate the activating transcription factor 4- (ATF4-) C/EBP-homologous protein- (CHOP-) CHAC1 cascade in ERS to induce ferroptosis in Burkitt's lymphoma, and the activation process may be related to ROS production [97]. Hong et al. discovered that ART can promote tumor necrosis factor- (TRAIL-) induced apoptosis via the p53-independent CHOP/p53 upregulated modulator of the apoptosis pathway in human colorectal cancer cell lines and pancreatic cancer cell lines in response to ferroptosis [98]. It is worth mentioning that in malignant glioma cells, activation of the ER minimizes cell injury. DHA can promote ferroptosis in malignant glioma cells. In addition, it can also inhibit ferroptosis by activating the protein kinase R-like ER kinase- (PERK-) ATF4-HSPA5 pathway in the ER by acting on GPX4. Thus, inhibition of any molecule in the protein PERK-ATF4-HSPA5 pathway could enhance the proferroptotic effect of DHA [47]. In this way, ART may serve as a more effective anticancer drug by regulating ferroptosis. Autophagosomes containing ferritin fuse with lysosomes to form autophagolysosomes. Ferritin is digested in autophagolysosomes, which results in the release of iron, and this process is called ferritinophagy [99, 100]. More importantly, Liu et al. found that ARTs are mainly located in the ER [101]. In summary, we investigated the possibility that artemisinin and its derivatives regulate ferroptosis through the ER (Figure 2).
2.4. Other Ways
2.4.1. Mitochondrial Metabolism
Ferroptosis can also be triggered in mitochondria. Lipid oxidation pathways in mitochondrial membranes are intrinsically linked to ferroptosis in an important way [102, 103]. Previous studies have shown that ARTs can affect the mitochondrial electron transport chain, thereby resulting in ROS formation [76, 104]. Qin et al. showed that ATS can induce a loss of mitochondrial transmembrane potential (ΔΨm) [105]. Therefore, we suggest that ARTs may induce ferroptosis by inducing ROS accumulation in mitochondria (Figure 2).
2.4.2. Xc-/GPX4 Pathway
Zhu et al. found that ART promotes ferritin degradation in lysosomes and regulates the system Xc-/GPX4 axis to induce ferroptosis [88]. Yi et al. found that the main mechanism by which DHA caused ferroptosis was downregulation of GPX4 and the accumulation of lipid ROS. All these changes will be reversed by ferrostatin-1, a specific ferroptosis inhibitor in glioblastoma [89]. A previous study reported that DHA decreased the expression of SLC7A11 and thus caused ferroptosis in lung cancer cells [106] (Figure 2).
2.4.3. p53 Pathway
Markowitsch et al. suggested that p53 may be a predictor for ART-induced ferroptosis in KTCTL-26 cells. Wang et al. demonstrated that p53 was involved in ferroptosis induced by ARM. Knockdown of p53 expression can suppress ARM-induced ferroptosis in hepatic stellate cells [107]. The results suggested that it was expected to become a new additional treatment option for patients with advanced or even sunitinib-resistant renal cell carcinoma [108] (Figure 2).
3. Summary
ART, as a class of compounds, has saved many lives due to its antimalarial effects [3]. In addition, ART has also been used to fight other diseases, among which the antitumor effect has been a hot topic of research in recent years. Various mechanisms of ART against tumors include apoptosis [8], necrosis [7], inhibition of tumor angiogenesis [7], and DNA damage [12–14]. Ferroptosis is a novel form of cell death characterized by lipid peroxidation and ROS production due to iron accumulation [19]. The antimalaria mechanism of ART, which includes iron accumulation and ROS production, has led researchers to wonder whether this approach applies to antitumor therapy [71, 73, 74]. Many studies have demonstrated that tumor tissues exhibit increased levels of ROS than normal tissues [109–111]. However, under sustained oxidative stress, tumor cells become well-adapted to such stress through a set of mechanisms, and they often have defects in cell death executioner mechanisms, which is one of the main causes of therapy resistance. Ferroptosis compared to apoptosis, a form of regulated necrosis driven by iron-dependent peroxidation of phospholipids, is regulated by cellular metabolism (metabolism of amino acids, lipids, and sugars), redox homeostasis, and various signaling pathways related to cancer. Therefore, ferroptosis exhibits a vulnerability to cell death. The sensitivity of ferroptosis to cell death is due to its involvement in a variety of biological processes [112, 113]. In addition, Viswanathan et al. suggested that ferroptosis overcomes cancer therapy resistance, which can transcend traditional approaches based on resistance mutations and driver oncogenes [114]. Therefore, ferroptosis may be a more effective treatment for cancer than to apoptosis.
ART is a ferroptosis inducer, and researchers have found that ART can inhibit tumor cell growth by regulating ferroptosis in a variety of malignancies. Although several articles have reported that ART can fight cancer by regulating ferroptosis, few articles have summarized the mechanism by which ART fights cancer by regulating ferroptosis. Combining the researchers' research results, we summarized ART anticancer through ferroptosis into three broad pathways: (1) interfering with cellular iron metabolism, (2) promoting ROS production, and (3) by activating endoplasmic reticulum stress, we found that ART can not only act as a ferroptosis inducer alone to induce the occurrence of ferroptosis but can also induce other forms of cell death in tumors in the process of inducing ferroptosis. More importantly, ART is readily available and inexpensive. It can also selectively kill tumor cells with less harm to normal cells [115]. ART-type drugs can promote the anticancer effects, thereby additive or synergistic effects [116]. These advantages make ART a promising antitumor drug.
ART has very promising clinical applications. ART acts not only on tumor cells alone in experiments but also in combination with a variety of drugs, including radiotherapy, natural products, photodynamic therapy, and recombinant proteins [16, 17]. Some researchers have accurately used exosomes and nanoparticles as tools to induce ferroptosis in tumor cells [117]. For example, Chen et al. developed carrier-free Fe3+-ART coordinated nanoparticles, which are more effective for the treatment of cancer in the future [118]. Additionally, Fei et al. obtained the same results by synthesizing nanomissiles carrying DHA for ROS generation and GSH exhaustion, thereby effectively targeting and killing tumor cells [119]. Various studies have shown that ART can be used as an in-depth inducer to induce ferroptosis in tumor cells and effectively shrink tumors [120–123]. In addition, some other ferroptosis-targeted drugs have been used in the clinical treatment of cancer. Cisplatin has been used in the clinical treatment of ovarian cancer [124] and pancreatic cancer [125]. Guo et al. reported that cisplatin, as a GSH inhibitor, induces ferroptosis and apoptosis in A549 and HCT116 cells [126]. Neratinib, as an HER-2 receptor tyrosine kinase inhibitor, has been used to treat breast cancer [127]. A previous study reported that neratinib promotes ferroptosis by regulating iron metabolism in vitro [128]. In addition, lapatinib has the same effect [129]. Sorafenib has been used to treat breast cancer, renal cell carcinoma, and hepatocellular carcinoma [130]. Sorafenib can also induce ferroptosis by suppressing the activity of system Xc- [112]. Sulfasalazine is widely used for immune diseases [131]. A previous study demonstrated that it can also inhibit ferroptosis by suppressing the activity of system Xc- [18]. Auranofin is commonly used in antirheumatoid arthritis. Yang et al. reported that high-dose auranofin can induce ferroptosis through inhibition of thioredoxin reductase activity [132]. Therefore, these drugs, as ferroptosis inducers, have potential effects in the treatment of cancer (Table 1). In addition, clinical trials have proven the tolerance and safety of ART. At present, ART has received approval for phase I/II clinical trials of cancer [133, 134]. Some clinical trials, including colorectal cancer [134], hepatocellular carcinoma [135], breast cancer [136, 137], and several intraepithelial neoplasias, are ongoing (http://www.clinicaltrials.gov). A few trial results have already been published, which highlighted the favorable tolerability of ART.
Table 1.
Drugs associated with ferroptosis.
| Drug | Target | Application | Reference |
|---|---|---|---|
| Cisplatin | GSH | Ovarian cancer, pancreatic cancer | [124, 125] |
| Neratinib | Iron metabolism | Breast cancer | [127] |
| Lapatinib | Iron metabolism | Breast cancer | [129] |
| Sorafenib | System Xc- | Renal cell carcinoma, hepatocellular carcinoma | [130] |
| Sulfasalazine | System Xc- | Immune diseases | [131] |
| Auranofin | Thioredoxin reductase | Rheumatoid arthritis | [132] |
However, there is still a long way to go before ART can be used as a clinical anticancer drug, and the main problems are focused on the following two aspects: (1) ART, as a kind of bioactive molecule, can induce ferroptosis in tumor cells. How is target identification used in ART? Why can it selectively induce tumor cell death? What about normal tissue? (2) The uniqueness of ferroptosis as a novel mode of cell death remains controversial, and the specific molecular mechanism of ferroptosis has not been investigated. Currently, the clinical use of these drugs still has limitations. ART is generally well-tolerated; however, some compounds induced by ferroptosis may result in tissue damage though the generation of lipid peroxidation. Therefore, long-term use of ART can lead to iron metabolic diseases, such as atherosclerosis [138], diabetes [139], myocardial dysfunction [140], and neuronal diseases [141].
To date, many articles have reported that ART has good anticancer effects, suggesting that ART-type drugs are an attractive class of cancer therapeutic candidates. In addition, this view is supported by clinical data. To provide more convincing evidence for the applicability of ART and its derivatives in cancer clinical drugs, more clinical trials should be carried out to further confirm that ART can be used as an effective anticancer drug in the future.
Acknowledgments
This study was financially supported by grants from the Science and Technology Strategic Cooperation Project of the Luzhou People's Government and Southwest Medical University (No. 2019LZXNYDJ34) and the Undergraduate Innovation and Entrepreneurship Training Program (S202010632282).
Contributor Information
Wenjun Wang, Email: wenjunwang1213@126.com.
Xiang Li, Email: lix2009@126.com.
Conflicts of Interest
The authors declare no competing financial interests.
Authors' Contributions
Xiang Li and Wenjun Wang were responsible for the study concept and design. Yingying Hu, Nan Guo, and Ting Yang drafted the manuscript. Wenjun Wang contributed to the interpretation of the figures and table. Xiang Li, Jianghong Yan, and Wenjun Wang provided a critical revision of the manuscript for important intellectual content. All authors read and approved the final version. Yingying Hu and Nan Guo contributed equally to this work.
References
- 1.Klayman D. Qinghaosu (artemisinin): an antimalarial drug from China. Science (New York, N.Y.) . 1985;228(4703):1049–1055. doi: 10.1126/science.3887571. [DOI] [PubMed] [Google Scholar]
- 2.Hien T. T., White N. J. Qinghaosu. Lancet . 1993;341(8845):603–608. doi: 10.1016/0140-6736(93)90362-K. [DOI] [PubMed] [Google Scholar]
- 3.Kannan R., Kumar K., Sahal D., Kukreti S., Chauhan V. S. Reaction of artemisinin with haemoglobin: implications for antimalarial activity. Biochemical Journal . 2005;385(2):409–418. doi: 10.1042/BJ20041170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang J., Tang W., Zhou R., et al. The new water-soluble artemisinin derivative SM905 ameliorates collagen-induced arthritis by suppression of inflammatory and Th17 responses. British Journal of Pharmacology . 2008;153(6):1303–1310. doi: 10.1038/bjp.2008.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li J., Casteels T., Frogne T., et al. Artemisinins target GABAA receptor signaling and impair α cell identity. Cell . 2017;168(1-2):86–100.e15. doi: 10.1016/j.cell.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li W. D., Dong Y. J., Tu Y. Y., Lin Z. B. Dihydroarteannuin ameliorates lupus symptom of BXSB mice by inhibiting production of TNF-alpha and blocking the signaling pathway NF-kappa B translocation. International Immunopharmacology . 2006;6(8):1243–1250. doi: 10.1016/j.intimp.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 7.Du J. H., Zhang H. D., Ma Z. J., Ji K. M. Artesunate induces oncosis-like cell death in vitro and has antitumor activity against pancreatic cancer xenografts in vivo. Cancer Chemotherapy and Pharmacology . 2010;65(5):895–902. doi: 10.1007/s00280-009-1095-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou C., Pan W., Xiao P. W., Tong S. C. Artesunate induces apoptosis via a Bak-mediated caspase-independent intrinsic pathway in human lung adenocarcinoma cells. Journal of Cellular Physiology . 2012;227(12):3778–3786. doi: 10.1002/jcp.24086. [DOI] [PubMed] [Google Scholar]
- 9.Zhao Y., Jiang W., Li B., et al. Artesunate enhances radiosensitivity of human non-small cell lung cancer A549 cells via increasing NO production to induce cell cycle arrest at G2/M phase. International Immunopharmacology . 2011;11(12):2039–2046. doi: 10.1016/j.intimp.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 10.Inhibition of angiogenesis in vivo and growth of Kaposi's sarcoma xenograft tumors by the anti-malarial artesunate. Biochemical Pharmacology . 2004;68(12):2359–2366. doi: 10.1016/j.bcp.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 11.Rasheed S. A. K., Efferth T., Asangani I. A., Allgayer H. First evidence that the antimalarial drug artesunate inhibits invasion and in vivo metastasis in lung cancer by targeting essential extracellular proteases. International Journal of Cancer Journal International Du Cancer . 2010;127(6):1475–1485. doi: 10.1002/ijc.25315. [DOI] [PubMed] [Google Scholar]
- 12.Berdelle N., Nikolova T., Quiros S., Efferth T., Kaina B. Artesunate induces oxidative DNA damage, sustained DNA double-strand breaks, and the ATM/ATR damage response in cancer cells. Molecular Cancer Therapeutics . 2011;10(12):2224–2233. doi: 10.1158/1535-7163.MCT-11-0534. [DOI] [PubMed] [Google Scholar]
- 13.Li P., Lam E., Roos W., Zdzienicka M., Kaina B., Efferth T. Artesunate derived from traditional Chinese medicine induces DNA damage and repair. Cancer Research . 2008;68(11):4347–4351. doi: 10.1158/0008-5472.CAN-07-2970. [DOI] [PubMed] [Google Scholar]
- 14.Wang B., Hou D., Liu Q., et al. Artesunate sensitizes ovarian cancer cells to cisplatin by downregulating RAD51. Cancer Biology & Therapy . 2015;16(10):1548–1556. doi: 10.1080/15384047.2015.1071738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lam N., Long X., Wong J., Griffin R., Doery J. Artemisinin and its derivatives: a potential treatment for leukemia. Anti-Cancer Drugs . 2019;30(1):1–18. doi: 10.1097/CAD.0000000000000697. [DOI] [PubMed] [Google Scholar]
- 16.Efferth T. Cancer combination therapies with artemisinin-type drugs. Biochemical Pharmacology . 2017;139:56–70. doi: 10.1016/j.bcp.2017.03.019. [DOI] [PubMed] [Google Scholar]
- 17.Kumar M., Yadav T., Khair R., Peters G., Yergeri M. Combination therapies of artemisinin and its derivatives as a viable approach for future cancer treatment. Current Pharmaceutical Design . 2019;25(31):3323–3338. doi: 10.2174/1381612825666190902155957. [DOI] [PubMed] [Google Scholar]
- 18.Dixon S. J., Lemberg K. M., Lamprecht M. R., et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell . 2012;149(5):1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wan S. Y., Stockwell B. R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chemistry & Biology . 2008;15(3):234–245. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dolma S., Lessnick S. L., Hahn W. C., Stockwell B. R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell . 2003;3(3):285–296. doi: 10.1016/S1535-6108(03)00050-3. [DOI] [PubMed] [Google Scholar]
- 21.Dixon S. J., Stockwell B. R. The role of iron and reactive oxygen species in cell death. Nature Chemical Biology . 2014;10(1):9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- 22.Yang W. S., Sriramaratnam R., Welsch M. E., et al. Regulation of ferroptotic cancer cell death by GPX4. Cell . 156(1-2):317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ursini F., Maiorino M., Valente M., Ferri L., Gregolin C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochimica et Biophysica Acta . 1982;710(2):197–211. doi: 10.1016/0005-2760(82)90150-3. [DOI] [PubMed] [Google Scholar]
- 24.Thomas J. P., Geiger P. G., Maiorino M., Ursini F., Girotti A. W. Enzymatic reduction of phospholipid and cholesterol hydroperoxides in artificial bilayers and lipoproteins. Biochimica et Biophysica Acta . 1990;1045(3):252–260. doi: 10.1016/0005-2760(90)90128-K. [DOI] [PubMed] [Google Scholar]
- 25.Parillo F., Sylla L., Palombi C., Monaci M., Stradaioli G. Immunocytochemical localisation of phospholipid hydroperoxide glutathione peroxidase in bull’s spermatogenic cells. Italian Journal of Animal Science . 2014;13(3) doi: 10.4081/ijas.2014.3483. [DOI] [Google Scholar]
- 26.Friedmann Angeli J. P., Schneider M., Proneth B., et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Free Radical Biology & Medicine . 2014;16(12):1180–1191. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen L., Hambright W. S., Na R., Ran Q. Ablation of the Ferroptosis Inhibitor Glutathione Peroxidase 4 in Neurons Results in Rapid Motor Neuron Degeneration and Paralysis. Journal of Biological Chemistry . 2015;290(47):28097–28106. doi: 10.1074/jbc.M115.680090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato H., Tamba M., Ishii T., Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins∗. The Journal of Biological Chemistry . 1999;274(17):11455–11458. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- 29.Bridges R. J., Natale N. R., Patel S. A. System xc− cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. British Journal of Pharmacology . 2012;165(1):20–34. doi: 10.1111/j.1476-5381.2011.01480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feng H., Stockwell B. R. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PLoS Biology . 2018;16(5, article e2006203) doi: 10.1371/journal.pbio.2006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dixon S. J., Patel D. N., Matthew W., et al. Pharmacological inhibition of cystine–glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife . 2014;3:p. e02523. doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu H., Yang C., Jian L., et al. Sulfasalazine-induced ferroptosis in breast cancer cells is reduced by the inhibitory effect of estrogen receptor on the transferrin receptor. Oncology Reports . 2019;42(2):826–838. doi: 10.3892/or.2019.7189. [DOI] [PubMed] [Google Scholar]
- 33.Louandre C., Marcq I., Bouhlal H., et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Letters . 2015;356(2):971–977. doi: 10.1016/j.canlet.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 34.Graham B. H., Craigen W. J. Genetic approaches to analyzing mitochondrial outer membrane permeability. Current Topics in Developmental Biology . 2004;59:87–118. doi: 10.1016/S0070-2153(04)59004-X. [DOI] [PubMed] [Google Scholar]
- 35.Yagoda N., Von Rechenberg M., Zaganjor E., et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature . 447(7146):865–869. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maldonado E. N., Sheldon K. L., DeHart D. N., et al. Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells. The Journal of Biological Chemistry . 2013;288(17):11920–11929. doi: 10.1074/jbc.M112.433847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eling N., Reuter L., Hazin J., Hamacher-Brady A., Brady N. R. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience . 2015;2(5):517–532. doi: 10.18632/oncoscience.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu Y., Xie Y., Cao L., et al. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Molecular & Cellular Oncology . 2015;2(4, article e1054549) doi: 10.1080/23723556.2015.1054549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feng H., Schorpp K., Jin J., et al. Transferrin receptor is a specific ferroptosis marker. Cell Reports . 2020;30(10):3411–3423.e7. doi: 10.1016/j.celrep.2020.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao M., Monian P., Quadri N., Ramasamy R., Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Molecular Cell . 2015;59(2):298–308. doi: 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lv H., Shang P. The significance, trafficking and determination of labile iron in cytosol, mitochondria and lysosomes. Metallomics . 2018;10(7):899–916. doi: 10.1039/C8MT00048D. [DOI] [PubMed] [Google Scholar]
- 42.Jiang L., Hickman J. H., Wang S. J., Gu W. Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle . 2015;14(18):2881–2885. doi: 10.1080/15384101.2015.1068479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang L., Kon N., Li T., et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature . 2015;520(7545):57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun X., Ou Z., Xie M., et al. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene . 2015;34(45):5617–5625. doi: 10.1038/onc.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun X., Ou Z., Chen R., et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology . 2016;63(1):173–184. doi: 10.1002/hep.28251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roh J. L., Kim E. H., Jang H., Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biology . 2017;11:254–262. doi: 10.1016/j.redox.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Very low intensity ultrasounds as a new strategy to improve selective delivery of nanoparticles-complexes in cancer cells. Journal of Experimental & Clinical Cancer Research . 2019;38(1):1–16. doi: 10.1186/s13046-018-1018-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao F., Vakhrusheva O., Markowitsch S., et al. Artesunate impairs growth in cisplatin-resistant bladder cancer cells by cell cycle arrest, apoptosis and autophagy induction. Cell . 2020;9(12):p. 2643. doi: 10.3390/cells9122643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ooko E., Saeed M. E., Kadioglu O., et al. Artemisinin derivatives induce iron-dependent cell death (ferroptosis) in tumor cells. Phytomedicine . 2015;22(11):1045–1054. doi: 10.1016/j.phymed.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 50.Aulbert E., Disselhoff W., Sörje H., Schulz E., Gericke D. Lysosomal accumulation of 67Ga-transferrin in malignant tumors in relation to their growth rate. European Journal of Cancer . 1980;16(9):1217–1232. doi: 10.1016/0014-2964(80)90181-4. [DOI] [PubMed] [Google Scholar]
- 51.Gatter K., Brown G., Trowbridge I., Woolston R., Mason D. Transferrin receptors in human tissues: their distribution and possible clinical relevance. Journal of Clinical Pathology . 1983;36(5):539–545. doi: 10.1136/jcp.36.5.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shterman N., Kupfer B., Moroz C. Comparison of transferrin receptors, iron content and isoferritin profile in normal and malignant human breast cell lines. Pathobiology : journal of immunopathology, molecular and cellular biology . 1991;59(1):19–25. doi: 10.1159/000163611. [DOI] [PubMed] [Google Scholar]
- 53.Judd W., Poodry C., Strominger J. Novel surface antigen expressed on dividing cells but absent from nondividing cells. The Journal of Experimental Medicine . 1980;152:1430–1435. doi: 10.1084/jem.152.5.1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sutherland R., Delia D., Schneider C., Newman R., Kemshead J., Greaves M. Ubiquitous cell-surface glycoprotein on tumor cells is proliferation-associated receptor for transferrin. Proceedings of the National Academy of Sciences of the United States of America . 1981;78(7):4515–4519. doi: 10.1073/pnas.78.7.4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ba Q., Zhou N., Duan J., et al. Dihydroartemisinin exerts its anticancer activity through depleting cellular iron via transferrin receptor-1. PLoS One . 2012;7(8, article e42703) doi: 10.1371/journal.pone.0042703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cao J. Y., Dixon S. J. Mechanisms of ferroptosis. Cellular and Molecular Life Sciences . 2016;73(11-12):2195–2209. doi: 10.1007/s00018-016-2194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin R., Zhang Z., Chen L., et al. Dihydroartemisinin (DHA) induces ferroptosis and causes cell cycle arrest in head and neck carcinoma cells. Cancer Letters . 2016;381(1):165–175. doi: 10.1016/j.canlet.2016.07.033. [DOI] [PubMed] [Google Scholar]
- 58.Greenshields A. L., Shepherd T. G., Hoskin D. W. Contribution of reactive oxygen species to ovarian cancer cell growth arrest and killing by the anti-malarial drug artesunate. Molecular Carcinogenesis . 2017;56(1):75–93. doi: 10.1002/mc.22474. [DOI] [PubMed] [Google Scholar]
- 59.Louandre C., Ezzoukhry Z., Godin C., et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. International Journal of Cancer . 2013;133(7):1732–1742. doi: 10.1002/ijc.28159. [DOI] [PubMed] [Google Scholar]
- 60.Skouta R., Dixon S. J., Wang J., et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. Journal of the American Chemical Society . 2014;136(12):4551–4556. doi: 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Torti S. V., Torti F. M. Iron and cancer: more ore to be mined. Nature Reviews Cancer . 2013;13(5):342–355. doi: 10.1038/nrc3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dowdle W. E., Nyfeler B., Nagel J., et al. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis _in vivo_. Nature Cell Biology . 2014;16(11):1069–1079. doi: 10.1038/ncb3053. [DOI] [PubMed] [Google Scholar]
- 63.Chen G. Q., Benthani F. A., Wu J., Liang D., Bian Z. X., Jiang X. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death and Differentiation . 2020;27(1):242–254. doi: 10.1038/s41418-019-0352-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang N. D., Tan S. H., Ng S., et al. Artesunate induces cell death in human cancer cells via enhancing lysosomal function and lysosomal degradation of ferritin∗. Journal of Biological Chemistry . 2014;289(48):33425–33441. doi: 10.1074/jbc.M114.564567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hou W., Xie Y., Song X., et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy . 2016;12:1425–1428. doi: 10.1080/15548627.2016.1187366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mancias J. D., Wang X., Gygi S. P., Harper J. W., Kimmelman A. C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature . 2014;509(7498):105–109. doi: 10.1038/nature13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Du J., Wang T., Li Y., et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radical Biology & Medicine . 2019;131:356–369. doi: 10.1016/j.freeradbiomed.2018.12.011. [DOI] [PubMed] [Google Scholar]
- 68.Ozben T. Oxidative stress and apoptosis: impact on cancer therapy. Journal of Pharmaceutical Ences . 2010;96(9):2181–2196. doi: 10.1002/jps.20874. [DOI] [PubMed] [Google Scholar]
- 69.Pandey A., Tekwani B., Singh R., Chauhan V. Artemisinin, an endoperoxide antimalarial, disrupts the hemoglobin catabolism and heme detoxification systems in malarial parasite. The Journal of Biological Chemistry . 1999;274(27):19383–19388. doi: 10.1074/jbc.274.27.19383. [DOI] [PubMed] [Google Scholar]
- 70.O’neill P. M., Barton V. E., Ward S. A. The molecular mechanism of action of artemisinin-the debate continues. Molecules . 2010;15(3):1705–1721. doi: 10.3390/molecules15031705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Efferth T. Willmar Schwabe Award 2006: antiplasmodial and antitumor activity of artemisinin - from bench to bedside. Planta Medica . 2007;73(4):299–309. doi: 10.1055/s-2007-967138. [DOI] [PubMed] [Google Scholar]
- 72.Gautam A., Ahmed T., Batra V., Paliwal J. Pharmacokinetics and pharmacodynamics of endoperoxide antimalarials. Current Drug Metabolism . 2009;10(3):289–306. doi: 10.2174/138920009787846323. [DOI] [PubMed] [Google Scholar]
- 73.Mercer A., Maggs J., Sun X., et al. Evidence for the involvement of carbon-centered radicals in the induction of apoptotic cell death by artemisinin compounds. The Journal of Biological Chemistry . 2007;282(13):9372–9382. doi: 10.1074/jbc.M610375200. [DOI] [PubMed] [Google Scholar]
- 74.Mercer A., Copple I., Maggs J., O'Neill P., Park B. The role of heme and the mitochondrion in the chemical and molecular mechanisms of mammalian cell death induced by the artemisinin antimalarials. The Journal of Biological Chemistry . 2011;286(2):987–996. doi: 10.1074/jbc.M110.144188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Efferth T., Briehl M. M., Tome M. E. Role of antioxidant genes for the activity of artesunate against tumor cells. International Journal of Oncology . 2003;23(4):1231–1235. doi: 10.3892/ijo.23.4.1231. [DOI] [PubMed] [Google Scholar]
- 76.Hamacher-Brady A., Stein H. A., Turschner S., et al. Artesunate activates mitochondrial apoptosis in breast cancer cells via iron-catalyzed lysosomal reactive oxygen species production. Journal of Biological Chemistry . 2011;286(8):6587–6601. doi: 10.1074/jbc.M110.210047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Efferth T., Oesch F. Oxidative stress response of tumor cells: microarray-based comparison between artemisinins and anthracyclines. Biochemical Pharmacology . 2004;68(1):3–10. doi: 10.1016/j.bcp.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 78.Horwedel C., Tsogoeva S. B., Wei S., Efferth T. Cytotoxicity of artesunic acid homo- and heterodimer molecules toward sensitive and multidrug-resistant CCRF-CEM leukemia cells. Journal of Medicinal Chemistry . 2010;53(13):4842–4848. doi: 10.1021/jm100404t. [DOI] [PubMed] [Google Scholar]
- 79.Valko M., Rhodes C. J., Moncol J., Izakovic M., Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chemico-Biological Interactions . 2006;160(1):1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 80.Shintoku R., Takigawa Y., Yamada K., Kubota C., Torii S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Science . 2017;108(11):2187–2194. doi: 10.1111/cas.13380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Efferth T., Olbrich A., Bauer R. mRNA expression profiles for the response of human tumor cell lines to the antimalarial drugs artesunate, arteether, and artemether. Biochemical Pharmacology . 2002;64(4):617–623. doi: 10.1016/S0006-2952(02)01221-2. [DOI] [PubMed] [Google Scholar]
- 82.Efferth T., Sauerbrey A., Olbrich A., et al. Molecular modes of action of artesunate in tumor cell lines. Molecular Pharmacology . 2003;64(2):382–394. doi: 10.1124/mol.64.2.382. [DOI] [PubMed] [Google Scholar]
- 83.Toth K., Spencer J., Tollefson A., et al. Cotton rat tumor model for the evaluation of oncolytic adenoviruses. Human Gene Therapy . 2005;16(1):139–146. doi: 10.1089/hum.2005.16.139. [DOI] [PubMed] [Google Scholar]
- 84.Wang Z., Hu W., Zhang J., Wu X., Zhou H. Dihydroartemisinin induces autophagy and inhibits the growth of iron-loaded human myeloid leukemia K562 cells via ROS toxicity. FEBS Open Bio . 2012;2(1):103–112. doi: 10.1016/j.fob.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhu H., Liao S., Shi J., et al. DJ-1 mediates the resistance of cancer cells to dihydroartemisinin through reactive oxygen species removal. Free Radical Biology & Medicine . 2014;71:121–132. doi: 10.1016/j.freeradbiomed.2014.03.026. [DOI] [PubMed] [Google Scholar]
- 86.Jia J., Qin Y., Zhang L., et al. Artemisinin inhibits gallbladder cancer cell lines through triggering cell cycle arrest and apoptosis. Molecular Medicine Reports . 2016;13(5):4461–4468. doi: 10.3892/mmr.2016.5073. [DOI] [PubMed] [Google Scholar]
- 87.Lemke D., Pledl H., Zorn M., et al. Slowing down glioblastoma progression in mice by running or the anti-malarial drug dihydroartemisinin? Induction of oxidative stress in murine glioblastoma therapy. Induction of oxidative stress in murine glioblastoma therapy, Oncotarget . 2016;7(35):56713–56725. doi: 10.18632/oncotarget.10723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhu S., Yu Q., Huo C., et al. Ferroptosis: a novel mechanism of artemisinin and its derivatives in cancer therapy. Current Medicinal Chemistry . 2020;28(2):329–345. doi: 10.2174/0929867327666200121124404. [DOI] [PubMed] [Google Scholar]
- 89.Yi R., Wang H., Deng C., et al. Dihydroartemisinin initiates ferroptosis in glioblastoma through GPX4 inhibition. Bioscience Reports . 2020;40(6) doi: 10.1042/BSR20193314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ishikawa C., Senba M., Mori N. Evaluation of artesunate for the treatment of adult T-cell leukemia/lymphoma. European Journal of Pharmacology . 2020;872, article 172953 doi: 10.1016/j.ejphar.2020.172953. [DOI] [PubMed] [Google Scholar]
- 91.Sporn M., Liby K. NRF2 and cancer: the good, the bad and the importance of context. Nature Reviews. Cancer . 2012;12(8):564–571. doi: 10.1038/nrc3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gorrini C., Harris I., Mak T. Modulation of oxidative stress as an anticancer strategy. Nature Reviews. Drug Discovery . 2013;12(12):931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 93.Yang W., Stockwell B. Ferroptosis: death by lipid peroxidation. Trends in Cell Biology . 2016;26(3):165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu S., Xu S., Wei R., Cui Z., Chen W. Keap1 cystenine 151 as a potential target for artemisitene-induced Nrf2 activation. BioMed Research International . 2019;2019:8. doi: 10.1155/2019/5198138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zhang Y., Li F., Song Y., et al. Numb and Numbl act to determine mammary myoepithelial cell fate, maintain epithelial identity, and support lactogenesis. The FASEB Journal . 2016;30(10):3474–3488. doi: 10.1096/fj.201600387R. [DOI] [PubMed] [Google Scholar]
- 96.Cao S. S., Kaufman R. J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxidants & Redox Signaling . 2014;21(3):396–413. doi: 10.1089/ars.2014.5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang N., Zeng G. Z., Yin J. L., Bian Z. X. Artesunate activates the ATF4-CHOP-CHAC1 pathway and affects ferroptosis in Burkitt's lymphoma. Biochemical and Biophysical Research Communications . 2019;519(3):533–539. doi: 10.1016/j.bbrc.2019.09.023. [DOI] [PubMed] [Google Scholar]
- 98.Hong S. H., Dae-Hee L., Young-Sun L., et al. Molecular crosstalk between ferroptosis and apoptosis: emerging role of ER stress-induced p53-independent PUMA expression. Oncotarget . 2017;8(70):115164–115178. doi: 10.18632/oncotarget.23046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Asano T., Komatsu M., Yamaguchi-Iwai Y., Ishikawa F., Mizushima N., Iwai K. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Molecular and Cellular Biology . 2011;31(10):2040–2052. doi: 10.1128/MCB.01437-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bogdan A. R., Miyazawa M., Hashimoto K., Tsuji Y. Regulators of iron homeostasis: new players in metabolism, cell death, and disease. Cell Death, and Disease, Trends Biochem Sci . 2016;41(3):274–286. doi: 10.1016/j.tibs.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu Y., Lok C. N., Ko B. C., Shum T. Y., Wong M. K., Che C. M. Subcellular localization of a fluorescent artemisinin derivative to endoplasmic reticulum. Organic Letters . 2010;12(7):1420–1423. doi: 10.1021/ol902890j. [DOI] [PubMed] [Google Scholar]
- 102.Wipf P., Xiao J., Jiang J., et al. Mitochondrial targeting of selective electron scavengers: synthesis and biological analysis of hemigramicidin−TEMPO conjugates. Journal of the American Chemical Society . 2005;127(36):12460–12461. doi: 10.1021/ja053679l. [DOI] [PubMed] [Google Scholar]
- 103.Ji J., Kline A., Amoscato A., et al. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nature Neuroscience . 2012;15(10):1407–1413. doi: 10.1038/nn.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Papanikolaou X., Johnson S., Garg T., Tian E., Heuck C. Artesunate overcomes drug resistance in multiple myeloma by inducing mitochondrial stress and non-caspase apoptosis. Oncotarget . 2014;5(12):4118–4128. doi: 10.18632/oncotarget.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Qin G., Wu L., Liu H., et al. Artesunate induces apoptosis via a ROS-independent and Bax-mediated intrinsic pathway in HepG2 cells. Experimental Cell Research . 2015;336(2):308–317. doi: 10.1016/j.yexcr.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 106.Yuan B., Liao F., Shi Z., et al. Dihydroartemisinin inhibits the proliferation, colony formation and induces ferroptosis of lung cancer cells by inhibiting PRIM2/SLC7A11 axis. Oncotargets and Therapy . 2020;13:10829–10840. doi: 10.2147/OTT.S248492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang L., Zhang Z., Li M., et al. P53-dependent induction of ferroptosis is required for artemether to alleviate carbon tetrachloride-induced liver fibrosis and hepatic stellate cell activation. IUBMB Life . 2019;71(1):45–56. doi: 10.1002/iub.1895. [DOI] [PubMed] [Google Scholar]
- 108.Markowitsch S., Schupp P., Lauckner J., et al. Artesunate inhibits growth of sunitinib-resistant renal cell carcinoma cells through cell cycle arrest and induction of ferroptosis. Cancers . 2020;12(11):p. 3150. doi: 10.3390/cancers12113150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang H., Ba S., Yang Z., et al. Graphene quantum dot-based nanocomposites for diagnosing cancer biomarker APE1 in living cells. ACS Applied Materials & Interfaces . 2020;12(12):13634–13643. doi: 10.1021/acsami.9b21385. [DOI] [PubMed] [Google Scholar]
- 110.Visconti R., Grieco D. New insights on oxidative stress in cancer. Current Opinion in Drug Discovery & Development . 2009;12(2):240–245. [PubMed] [Google Scholar]
- 111.Ramdzan Z. M., Vickridge E., Faraco C. C. F., Nepveu A. CUT domain proteins in DNA repair and cancer. Cancers (Basel) . 2021;13(12):p. 2953. doi: 10.3390/cancers13122953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Stockwell B. R., Friedmann Angeli J. P., Bayir H., et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell . 2017;171(2):273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jiang X., Stockwell B. R., Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nature Reviews. Molecular Cell Biology . 2021;22(4):266–282. doi: 10.1038/s41580-020-00324-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Viswanathan V. S., Ryan M. J., Dhruv H. D., et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature . 2017;547(7664):453–457. doi: 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lai H., Sasaki T., Singh N., Messay A. Effects of artemisinin-tagged holotransferrin on cancer cells. Life Sciences . 2005;76(11):1267–1279. doi: 10.1016/j.lfs.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 116.Drenberg C., Buaboonnam J., Orwick S., et al. Evaluation of artemisinins for the treatment of acute myeloid leukemia. Cancer Chemotherapy and Pharmacology . 2016;77(6):1231–1243. doi: 10.1007/s00280-016-3038-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yu M., Gai C., Li Z., Ding D., Li W. Targeted exosome-encapsulated erastin induced ferroptosis in triple negative breast cancer cells. Cancer ence . 2019;110(10):3173–3182. doi: 10.1111/cas.14181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chen J., Wang X., Zhang Y., et al. A redox-triggered C-centered free radicals nanogenerator for self-enhanced magnetic resonance imaging and chemodynamic therapy. Biomaterials . 2021;266, article 120457 doi: 10.1016/j.biomaterials.2020.120457. [DOI] [PubMed] [Google Scholar]
- 119.Fei W., Chen D., Tang H., et al. Targeted GSH-exhausting and hydroxyl radical self-producing manganese-silica nanomissiles for MRI guided ferroptotic cancer therapy. Nanoscale . 2020;12(32):16738–16754. doi: 10.1039/D0NR02396E. [DOI] [PubMed] [Google Scholar]
- 120.Vandewynckel Y. P., Laukens D., Geerts A., et al. Therapeutic effects of artesunate in hepatocellular carcinoma: repurposing an ancient antimalarial agent. European Journal of Gastroenterology & Hepatology . 2014;60(1):S85–S85. doi: 10.1016/S0168-8278(14)60218-X. [DOI] [PubMed] [Google Scholar]
- 121.Jeong D., Song H., Lim S., et al. Repurposing the anti-malarial drug artesunate as a novel therapeutic agent for metastatic renal cell carcinoma due to its attenuation of tumor growth, metastasis, and angiogenesis. Oncotarget . 2015;6(32):33046–33064. doi: 10.18632/oncotarget.5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kast R., Boockvar J., Brüning A., et al. A conceptually new treatment approach for relapsed glioblastoma: coordinated undermining of survival paths with nine repurposed drugs (CUSP9) by the International Initiative for Accelerated Improvement of Glioblastoma Care. Oncotarget . 2013;4(4):502–530. doi: 10.18632/oncotarget.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Augustin Y., Krishna S., Kumar D., Pantziarka P. The wisdom of crowds and the repurposing of artesunate as an anticancer drug. Ecancer medical science . 2015;9:p. ed50. doi: 10.3332/ecancer.2015.ed50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Galluzzi L., Senovilla L., Vitale I., et al. Molecular mechanisms of cisplatin resistance. Oncogene . 2012;31(15):1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 125.Reni M., Cereda S., Galli L. PEFG (cisplatin, epirubicin, 5-fluorouracil, gemcitabine) for patients with advanced pancreatic cancer: the ghost regimen. Cancer Letters . 2007;256(1):25–28. doi: 10.1016/j.canlet.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 126.Guo J., Xu B., Han Q., et al. Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Research and Treatment . 2018;50(2):445–460. doi: 10.4143/crt.2016.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhu J., Xiong Y., Zhang Y., et al. The molecular mechanisms of regulating oxidative stress-induced ferroptosis and therapeutic strategy in tumors. Oxidative Medicine and Cellular Longevity . 2020;2020 doi: 10.1155/2020/8810785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nagpal A., Redvers R. P., Ling X., et al. Neoadjuvant neratinib promotes ferroptosis and inhibits brain metastasis in a novel syngeneic model of spontaneous HER2(+ve) breast cancer metastasis. Breast Cancer Research . 2019;21(1):p. 94. doi: 10.1186/s13058-019-1177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Qiu Y., Cao Y., Cao W., Jia Y., Lu N. The application of ferroptosis in diseases. Pharmacological Research . 2020;159, article 104919 doi: 10.1016/j.phrs.2020.104919. [DOI] [PubMed] [Google Scholar]
- 130.Escudier B., Worden F., Kudo M. Sorafenib: key lessons from over 10 years of experience. Expert Review of Anticancer Therapy . 2019;19(2):177–189. doi: 10.1080/14737140.2019.1559058. [DOI] [PubMed] [Google Scholar]
- 131.Plosker G. L., Croom K. F. Sulfasalazine. Drugs . 2005;65(13):1825–1849. doi: 10.2165/00003495-200565130-00008. [DOI] [PubMed] [Google Scholar]
- 132.Yang L., Wang H., Yang X., et al. Auranofin mitigates systemic iron overload and induces ferroptosis via distinct mechanisms. Signal Transduction and Targeted Therapy . 2020;5(1):p. 138. doi: 10.1038/s41392-020-00253-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ericsson T., Blank A., von Hagens C., Ashton M., Äbelö A. Population pharmacokinetics of artesunate and dihydroartemisinin during long-term oral administration of artesunate to patients with metastatic breast cancer. European Journal of Clinical Pharmacology . 2014;70(12):1453–1463. doi: 10.1007/s00228-014-1754-2. [DOI] [PubMed] [Google Scholar]
- 134.Krishna S., Ganapathi S., Ster I., et al. A randomised, double blind, placebo-controlled pilot study of oral artesunate therapy for colorectal cancer. Placebo-Controlled Pilot Study of Oral Artesunate Therapy for Colorectal Cancer, EBioMedicine . 2015;2(1):82–90. doi: 10.1016/j.ebiom.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Deeken J., Wang H., Hartley M., et al. A phase I study of intravenous artesunate in patients with advanced solid tumor malignancies. Cancer Chemotherapy and Pharmacology . 2018;81(3):587–596. doi: 10.1007/s00280-018-3533-8. [DOI] [PubMed] [Google Scholar]
- 136.von Hagens C., Walter-Sack I., Goeckenjan M., et al. Long-term add-on therapy (compassionate use) with oral artesunate in patients with metastatic breast cancer after participating in a phase I study (ARTIC M33/2) Phytomedicine : international journal of phytotherapy and phytopharmacology . 2019;54:140–148. doi: 10.1016/j.phymed.2018.09.178. [DOI] [PubMed] [Google Scholar]
- 137.König M., von Hagens C., Hoth S., et al. Erratum to: investigation of ototoxicity of artesunate as add-on therapy in patients with metastatic or locally advanced breast cancer: new audiological results from a prospective, open, uncontrolled, monocentric phase I study. Cancer Chemotherapy and Pharmacology . 2016;77(6):p. 1321. doi: 10.1007/s00280-016-3023-9. [DOI] [PubMed] [Google Scholar]
- 138.Dabbagh A. J., Shwaery G. T., Keaney J. F. J., Frei B. Effect of iron overload and iron deficiency on atherosclerosis in the hypercholesterolemic rabbit. Arteriosclerosis, Thrombosis, and Vascular Biology . 1997;17(11):2638–2645. doi: 10.1161/01.ATV.17.11.2638. [DOI] [PubMed] [Google Scholar]
- 139.Huang J., Jones D., Luo B., et al. Iron overload and diabetes risk: a shift from glucose to fatty acid oxidation and increased hepatic glucose production in a mouse model of hereditary hemochromatosis. Diabetes . 2011;60(1):80–87. doi: 10.2337/db10-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Elena G., Stefania R., Ilona R., Paolo B., Gaetano C. Iron-induced damage in cardiomyopathy: oxidative-dependent and independent mechanisms. Oxidative Medicine and Cellular Longevity . 2015;2015 doi: 10.1155/2015/230182.230182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dominic H., Scott A., Ashley B., Peng L. A delicate balance: iron metabolism and diseases of the brain. Frontiers in Aging Neuroscience . 2013;5:p. 34. doi: 10.3389/fnagi.2013.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]