

CONSPECTUS
Methods for detecting and quantifying disease biomarkers in biofluids with high specificity and sensitivity play a pivotal role in enabling clinical diagnostics, including point-of-care tests. The most widely used molecular biomarkers include proteins, nucleic acids, hormones, metabolites, and other small molecules. While numerous methods have been developed for analyzing biomarkers, most techniques are challenging to implement for clinical use due to insufficient analytical performance, high cost, and/or other practical shortcomings. For instance, the detection of cell-free nucleic acid (cfNA) biomarkers by digital PCR and next generation sequencing (NGS) requires time-consuming nucleic acid extraction steps, often introduces enzymatic amplification bias, and can be costly when high specificity is required. While several amplification-free methods for detecting cfNAs have been reported, these techniques generally suffer from low specificity and sensitivity. Meanwhile, the quantification of protein biomarkers is generally performed using immunoassays such as enzyme-linked immunosorbent assay (ELISA); the analytical performance of these methods is often limited by the availability of antibodies with high affinity and specificity, as well as the significant nonspecific binding of antibodies to assay surfaces. To address the drawbacks of existing biomarker detection methods and establish a universal diagnostics platform capable of detecting different types of analytes, we have developed an amplification-free approach, named single-molecule recognition through equilibrium Poisson sampling (SiMREPS), for the detection of diverse biomarkers with arbitrarily high specificity and single-molecule sensitivity. SiMREPS utilizes the transient, reversible binding of fluorescent detection probes to immobilized target molecules to generate kinetic fingerprints that are detected by single-molecule fluorescence microscopy. Analysis of these kinetic fingerprints enables near-perfect discrimination between specific binding to target molecules and any nonspecific binding. Early proof-of-concept studies demonstrated the in vitro detection of miRNAs with limits of detection (LOD) of approximately 1 fM and >500-fold selectivity for single-nucleotide polymorphisms. The SiMREPS approach was subsequently expanded to the detection of rare mutant DNA alleles from biofluids at mutant allele fractions as low as 1 in 1 million, corresponding to a specificity of >99.99999%. Recently, SiMREPS was generalized to protein quantification using dynamically binding antibody probes, permitting LODs in the low-femtomolar to attomolar range. Finally, SiMREPS has been demonstrated to be suitable for the in situ detection of miRNAs in cultured cells, the quantification of small-molecule toxins and drugs, and the monitoring of telomerase activity at the single-molecule level. In this Account, we discuss the principles of SiMREPS for the highly specific and sensitive detection of molecular analytes, including considerations for assay design. We discuss the generality of SiMREPS for the detection of very disparate analytes, and provide an overview of data processing methods, including the expansion of dynamic range using super-resolution analysis and the improvement of performance using deep learning algorithms. Finally, we describe current challenges, opportunities, and future directions for the SiMREPS approach.
Graphical Abstract
1. INTRODUCTION
The detection and/or quantification of disease biomarkers such as proteins, nucleic acids, hormones, enzymes, peptides and metabolites at low concentrations in complex biological samples is crucial in a variety of clinical settings, including the early detection of disease5, assessment of response to therapy6, and prognosis of disease relapse7. For instance, prostate specific antigen (PSA) found at femtomolar levels in human serum has emerged as an important biomarker for prostate cancer recurrence after radical prostatectomy8. In addition, cell-free nucleic acids (cfNAs) such as circulating tumor DNA (ctDNA) and microRNA (miRNA) found in biofluids have been increasingly used as biomarkers in so-called liquid biopsies for the early detection of cancers and minimal residual disease9. While the performance of current methods suffices for some clinically important biomarkers, it is challenging to simultaneously achieve high analytical performance with a simple work- flow and at low cost. Furthermore, with a few exception like single molecule arrays (Simoa)10 and single cell multi-omics methods11, most techniques do not provide a unified platform for the sensitive quantification of DNA, RNA, protein, and small-molecule biomarkers, necessitating diverse sample handling and measurement methods that complicate analysis.
Recently, our lab has developed an approach called single-molecule recognition through equilibrium Poisson sampling (SiMREPS) for the detection and quantification of diverse disease biomarkers with ultrahigh specificity and sensitivity1,2,12,13. SiMREPS utilizes the transient and reversible binding of fluorescent detection probes to immobilized target molecules; this repetitive binding is detected at the single-molecule level to generate kinetic fingerprints that permit the differentiation between specific binding (to target molecules) and nonspecific background binding with high confidence. Figure 1A shows a simplified view of the detection of an analyte via SiMREPS along with the resulting distinct single-molecule kinetic fingerprints originating from the specific binding of fluorescent probe (FP) to the correct target molecule, nonspecific binding to spurious targets, or background binding (i.e., capture probes, assay surfaces). The transient binding and dissociation of probes at equilibrium in a defined observation window can be modeled as a Poisson process wherein the expected number of observed binding events per target molecule becomes more sharply defined (i.e., more deterministic) with longer observation. Thus, with increasing acquisition time, a better separation is obtained between the distribution of the number of binding and dissociation events (Nb+d) for specific and nonspecific binding (Figure 1B). To date, SiMREPS has been successfully demonstrated to detect molecular analytes as diverse as miRNAs1,14, ctDNAs2,15, proteins3 and small molecules16 with high specificity and sensitivity (see Section 3, and Table S1). This uncommon ability to detect and accurately count such a broad range of analytes at the single-molecule level suggests great potential for SiMREPS as a generalized platform for biomarker diagnostics.
Figure 1.
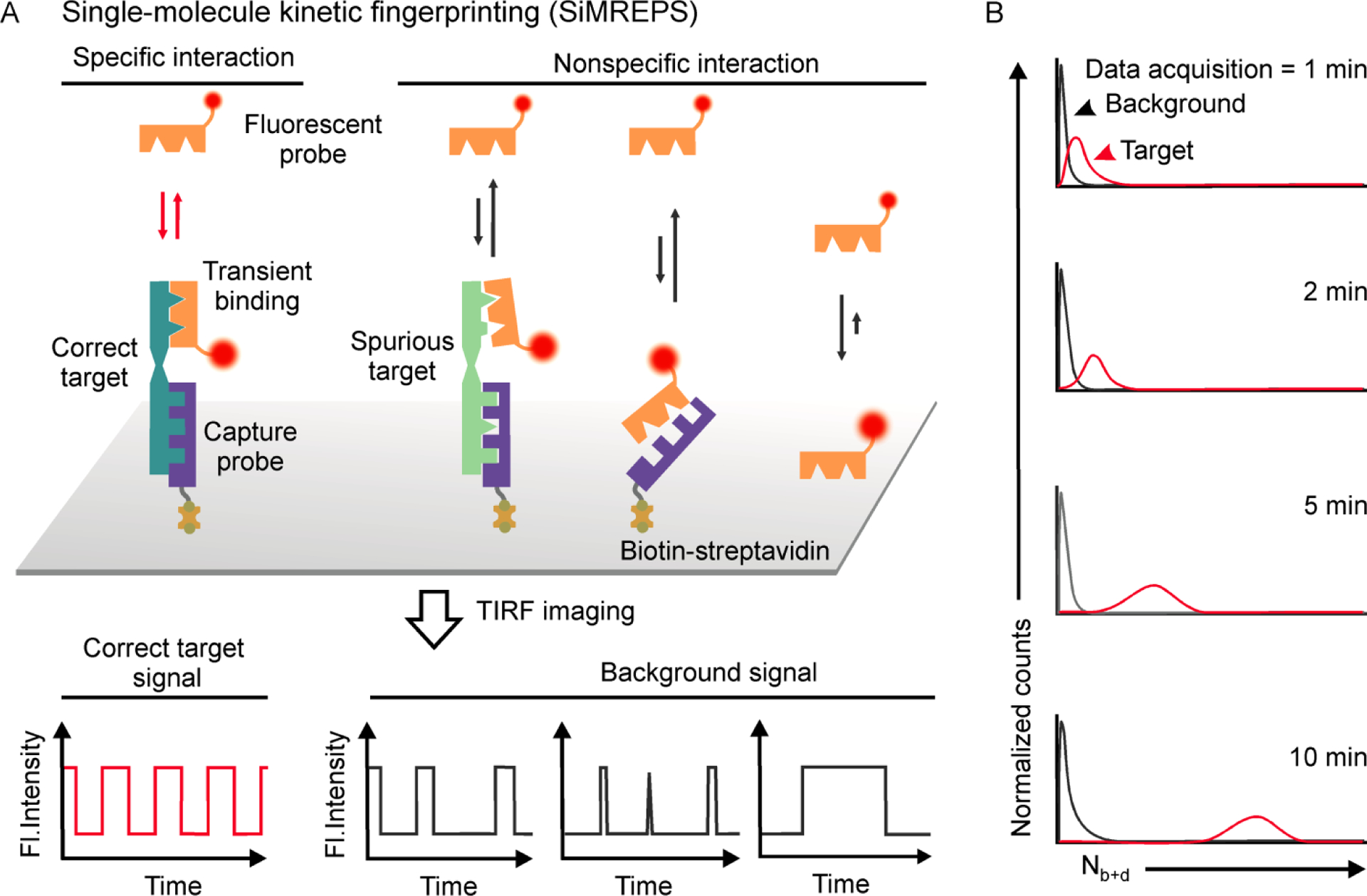
Schematic of the principle of single-molecule kinetic fingerprinting (SiMREPS). (A) SiMREPS uses the transient and reversible binding of low-affinity fluorescent probes to immobilized target molecules to generate distinct kinetic fingerprints that permit high- confidence differentiation between specific binding to correct target and nonspecific background binding. Probe binding and dissociation to single molecules is observed in real time by TIRF microscopy. (B) Predicted distribution of the number of binding and dissociation (Nb+d) events as a function of time. With increasing standard acquisition time, a better separation is obtained between specific and nonspecific or background binding.
A wide variety of innovative methods have been developed for detection of DNA, miRNA, protein, and small molecules with variable analytical performance, either in ensemble or single-molecule assay formats13,17–20. The gold-standard methods for detecting nucleic acids include polymerase chain reaction (PCR) and next-generation sequencing (NGS)19. PCR-based detection methods rely on enzymatic amplification steps, in which a small number of target nucleic acid molecules in the sample are exponentially amplified for increasing sensitivity. For instance, digital PCR (dPCR) amplifies and quantifies target molecules by partitioning them into individual wells or droplets and allows for absolute target quantification (Figure 2A, top panel). Although dPCR has extremely high sensitivity21, PCR-based detection methods suffer from several drawbacks, including the possibility of heat-induced chemical damage2,22, amplification bias, inefficient amplification of short nucleic acids (e.g. miRNAs23) and interference from PCR inhibitors24. Recently, optimized NGS has become popular for the high-throughput sequencing of nucleic acids in a complex mixture, for screening and early detection of cancer19,25. However, achieving high sensitivity and specificity with NGS requires high sequencing depth26 to correct amplification and readout errors, which is time consuming and often increases cost27. In contrast to the above methods, SiMREPS entirely eliminates amplification steps and the errors associated with them, enabling more straightforward sample preparation while achieving very high intrinsic analytical specificity to the detection of a small number of targets (see Table S2 for advantages and limitations of SiMREPS) through direct fingerprinting of each molecule (Figure 2A, bottom panel).
Figure 2.
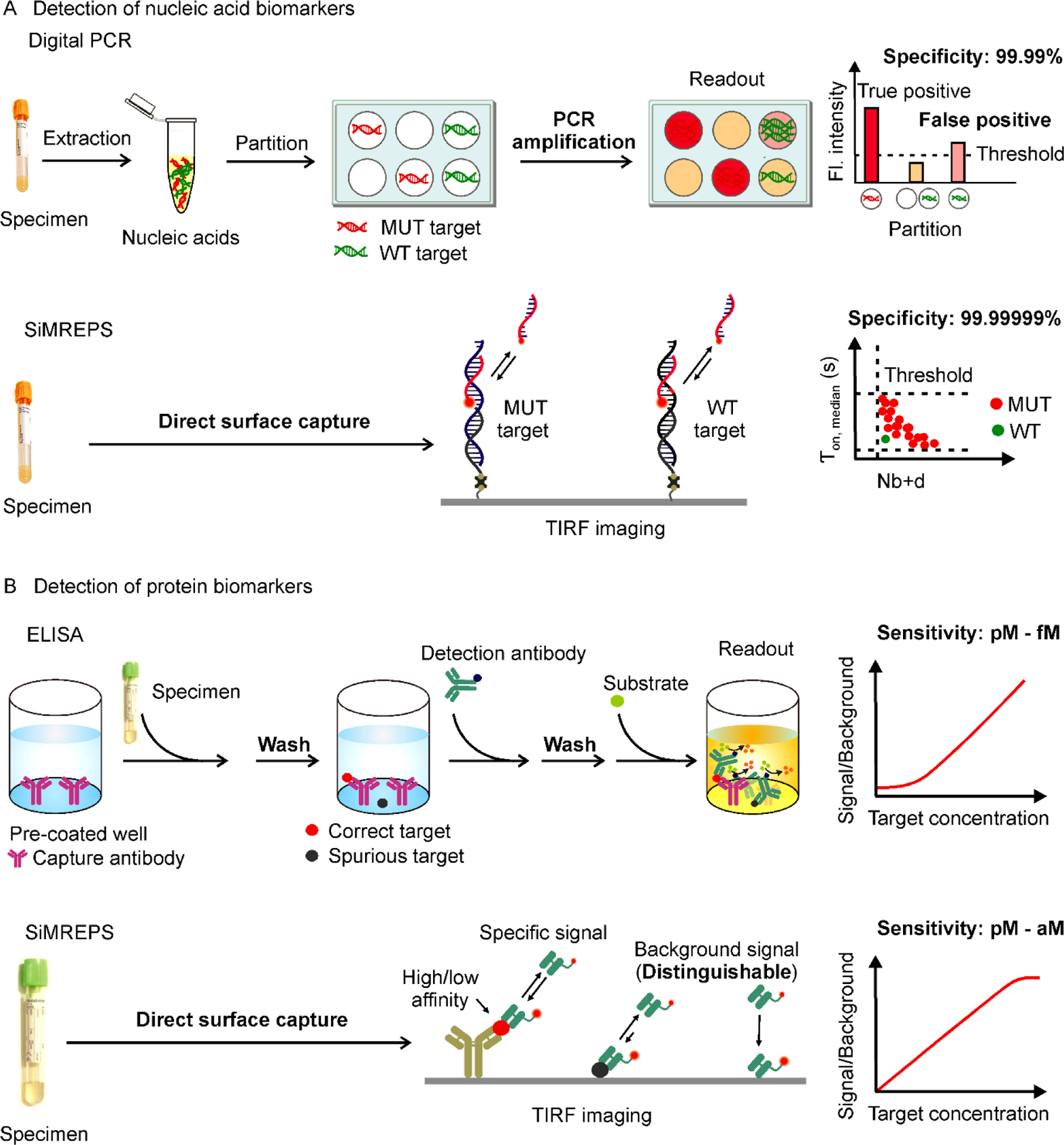
Comparison of conventional and SiMREPS approaches for the detection of nucleic acids and proteins. (A) Comparison between digital PCR and SiMREPS for detection of mutant (MUT) DNA alleles. Digital PCR is limited by its specificity due to heat-induced chemical modification of nucleobases and amplification bias that can generate false positive signals in wildtype (WT) DNA. SiMREPS is an amplification free single-molecule kinetic fingerprinting approach that utilizes transient interaction of detection probe to achieve arbitrarily high discrimination between closely related nucleic acid sequences. (B) Comparison between ELISA and SiMREPS for detection of proteins. ELISA utilizes laborious multistep stringent washing protocols, and suffers from its lower sensitivity, and dynamic range because of high background signals generated by nonspecific interaction of proteins with assay surface. SiMREPS uses a direct wash-free protocol for highly sensitive and specific detection of proteins with broader dynamic range because of its ability to suppress background signals applying kinetic thresholds.
To detect protein biomarkers, enzyme-linked immunosorbent assay (ELISA)28 has long been the preferred technique in clinical research laboratories and hospitals29. One of the most high-sensitivity and -specificity ELISA formats, sandwich ELISA30, utilizes a pair of antibodies to capture and detect protein targets. The specificity of detection is enhanced by the dual recognition by two high-affinity antibodies that bind distinct epitopes of the same antigen. However, the selection and optimization of a pair of high-affinity antibodies for specific protein biomarkers is time-consuming and costly31. Moreover, nonspecific binding of the detection antibody to other matrix components or to the assay surface gives rise to significant and variable background signals even in the absence of the antigen, limiting the sensitivity and dynamic range of conventional ELISA (Figure 2B, top panel).
Consequently, conventional ELISA lacks the sensitivity to detect the sub-picomolar concentrations of many protein biomarkers in human serum in the early stages of disease32. The development of immuno-PCR assays and digital ELISA or single molecule array (Simoa) has enabled the detection of several proteins with LODs in the femtomolar-to-attomolar range; however, these methods require complex workup procedures such as stringent washing steps and enzymatic amplification, and still require two compatible high-affinity antibodies per target33,34.
Recently, wash-free protein quantification methods have been reported such as nanoswitch-linked immunosorbent assay (NLISA)35, linker-mediated immunoassay (LMI)36, and programmable nucleic acid nanoswitches37, but these techniques lack the sensitivity of digital ELISA38. In contrast, protein SiMREPS enables a one-step, no-wash approach that uses direct kinetic fingerprinting to distinguish specific signal from nonspecific binding to single molecules, and achieves LODs in the femtomolar-to-attomolar concentration range using low-affinity detection probes3. Protein SiMREPS achieves a linear dynamic range of about 3.5 orders of magnitude when employing super-resolution analysis3, which is larger than that of conventional ELISAs (analog) (Figure 2B, bottom panel), and comparable to that of digital ELISA (Simoa)38. This wide dynamic range is advantageous given the broad concentration range (attomolar to picomolar) exhibited by protein biomarkers in biofluids32.
In this Account, we first introduce the working principles of SiMREPS, as well as the most important parameters in obtaining kinetic fingerprints useful for the high-specificity detection of analytes of interest. We then discuss the generality of SiMREPS for the detection and quantification of diverse analytes including nucleic acids, proteins, and small molecules. Next, we provide an overview of standard SiMREPS data analysis, as well as recently developed data processing methods that exploit super-resolution localization and deep learning4. Lastly, we suggest possible future advances of SiMREPS and its application to ever-broader scientific and clinical questions.
2. PRINCIPLES AND METHODS OF SINGLE-MOLECULE KINETIC FINGERPRINTING
2.1. SiMREPS principles and assay design
In 2006, Hochstrasser and colleagues introduced the pointillist super-resolution imaging technique PAINT (points accumulation for imaging in nanoscale topology), which relies on the repetitive interrogation of nanoscale structures by transiently binding dye molecules39. Subsequently, Jungmann et al.40 adapted this concept to transiently binding oligonucleotide fluorescent probes, giving rise to a family of methods known as DNA-PAINT41. Taking inspiration from these methods, SiMREPS employs the transient binding of FPs not for the imaging of nanoscale features, but to generate distinctive temporal patterns (kinetic fingerprints) for the high-confidence detection of single molecule analytes. Typically, SiMREPS employs TIRF microscopy to suppress background fluorescence from the freely diffusing FPs present in the imaging solution, thus permitting single-molecule detection at or near the surface of a slide or coverslip. The repeated binding of FPs to individual analyte molecules can be modeled as a Poisson process with random arrival times of individual FPs, but a well-defined mean number (μ) of binding and dissociation events (Nb+d) per target molecule for a given observation time, and a standard deviation (σ) proportional to (and theoretically equal to) √Nb+d13,42. As a result, the coefficient of variation (CV = σ/μ) decreases as Nb+d increases13, implying that any kinetic difference between specific and nonspecific binding, no matter how small, can be resolved with a sufficiently long observation period (Figure 1). At room temperature in 4×PBS (phosphate buffered saline) buffer and using oligonucleotide FPs 8–10 nucleotides (nt) in length, a 10-minute interrogation time is sufficient for discriminating even single nucleotide variants (SNVs) in RNA or DNA1,2.
In principle, any analyte of interest that can be stably bound to a surface and probed repeatedly is a candidate for SiMREPS. The basic requirements of a typical assay include a passivated solid substrate (typically glass or fused silica), a surface-immobilized capture probe (CP), and a FP (Figure 1). The surface is usually functionalized with m-PEG, biotin-PEG and streptavidin, both to provide passivation against excessive nonspecific binding and to immobilize biotinylated CPs. The CP may be, for example, a biotinylated DNA or locked nucleic acid (LNA) strand complementary to part of a target DNA or RNA sequence1,2, or a biotinylated antibody with strong affinity to a particular epitope of a target protein3. The FP may be a fluorescently labeled DNA strand of 8–10 nucleotides in length (in the case of nucleic acid analytes), or a fluorescently labeled detection antibody with a KD typically in the range of ~10–600 nM (for proteins). A well-chosen FP has both rapid binding and dissociation kinetics, thus quickly generating kinetic fingerprints with large values of Nb+d to achieve sufficient specificity in the shortest possible observation time.
Movies of FPs interacting with all immobilized targets within a microscopic field of view (FOV) in a defined observation time window (1–10 min, exposure time 0.1–1 s per frame) are recorded using TIRF and an Electron Multiplying CCD (EMCCD) or scientific complementary metal-oxide-semiconductor (sCMOS) camera, and then analyzed using custom MATLAB scripts (see Section 4 for more details). Fluorescent intensity-versus-time traces of single molecules are extracted and their kinetics are analyzed to distinguish targets from non-targets with high specificity1,2,12.
2.2. Assay chip preparation
In principle, the SiMREPS concept is compatible with any sample geometry that permits observation of single FP binding under relatively low-oxygen conditions. In practice, the sample chamber design varies depending on the type of microscope used (i.e., prism-type or objective-type TIRF) as well as the desired throughput and sensitivity13. Objective-type TIRF permits an open-top chip design and requires only a single substrate functionalized for sample immobilization (i.e., a glass coverslip); sample wells are constructed by cutting pipette tips or 3D printed wells and attaching them to passivated coverslips. In contrast, prism-type TIRF usually requires placing the sample cell between a prism and an objective lens; in this case, closed flow cells sandwiched between a passivated microscope slide (fused silica or glass) and a glass coverslip are preferred. The coverslips or slides are functionalized with an aminosilane followed by a mixture of succinimide esters of biotin-PEG and methoxy-PEG at a certain ratio (e.g., 1:10 or 1:100), and further passivated by disulfosuccinimidyl tartrate to quench the unreacted amine groups. Subsequently, the surface is coated with streptavidin to permit immobilization of biotinylated CPs. In the case of in situ analyte SiMREPS detection within cells (e.g., miRNAs14), objective-type TIRF is used together with glass-bottom cell culture dishes. Cellular fixation is performed using treatment with paraformaldehyde or 1–ethyl–3–(3–dimethylaminopropyl) carbodiimide (EDC). The fixed cells are ethanol permeabilized prior to imaging43.
2.3. Sample preparation and assay conditions
With no need for enzymatic amplification, SiMREPS assays have shown robust performance in a variety of buffers and minimally treated crude biofluids. Detailed sample preparation and assay protocols are described elsewhere12. Briefly, dsDNA samples require a short denaturation (e.g., heating to 80–95°C for 3 min) in the presence of carrier oligonucleotides (e.g., dT10) and cooling to room temperature before analysis2. For direct capture of miRNAs from serum or cell extract, samples can be pretreated with SDS and proteinase K1. Protein analytes have been directly captured from 1% or 25% serum, and can be detected without washing away excess serum or detection probe3. Notably, like other techniques utilizing passive surface capture, the sensitivity of SiMREPS is limited by analyte diffusion to the surface and by the capture kinetics (Table S2), typically yielding capture efficiencies of ~1%3. Nevertheless, limits of detection <10 fM are typical.
The imaging buffers for most SiMREPS assays contained 25–100 nM of FP in 1to 4×PBS buffer. To prolong the usable lifetime (i.e., reduce the photobleaching rate) of fluorophores for more accurate and reproducible kinetic fingerprinting, an oxygen scavenger system comprising 3,4-dihydroxybenzoate, protocatechuate dioxygenase, and Trolox is typically added. In protein-SiMREPS assays, Tween 20 is often added to the imaging buffer to reduce nonspecific binding of FPs to the imaging surface. To achieve the desired repetitive binding of FPs to targets yielding reproducible kinetic fingerprints distinct from background, it is important to control the imaging temperature (±2 °C) and the ionic strength.
3. APPLICATION OF SiMREPS TO THE QUANTIFICATION OF DIVERSE BIOMARKERS
3.1. SiMREPS detection of nucleic acids
Cell-free nucleic acids (cfNAs) such as ctDNA, mRNA, and non-coding RNAs (i.e., miRNAs) found in the biofluids of cancer patients have emerged as established or potential biomarkers9. Since changes in the levels of ctDNA reflect tumor burden and malignant progression, ctDNAs are increasingly employed as biomarkers in liquid biopsies of cancer. As an example, the Cobas EGFR Mutation Test v2 for EGFR alterations has been approved for use as a companion diagnostic for the selection of therapies in non-small cell lung cancer (NSCLC)44. Additionally, the expression levels of miRNAs are frequently dysregulated in tumor development, raising the possibility of using circulating miRNAs as biomarkers45. For example, miR-21 and miR-125b are deregulated in NSCLC45. Several potential cfNA cancer biomarkers have been discussed in recent reviews9,45.
Highly specific and sensitive detection of rare mutant DNA alleles in biofluids is challenging because the allelic frequency of ctDNA is often quite low, frequently <1% even in advanced (e.g., Stage IV) cancers46. Accurate detection of ctDNA therefore requires high specificity for the mutant allele. To this end, we recently demonstrated the ability of SiMREPS to detect two NSCLC-related EGFR mutations—an exon 19 deletion and the T790M (c.2369C>T) point mutation—with extremely high specificity in dsDNA without PCR amplification2. Each of the SiMREPS assays used a mutant (MUT)-specific 8-nt oligonucleotide FP to discriminate between specific binding to MUT molecules and nonspecific interactions with spurious or wild-type (WT) nucleic acid sequences (Figure 3). Detailed guidelines for designing SiMREPS FPs have been discussed1,2,12. Briefly, the maximum theoretical discrimination factor, , where ΔΔG0 is the difference in the Gibbs free energy of hybridization of an FP with MUT and of the same FP with WT DNA target, was calculated for various candidate FPs using the web software NUPACK48, and the FPs with the largest values of Qmax,therm were empirically tested for suitability (i.e., rapid kinetics) in SiMREPS assays.
Figure 3.
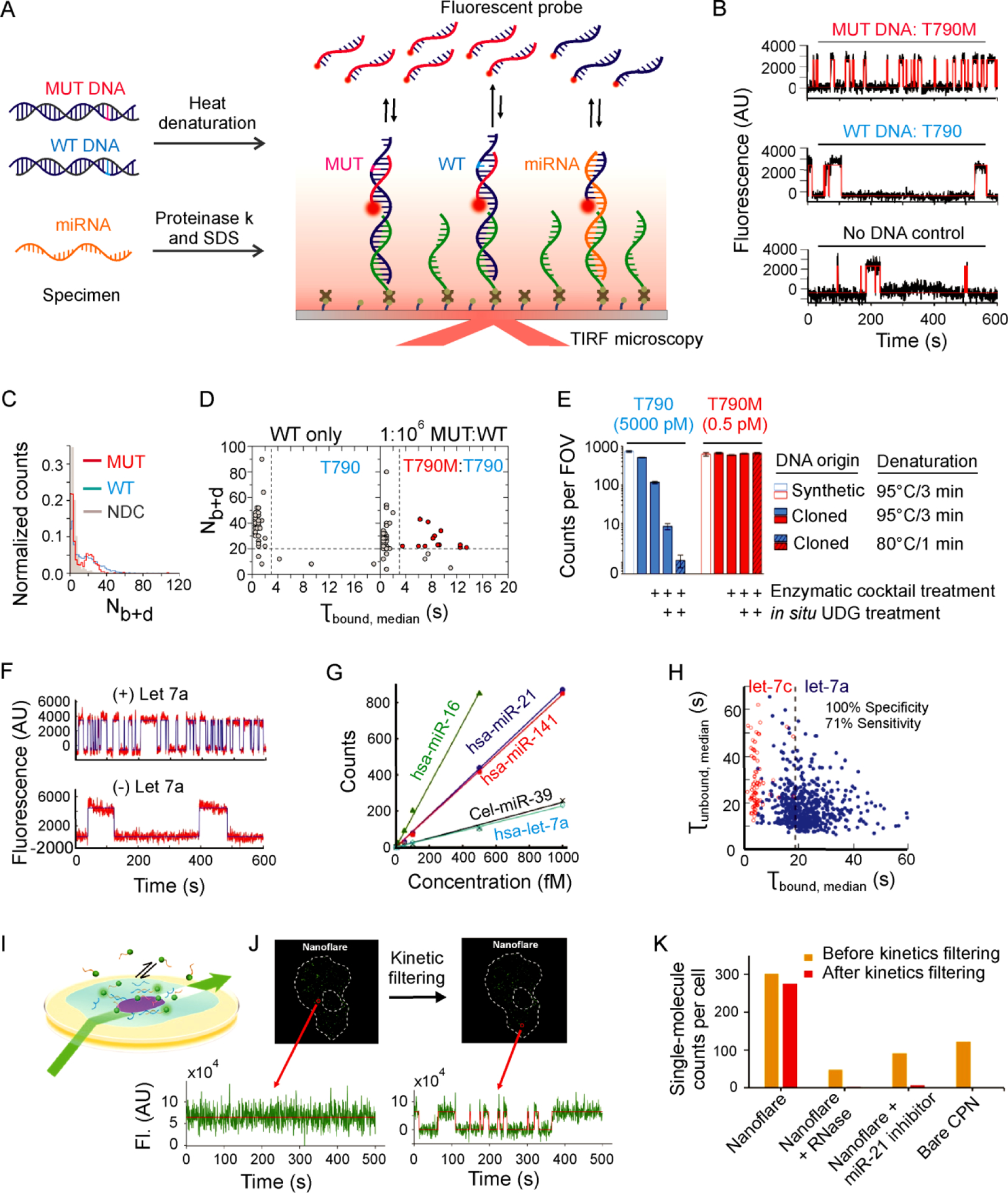
Highly specific and sensitive detection of nucleic acid biomarkers with single-molecule kinetic fingerprinting (SiMREPS). (A) Experimental scheme for SiMREPS assays of DNA and miRNA. (B) Representative single-molecule kinetic traces for MUT DNA (top), WT DNA (middle), and a no-DNA control (bottom) using an FP specific for the EGFR mutation T790M (c.2369C>T). (C) Histogram comparing the number of binding and dissociation events (Nb+d) observed per single-molecule trace for a no-DNA control (NDC), T790 (WT, 50 nM), and T790M (MUT, 50 fM). (D) Kinetic thresholding based primarily on Nb+d and τbound,median distinguishes between samples containing WT only and a 1:106 mixture of MUT and WT sequence. (E) Varying heat denaturation conditions and enzymatic treatments of T790 (WT, blue) and T790M (MUT, red) demonstrate the impact of spontaneous heat-induced cytosine deamination on specificity. B-E reproduced with permission from ref 2, Copyright 2018, American Chemical Society. (F) Representative single-molecule kinetic traces for in vitro detection of miRNA (hsa-let-7a). (G) Standard curves for in vitro detection of five different miRNAs. (H) Dwell time analysis enables high-confidence discrimination between let-7a and let-7c. F-H reproduced with permission from ref 1, Copyright 2015, Springer Nature. (I) Experimental scheme for HILO imaging of single cells using a miR-21-specific nanoflare SiMREPS probe. (J) Time traces illustrating the ability to distinguish single miR-21 molecules from background binding in a single A549 cell. (K) Apparent single-molecule counts from SiMREPS assays of miR-21 under different experimental conditions with and without kinetic filtering. I-K reproduced with permission from ref 14, Copyright 2019, American Chemical Society.
To permit the surface capture of single-stranded target molecules for detection of EGFR mutations by SiMREPS, the target dsDNA was subjected to gentle thermal denaturation (at 80 °C, to minimize spontaneous deamination of cytosine to uracil, observed to be suffered by PCR22) in the presence of a carrier oligonucleotide (dT10) at high molar excess to substantially reduce reannealing (Figure 3A).
The kinetic fingerprints generated by the transient binding of the optimized MUT-specific FP effectively distinguished among MUT, WT, and no-DNA controls with an acquisition time of 10 min (Figure 3B–C). Both the exon 19 deletion and the T790M mutation were detected at an allelic fraction as low as 0.0001% (1 MUT molecule in 1 million WT molecules). Notably, the assay for the point mutation T790M exhibited an apparent specificity of 99.99999% and apparent discrimination factor Qapp of 1.1×107, which is ~2,600 times greater than the maximum thermodynamic discrimination factor estimated from Gibbs free energy calculations by NUPACK (Figure 3D)2. This achievement attests to the power of kinetic fingerprinting, which – unlike single-measurement thermodynamic discrimination – reaches arbitrarily high specificity by repeated evaluation over an arbitrarily long-time window. Particularly high specificity for the C>T mutation T790M arose from minimizing the heat-induced deamination of cytosine to uracil that converts WT EGFR into a MUT-like sequence, as well as by the enzymatic removal of damaged DNA bases by treatment with UDG (uracil-DNA glycosylase) after denaturation and surface capture (Figure 3E).
For in vitro detection of miRNAs by SiMREPS, LNA-modified capture probes 9–11 nt in length were employed to capture miRNA targets from buffer or biofluids pretreated with proteinase K and SDS to protect against RNase activity (Figure 3A). A FP 9–10 nt in length generated distinct single-molecule kinetic fingerprints for specific binding to the target miRNA and nonspecific binding (Figure 3F). The generality of this approach was evaluated by detecting four human miRNAs (hsa-let-7a, hsa-miR-21, hsa-miR-16 and hsa-miR-141) and one miRNA from Caenorhabditis elegans (cel-miR-39) with a dynamic range spanning two to three orders of magnitude and a LOD of approximately 1 fM1 (Figure 3G). The ability to discriminate between single-nucleotide variants was demonstrated by comparing the kinetic fingerprints generated by the FP for let-7a in the presence of either let-7a or let-7c in buffer (Figure 3H); detection of let-7 family members was also demonstrated in cell extract1.
Finally, in situ detection of miRNA by SiMREPS within fixed, permeabilized eukaryotic cells was demonstrated by Li et al.14 using HILO microscopy (Figure 3I). Kinetic fingerprinting enabled strong discrimination between specific and nonspecific binding of a FP for miR-21 (Figure 3J), permitting the single-molecule counting of miRNAs in single cells in situ (Figure 3K). Compared to single-molecule fluorescence in situ hybridization (smFISH)43,49, which typically requires dozens of FPs binding to the same long RNA for achieving discrimination from spurious FP signal, SiMREPS provides a means of detecting smaller nucleic acids including miRNAs with high accuracy and low risk of photobleaching using a nanoflare. However, the higher background autofluorescence, potentially high intracellular concentrations of miRNAs, and potential masking by proteins or other binding partners may still pose challenges to the accurate quantification of miRNAs in cells by SiMREPS. The use of expansion microscopy50 as well as super-resolution data analysis2 might solve these problems.
3.2. SiMREPS detection of protein biomarkers
Proteins are involved in many biological processes and are useful biomarkers to differentiate between healthy and diseased states in clinical diagnostics51. Mutated or misfolded proteins are associated with multiple diseases, such as Alzheimer’s, and Parkinson’s diseases52. Uncontrolled protein expression leads to increased levels of specific proteins in blood that are associated, e.g., with different types of cancer53. Thus, the sensitive and accurate quantification of proteins in human biofluids could be critical for the early-stage diagnosis of disease.
For protein detection, the surface-immobilized antigen is allowed to interact transiently and repeatedly with a fluorescent detection probe to generate kinetic fingerprints characteristic of specific binding to the antigen (Figure 4A–D)3. In contrast to SiMREPS detection of nucleic acids, in which synthetic oligonucleotides can be readily designed for use as FPs, protein-SiMREPS employs a fluorescently labeled detection antibody (typically a monovalent Fab fragment with fast dissociation kinetics) as the FP. Thus, the successful development of a SiMREPS assay for proteins depends on the availability of an antibody with suitable kinetics.
Figure 4.
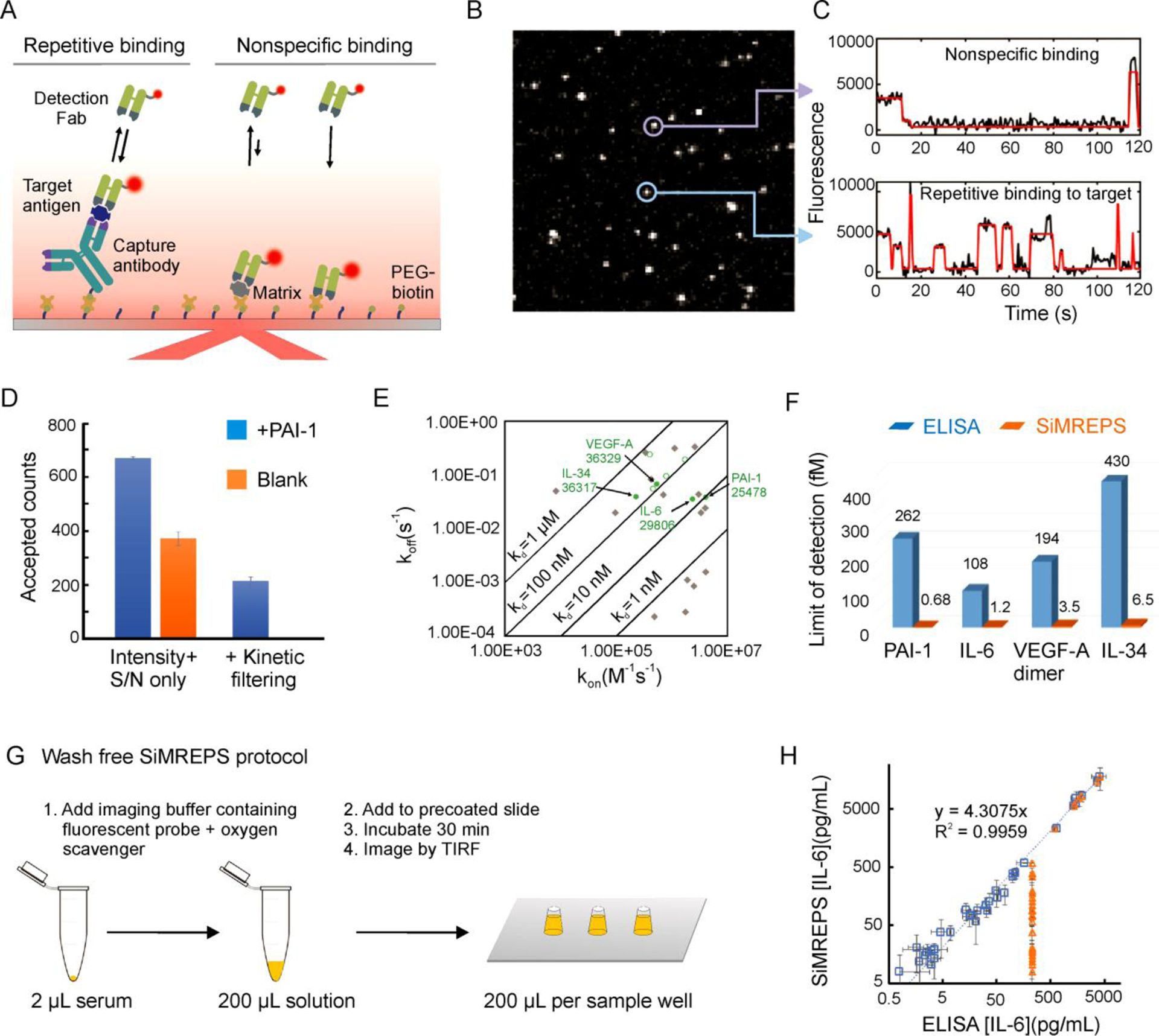
High-confidence detection and counting of single protein molecules by SiMREPS. (A) Experimental scheme for the detection of target protein antigens by SiMREPS. (B) Single movie frame of a representative microscope FOV; the bright puncta represents single FPs bound at or near the coverslip surface. (C) Representative intensity-versus-time traces showing the distinct kinetic fingerprints of non-specific binding (top) and repetitive binding to the target antigen (bottom). (D) Impact of kinetic filtering on the number of accepted counts in animal serum samples with and without the spiked-in antigen PAI-1. (E) Scatter plot of binding (kon) and dissociation (koff) rate constants (determined from BLI or SPR measurements) of candidate detection Fabs, with their success or failure as SiMREPS probes at room temperature indicated by color (not suitable: gray diamonds; suitable: green circles; suitable and chosen for final assays: filled green circles). (F) Bar graph showing the superior sensitivity of SiMREPS (orange bars) compared to ELISA (blue bars) for the same antigens. (G) Wash-free SiMREPS protocol for quantifying IL-6 in serum. (H) Correlation plot of endogenous IL-6 measurements by the wash-free protocol in 34 patient-derived serum samples by SiMREPS (100-fold dilution of all samples) and ELISA with variable dilution factors (4-fold dilution, closed blue squares; 64-fold dilution, open blue squares) or ELISA with 100-fold dilution of all samples (orange triangles).
Fortuitously, in vitro selection methods permit the selection of antibodies with sufficiently rapid dissociation kinetics for use as FPs in SiMREPS. Recombinant monovalent Fab antibodies against a target antigen can be isolated, e.g., from the HuCAL PLATINUM library, which comprises 45 billion fully human antibody clones that can be screened for antigen binding using phage display and its variants54. To facilitate the selection of Fab clones with suitably fast kinetics for SiMREPS, a modified strategy was developed to allow enrichment for clones with high off-rates3. ELISA hits from each panning showing binding to antigen were subjected to a high-throughput off-rate screening55 using Bio-layer Interferometry (BLI) and/or Surface Plasmon Resonance (SPR)55,56; clones with the highest off-rates were sequenced to identify unique Fabs suitable for SiMREPS.
Screening of different in vitro-selected Fabs against target antigens IL-6, PAI-1, VEGF-A and IL-34 by SiMREPS showed that the most useful probes exhibit rate constants of association (kon) in the range of 0.5–5 × 106 M−1 s−1 and rate constants of dissociation (koff) in the range of 0.05–0.5 s−1 in PBS at 25–30 °C, corresponding to KD values of 10–600 nM (Figure 4E)3, similar to the most useful rate constants for SiMREPS detection of nucleic acids15. Encouragingly, 50% of the Fabs that were in vitro-selected for high off-rates were found to be suitable as SiMREPS probes. Furthermore, it was found that the kinetics of FP interaction with the antigen could easily be manipulated in SiMREPS measurements by modifying assay temperature and/or salt concentration3.
Since SiMREPS can filter out the signal arising from nonspecific binding based on its kinetic profile (Figure 4C), it removes the background “floor” and achieves higher sensitivity than ELISA and other conventional techniques3. For the four antigens tested, SiMREPS achieved LODs in the low-femtomolar to attomolar range, or 55- to 383-fold lower than commercial ELISA kits for the same antigens (Figure 4F), suggesting that SiMREPS may facilitate the detection of trace protein biomarkers at the earliest stages of disease. Moreover, SiMREPS was shown to be amenable to a wash-free protocol (Figure 4G), meaning that no buffer exchanges are required after the addition of the antigen mixture to the sample well.
In addition to its simpler sample handling requirements, this wash-free SiMREPS assay was more sensitive than ELISA for the measurement of endogenous IL-6 in serum from CAR-T cell therapy patients (Figure 4H). Finally, since SiMREPS requires only one tightly binding antibody (the CP), and since weakly binding FPs are easily found by, e.g., in vitro selection, this approach may prove compatible with antigens for which no high-quality antibody pairs for sandwich ELISA are available.
3.3. SiMREPS detection of toxins and other small molecules
In addition to large biomolecules like proteins and nucleic acids, small molecules such as vitamins, hormones, metabolites, intracellular messengers, and cofactors also play an important role in assessing disease etiology and treatment efficacy57. For instance, the concentration of adenosine increases in the plasma of patients with Congestive Heart Failure (CHF)58, and circulating ATP has emerged as a biomarker of cognitive impairment in HIV59.
Aptamers are single-stranded RNA or DNA generated by in vitro selection or systematic evolution of ligands by exponential enrichment (SELEX)60, and provide a promising approach for the specific detection of small molecules. However, their sensitivity and specificity are often limited by high KD61 as well as the difficulty of completely suppressing signal in the absence of analyte.
Recently, Weng et al.,16 presented a possible strategy to overcome these challenges, demonstrating the ultrasensitive and specific detection of adenosine biomarkers by combining aptamers with SiMREPS (Figure 5A). In this approach, similar to Single Molecule Kinetic Analysis of RNA Transient Structure (SiM-KARTS)62, the specific binding of an adenosine target with a surface-immobilized hairpin-shaped aptamer induces a conformational change in the aptamer to expose a hairpin stem that transiently and repetitively interacts with FPs (Figure 5A). High-accuracy discrimination of the ligand-bound and ligand-free states was achieved by monitoring the interactions for 15 min under TIRF microscopy, resulting in virtually zero-background measurements of the small-molecule analyte (Figure 5B & C). The LOD for adenosine spiked into chicken meat extract was 0.3 pM (Figure 5D), which is superior to recently reported aptasensors63. The aptamer-based SiMREPS approach also exhibited high specificity, showing little interference from other small-molecule ligands (Figure 5E).
Figure 5.
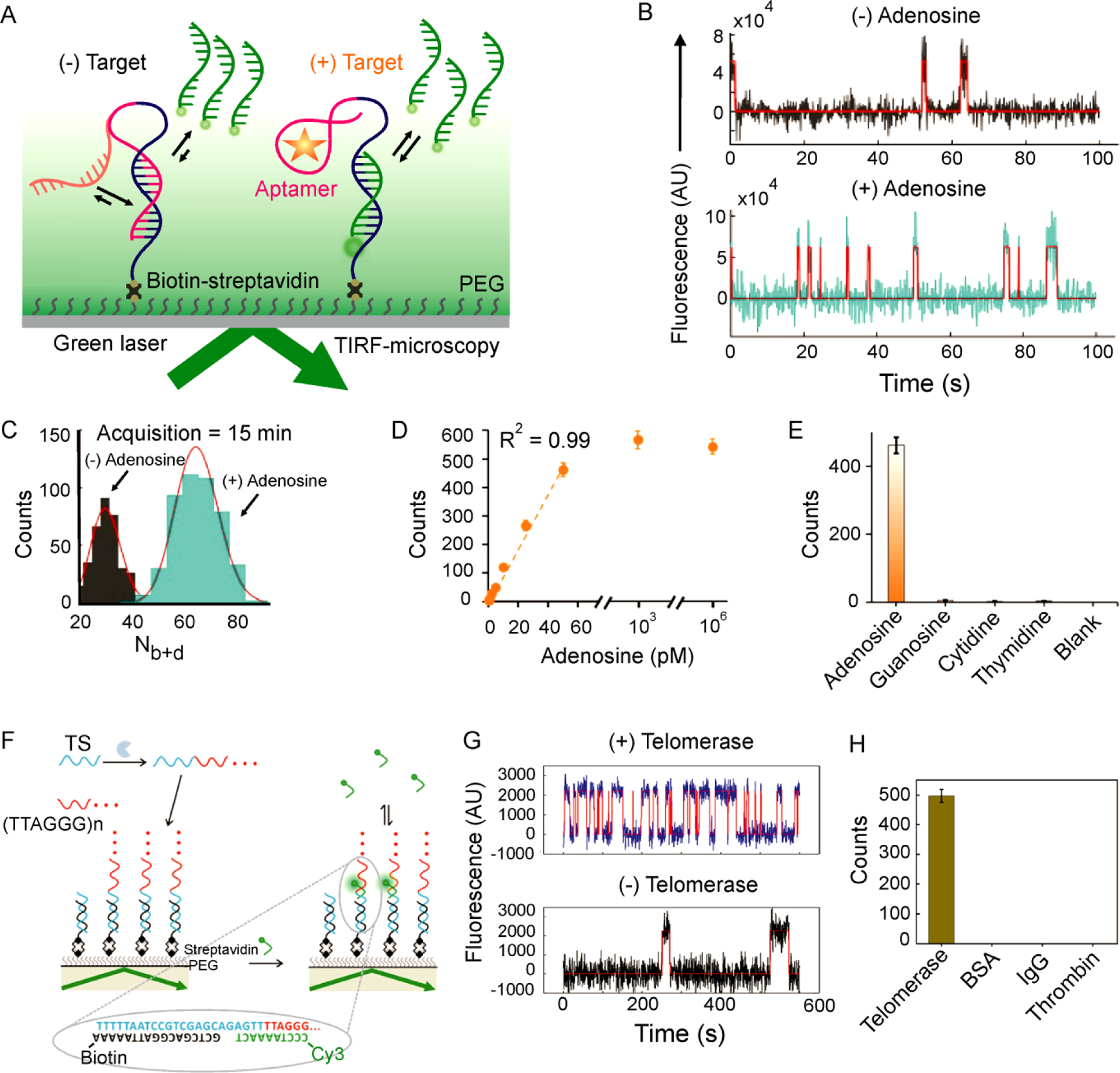
Detection of small molecules and monitoring enzyme activity using SiMREPS. (A) Experimental scheme showing the use of SiMREPS to probe the state of an aptamer for the high-sensitivity detection of small molecules by TIRF-microscopy. (B) Representative intensity-versus-time traces in the absence and presence of adenosine (50 pM). (C) Histograms of Nb+d in the absence (gray) or presence (cyan) of adenosine (50 pM). (D, E) Standard curve (D) and selectivity (E) of adenosine detection. A-E reproduced with permission from ref 16, Copyright 2019, American Chemical Society. (F) Experimental scheme for the detection of telomerase activity at the single-molecule level using SiMREPS. (G) Single-molecule kinetic traces in the presence (top) and absence (bottom) of telomerase activity. (H) The single-molecule assay showed a response in the presence of telomerase but not for other proteins. F-H reproduced with permission from ref 65, Copyright 2017, American Chemical Society.
The aptamer based SiMREPS (Figure 5A) was further tested by detecting two additional small molecule toxins such as acetamiprid and PCB-7716. The LODs for acetamiprid and PCB-77 were determined to be 0.35 pM and 0.72 pM, respectively16, approximately 3- and 70-times lower than recently reported biosensors63,64. SiMREPS thus significantly improves the performance of aptamers in the detection of diverse small-molecule analytes. However, the generality of the SiMREPS approach is limited by the availability of suitable aptamers (see Table S2).
3.4. Monitoring of enzyme activity with SiMREPS
Given its sensitivity to small chemical differences in single molecules, SiMREPS provides an interesting means to monitor the activity of enzymes. For example, Su et al.65 employed SiMREPS to monitor the activity of telomerase (Figure 5F), an enzyme that plays a critical role in maintaining chromosomal integrity and is over expressed in approximately 90% of all malignant tumors66. Telomerase activity was monitored in vitro by observing the dynamic binding of a short DNA FP with telomerase reaction products (the repeated sequence TTAGGG) (Figure 5F), yielding a distinct kinetic signature from background binding (Figure 5G). With this method, the activity of telomerase extracted from as few as 10 cancer cells was detected65; in contrast, no such signal was detected in the presence of proteins other than telomerase (Figure 5H).
4. SiMREPS DATA ANALYSIS AND PROCESSING
4.1. Idealization and kinetic analysis of single-molecule intensity traces
Standard SiMREPS analysis is performed by generating single-molecule intensity-versus time traces from TIRF microscopy videos, characterizing the kinetics of FP binding within each trace using hidden Markov modeling (HMM)67, and either rejecting or accepting each trace as evidence of the presence of a single analyte molecule by enforcing minimum and/or maximum value thresholds for several criteria3,12. The typical criteria used to distinguish traces containing specific binding to the analyte from those containing only nonspecific binding include12:
Signal intensity
Signal-to-noise ratio
Number of binding and dissociation events per trace (Nb+d)
Median lifetime in the bound (τbound,median) and unbound (τunbound,median) states
Maximum individual dwell time in the bound and unbound states
Thresholds for the above parameters are usually set empirically by comparing positive (e.g., in the presence of ~1 pM target) to negative (i.e., matrix-only) control experiments, and choosing thresholds that minimize false positives and maximize true positives.
While this standard approach has the advantages of simplicity and transparency, it has two main drawbacks: it is a diffraction-limited analysis method, making it challenging to apply to fields of view with very densely captured analytes (e.g., >1 molecule per 10-μm2 area); and its output is influenced by the quality of the HMM fitting. To address these limitations, we recently developed super-resolution2 and deep learning4-based analytic pipelines, respectively.
4.2. Super-resolution analysis
At high concentrations (e.g., > 1 pM), multiple analyte molecules may be captured within a single diffraction-limited68 region. Consequently, the emission of fluorescent probes binding to multiple distinct analyte molecules will overlap, making it difficult or impossible to analyze binding kinetics accurately and placing an upper limit on the dynamic range of SiMREPS measurements performed with standard diffraction-limited analysis2.
To overcome this challenge, we developed a super-resolution approach to the analysis of SiMREPS data2 inspired by microscopy methods40. However, unlike conventional super-resolution microscopy, our approach performs sub-pixel localization using the frame-to-frame changes in fluorescence intensity rather than raw intensity, permitting the analysis of fields of view with very dense probe binding (Figure 6). Hierarchical clustering is used to identify groups of probe binding and dissociation events to the same analyte molecule; these clusters are then subjected to kinetic thresholding analysis similar to that performed in HMM analysis2. This approach extends the dynamic range by approximately two orders of magnitude for both nucleic acid2 and protein3 SiMREPS measurements.
Figure 6.
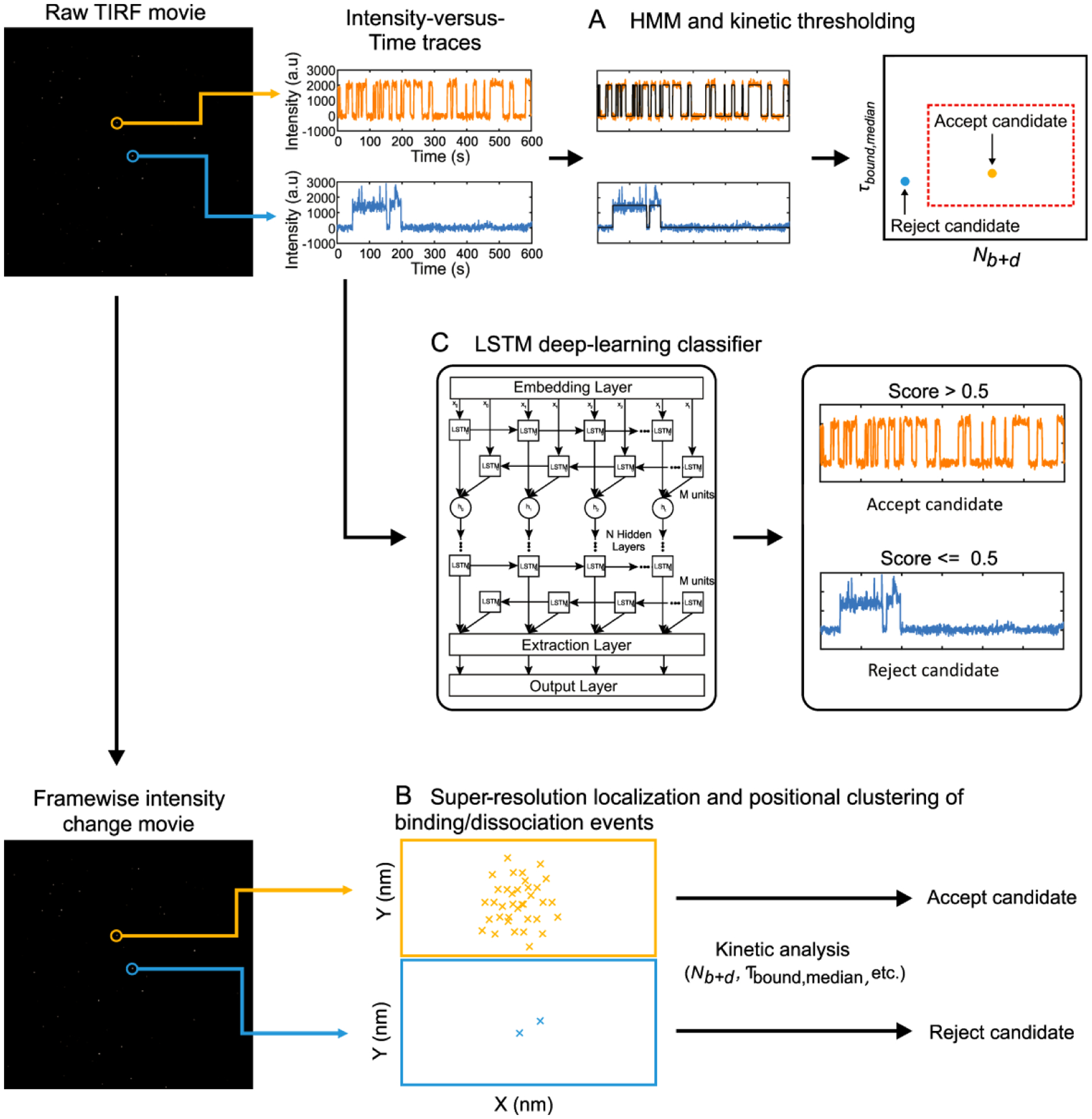
Approaches to SiMREPS data analysis. A TIRF microscopy movie is used to generate single-molecule time traces. These traces are then subjected to HMM and kinetic analysis (A) and accepted or rejected as kinetic fingerprints of analyte molecules. Alternatively, higher dynamic range can be achieved by performing a frame-by-frame subtraction to yield a framewise intensity change movie, which is then analyzed by super-resolution localization methods (B) to identify clusters of binding events indicative of the presence of analyte molecules. As a third alternative, the single-molecule traces are passed to an LSTM deep-learning classifier that was previously trained using control experiments (C) to score and reject or accept each trace.
4.3. Deep learning for fast, automated, and accurate analysis of SiMREPS data
HMM and super-resolution analysis rely on the fitting of models to naturally noisy raw data, which yield occasional false positives due to, for example, interpretation of baseline noise or photophysical blinking as binding transitions. As a result, strict thresholds for signal-to-noise and kinetics often must be employed to avoid these errors, which in turn results in rejection of some true positives and, hence, lower sensitivity. To overcome these shortcomings, we recently developed a deep learning-based method for SiMREPS data analysis (Figure 6)4.
Deep recurrent neural network (RNN) methods have been effectively used for learning sequential biological information69,70. Long short-term memory (LSTM) is a modification of the RNN architecture to learn long-range dependencies in sequential data71, making it suitable for kinetic analysis of SiMREPS traces. We therefore developed an LSTM-based deep learning approach for automated classification of SiMREPS traces, and found it to yield both higher sensitivity and specificity than HMM-based methods in measurements of the EGFR point mutation T790M4. It can be further adapted by transfer learning on new datasets4, suggesting an important future role for artificial intelligence approaches in further streamlining SiMREPS based molecular diagnostics.
5. CONCLUSIONS AND FUTURE DIRECTIONS
SiMREPS is a unique analytical approach that permits the amplification-free detection of single molecules with high sensitivity and specificity using kinetic fingerprinting with transiently binding probes. Due to its lack of analyte-specific chemistry or enzymatic steps, it provides a comprehensive platform for the detection of diverse molecular biomarkers including nucleic acids, proteins, and small molecules. Since its kinetic fingerprinting provides exquisite sensitivity to even minute chemical differences (e.g., single-nucleotide variations or mutations), SiMREPS may offer a future means of detecting epigenetic, epitranscriptomic, and post-translational modifications with single-site and single-molecule sensitivity. Through spatial patterning (e.g., in a microarray or through a water-in-oil droplet emulsion) and/or color encoding, a diverse panel of disease biomarkers could be detected in parallel on a single instrument platform. Similarly, combining in situ SiMREPS with expansion microscopy may have potential for single-cell multi-omics11.
Although standard diffraction-limited analysis methods for SiMREPS already provide very high specificity, the newly developed super-resolution and deep-learning approaches increase the dynamic range and sensitivity of this technique. Future developments, particularly utlilizing deep learning, may increase data analysis pipeline efficiency by operating directly on raw movies. Recent publications have shown the attention-based networks72 and 2D convolutional-based neural networks73 outperform traditional RNN/LSTM models for processing sequential data. Convolution-based approaches could use raw movie data as input, using less hardware resources, eliminating data preprocessing steps, and yielding potentially more accurate classification results.
While the sensitivity of SiMREPS already rivals or surpasses leading techniques for protein and small-molecule detection, its sensitivity for nucleic acids still falls somewhat short of PCR-based amplification approaches. This is not due to an intrinsic sensitivity limit (SiMREPS can detect single molecules), but because it is challenging to transport analytes to a surface and image them with ~100% efficiency. As a result, < 0.1% of the analyte molecules present in a sample are detected in a typical measurement. Methods to actively pre-concentrate analytes or actively transport them to the imaging surface, as well as optics that permit measurement over a wider field of view, may therefore improve LODs by a factor of 100 or more. Finally, the development of a dedicated, affordable instrument will render the technique accessible to a broad set of scientific and clinical laboratories.
Supplementary Material
ACKNOWLEDGMENT
The authors thank to aLight Sciences and Bio-Rad Laboratories, Inc. for financial support and providing antibodies, animal serum, and recombinant antigens. We thank Mingde Zhu for performing the SPR experiments for determining the kinetics of antibodies; Amanda Mazzoli and Ryan Lindstrom for clinical research coordination; and Jenny Barabas and Mary Olesnavich for technical assistance in specimen processing. We also wish to thank J.D. Hoff from the SMART Center for training, technical advice, and use of the objective-type TIRF microscope.
Funding Sources
Michigan Economic Development Corporation MTRAC for Life Sciences grant to N.G.W. and M.T. and A.J-B. The James Selleck Bower Permanently Endowed Innovative Promise Funds for Cancer Research, Cellular Cancer Biology Imaging Research (CCBIR) Program Pilot Grant from the University of Michigan, Ann Arbor (U-M) Rogel Cancer Center, Pilot grants from the U-M MCubed 2.0 program, and a Fast Forward GI Innovation Fund to N.G.W. and M.T. National Institute of Health (NIH) grants R21 CA204560 and R33 CA229023 to N.G.W and M.T. K.M. was supported by NIH T-32-GM132046 and a U-M Rackham Merit Fellowship. We also acknowledge NSF MRI-ID grant DBI-0959823 (to N.G.W.) for seeding the Single Molecule Analysis in Real-Time (SMART) Center, whose equipment was used for some of the studies with support from J.D. Hoff.
The authors declare the following competing financial interests: The University of Michigan and Bio-Rad Laboratories, Inc. have filed patent applications on technologies described herein, on which A.J.-B., M.T. and N.G.W. are listed as inventors. A.J.-B., M.T. and N.G.W. are co-founders of aLight Sciences, which sponsored part of this work and seeks to commercialize the SiMREPS technology.
Biographies
Shankar Mandal received his B.S. in Chemistry and M.S. in Organic Chemistry from University of Dhaka, Bangladesh. He earned his Ph.D. in Chemistry from Kent State University, USA. Currently, he is a postdoctoral research fellow in the Walter laboratory at the U-M. His research interests focus on development of biosensors for analyzing cancer-specific biomarkers using SiMREPS.
Zi Li received her B.S. in Material Chemistry Ocean from University of China. She earned her Ph.D. in Chemistry from Kansas State University, USA. Currently, she is a postdoctoral research fellow in the Walter laboratory at the U-M, working on developing ultrasensitive assays for analyzing biomarkers in biofluids using single molecule fluorescence microscopy techniques.
Tanmay Chatterjee received his B.S. and M.S. degrees from University of Burdwan, India. He earned his Ph.D. from the Indian Institute of Science Education and Research, India. He is currently a postdoctoral research fellow in the Walter laboratory at the U-M. His current research focus is on ultra-sensitive protein detection by SiMREPS.
Kunal Khanna received his B.S. at Hunter college and worked as a postbac at SUNY Downstate. He is currently a Ph.D. student in the Walter laboratory at the U-M. His current research focuses on developing biosensors for ultrafast detection of nucleic acids biomarkers using SiMREPS.
Karen Montoya received her B.S. from the University of California and her M.S. in Chemistry from the U-M, Ann Arbor. Currently she is a Ph.D. researcher in the Walter laboratory at the U-M. Her research interests focus on enhancing our understanding of individual cells to detect molecular profiles associated with disease utilizing microscopy techniques.
Liuhan Dai received his B.S. in Chemical Biology from Nankai University, China. He is currently a Ph.D. student in the Walter laboratory at the U-M. His research interests focus on detection of methylated DNA biomarkers using SiMREPS.
Chandler Petersen received his B.S. in computer science from Oregon State University, USA. He spent a year at the U-M in the Walter laboratory. He is currently a Ph.D. student in the Paul G. Allen Computer Science and Engineering at the University of Washington. His research interests include molecular programming, natural computing, and unconventional computation.
Lidan Li received her B.S. in Pharmaceutical Engineering from Beijing University of Chemical Technology (BUCT), China. She worked as a visiting scholar in the Walter laboratory at the U-M. Currently, she is a Ph.D. student in Chemical Engineering and Technology at BUCT. Her research interests focus on enzyme operated DNA nanotechnology in detection and imaging methods.
Muneesh Tewari completed his undergraduate studies in Biochemistry at Case Western Reserve University, and his MD/PhD degrees at the U-M in 1997. During his PhD he discovered the apoptosis effector known as Caspase-3. After Internal Medicine residency at U-M, he obtained medical oncology training at the Dana-Farber Cancer Institute and postdoctoral training in systems biology at Harvard Medical School. In 2005, he began his independent career at the Fred Hutchinson Cancer Research Center in Seattle, where his lab discovered extracellular microRNAs as a new class of biomarkers. In 2014, he moved back to U-M to continue his research at the intersection of systems biology, disease biomarkers, and new technology.
Alexander Johnson-Buck received his B.A. from Northern Michigan University. He earned his Ph.D. in Chemistry from the U-M, using single-molecule fluorescence microscopy to characterize DNA nanomachines and co-inventing SiMREPS. He then performed postdoctoral research at the Dana-Farber Cancer Institute, where he developed the first synthetic single-molecule clocks. He subsequently returned to the U-M as a Research Assistant Professor, where he worked on biomarker assay technology development. He is currently the Chief Scientific Officer and a co-founder of aLight Sciences.
Nils G. Walter received his “Vordiplom” (B.S.) and “Diploma” (Masters) from the Technical University of Darmstadt performing research on a protein dehyrogenase enzyme. He earned his Dr. Ing. while studying molecular evolution using fluorescence techniques at the Max-Planck-Institute for Biophysical Chemistry, Göttingen. For his postdoctoral studies, he turned to RNA enzymes at the University of Vermont. He is currently the Francis S. Collins Collegiate Professor of Chemistry, Biophysics, and Biological Chemistry at the U-M, focusing on RNA through the lens of single molecule fluorescence. He founded the Single Molecule Analysis in Real-Time (SMART) Center, co-founded and co-directs the Center for RNA Biomedicine, and directs the Microscopy Core of BRCF at the U-M.
REFERENCES
- (1).Johnson-Buck A; Su X; Giraldez MD; Zhao MP; Tewari M; Walter NG: Kinetic Fingerprinting to Identify and Count Single Nucleic Acids. Nat. Biotechnol 2015, 33, 730–732. [DOI] [PMC free article] [PubMed] [Google Scholar]; The kinetic fingerprinting approach, named single-molecule recognition through equilibrium Poisson sampling (SiMREPS) for the amplification-free, high-confidence, single-molecule detection of synthetic and endogenous miRNAs in both buffer and biofluids.
- (2).Hayward SL; Lund PE; Kang Q; Johnson-Buck A; Tewari M; Walter NG: Ultraspecific and Amplification-Free Quantification of Mutant DNA by Single-Molecule Kinetic Fingerprinting. J. Am. Chem. Soc 2018, 140, 11755–11762. [DOI] [PMC free article] [PubMed] [Google Scholar]; The single-molecule kinetic fingerprinting approach detects cancer mutation EGFR T790M (a single C→ T substitution) and EGFR exon19 deletion mutation with a specificity of >99.99999%, surpassing even the leading PCR-based methods and enabling detection of 1 mutant molecule in a background of at least 1 million wild-type molecules.
- (3).Chatterjee T; Knappik A; Sandford E; Tewari M; Strong WB; Thrush EP; Oh KJ; Liu N; Walter NG; Johnson-Buck A: Direct Kinetic Fingerprinting and Digital Counting of Single Protein Molecules. Proc. Natl. Acad. Sci. U.S.A 2020, 117, 22815–22822. [DOI] [PMC free article] [PubMed] [Google Scholar]; Direct, digital counting of clinically relevant single protein molecules with femtomolar-to-attomolar sensitivity. The method utilizes surface captured high-affinity antibody combined with kinetic fingerprinting using a dynamically binding, low-affinity fluorescent antibody fragment that differentiates between specific and nonspecific binding with high confidence at the single-molecule level.
- (4).Li J; Zhang L; Johnson-Buck A; Walter NG: Automatic Classification and Segmentation of Single-Molecule Fluorescence Time Traces with Deep Learning. Nat. Commun 2020, 11, 5833. [DOI] [PMC free article] [PubMed] [Google Scholar]; Deep learning to develop a rapid, automatic SMFM trace selection that improves the sensitivity and specificity of single-molecule kinetic fingerprinting assays.
- (5).Wulfkuhle JD; Liotta LA; Petricoin EF: Proteomic Applications for the Early Detection of Cancer. Nat. Rev. Cancer 2003, 3, 267–275. [DOI] [PubMed] [Google Scholar]
- (6).Lewis JD: The Utility of Biomarkers in the Diagnosis and Therapy of Inflammatory Bowel Disease. Gastroenterology 2011, 140, 1817–U160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Cho WCS; Yip TTC; Yip C; Yip V; Thulasiraman V; Ngan RKC; Yip TT; Lau WH; An JSK; Law SCK; Cheng WW; Ma VWS; Lim CKP: Identification of Serum Amyloid a Protein as a Potentially Useful Biomarker to Monitor Relapse of Nasopharyngeal Cancer by Serum Proteomic Profiling. Clin. Cancer Res 2004, 10, 43–52. [DOI] [PubMed] [Google Scholar]
- (8).Thaxton CS; Elghanian R; Thomas AD; Stoeva SI; Lee JS; Smith ND; Schaeffer AJ; Klocker H; Horninger W; Bartsch G; Mirkin CA: Nanoparticle-Based Bio-Barcode Assay Redefines “Undetectable” PSA and Biochemical Recurrence After Radical Prostatectomy. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 18437–18442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Schwarzenbach H; Hoon DSB; Pantel K: Cell-Free Nucleic Acids as Biomarkers in Cancer Patients. Nat. Rev. Cancer 2011, 11, 426–437. [DOI] [PubMed] [Google Scholar]
- (10).Wang X; Walt DR: Simultaneous detection of small molecules, proteins and microRNAs using single molecule arrays. Chem. Sci 2020, 11, 7896–7903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hu Y; An Q; Sheu K; Trejo B; Fan S; Guo Y: Single Cell Multi-Omics Technology: Methodology and Application. Front. Cell Dev. Biol 2018, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Johnson-Buck A; Li JM; Tewari M; Walter NG: A Guide to Nucleic Acid Detection by Single-Molecule Kinetic Fingerprinting. Methods 2019, 153, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chatterjee T; Li Z; Khanna K; Montoya K; Tewari M; Walter NG; Johnson-Buck A: Ultraspecific Analyte Detection by Direct Kinetic Fingerprinting of Single Molecules. Trac, Trends Anal. Chem 2020, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Li LN; Yu YJ; Wang CS; Han QQ; Su X: Transient Hybridization Directed Nanoflare for Single-Molecule miRNA Imaging. Anal. Chem 2019, 91, 11122–11128. [DOI] [PubMed] [Google Scholar]
- (15).Su X; Li LD; Wang SS; Hao DD; Wang L; Yu CY: Single-Molecule Counting of Point Mutations by Transient DNA Binding. Sci. Rep 2017, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Weng R; Lou ST; Li LD; Zhang Y; Qiu J; Su X; Qian YZ; Walter NG: Single-Molecule Kinetic Fingerprinting for the Ultrasensitive Detection of Small Molecules with Aptasensors. Anal. Chem 2019, 91, 1424–1431. [DOI] [PubMed] [Google Scholar]
- (17).Akkilic N; Geschwindner S; Höök F: Single-molecule biosensors: Recent advances and applications. Biosen. Bioelectron 2020, 151, 111944. [DOI] [PubMed] [Google Scholar]
- (18).Ma F; Li Y; Tang B; Zhang C. y.: Fluorescent Biosensors Based on Single-Molecule Counting. Acc.Chem. Res 2016, 49, 1722–1730. [DOI] [PubMed] [Google Scholar]
- (19).Postel M; Roosen A; Laurent-Puig P; Taly V; Wang-Renault SF: Droplet-based digital PCR and next generation sequencing for monitoring circulating tumor DNA: a cancer diagnostic perspective. Expert Rev Mol Diagn 2018, 18, 7–17. [DOI] [PubMed] [Google Scholar]
- (20).Cohen L; Walt DR: Single-Molecule Arrays for Protein and Nucleic Acid Analysis. Annu Rev Anal Chem 2017, 10, 345–363. [DOI] [PubMed] [Google Scholar]
- (21).Milbury CA; Zhong Q; Lin J; Williams M; Olson J; Link DR; Hutchison B: Determining lower limits of detection of digital PCR assays for cancer-related gene mutations. Biomol Detect Quantif 2014, 1, 8–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chen GL; Mosier S; Gocke CD; Lin MT; Eshleman JR: Cytosine Deamination Is a Major Cause of Baseline Noise in Next-Generation Sequencing. Mol. Diagn. Ther 2014, 18, 587–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Benes V; Castoldi M: Expression Profiling of MicroRNA Using Real-Time Quantitative PCR, How to Use It and What Is Available. Methods 2010, 50, 244–249. [DOI] [PubMed] [Google Scholar]
- (24).Schrader C; Schielke A; Ellerbroek L; Johne R: PCR Inhibitors - Occurrence, Properties and Removal. J. Appl. Microbiol 2012, 113, 1014–1026. [DOI] [PubMed] [Google Scholar]
- (25).Chen M; Zhao H: Next-generation sequencing in liquid biopsy: cancer screening and early detection. Human Genomics 2019, 13, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Li BT; Janku F; Jung B; Hou C; Madwani K; Alden R; Razavi P; Reis-Filho JS; Shen R; Isbell JM; Blocker AW; Eattock N; Gnerre S; Satya RV; Xu H; Zhao C; Hall MP; Hu Y; Sehnert AJ; Brown D; Ladanyi M; Rudin CM; Hunkapiller N; Feeney N; Mills GB; Paweletz CP; Janne PA; Solit DB; Riely GJ; Aravanis A; Oxnard GR: Ultra-deep next-generation sequencing of plasma cell-free DNA in patients with advanced lung cancers: results from the Actionable Genome Consortium. Ann Oncol 2019, 30, 597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).KA. W: DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program, GSP 2020.
- (28).Engvall E; Perlmann P: Enzyme-Linked Immunosorbent Assay (ELISA) Quantitative Assay of Immunoglobulin-G. Immunochemistry 1971, 8, 871–874. [DOI] [PubMed] [Google Scholar]
- (29).Tosswill JHC; Ridley DS: An Evaluation of the ELISA for Schistosomiasis in a Hospital Population. Trans. R. Soc. Trop. Med. Hyg 1986, 80, 435–438. [DOI] [PubMed] [Google Scholar]
- (30).Gosling JP: A Decade of Development in Immunoassay Methodology. Clin. Chem 1990, 36, 1408–1427. [PubMed] [Google Scholar]
- (31).del Campo M; Jongbloed W; Twaalfhoven HAM; Veerhuis R; Blankenstein MA; Teunissen CE: Facilitating the Validation of Novel Protein Biomarkers for Dementia: an Optimal Workflow for the Development of Sandwich Immunoassays. Front. Neuro 2015, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Geyer PE; Holdt LM; Teupser D; Mann M: Revisiting Biomarker Discovery by Plasma Proteomics. Mol. Syst. Biol 2017, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Rissin DM; Kan CW; Campbell TG; Howes SC; Fournier DR; Song L; Piech T; Patel PP; Chang L; Rivnak AJ; Ferrell EP; Randall JD; Provuncher GK; Walt DR; Duffy DC: Single-Molecule Enzyme-Linked Immunosorbent Assay Detects Serum Proteins at Subfemtomolar Concentrations. Nat. Biotechnol 2010, 28, 595–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Perez-Ruiz E; Decrop D; Ven K; Tripodi L; Leirs K; Rosseels J; de Wouwer MV; Geukens N; De Vos A; Vanmechelen E; Winderickx J; Lammertyn J; Spasic D: Digital ELISA for the Quantification of Attomolar Concentrations of Alzheimer’s Disease Biomarker Protein Tau in Biological Samples. Anal. Chim. Acta 2018, 1015, 74–81. [DOI] [PubMed] [Google Scholar]
- (35).Hansen CH; Yang D; Koussa MA; Wong WP: Nanoswitch-linked immunosorbent assay (NLISA) for fast, sensitive, and specific protein detection. Proc Natl Acad Sci U S A 2017, 114, 10367–10372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Smith LD; Willard MC; Smith JP; Cunningham BT: Development of a Linker-Mediated Immunoassay Using Chemically Transitioned Nanosensors. Anal. Chem 2020, 92, 3627–3635. [DOI] [PubMed] [Google Scholar]
- (37).Porchetta A; Ippodrino R; Marini B; Caruso A; Caccuri F; Ricci F: Programmable Nucleic Acid Nanoswitches for the Rapid, Single-Step Detection of Antibodies in Bodily Fluids. J.Am. Chem. Soc 2018, 140, 947–953. [DOI] [PubMed] [Google Scholar]
- (38).Rissin DM; Fournier DR; Piech T; Kan CW; Campbell TG; Song L; Chang L; Rivnak AJ; Patel PP; Provuncher GK; Ferrell EP; Howes SC; Pink BA; Minnehan KA; Wilson DH; Duffy DC: Simultaneous detection of single molecules and singulated ensembles of molecules enables immunoassays with broad dynamic range. Anal Chem 2011, 83, 2279–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Sharonov A; Hochstrasser RM: Wide-field Subdiffraction Imaging by Accumulated Binding of Diffusing Probes. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 18911–18916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Jungmann R; Steinhauer C; Scheible M; Kuzyk A; Tinnefeld P; Simmel FC: Single-Molecule Kinetics and Super-Resolution Microscopy by Fluorescence Imaging of Transient Binding on DNA Origami. Nano Lett 2010, 10, 4756–4761. [DOI] [PubMed] [Google Scholar]
- (41).Auer A; Strauss MT; Schlichthaerle T; Jungmann R: Fast, Background-Free DNA-PAINT Imaging Using FRET-Based Probes. Nano Lett 2017, 17, 6428–6434. [DOI] [PubMed] [Google Scholar]
- (42).Floyd DL; Harrison SC; van Oijen AM: Analysis of Kinetic Intermediates in Single-Particle Dwell-Time Distributions. Biophys. J 2010, 99, 360–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Pena JTG; Sohn-Lee C; Rouhanifard SH; Ludwig J; Hafner M; Mihailovic A; Lim C; Holoch D; Berninger P; Zavolan M; Tuschl T: miRNA in Situ Hybridization in Formaldehyde and EDC-fixed Tissues. Nat. Methods 2009, 6, 139–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Gazdar AF: Activating and Resistance Mutations of EGFR in Non-Small-Cell Lung Cancer: Role in Clinical Response to EGFR Tyrosine Kinase Inhibitors. Oncogene 2009, 28, S24–S31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Esteller M: Non-Coding RNAs in Human Disease. Nat. Rev. Genet 2011, 12, 861–874. [DOI] [PubMed] [Google Scholar]
- (46).Taniguchi K; Uchida J; Nishino K; Kumagai T; Okuyama T; Okami J; Higashiyama M; Kodama K; Imamura F; Kato K: Quantitative Detection of EGFR Mutations in Circulating Tumor DNA Derived from Lung Adenocarcinomas. Clin. Cancer Res 2011, 17, 7808–7815. [DOI] [PubMed] [Google Scholar]
- (47).Zhang DY; Chen SX; Yin P: Optimizing the Specificity of Nucleic Acid Hybridization. Nat. Chem 2012, 4, 208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Caltech: NUPACK Analysis. 2007.
- (49).Lyubimova A; Itzkovitz S; Junker JP; Fan ZP; Wu XB; van Oudenaarden A: Single-Molecule mRNA Detection and Counting in Mammalian Tissue. Nat. Protoc 2013, 8, 1743–1758. [DOI] [PubMed] [Google Scholar]
- (50).Asano SM; Gao R; Wassie AT; Tillberg PW; Chen F; Boyden ES: Expansion Microscopy: Protocols for Imaging Proteins and RNA in Cells and Tissues. Curr Protoc Cell Biol 2018, 80, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Jain Sachin GV, Naseem Sania: Acute-Phase Proteins: As Diagnostic Tool. J. Pharm. BioAllied Sci 2011, 3, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Selkoe DJ: Cell Biology of Protein Misfolding: The Examples of Alzheimer’s and Parkinson’s Diseases. Nat. Cell Biol 2004, 6, 1054–1061. [DOI] [PubMed] [Google Scholar]
- (53).Cohen JD; Li L; Wang YX; Thoburn C; Afsari B; Danilova L; Douville C; Javed AA; Wong F; Mattox A; Hruban RH; Wolfgang CL; Goggins MG; Dal Molin M; Wang TL; Roden R; Klein AP; Ptak J; Dobbyn L; Schaefer J; Silliman N; Popoli M; Vogelstein JT; Browne JD; Schoen RE; Brand RE; Tie J; Gibbs P; Wong HL; Mansfield AS; Jen J; Hanash SM; Falconi M; Allen PJ; Zhou S; Bettegowda C; Diaz LA; Tomasetti C; Kinzler KW; Vogelstein B; Lennon AM; Papadopoulos N: Detection and Localization of Surgically Resectable Cancers with a Multi-Analyte Blood Test. Science 2018, 359, 926–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Prassler J; Thiel S; Pracht C; Polzer A; Peters S; Bauer M; Nörenberg S; Stark Y; Kölln J; Popp A; Urlinger S; Enzelberger M: HuCAL PLATINUM, a Synthetic Fab Library Optimized for Sequence Diversity and Superior Performance in Mammalian Expression Systems. J. Mol. Biol 2011, 413, 261–278. [DOI] [PubMed] [Google Scholar]
- (55).Ylera F; Harth S; Waldherr D; Frisch C; Knappik A: Off-Rate Screening for Selection of High-Affinity Anti-Drug Antibodies. Anal. Biochem 2013, 441, 208–213. [DOI] [PubMed] [Google Scholar]
- (56).Bio-Rad Laboratories, I.: ProteOn Sensor Chips User Menu.
- (57).Wishart DS: Chapter 3: Small Molecules and Disease. Plos Computational Biology 2012, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Funaya H; Kitakaze M; Node K; Minamino T; Komamura K; Hori M: Plasma Adenosine Levels Increase in Patients with Chronic Heart Failure. Circulation 1997, 95, 1363–1365. [DOI] [PubMed] [Google Scholar]
- (59).Velasquez S; Prevedel L; Valdebenito S; Gorska AM; Golovko M; Khan N; Geiger J; Eugenin EA: Circulating Levels of ATP is a Biomarker of HIV Cognitive Impairment. Ebiomedicine 2020, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Tuerk C; Gold L: Systematic Evolution of Ligands by Exponential Enrichment - RNA Ligands to Bacteriophage-T4 DNA-Polymerase. Science 1990, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- (61).Chang AL; McKeague M; Liang JC; Smolke CD: Kinetic and Equilibrium Binding Characterization of Aptamers to Small Molecules using a Label-Free, Sensitive, and Scalable Platform. Anal. Chem 2014, 86, 3273–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Rinaldi AJ; Lund PE; Blanco MR; Walter NG: The Shine-Dalgarno Sequence of Riboswitch-Regulated Single mRNAs Shows Ligand-Dependent Accessibility bursts. Nat. Commun 2016, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Madianos L; Tsekenis G; Skotadis E; Patsiouras L; Tsoukalas D: A Highly Sensitive Impedimetric Aptasensor for the Selective Detection of Acetamiprid and Atrazine Based on Microwires Formed by Platinum Nanoparticles. Biosens. Bioelectron 2018, 101, 268–274. [DOI] [PubMed] [Google Scholar]
- (64).Cheng RJ; Liu SY; Shi HJ; Zhao GH: A Highly Sensitive and Selective Aptamer-based Colorimetric Sensor for the Rapid Detection of PCB 77. J. Hazard. Mater 2018, 341, 373–380. [DOI] [PubMed] [Google Scholar]
- (65).Su X; Li ZH; Yan XZ; Wang L; Zhou X; Wei L; Xiao LH; Yu CY: Telomerase Activity Detection with Amplification-Free Single Molecule Stochastic Binding Assay. Anal. Chem 2017, 89, 3576–3582. [DOI] [PubMed] [Google Scholar]
- (66).Kim NW; Piatyszek MA; Prowse KR; Harley CB; West MD; Ho PLC; Coviello GM; Wright WE; Weinrich SL; Shay JW: Specific Association of Human Telomerase Activity with Immortal Cells and Cancer. Science 1994, 266, 2011–2015. [DOI] [PubMed] [Google Scholar]
- (67).Bronson JE; Fei JY; Hofman JM; Gonzalez RL; Wiggins CH: Learning Rates and States from Biophysical Time Series: A Bayesian Approach to Model Selection and Single-Molecule FRET Data. Biophys. J 2009, 97, 3196–3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).E. A: Beiträge zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung. Archiv fuer Mikroskopische Anatomie 1873, 9, 413–468. [Google Scholar]
- (69).Ouyang W; Aristov A; Lelek M; Hao X; Zimmer C: Deep Learning Massively Accelerates Super-Resolution Localization Microscopy. Nat. Biotechnol 2018, 36, 460-+. [DOI] [PubMed] [Google Scholar]
- (70).Wu ACY; Rifkin SA: Aro: a Machine Learning Approach to Identifying Single Molecules and Estimating Classification Error in Fluorescence Microscopy Images. BMC Bioinf. 2015, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Hochreiter S; Schmidhuber J: Long Short-Term Memory. Neural Comput. 1997, 9, 1735–1780. [DOI] [PubMed] [Google Scholar]
- (72).Vaswani A; Shazeer N; Parmar N; Uszkoreit J; Jones L; Gomez AN; Kaiser L; Polosukhin I: Attention Is All You Need. In NIPS’17: Proceedings of the 31st International Conference on Neural Information Processing Systems; Guyon I, Luxburg UV, Bengio S, Wallach H, Fergus R, Vishwanathan S, Garnett R, Eds.; Curran Associates Inc., 57 Morehouse Lane, Red Hook, NY, United States, 2017; Vol. 30; pp 6000–6010. [Google Scholar]
- (73).Elbayad Maha, B. L, Verbeek Jakob: Pervasive Attention: 2D Convolutional Neural Networks for Sequence-to-Sequence Prediction. 2018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.