

Abstract
Building structural models of entire cells has been a long-standing cross-discipline challenge for the research community, as it requires an unprecedented level of integration between multiple sources of biological data and enhanced methods for computational modeling and visualization. Here, we present the first 3D structural models of an entire Mycoplasma genitalium (MG) cell, built using the CellPACK suite of computational modeling tools. Our model recapitulates the data described in recent whole-cell system biology simulations and provides a structural representation for all MG proteins, DNA and RNA molecules, obtained by combining experimental and homology-modeled structures and lattice-based models of the genome. We establish a framework for gathering, curating and evaluating these structures, exposing current weaknesses of modeling methods and the boundaries of MG structural knowledge, and visualization methods to explore functional characteristics of the genome and proteome. We compare two approaches for data gathering, a manually-curated workflow and an automated workflow that uses homologous structures, both of which are appropriate for the analysis of mesoscale properties such as crowding and volume occupancy. Analysis of model quality provides estimates of the regularization that will be required when these models are used as starting points for atomic molecular dynamics simulations.
Keywords: whole cell modeling, computational modeling, nucleoid structure, scientific visualization, Mycoplasma genitalium
Graphical Abstract
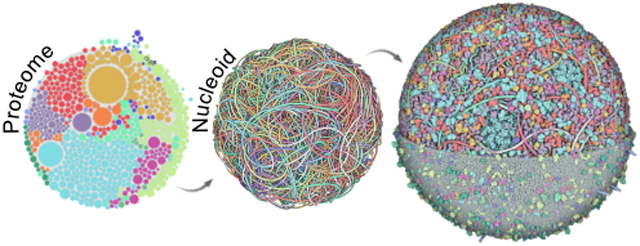
INTRODUCTION
Whole-Cell (WC) modeling is a “grand challenge of the 21st century” [1] and holds great promise to transform bioscience, bioengineering, and medicine [2]. Extraordinary advances in both experimental and computational structural biology have led to an exponential increase in our structural understanding of cells and cell components. High-resolution imaging [3] and cryo-electron microscopy [4] have provided experimental access to the atomic structures of large protein complexes. The confluence of data accessibility and advances in computational resources have opened the door to structural investigations of entire cells and the possibility to build structural models of an entire cell seems now within reach [5]. However, experimental methods alone have not been able to provide a unified picture of biomolecular structure, dynamics, and function across the entire range of scales from the molecular to the cellular level. Modeling can fulfill this role via mesoscale methods that incorporate experimental data from multiple sources and different resolution levels, providing a synergistic integrated insight that is not available from the individual pieces [6].
Because of the complexity of cellular environments, numerous challenges remain to achieve 3D Whole-Cell (3D-WC) models that capture a snapshot of the structure, location, orientation, and interactions of all macromolecules and membranes in a cell. Our ability to build molecular models of entire cells depends on an adequate understanding of the biological mesoscale, from the atomic to the cellular level [7]. Cells are heterogeneous crowded environments, packed with components different in size and shape, with variable concentrations and modes of interacting with each other. Assembling and curating the necessary data remains a major challenge for building 3D models of cellular environments [8, 9]. At the minimum, information about the system composition, concentration of each species, molecular interactions, and structural information are needed [10]. We immediately face two challenges when gathering this data: (1) the current state of knowledge has many gaps, and (2) the information is scattered across many resources.
Despite the growing body of experimental data, structural coverage at the molecular scale is still far from complete [5, 11]. High-quality 3D protein structures with genome-scale coverage are still scarce for any organism. Computational structure prediction can fill the gap and, in many cases, does so quite well [12, 13]. Recent successes from deep learning methods AlphaFold2 [14, 15] and RoseTTAFold [16] are particularly encouraging. However, the accuracy of predicted structures varies and similarly, proteomics and interactomics information is often highly domain specific and may show considerable variation across studies.
The internet provides instant access to diverse biological information, which is both a blessing and a curse. For example, the recent “Database issue” of Nucleic Acids Research features almost 200 biological databases [17]. Collectively, these databases contain much of the data required for WC modeling. However, the large number of siloed databases underscores the current reality that the scattering of biological data across multiple databases, each with their own conventions for formatting and disseminating their data, is one of the biggest bottlenecks to modeling [18]. As a result, substantial effort is still required to integrate the data required for WC modeling. Fortunately, structural and sequence data are becoming increasingly interconnected through mature global resources such as the worldwide Protein Data Bank [19] and UniProt [20]. Three efforts to integrate other types of data required for WC modeling include the Omics Discovery Index [21], Datanator [22], and EcoCyc [23].
Finally, once we have defined, gathered, and curated this large amount of heterogeneous data needed for WC modeling, 3D-WC models pose significant challenges for construction, visualization, navigation, analysis, and dissemination. Most molecular modeling and graphic tools have been designed to handle a limited number of instances, such as individual protein structures or single-molecule trajectories, which are far below the content of cellular systems containing millions of particles. Researchers are approaching this in multiple ways, ranging from using traditional tools to manually construct models (for example, models of cancer cell surfaces and extracellular matrix [24]) to fully procedural methods for constructing model instances that are consistent with the body of available data (as in our HIV-1 virion models [25]). For a wider perspective on these challenges, see [5, 6, 8, 26].
Small bacteria with limited genomes such as Mycoplasma are achievable targets for 3D-WC modeling. One of the most promising starting points for 3D-WC modeling is the compartmentalized, molecular-scale WC model of Mycoplasma genitalium (WC-MG) [27], one of the smallest members of the Mycoplasma genus [28]. The WC-MG model represents the function of all annotated gene products in 28 subcellular processes such as metabolism, DNA replication, transcription, and translation. Together, this enables the model to capture a variety of phenotypes throughout the life cycle of MG, such as the variation in the growth of individual cells and the essentiality of most genes. The WC-MG model was based on over 1,900 experimentally-determined parameters integrated from over 900 publications.
The systems-based approach of the WC-MG model accounted for only limited aspects of the structure of MG. To model the detailed molecular structure of cells, a number of approaches have been explored. These have included simulating portions of soluble cytoplasm [29–33] and mesoscale models for portions of many different organisms, including Escherichia coli cytoplasm [30], M. genitalium cytoplasm [34, 35], bacterial nucleoids [36–38], ribosomes and genome in a minimal cell [39], influenza virus [40, 41], a synaptic vesicle [42], HIV in its physiological context [25], a synaptic bouton [43], and a photosynthetic chromatophore vesicle from a purple bacterium [44]. The ability to build an atomistic model of a whole bacterial cell is still an unrealized and challenging goal for the structural biology community.
Here, we describe the first 3D-WC models of a MG cell (3D-WC-MG). This work addresses the central hypothesis: “Is it possible to create and visualize a comprehensive model of a whole cell with individual macromolecular detail, integrating available experimental data?” Our work integrates data from WC-MG systems biology simulations, experimentally-determined biomolecular structures, structure prediction tools, and bacterial nucleoid modeling tools, using a new toolkit of data curation tools, modeling methods and molecular graphics methods (presented here). With these tools, we generated 3D structural models of an entire bacterial cell at defined time points in the life cycle, which include all MG proteins, nucleic acids, and the cell membrane. As with the WC-MG work, the cell shape is simplified and the details of the attachment organelle are omitted in the current model. This report describes key challenges and solutions to building these types of 3D-WC mesoscale models. With our current toolkit, we are able to generate mesoscale models interactively, allowing exploration of multiple time points from WC-MG simulations. Our toolkit also enables us to generate multiple structural models for each time point, each of which is consistent with the current state of knowledge for the integrated input data. These ensembles of structural models, which capture different configurations of the position and orientation of each protein, allow us to explore the potential variations in the structural organization of individual cells at a given time point. The integrative modeling approach developed for MG can serve as a framework to build new 3D-WC models for more complex organisms.
RESULTS
Overview
This report presents a full workflow for integrating and curating structural data and using it to create and visualize a whole cell structural model of a bacterial cell (Figure 1). The overarching approach was developed in the first version of CellPACK [45]: all molecular “ingredients” are defined and individually modeled, then a “recipe” defines the number, location, and interactions of these ingredients, which is used to specify construction of a “model” of the cell. In the current work, we have developed a curated workflow for generating and assessing structural models of all of the MG macromolecular ingredients using a combination of experimental structures and homology models. A specialized tool, LatticeNucleoid [37], is used to generate the nucleoid, which is currently treated as one large ingredient in the models. The online tool Mesoscope [9] is then used to author and curate a recipe, in the current study, using information from specific timepoints from WC-MG simulations. Finally, the recipe is exported to CellPACKgpu, a gpu-accelerated version of CellPACK that allows construction, optimization and visualization of the full structural model. A variety of 3rd party methods are incorporated into the basic process for specialized tasks, including use of the LipidWrapper algorithm [46] to generate structures of membranes and the NVIDIA Flex library (developer.nvidia.com/flex) for local optimization of packing and interactions. During construction, a coarse-grain representation is used, and each ingredient is comprised of a collection of beads of 17Å radius. During optimization, each ingredient (molecule or molecular assembly) is treated as a rigid body. Atomic models are then generated by overlaying the original atomic models of the ingredients onto this coarse-grain model.
Figure 1.
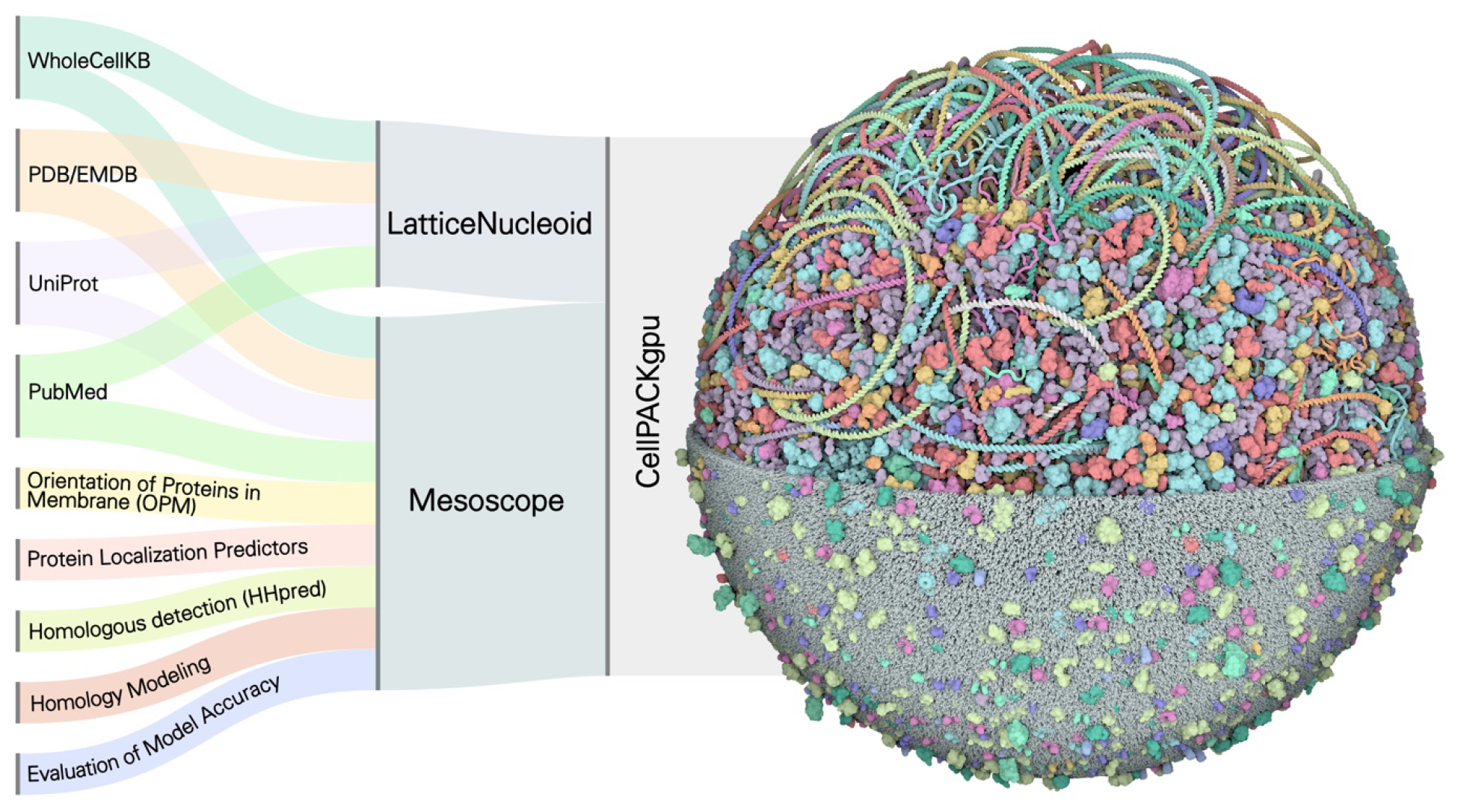
Data integration to generate computational models of entire M. genitalium cells. (Left) Multiple data sources were used to collect information about all MG ingredients. Identities and abundances of all molecules were taken from the WholeCellKB, together with the data on the genome, and the transcriptome. Structures were retrieved from structural databases or homology modeled. Protein location and membrane orientation were either computationally predicted or downloaded from OPM. Further details on oligomeric states or nucleic acid interactions were retrieved in the literature or UniProt. A script-based workflow assembled these diverse sources of information to generate a spreadsheet describing functions, structural models, gene position and interaction of each molecule. (Center) The nucleoid is based on the MG gene sequence and position and built with the coarse-grain method LatticeNucleoids. The online tool Mesoscope is used to curate the information and generate the CellPACK “recipe”. (Right) Sequence, recipe, and functional data are then integrated into CellPACKgpu, where complete models can be interactively constructed and visualized. In this image, the molecules in the model are successively clipped to show the membrane at the bottom, cytoplasm in the center, and only the nucleoid at the top. Molecules and genes are colored by function as in Figure 2. Please see the Methods section for a full description of each of these methods with citations.
Computer-assisted Curation Workflow for Molecular Ingredient Modeling
Information about MG molecular content, abundance, and localization was obtained from the WC-MG model [27] and its related web-based databases WholeCellKB [47], WholeCellSimDB [48] and WholeCellViz [49]. WholeCellKB is an extensive database that collects detailed information about every species described in the MG-WC model, including the structure and function of each gene, protein, reaction and pathway. The database is also extensively cross-referenced to existing resources including BioCyc, KEGG, BRENDA, UniProt and GeneBank. Based on information gathered for these studies, structural models of MG need to account for a circular genome of 580,076 base pairs that encodes 525 genes, including 482 mRNA, 3 rRNA, 4 sRNA, and 36 tRNA. A coarse-grain nucleoid model was generated with an enhanced version of LatticeNucleoids [37], including a 10 basepairs/bead representation of DNA, nascent RNA, and free RNA, and single beads for transcribing RNA polymerase, translating ribosomes, and nucleoid-associated proteins.
The ingredient-modeling workflow is designed to weigh several conflicting requirements to achieve the primary goal of providing structures for all MG ingredients. We found that there is no single method for generating this collection of ingredients. Experimental structures of MG proteins are ideal, but are only available for a handful of MG proteins. Homology modeling methods, including the recent advances embodied in AlphaFold2, provide models with proper chain length and sequence, but are currently effective for well-folded monomeric proteins, which comprise less than half of the MG ingredient list (216 act as monomers, 116 act as homomeric oligomers, and 150 are part of heteromeric assemblies. In preliminary reports, AlphaFold2 is also showing promise for prediction of oligomeric structures, but as of the time of writing, homologous structures are the only tenable solution for providing effective models for most biological assemblies. These homologous structures, such as the use of ribosome structures from related bacteria, necessarily require acceptance of some miss-match of chain length and sequence. Overall, we found that a combination of experimentally-determined structures, experimental structures of homologs, and homology modeled structures is currently required to generate a comprehensive list that honors known biological assemblies.
We developed a supervised workflow of these methods to streamline the creation of a manually-curated set of ingredients, designing quality measures to weight trade-offs between the advantages and disadvantages of the different methods (see Figure 9 in Methods). Given the ongoing goal of automating the entire modeling process, we also created an automated workflow to evaluate the difference between using the manually-supervised hybrid system for data gathering (the curated recipe) versus using only homologous experimental structures (the auto recipe). The final set of curated ingredients is presented in Figure 2.
Figure 9.
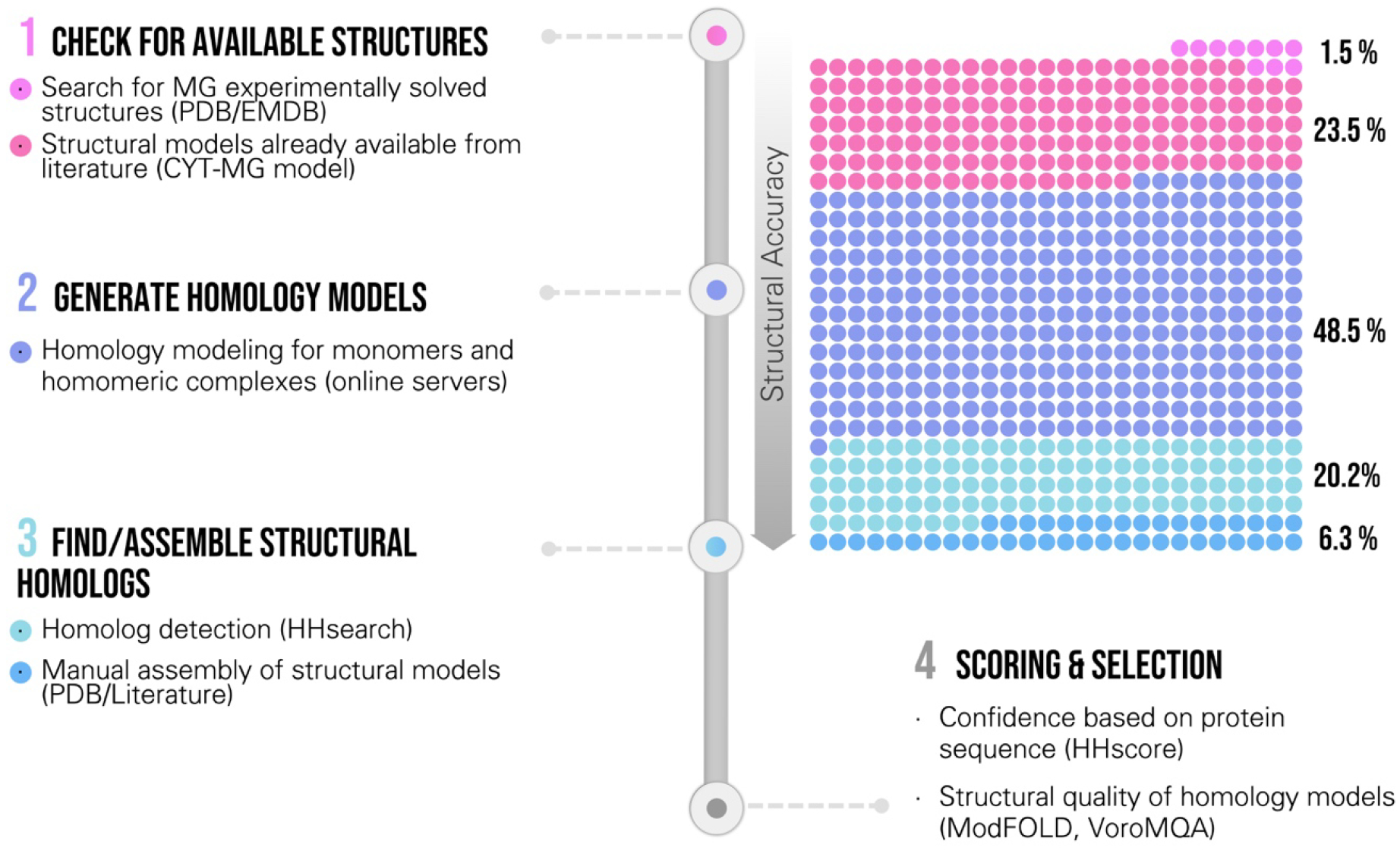
Workflow for generating structural models of the MG proteome. The general workflow involves four major steps. 1) Check for available structures, including experimentally-determined MG proteins and structural models available in literature (CYT-MG model [34]); then verify that these available structures are consistent with the oligomeric state and protein localization of the WC-MG model. 2) Generate multiple homology models for all ingredients that lack a structural representation. Homology models are generated for monomers and homo-oligomers. 3) Assign a homologous structure to hetero-oligomeric ingredients and to ingredients where homology modeling failed to provide reliable models. When a single homologous structure did not cover the entire complex ingredient composition, multiple homologous structures were assembled manually in a single model. 4) Homology models for each ingredient were evaluated and the best models were included in the recipe. All ingredients were assigned a confidence score based on their sequence (HHscore).
Figure 2.
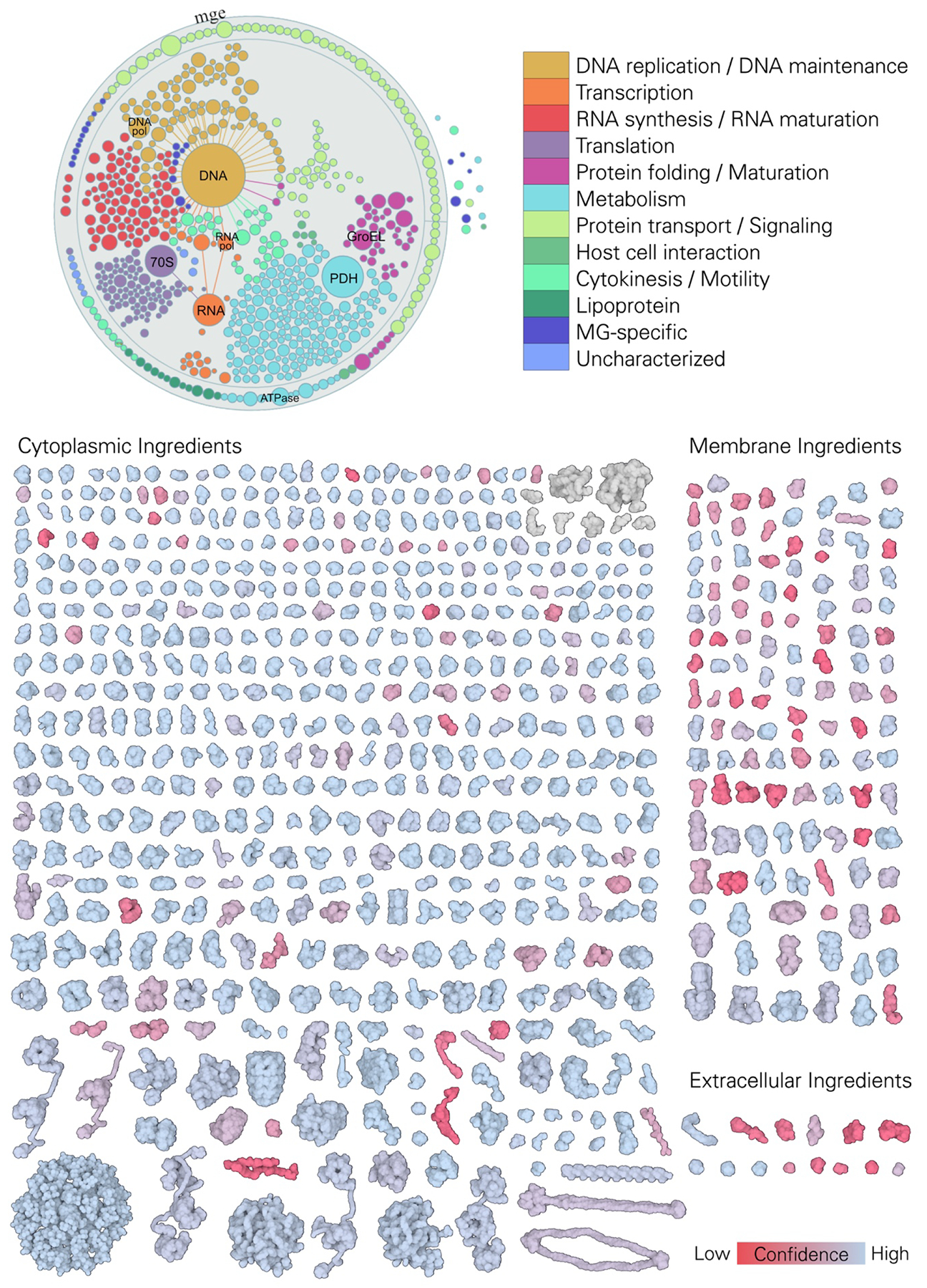
3D-WC-MG recipe. Top, graphical visualization of the proteome of MG, separated and colored by function, displayed with Mesoscope, our tool for integration and curation of mesoscale data (presented in more detail in Figure 10). Bottom, reconstructed proteome of MG displayed with atom radius scaled by a factor of three and colored by structure confidence score; RNA molecules are colored in grey.
The generation of all homology models, literature review, and selection of homologous protein structures for the curated recipe took eight months of effort by the first author. For the automated recipe, it took three weeks to assign all the structures, given that part of the literature review had already been done. Both recipes went through several rounds of revision, which were equally time consuming. Laborious tasks included researching details of oligomeric states, particularly when disagreements were found between the various sources of structures, defining binding modes for the DNA-binding proteins, and defining appropriate orientations for membrane-bound proteins.
The auto recipe uses homologous structures available in the PBD/EMDB as structural models for all protein ingredients. Compared to the curated recipe, the models in the auto recipe present more outliers both with low structural coverage and overly-high structural coverage. In both recipes, the majority of the monomeric models show good coverage: in 69.5% of the automated set and 86.7% of the curated set, the modeled structure covers between 75% and 125% of the protein sequence (Figure 3). These well-covered structures are primarily cytoplasmic proteins (88.6% in the auto and 84.2% in the curated recipe), which are either stand-alone monomers or part of complexes. Roughly half of the models with sub-optimal coverage in both recipes are membrane proteins, which are typically less structurally characterized than cytoplasmic proteins due to technical challenges in experimental methods. Homology models comprise the majority of the curated recipe, thus models in this set tend to have excellent coverage, with only few outliers showing low (4.4%) or excess (1%) coverage. In contrast, auto models are all homologs coming from various organisms, therefore they are more likely to have sequence length distant from MG proteins. In the auto set, the majority of the outliers present low (10.8%) structural coverage, while a handful (5.2%) of models are up to 4-fold larger than the actual protein. Proteins involved in cell motility, transport and signaling, lipoproteins and MG-specific proteins were the most challenging ones for finding reliable structural homologs.
Figure 3.
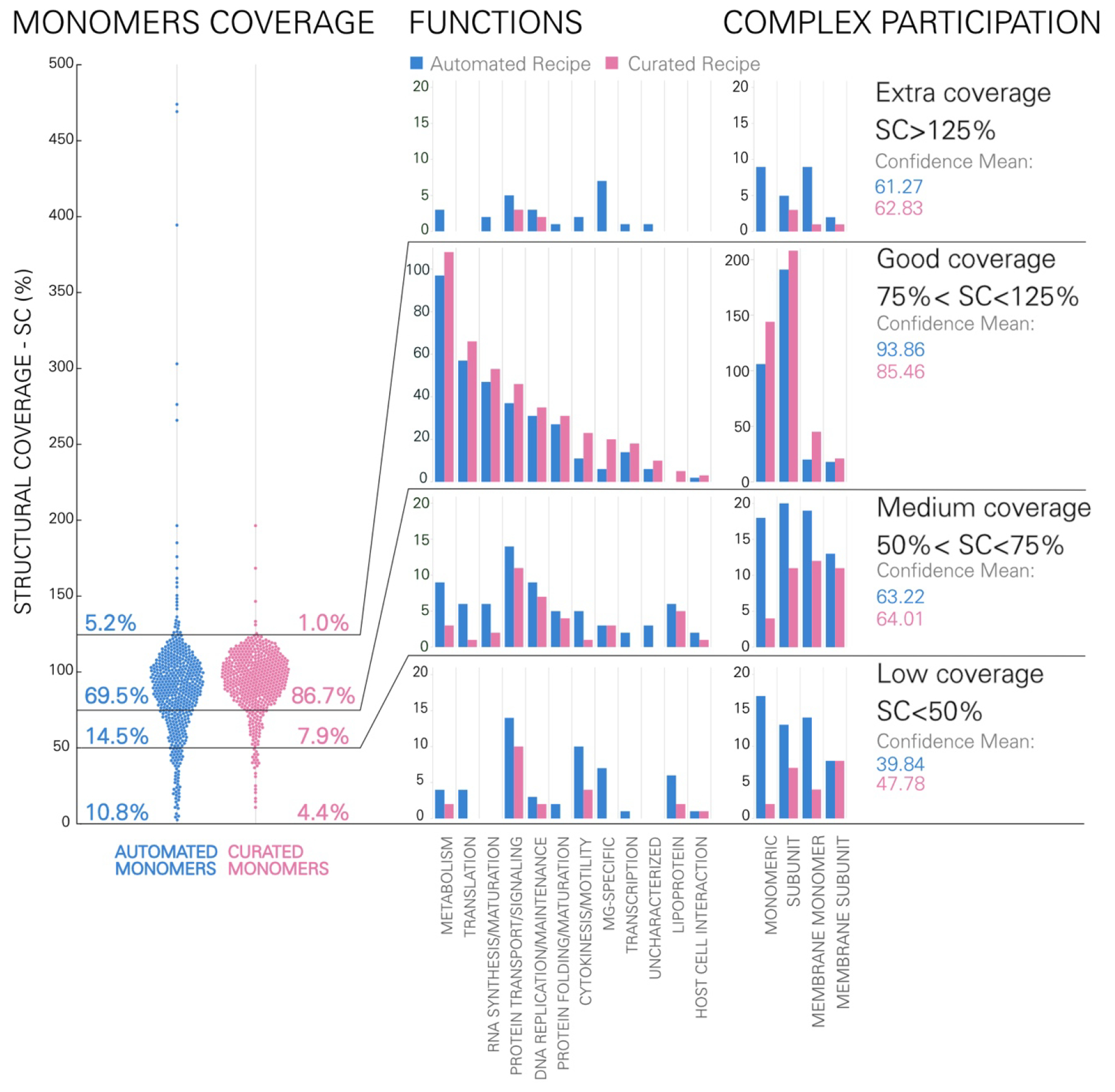
Structural coverage for models of monomeric proteins in the auto (blue) and curated (pink) data sets. At left, the structural coverage shows that most modeled protein chains are roughly the same length as the actual protein sequence, with fewer outliers in the curated set. Structural coverage is calculated as the ratio between the model length and the MG protein length expressed as a percentage. At right, histograms separate proteins in the confidence intervals by function and by oligomeric state or location. The confidence mean is the mean confidence score (HHscore) among monomers in the same group.
One of the most remarkable differences between the curated and auto recipes is the structural representation of macromolecular complexes. For many MG complexes, the subunit composition of homologs used in the auto recipe recapitulates the predicted MG complex composition used in WC-MG simulations. However, for large and dynamic complexes, finding a complete structure in an automated way is still challenging. In several cases, such as pyruvate dehydrogenase complex (PDH), structural maintenance of chromosomes complex (SMC), DnaA oligomeric structures, NADH oxidase, and others, the automated workflow was only able to find partial structures for the complex or structures with different oligomeric compositions than the predicted one (described in more detail below). The curated recipe, on the other hand, included human supervision in the workflow, which led to a more realistic structure but was also more time consuming.
Taken together, these observations underscore the limits of our structural knowledge with certain classes of molecules and the lack of effective automatic approaches to model macromolecular assemblies. Fortunately, the flexibility of our workflow allows facile replacement with improved models as they become available.
Structural Models of an Entire Cell
Given the complexity of 3D-WC modeling, we focused on modeling the first phase of the cell cycle when one copy of the chromosome is present, which corresponds to roughly 23% of the cell cycle in wild-type simulations. The cell shape is approximated as a sphere, and the attachment organelle is omitted; we are currently developing methods to incorporate data from cryo-electron tomography to incorporate these ultrastructures. We selected three representative time points in this cell cycle phase from a single WC-MG simulation for further analysis: (1) Frame 149 s, the cell at the very beginning of the cell cycle, (2) Frame 1184 s, the cell after ~20 minutes when at least 90% of the chromosome has been bound at least once by a protein (typically RNA polymerase), and (3) Frame 6973 s, the last frame before the start of DNA replication (Table 1). Note, the WC-MG model is a stochastic model, and the selected WC-MG simulation is a single trajectory of this model which represents the life cycle of a single cell. Thus, the selected frames characterize single cells, rather than average properties of an entire population of MG cells. In these three frames, cell size and ingredient count increased over time, with the cytoplasm being consistently the most crowded compartment. In addition, two variants of the recipes (auto and curated as described above) were generated at each of the three chosen frames, resulting in six types of models that were constructed and visually explored with CellPACKgpu.
Table 1: Summary of Model Parameters.
Values correspond to three selected time points of the first cell cycle phase of one representative simulation of the WC-MG model.
| Frame 149 s | Frame 1184 s | Frame 6973 s | |
|---|---|---|---|
| cell radius (nm) | 144.47 | 147.97 | 151.19 |
| cytoplasmic monomers | 17110 | 17563 | 21062 |
| DNA bound monomers | 2 | 4 | 6 |
| membrane monomers | 3934 | 4007 | 4672 |
| extracellular monomers | 331 | 334 | 409 |
| cytoplasmic complexes | 3949 | 4058 | 4604 |
| DNA bound complexes | 198 | 202 | 181 |
| membrane complexes | 550 | 556 | 610 |
| extracellular complexes | 0 | 0 | 0 |
| 70s ribosomes | 69 | 74 | 79 |
| RNA polymerases | 77 | 76 | 86 |
| mRNAs | 69 | 66 | 85 |
| rRNAs | 17 | 17 | 19 |
| sRNAs | 23 | 27 | 33 |
| tRNAs | 1653 | 1702 | 1945 |
| volume fraction protein, curated (1) | 0.144 | 0.137 | 0.147 |
| volume fraction protein, auto | 0.137 | 0.130 | 0.138 |
calculated using atomic structures from recipes, assuming a protein density of 1.35 g/cm3 and adding 8% mass percent of hydrogen atoms.
The ingredients could be successfully packed and optimized in the cell volume for all three time points, and both for the auto and curated recipes (Figure 4). Overall, the models are quite similar. The volume occupancy is similar across the three time points, with a fractional volume of ~0.14 protein and ~0.034 nucleic acid. Volume occupancy was also similar when comparing the auto and curated recipes, with auto models having total protein volume about 5% less than the curated models (Table 1, bottom). This is an encouraging result, indicating that for analysis of overall mesoscale properties such as crowding and volume occupancy, the simpler automated approach may be sufficient. The major differences between the models emerge in the details. As described below, we used coloration and selective display to highlight functional features of individual entities within the models, and these lead to quite different impressions when comparing the individual time points or auto/curated techniques. For example, the auto structure for PDH does not account for the assembled nature of the complex and would not be useful for any study addressing substrate channeling, and the auto recipe does not account for the shape of SMC and thus its role in DNA packaging (Figure 4). These observations also apply to use of models in education and outreach settings. Both the auto and curated models give an acceptable visual impression of the overall density and distribution of molecules in the cell, but the auto recipe will not be as useful when presenting the detailed structure and function of individual molecules.
Figure 4.
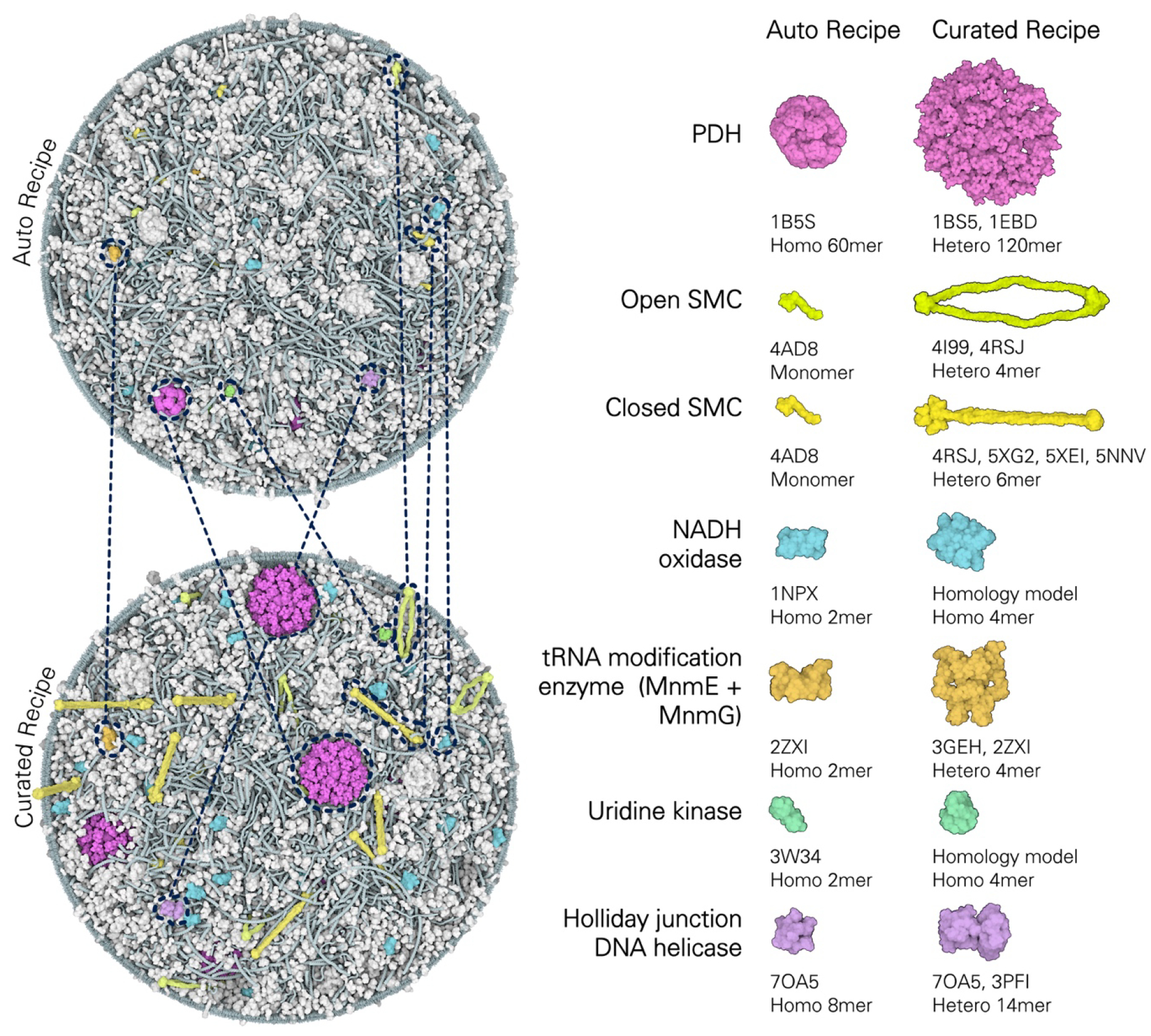
Comparison between the auto and curated recipes. Left panel: auto (top) and curated (bottom) 3D-WC models for Frame 6973 s. Selected structures corresponding to the same ingredient in both recipes are circled and colored as in the right panel. Right panel: representative examples of ingredients with notable differences in their oligomeric organization between the two recipes. PDH: in the auto recipe, 1B5S represented a multienzyme complex, the model includes only the 60meric dihydrolipoyl acetyltransferase core E2; in the curated recipe, we used a PDH reconstruction composed by 60E2+60E3, where E3 (PDBid 1EBD) monomers represent the two most likely locations of a given E3 at any given time [71]. SMC: in the auto recipe, monomeric 4AD8 represented both open and closed conformations of SMC, in the curated recipe 4I99 and 4RSJ were manually assembled to represent the open conformation of SMC, while 4RSJ, 5XG2, 5XEI and 5NNV were assembled to represent the closed conformation of SMC [83]. NADH oxidase and uridine kinase enzymes are represented by the homodimers 1NPX and 3W34 in the auto recipe, respectively, while in the curated we used homology models that recapitulate their predicted tetrameric organization. Holliday junction DNA helicase (ruvA+ruvB complex): in the auto recipe is represented by 7OA5, only covering ruvA, in the curated model was manually assembled using 7OA5 for ruvA and 3PFI for ruvB.
DNA is modeled using a lattice-based approach that builds supercoils based on locations of RNA polymerase transcription complexes. The simple Flex approach used to optimize the model uniformly extends local regions of the DNA model, which leads to some unwinding of the supercoiling from the original lattice model. More sophisticated models that incorporate torsional constraints on neighboring beads, due both to the local characteristics of the double helix and interactions of protein, will be required to more accurately model the superhelical properties of the DNA in these models [50].
Since the resulting model specifies the compartment and orientation of each ingredient, models with different levels of detail may be generated, from coarse-grain bead models to models with atomic detail. In these 3D-WC models, the molecules are treated as rigid bodies, which imposes several limitations on the quality of the final models: interactions within molecules (such as with cofactors) and local conformations of loops and domains are frozen in the state captured in the ingredient coordinate files, and local atomic details at sites of interaction between instances of these ingredients are not specifically optimized. The resulting models are acceptable for visual applications, such as in education and outreach and as thinking tools in research, and also for simulation of bulk properties of cells, such as simulated cryoelectron tomograms or generation of contact maps. However, the rigid-body approach could potentially cause problems if these models are used as the starting points for simulation, since the detailed sites of interaction would need to be optimized. As shown in Figure 5, many molecules have steric clashes with neighbors in the initial models, but most of these clashes are resolved in the optimized relaxed models (see “Optimization of Models” in Methods for details on overlap calculation). After optimization using the Standard Protocol with 17Å beads, roughly a third of molecules have small clashes with a neighboring molecule using a stringent overlap threshold of 0.0 nm (no clashes), but for large clashes with bead overlap of 10Å, this is reduced to less than 2%. After the second round of optimization with beads of expanded radius (the Expanded Radius Protocol with 21Å beads), only 1–2% of ingredients show clashes with the stringent 0.0 nm threshold.
Figure 5.
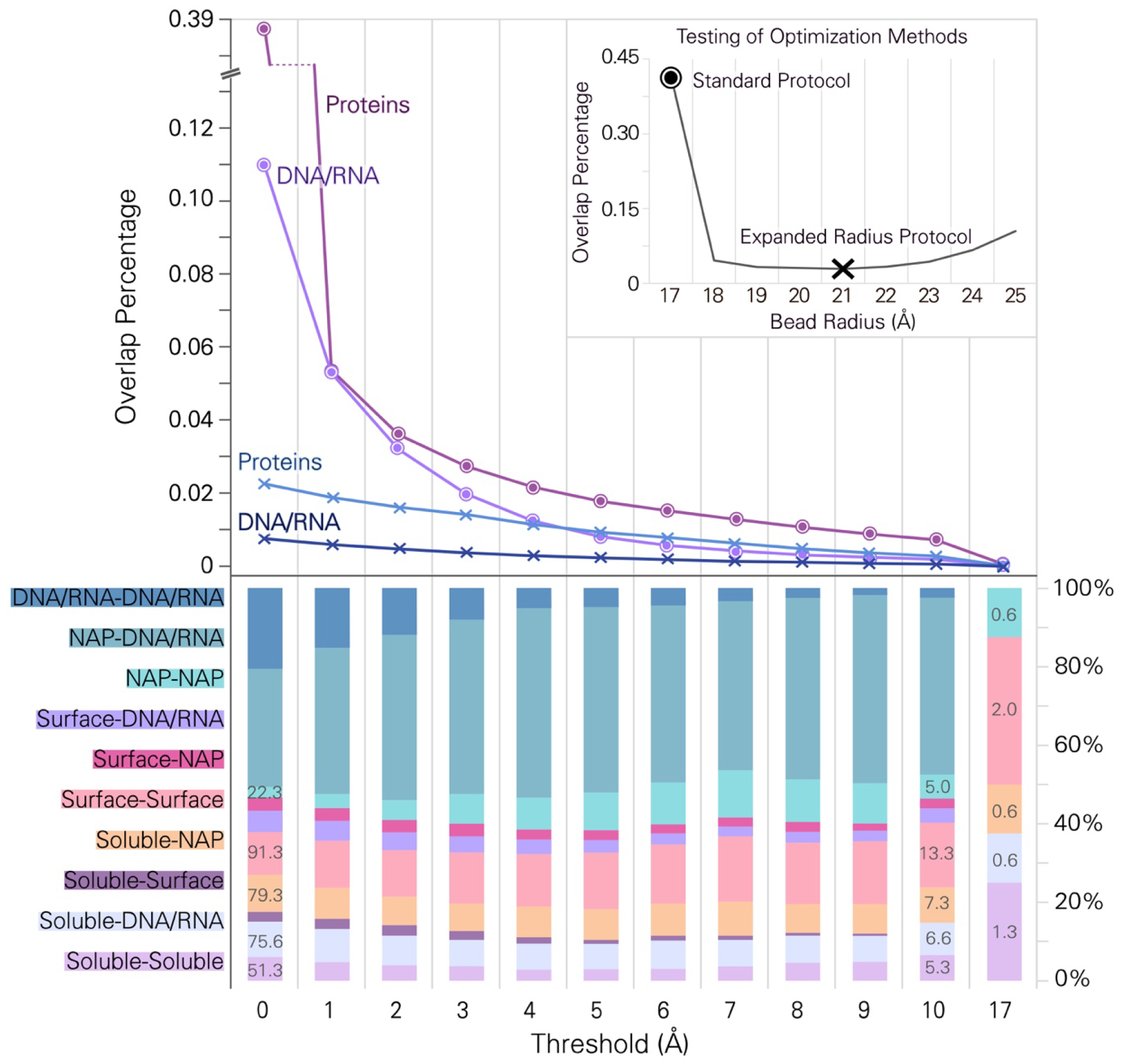
Clashes analysis. The graph plots the percentage of protein and fiber overlaps (vertical axis) against the threshold that defines the clash (horizontal axis) after model relaxation. Each curve is the average of the analysis at three time points (Frames 149, 1184, 6973). Violet/purple lines refer to overlaps between proteins or DNA/RNA calculated using the “Standard Protocol”, blue/cobalt lines refer to overlaps calculated using the “Expanded Radius Protocol”. Molecules were represented as collections of beads and the “threshold” is amount of overlap between beads used to define a clash: a “threshold” value of 0 scores a clash when beads show any intersection, and a “threshold” value of 17Å scores a clash when beads overlap by more than one bead radius. The inset graph shows results of optimization using expanded radii, showing the percentage of overlap (vertical axis) when bead radii of 17Å to 25Å (horizonal axis) are used during optimization. A bead radius equal to 17Å is considered the Standard Protocol for relaxation, 21Å bead radius is the optimal bead radius for relaxation for an Expanded Radius Protocol. The lower charts illustrate the types of interactions that are responsible for clashes after the Expanded Radius Protocol. The numbers on the bar refer to the average number of clashes in that category. “Soluble” indicates cytoplasmic proteins, “Surface” indicates membrane proteins, “NAP” indicates DNA/RNA binding proteins, “DNA/RNA” indicates fiber-like ingredients like DNA or RNA.
It is also interesting to note that roughly half of the clashes are found in interactions with and between nucleic acid ingredients, underscoring the challenge of optimizing local structural details of fibrous molecules that fill large spaces within the cell. However, at a threshold of one bead radius (17Å), only 8 clashes remain in these optimized models, scattered between different types of ingredients and not including any nucleic acid clashes. The magnitude of this threshold (~17Å or less than one bead radius) gives an estimate of the level of additional optimization that would be necessary if these models are used as starting points for detailed molecular dynamics simulations at the atomic level. This analysis also gives a measure of the small errors that may be encountered when these models are used to evaluate mesoscale properties such as volume occupancy or crowding.
Visual Exploration of the Model
The three time points differ from each other in terms of cell size, gene expression, chromosome exploration and protein content, so a nimble and flexible approach to visualization is essential for exploring and understanding the many properties of these models. To highlight these differences, coloring may be specified in Mesoscope and used within CellPACKgpu to explore genomic and proteomic properties of interest. For example, Figure 6 takes its lead from the WC-MG study, presenting derived properties explored during the simulation: regions explored by RNA polymerase are highlighted on the genome model with coloring, showing the progressive chromosome exploration over the selected time points, and coloring based on gene expression levels highlights highly-expressed genes at specific time points.
Figure 6.
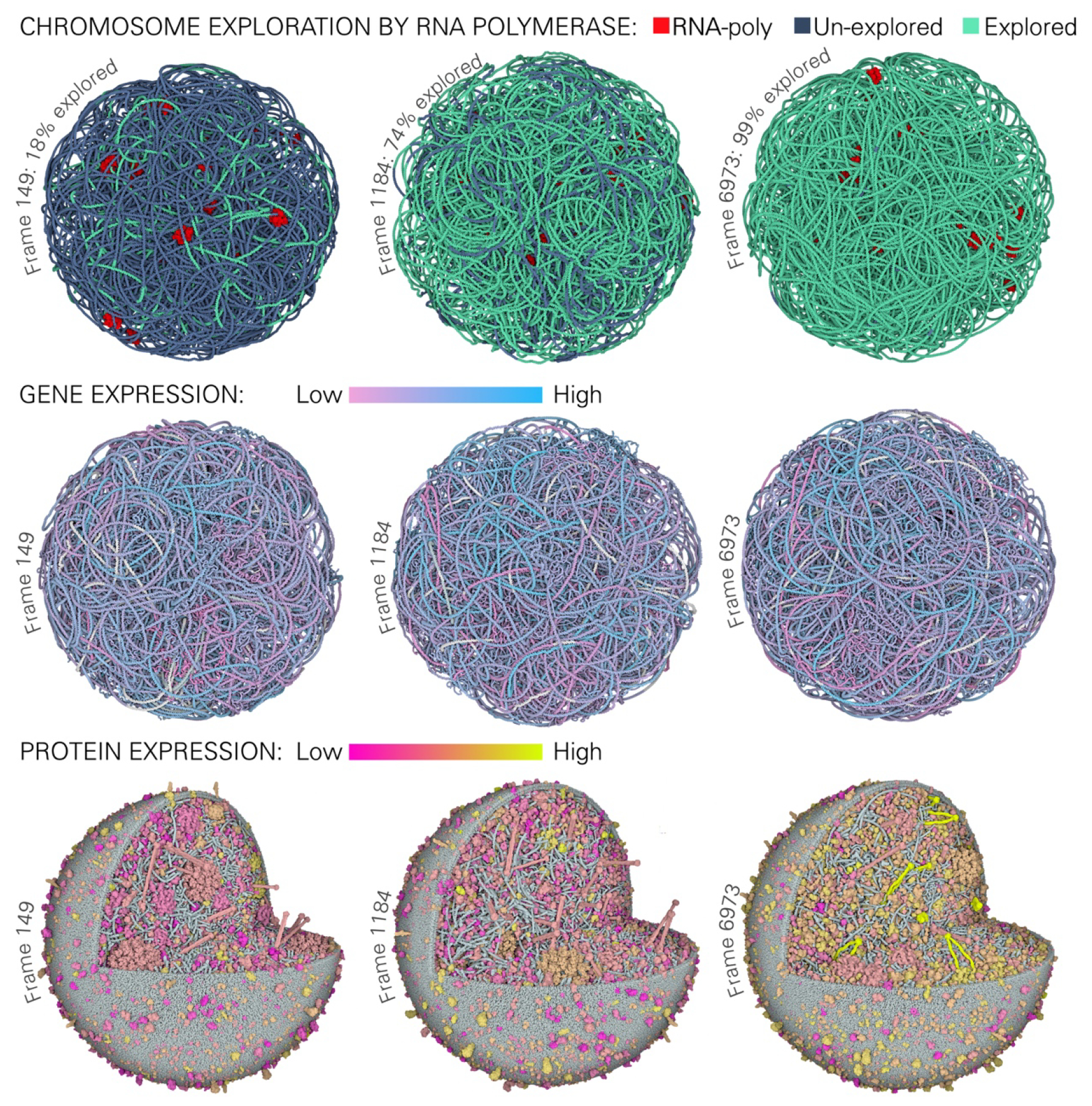
Visualization of time-dependent properties in instances of 3D-WC-MG models at three time points. Chromosome Exploration shows regions of the nucleoid that have been explored (blue) or have not yet been explored (green) by RNA polymerase since the last replication. RNA polymerases at the time point are in red. Gene Expression: each gene is colored by its expression level normalized by the mean copy number across the whole simulation. Pink indicates low expression and light blue indicates high expression, white indicates non-coding regions. Protein Expression: each protein monomer or complex is normalized to its mean copy number across the whole simulation; yellow indicates low abundance; magenta indicates high abundance.
We have exposed multiple parameters of interest within Mesoscope that may be used for this type of coloring of model ingredients. Color palettes can be generated in Mesoscope and imported in CellPACKgpu as JSON files, or colors can be assigned directly through CellPACK. Color palettes can be generated both as gradients based on continuous variables, or based on discrete ingredient properties. For example, in the upper left panel of Figure 7, structural models have been colored by category, depending on the source of structural data. The color scheme shows immediately how the majority of structures derive from homology modeling, either in-house or from an existing model of MG cytoplasm (CYT-MG) [34], while experimentally-determined structures of MG proteins are few and mostly surface proteins. This reflects the primary interest of MG structural biologists towards studying proteins involved in cell motility or immune response. In this way, this coloring choice provides visual feedback on the lack of structural data available for other MG protein classes.
Figure 7.
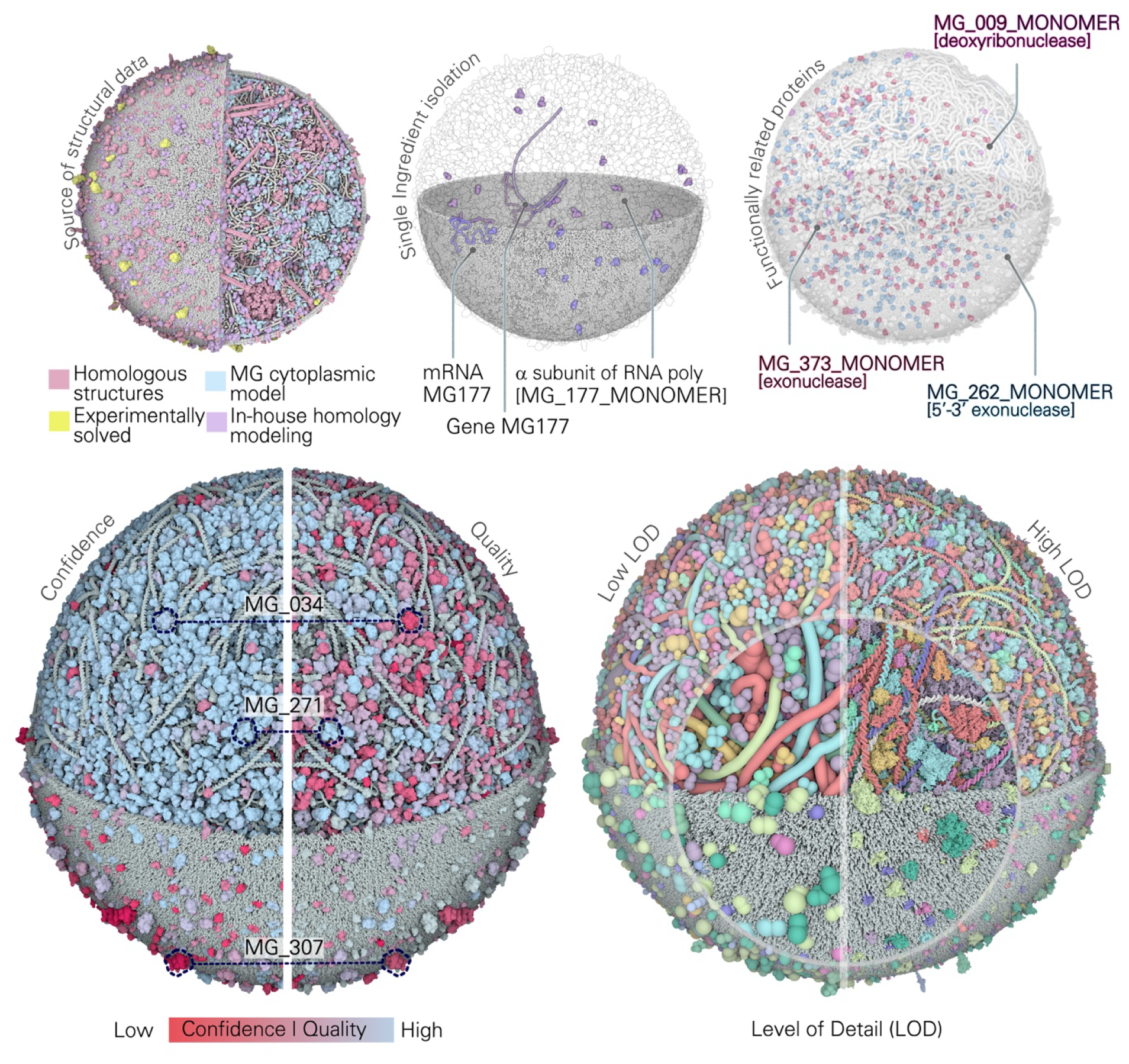
Methods for functional visualization of 3D-WC models. Coloring by user-defined properties: ingredients colored based on the source of the structural model (top left). Isolation of single ingredients and ghosting: gene MG177 and its corresponding protein and mRNA isolated in the context of the whole 3D-WC-MG model (top center). Highlight ingredients with specific biological functions: DNA processing proteins MG_262_MONOMER (violet), MG_009_MONOMER (blue), MG_373_MONOMER (magenta) highlighted in the context of the whole 3D-WC-MG model (top right). (Bottom left) Coloration is used to compare confidence and quality measures, such as in MG_034_DIMER, MG_271_MONOMER and MG_307_MONOMER. (Bottom right) Models are rendered with the two extreme levels of detail: on the right (low LOD4) ingredients are represented as beads defined in Mesoscope, while on the right (high LOD0) each atom is represented by a sphere (a portion of each representation is magnified in the inset). The rendering dynamically switches to appropriate level of detail depending on the distance to the camera, or users may select a desired level of detail. All figures represent Frame 149 s. Soluble extracellular proteins were excluded from the visualizations for clarity.
Additionally, the usage of homologs to represent several macromolecular complexes (e.g., PDH, SMC, replisome proteins), reflects the current limits of homology modeling. Properties like confidence or model quality, which can be expressed as a range of values, are effectively displayed with color gradients (Figure 7, lower-left). For example, the confidence palette shows that we have less confidence in the membrane protein structures compared to the cytoplasmic ones. Gradients can be used to make comparisons between different properties. In Figure 7 lower-left panel, we compared confidence and quality estimates, observing that for well-studied proteins confidence and quality scores tend to be aligned, being both on the high end of the value range (MG_271_MONOMER). Similarly, proteins with few homologs in the PDB (aka low confidence) tend to have low structural quality scores (MG_307_MONOMER). On the other hand, low-quality scores can also be associated with proteins with high confidence (MG_034_DIMER). This circumstance is observed mostly with complex homologous structures, where the quality evaluates subunit interfaces, which are still challenging to accurately predict even if decent templates are available. For all of these examples, the color comparison allowed us to better understand the relationship between our metrics and to identify critical cases.
Other visualization tools can improve the interpretability of these complex models. Single ingredients, genes, proteins and transcripts can be viewed in ‘isolation mode’ in CellPACK (Figure 7, upper-middle). This feature allows viewers to instantly get a sense of the concentration of a specific ingredient and to see if a gene is being transcribed and where its mRNA can be found in that specific time frame. The concept can be applied to groups of ingredients sharing a specific characteristic (e.g., same functional category, compartment, or binding partners, Figure 7 upper-right). These kinds of visualizations help focus attention on desired classes of molecules and identify them in the context of the cellular environment. Finally, CellPACKgpu automatically adjusts the level of detail of all ingredients based on the camera position, showing finer atomic detail when portions of the model are viewed close-up, and coarser representations when the entire model is being viewed. However, viewers may also manually tune the magnification if desired. Currently seven levels are available: (LOD0) all atoms; (LOD1–3) three K-means clusterings at 1.5%, 1.0% and 0.5% of the total number of atoms [51]; (LOD4) the low resolution minimal representation prepared in Mesoscope; and (LOD5) the 17.0Å spheres prepared in Mesoscope and used in Flex. LOD4 and LOD0 are shown in Figure 7, lower-right.
DISCUSSION
This study is a proof of principle, encompassing the entire process of researching, building and visualizing 3D models of an entire cell. By far, the most time consuming and heterogenous aspect of this process is the first step--gathering and curating data to support the modeling. In the current study, we strongly leveraged the extensive work that went into the WC-MG model, which provided directly-usable data for many of the parameters, including gene IDs, abundances of gene products, interactions and localizations. The major tasks that remained were to generate convincing models of the hundreds of ingredients and to implement methods to distribute them appropriately based on parameters from specific time points in the network model.
Of course, this is a very active field of research, and if we were to start this project today, we would take advantage of all the new technologies and data integration tools that are now accessible to the scientific community. For example, in our work, we explored multiple homology modeling tools, because at the beginning of this journey there was not a single accessible method that was universally considered the best in the modeling community. AlphaFold2 or RoseTTAFold are currently the best available methods for predicting protein structures, but have become available only in July 2021. ColabFold notebooks [52], which make AlphaFold2 accessible to non-expert modelers, became available in August 2021. Platforms that promote data integration like 3D-Beacons Network (https://www.ebi.ac.uk/pdbe/pdbe-kb/3dbeacons/) were announced in September 2021. Using fewer tools for modeling would definitely streamline model generation. However, despite the incredible progress made by homology modeling, the folding problem has not been completely solved. Even with these new technologies, compiling a comprehensive, curated recipe would still take months for the revisions and literature search, especially to assign structures for macromolecular complexes.
Our resulting workflow incorporates several design features that made the entire enterprise possible, and provides a template for future work as we incorporate new technologies. First, Mesoscope was implemented as part of this effort and played an essential role in the assembly and curation of the recipe. Data management quickly becomes intractable for a system of this size, and Mesoscope provided a user-friendly and centralized resource to manage the many prototype recipes that were generated over the life of the project. Second, we strived to create the most accurate set of ingredients that was possible with current tools, which required extensive manual curation. Throughout this work, we kept automation in mind, with the hope of progressively moving towards automated workflows as search and prediction methods continually improve. We envision the model generation step will become faster and more accurate in the future, thanks to the application of deep learning methods that are revolutionizing the homology modeling field, as demonstrated by the recent successes of AlphaFold2 or RoseTTAFold. As these become available, the flexibility of our workflow allows us to rapidly update structural models through Mesoscope. Finally, we decided early in development to focus on interactive approaches to 3D modeling of the cell and to visualization, to allow nimble exploration of multiple approaches both to the building of models and exploration of hypotheses based on these models. This performance opens the door to exploring pleomorphic systems and/or multiple instances of each system.
Challenges for Ingredient Modeling and Scoring
Many challenges were encountered during the process of building a full set of macromolecular ingredients for the models. Oligomeric assemblies posed the most persistent challenges, which were ultimately solved by brute-force manual curation and modeling of each case. This is obviously a challenge for the entire field: we compared oligomeric state predictions, functional annotations and protein localization between the WC-MG model and a previous atomic model of MG cytoplasm (CYT-MG [33]), and for the vast majority of MG macromolecular complexes, stoichiometry and subunit composition were not experimentally determined, resulting in some disagreements. These differences are presumably the consequence of different methods for assigning oligomeric states: in the WholeCellKB-MG database, the multimeric assembly of protein complexes was reconstructed based on a consensus of curated databases, while in the CYT-MG model, it was inferred from template structures used for the modeling. We chose to rely primarily on the WholeCellKB-MG database and check UniProt and homolog stoichiometries in cases with disagreements. For example, ingredient MG_034_DIMER, a thymidine kinase, originally predicted to be a dimer, is more likely to acquire a tetrameric assembly according to both UniProt and the model from CYT-MG. On the other hand, the ribose-phosphate pyrophosphokinase MG_058_HEXAMER was considered a trimer in the CYT-MG, while UniProt and its closest homologs suggest a hexameric organization. Details on how the disagreements were handled in each case are provided in the “Comments” column of Supplementary Table S1. This is doubly challenging for hetero-complexes, such as the PTS system and ABC transporters, where we could find no efficient automated methods and manual structural assembly was required.
Given the many available approaches for finding homologous molecules and generating homology models, we quickly came to the realization that we needed good metrics for model validation and selection. Our current approach draws directly from the homology modeling community, leveraging their extensive experience with evaluating entries to modeling challenges. Our metrics, however, are still qualitative and human guidance is required in some cases to pick the best model. This was the case for MG_185_MONOMER, a putative lipoprotein with a high confidence score (HHscore>99), suggesting high probability to get a reliable model. However, the quality measures calculated on the five homology models indicated low quality and assigned a higher score to models with long artificial loops. Manual intervention picked the model with a lower quality score that, despite providing less structural coverage, was more similar to its closest structural homolog (Figure 8).
Figure 8.
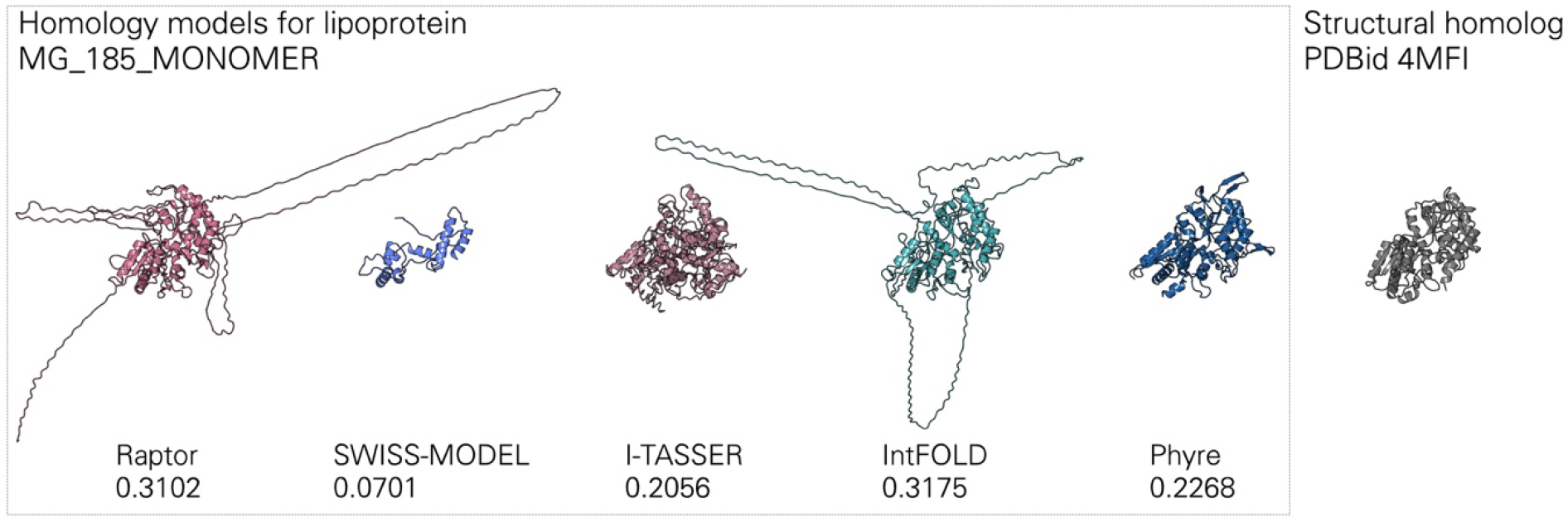
Challenging modeling case that required manual curation, underscoring the limitations of current quality metrics. Five homology models obtained by submitting the MG_185_MONOMER sequence to RaptorX, IntFOLD5, SWISS-MODEL, I-TASSER and Phyre2 and their relative model quality scores. Models with the highest scores display artificially-extended loops, due to their greater sequence coverage. Models with the lowest scores are the most similar to the closest homolog 4MFI. After manual review the model from Phyre was selected. Figures were generated with Protein Imager [84].
We have also found that we needed to use different scoring methods for monomers and complexes. Historically, Estimates of Model Accuracy (EMA) methods were developed for single chain targets, producing a plethora of methods to efficiently assess the quality of monomeric models, among which we chose ModFOLD7. However, most EMA methods are not capable of assessing the quality of oligomeric assemblies because they typically do not account for interactions among subunits. Following the example of CAPRI and recent CASP experiments [53–55], we decided to use a method, VoroMQA, that evaluates protein interfaces as a selection metric for oligomeric homology models. We found that human supervision was still required for oligomeric model selection, especially when homologs were scarce. This is in line with the experiences of other groups participating in the recently introduced ‘Assembly’ category in CASP experiments, where the human factor was often key for the success of their oligomeric modeling methods [53, 54].
Certain classes of molecules continue to pose challenges to structural biologists, so structural information is scarce for modeling of MG ingredients. These include membrane-spanning proteins, lipoproteins, and proteins that interact with nucleic acids. Within the model, we need to specify both the structure of the ingredient and its interaction/orientation with the membrane or nucleic acid. Perhaps this is an indication, in part, for why it is difficult to determine structures of these molecules.
Finally, the intrinsic complexity of biology causes continual challenges, but the process of chasing down solutions to these challenges forces us to repeatedly admire the solutions that nature finds to functional problems. These challenges are numerous and diverse, and typically require customization of some aspect of the modeling workflow. In the 3D-WC-MG model, these included assemblies with dynamic, flexible interactions such as the replisome complexes, UvrA/UvrB incision complex, RuvA/RuvB Holliday junction, elongation factors Tu and Ts, FtsZ ring, SMC, pyruvate dehydrogenase, and DnaA complexes.
Challenges for Mesoscale Modeling and Visualization
Our current paradigm for modeling places rigid-body ingredients randomly in soluble or membrane spaces to generate a draft model with atomic detail. The models are highly effective in visual applications such as education and outreach, where the goal is to give an impression of the overall ultrastructure, crowding, and heterogeneity of molecules within the cellular environment. As we move towards full atomic modeling, two major challenges to the current workflow must be overcome: lipids and DNA/protein interactions. Lipid membrane modeling is a field that requires lifetimes of dedicated work, so we rely on 3rd party experts to provide the current state-of-the-art. For this reason, we currently use LipidWrapper [46], which is a popular method that provides interactive performance. DNA/protein interactions rely on experimental or modeled structures of the complex, and CellPACKgpu currently does a simple overlap of the protein position on the site in the genome. A topic for future development will be to incorporate a more detailed representation of the interaction into the Flex optimization, to resolve small clashes that result from improper local structure of the DNA at the site of interaction.
Once these models are constructed, visualization itself poses a second grand challenge. These models are complex and heterogeneous, and biologists studying these systems will need to explore many properties of the model at many scale levels. We have worked for many years to develop methods to improve comprehension of mesoscale scenes, both through using traditional artistic approaches [10], and through collaboration with computer graphics experts to develop advanced methods for interactive visualization. Since CellPACKgpu is built upon cellVIEW [51], we have access to numerous effective methods for tailoring a visualization for specific needs. First, several methods for clipping the scene with custom primitives (selectively hiding specific 3D regions) allow facile exploration of features within the interior of models. For example, Figure 1 uses two parallel clipping planes to progressively remove portions of the models, Figure 6 includes several examples of a cube clipping primitive to remove a quadrant of the model. Other options include “ghosting,” where clipped portions of the model are rendered as transparent to retain the context of the feature being examined, and level-of-detail control, changing the detail of each ingredient interactively based on the size that it is displayed on the screen (Figure 7).
The magnitude of these models is pushing the current boundaries of what is possible with modeling and visualization. CellPACKgpu is currently capable of modeling and visualizing up to 50 million proteins--for reference there are ~250 million hemoglobin in one red blood cell and ~27000 proteins in the 3D-WC-MG model. However, using Flex for relaxation reduces that number to ~1 million beads which represents twice the size of the MG model, putting an E. coli cell within reach in the near future. While the boundary of visualization could be pushed with a more advanced culling and level-of-detail approach, the relaxation is more challenging and will require a divide-and-conquer approach as a next step, where sub-volumes will be relaxed in parallel or in serial. While CellPACKgpu is the only current software capable of rendering systems up to 50 million proteins, we can export smaller models and visualize them in a regular molecular viewer. For example, exporting an all-atom version of the MG model in CIF format results in a ~8Gb file which is impossible to load in any viewer. To avoid this problem, we implemented an exporter that relies on the CIF symmetric operator that applies a transformation matrix to individual molecules. This reduces the final all-atom file to ~240Mb, which can then be loaded directly even in web-based viewers such as Molstar. The same idea is applied to the lipid bilayer: we export the atomic description of a small number of lipids associated with the transformation matrix that tiles them on the mesh triangles defining the compartment, generating a file of ~12Mb. With these formats, users are able to import and explore complex models as large as the MG model using commonly-available computer hardware.
Future Directions
The primary motivation for this modeling work is to provide a mechanism for hypothesis generation in the classic mode of scientific visualization: as a mechanism for promoting thought and inviting exploration in research and educational settings [56]. This builds on our previous mesoscale visualization efforts by providing a quantitative exploration of mesoscale structure and function of cells, with direct user-accessible connections to the experimental sources underlying the model. Based on anecdotal responses to our decades of work on artistic approaches to mesoscale imagery, these types of visualizations change the way that viewers understand the cell and help promote an approach that incorporates the cellular context into an understanding of biomolecular function [6, 10].
In related work, we have also begun the first steps towards applying these models for the exploration of several derived properties that relate directly to experimental mesoscale structural observations. For example, we are exploring the simulation and interpretation of super-resolution micrographs of HIV spike distribution [25] and prototyping ways to interactively interpret cryo-electron maps of mitochondria [57], and we have used nucleoid models as a way to simulate contact maps such as those obtained from HiC experiments [37]. The preliminary proof-of-principle models presented here potentially open the door to a continued dialog with experiment, incorporating additional levels of experimental structural and tomographic data into construction of the model and providing direct mechanisms to simulate images and derived properties such as interaction maps for comparison with these experimental data.
We also see these models as providing the starting points for more advanced molecular simulations. Our use of LAMMPS and Flex for relaxation of models shows that this is currently feasible, at least in the context of coarse-grained representations. The CellPACK binary file along with the recipe file, as well as the CIF files, are designed for facile input into other methods. For example, we have developed a simple script for Moltemplate [50] to perform dynamics simulations based on a CellPACK model using the LAMMPS molecular dynamics engine. Our NVIDIA Flex representation is consistent with LAMMPS (all proteins are rigid bodies made of one atom type of radius 17.0Å and fibers are polymer chains of atom beads), and LAMMPS trajectories record the position of all beads. In anticipation of future dynamics simulations based on mesoscale models, we prototyped a parallel function to play back simulations, that computes the position/rotation of rigid bodies based on the local bead position using singular value decomposition methods.
CONCLUSIONS
This study is a proof of principle for mesoscale modeling of the macromolecular structure of an entire bacterial cell. To our knowledge, this is the first structural model of an entire cell at macromolecular detail, and this report is intended as a worked example of how a whole cell structural model can be constructed with the current state of knowledge and current technologies. Since the study built on detailed information on molecular content and abundance from WC simulations, the bulk of the effort was expended in generating and validating structures for each component. Mesoscope played an essential role in managing and curating the complex recipe of macromolecular ingredients, and a multi-step workflow was needed to build and visualize the multiple environments of the cell (cell membrane, nucleoid, soluble cytoplasm). We hope that other groups will use this large and complex model to benchmark their own visualization tools, as a way to reflect on their data and as a thinking tool as the entire community works towards simulation of systems of this magnitude and heterogeneity.
Data Availability
The CellPACK binaries dedicated to the MG modeling, the source code and scripts for generating LatticeNucleoid MG nucleoid models, representative models (in different file formats including PDB, CIF, binary), ingredient modeling notes and scripts for the modeling workflow, and tables and structure files for the 3D-WC-MG recipes are freely available on GitHub at: https://github.com/ccsb-scripps/MycoplasmaGenitalium. Mesoscope is available online at mesoscope.scripps.edu/beta, and examples of the Frame 149 s curated and auto models are available in the Load→New Recipe→From Examples menu. Note that these examples do not include lipids, which seriously impact performance of the Mol* visualization.
MATERIALS & METHODS
Data Sources and Adaptations from the WC-MG Computational Model
WholeCellKB was used as the main data source to infer genomics (chromosome sequence; transcription units organization; location, length, direction and essentiality of each gene, Supplementary Table S2) and proteomics (protein content, functional annotations; localization, length, molecular weight, signal sequence, macromolecular complexation and stoichiometry, DNA binding properties and DNA footprint of each protein species, Supplementary Table S1) information. WholeCellSimDB [48] wild-type set #1–1 (www.wholecellsimdb.org/simulation/1) was used to retrieve data for the 3D MG nucleoid model: ribosome status (translating or stalled), ribosome position along the mRNA/tmRNA templates, RNA polymerase chromosomal occupancy, length of transcripts, length of nascent polypeptide chains. The simulation file also contained information on protein concentrations and cell size at each point of the simulated cell cycle. WholeCellViz [49] JSON files (www.wholecellviz.org/getSeriesData.php?sim_id=2011_10_19_02_53_45_1&class_name=Chromosome&attr_name=monomerBoundSites; www.wholecellviz.org/getSeriesData.php?sim_id=2011_10_19_02_53_45_1&class_name=Chromosome&attr_name=complexBoundSites; these JSON files are a different format for the results of the same simulation) were used to define DNA-binding protein occupancy of each nucleotide of each strand at different time points. In order to easily reconcile our physical model with WC-MG simulation, we maintained the name convention developed by Karr and colleagues. Protein monomers are named MG_GeneID_MONOMER, while the name of the complexes reflects their macromolecular composition.
A number of adaptations and approximations from the WC-MG computational model were applied to construct the 3D-WC-MG models. (1) MG cells typically present a protruding adhesion structure termed the terminal organelle. The WC-MG model approximated MG cells as a cylindrical rod with hemispherical caps. Our 3D-WC-MG models further approximated MG cells as spheres with identical volume since the cylindrical portion of the cell was minimal at the three selected time points. (2) The WC-MG computational model accounted for five cellular compartments: cytosol, membrane, extracellular space and terminal organelle specific compartments, terminal cytosol and terminal organelle membrane, and extracellular space. Since the terminal organelle was not modeled in the 3D-WC-MG model, we lumped the protein species from the terminal cytosol and terminal membrane cytosol into the cytosol and membrane compartments, respectively. The monomeric components of membrane complexes and secreted complexes were assigned to the cytosol compartment. (3) In this model we did not model ions, solvent or metabolites. (4) A single structural model was used in cases where WholeCellKB contained separate entries for multiple oxidation states or multiple states of bound ligands (i.e. reduced/oxidized thioredoxin or the acyl carrier protein bound to different fatty acids). (5) In the time points considered in this work, the vast majority of the proteins are in the mature or bound state. The WC-MG model captured additional states (e.g., inactivated, misfolded, damaged RNAs and proteins). However, the occupancy of these states was low. As a result, the 3D-WC-MG models represented these states identically to the mature states.
Structural Data for MG Biomolecules
Macromolecular structures came from four overall ‘categories’: 1) experimental MG protein structures, 2) modeled structures from a previous MG cytoplasmic model, 3) homology models produced ‘in-house’, and 4) homologous structures from experimental databases (PDB/EMDB).
Experimental MG structures.
Experimental structures were available for only fourteen MG genes. Ten of these experimental structures were used in our spatial model for the following gene products: MG396 (RpiB, 6MU0), MG027 (NusB, 1Q8C), MG191 and MG192 (P140 and P110, 6RUT), MG281 (Protein M, 4NZR), MG289 (p37, 3MYU), MG305 (DnaK, 5OBU), MG354 (uncharacterized membrane-anchored protein, 1TM9), MG438 (S-protein, 1YDX), and MG491 (uncharacterized terminal organelle, 4XNG). The other four genes are only partially captured by experimentally-determined structures: MG200 (DnaJ-like, 4DCZ), MG238 (tig, 1HXV), MG301 (GapA, 7JWK), and MG469 (DnaA, 2JMP). For these genes, we used homology models of the full-length proteins.
MG cytoplasmic model.
In previous work, Feig and colleagues modelled a portion of MG cytoplasm in atomic detail [34], referred to here as CYT-MG. The authors used homology modeling to generate a total of 128 models, and after review, we integrated structures of 55 complexes and 29 monomers from CYT-MG into our curated recipe. In cases where there were disagreements on the oligomeric state between WC-MG and CYT-MG, we considered the following sources of information (in order of priority): 1) interactions annotated in UniProt; 2) stoichiometry consensus of homologous structures in the PDB found with HHpred [58, 59]; and 3) primary literature describing similar proteins.
Homology modeling.
Sequences of monomeric proteins were submitted to Phyre2 [60], SWISS-MODEL [13], IntFOLD5 [61], RaptorX [62] and I-TASSER [63]. Homomeric complex structures were predicted by submitting sequences of subunits to SWISS-MODEL and GalaxyHomomer [64] web servers. Hetero-oligomers were not modeled with automatic methods and were assigned manually (see below).
Structural homologs.
Representative homologs for each ingredient were selected with HHpred, a method for remote homology detection [58, 59]. Standard HHpred searches were performed using precompiled PDB_mmCIF70 as the target database, containing PDB sequences clustered at 70% maximum pairwise sequence identity. For monomeric sequences, the top hit obtained in a HHpred run was automatically selected as the reference homologous structure for that specific monomer. For oligomers, amino acid sequences for each subunit were used as queries for HHpred search. A representative structural homolog was assigned to every hetero-complex after a manual evaluation of HHpred results of each subunit and primary literature search.
Manually assembled structures.
Solving the complete structures of large hetero-oligomeric complexes remains a demanding task for structural biology. For several heteromeric complex ingredients, fully assembled structures were not available on the PDB/EMDB. However, partial complex structures or individual subunits of homologous proteins were available. In these cases, separate PDB entries were manually assembled into full complexes with PyMOL (www.pymol.org). Coordinates of separate PDB entries were aligned manually in PyMOL and exported as single pdb files. Depending on the case, complex assembly was based on available experimental data, primary sources of literature, and subunit composition described in the WholeCellKB. Assemblies of the replisome components, SMC, and PDH were found in the literature (see Molecular Definition and Modeling in the Results section). Details on how each structure was assembled can be found in Supplementary Table S1, column ‘Comments’.
Curated Workflow for Structural Modeling
We gathered structural data from different sources to represent every protein species in the WC-MG model. The general workflow for how structural data was inferred is illustrated in Figure 9. This semi-automated workflow was designed to integrate data from inhomogeneous sources and includes three progressive tiers. The first tier includes structural information that is currently available from published sources. This includes MG protein structures that have been experimentally determined, which comprise less than 3% of the proteome, and homology models from a simulation of MG cytoplasm, termed CYT-MG here [34]. In the second tier, we use homology modeling to predict structures. The accuracy of homology modeling has seen dramatic improvements in recent years, however, the ‘folding problem’ has not been entirely addressed and current homology modeling methods can be unreliable for challenging targets (i.e. proteins with no homologs with determined structures or complex oligomeric assemblies) [65]. The final tier uses homologous structures when experimental structures or homology models are not available, chosen from related organisms and performing similar functions. Homologous structures are also used for hetero-oligomeric complexes. A list of the data sources used for every protein can be found in Supplementary Table S1.
All of this information is compiled into a “recipe” that defines structures, abundances, locations, and interactions for each molecular “ingredient” that will be included in the final model. The 3D-WC-MG recipe includes 683 protein ingredients, comprising 482 protein monomers and 201 protein complexes. The complexes include 159 unique entities and 42 macromolecular complexes with GDP, GTP, ATP, ADP (e.g., DnaA protein bound to ATP), lipids, RNA (e.g., ribosome) or oxidized forms (e.g., oxidized thioredoxin).
For challenging targets, homology modeling provides only an approximation of the actual protein structure and there are currently many methods available for use. Therefore, we produced several homology models for each ingredient using different freely available web-servers. The aim was to test a general workflow that could be accessible and usable even by non-experts. Automatic homology modeling methods were selected based on their performance in CAMEO [66, 67] and CASP [65], the possibility to perform batch submission, their response time, and their accessibility. We ultimately chose five methods for monomers and two methods for homomers, as described above. All structure predictions were downloaded, subjected to automatic quality assessment and manual supervision to select the best model for each ingredient. Monomers forming homomeric complexes were represented by a single chain of the complex model. All methods tended to perform better when the confidence score (HHscore) on the input sequence was above 80.
Homologs were chosen from the PDB/EMDB based on HHpred [58, 59], a method for sequence database searching and structure prediction that has been employed as a metric for target classification in CASP assessments. Homologs were used in two circumstances: for 35 oligomers and for 18 monomers for which homology modeling did not produce meaningful results. Currently, no consistent automated computational method is available for quaternary protein modeling [53], especially for hetero-oligomeric assemblies. Thirty-six complexes were manually assembled in the curated WC-MG recipe. This approach was used to build 12 complexation states of replication initiator factor DnaA, 8 complexation states of the cell division protein FtsZ, the PTS system, tRNA uridine 5-carboxymethylaminomethyl modification enzyme, the chromosome segregation protein SMC, topoisomerase IV and the phosphate ABC transporter. Putative assemblies of transient complexes like the DNA-directed DNA polymerase holoenzyme, the replisome and the components of the replication fork were provided by Dr. Jacob Lewis and are based on E. coli experimental data [68, 69]. The hypothetical structure of replication initiator DnaA bound to dsDNA was provided by the authors of the DnaA-ssDNA structure [70]. A reconstruction of the multienzyme complex pyruvate dehydrogenase was kindly supplied by the authors of the icosahedral pyruvate dehydrogenase structure from Bacillus stearothermophilus [71, 72].
Localization
In the WC-MG computational model, the localization for each protein ingredient was assigned based on the consensus among several sources, including computational predictions, databases and primary literature [27]. In several cases, protein localization was not consistent between the WC-MG computational model and the CYT-MG cytoplasmic model, as documented in Supplementary Table S3. These cases were reviewed by combining database annotations (UniProt), primary literature, protein localization predictors (BUSCA [73], SOSUI [74], Psortb [75]), and signal peptide detectors (SignalP-5.0 [76], Phobius [77]) to determine the localization of these gene products in the 3D-WC-MG model.
Confidence, Quality Assessment and Model Selection, Automated Recipe
Since heterogeneous sources were used to infer structural models for our 3D-WC-MG model, it was challenging to define a unique metric to compare all the structures. We assigned a confidence score to both monomers and complexes based on a feature common to all ingredients: their sequence. The confidence score, derived from HHpred results, indicates the likelihood to find a homologous PDB structure for a specific ingredient. Proteins with high sequence similarity and structural coverage are likely to share the same fold. Plus, the availability of homologous structures is critical for template-based homology modeling, making the confidence score an indirect indicator for the quality of the predicted models. Structures obtained via homology modeling were assigned an additional score, based on the quality of the 3D protein models. The quality score was used to pick the best model for each ingredient among the models predicted by multiple servers. For quality estimates and model selection, we chose two servers within the top performing in the quality assessment category in CAMEO and CASP experiments: ModFOLD7 [78] for monomers and VoroMQA [79] for oligomers.
Confidence score.
The confidence score (HHscore) was calculated by using the protein sequence of each monomeric ingredient as a query for a HHpred search against the sequences of all the PDB entries available at the time of writing. The HHscore was calculated as the product of the raw HHpred probability of the top hit and the alignment coverage of the query. For monomeric ingredients, the HHpred score was derived from the HHpred results. Instead, the HHpred score assigned to complexes was calculated as a weighted average among HHpred scores of the monomers composing a specific multimer and their predicted stoichiometry.
Quality assessment and model selection.
For each monomeric ingredient, all structural models generated by Phyre2, SWISS-MODEL, RaptorX, IntFOLD5 or I-TASSER were submitted to ModFOLD7, along with their sequences. The model with the highest global ModFOLD7 score was selected to represent that ingredient in the WC-MG curated recipe. In the case of oligomeric ingredients, all complex models by SWISS-MODEL and GalaxyHomomer were analyzed with VoroMQA and ranked based on the quality of their protein-protein interfaces. Oligomeric models with the highest interface quality scores were selected for the final recipe. VoroMQA calculations were run in batch on a local machine. Single monomeric subunits of oligomeric models were scored with ModFOLD7. ModFOLD7 calculations were run in batch by the server developers. All the automatically-selected structures were manually inspected using PyMol. In several cases manual inspection led to the selection of models originally discarded by the quality measurements. If all the homology models displayed low quality, a homologous structure was selected instead.
Automated structure workflow.
The auto recipe used homologous structures for all 3D-WC-MG ingredients. It was generated to compare the results of the semi-automated workflow for data gathering with a more automated and less time-consuming approach. A homologous structure was assigned to each ingredient based on HHpred search. Structural homologs selection for monomers and oligomers was as described in the ‘Structural Homologs’ paragraph above.
Recipe Curation with Mesoscope
After collecting structural information for all the ingredients, the data was assembled into a full recipe in Mesoscope (mesoscope.scripps.edu/beta) [9]. Mesoscope is a web-based tool developed to streamline the laborious tasks of data gathering, integration and curation for structural mesoscale models. Development of advanced features in Mesoscope were driven by the current study, including importing custom data through spreadsheets, tailoring displays based on properties, different approaches for generating coarse-grain models to speed the model computations, and exposing multiple properties of ingredients and models for use in visualization.
Both the curated and auto recipes were drafted as CSV files using a set of customized scripts to compile key information on all ingredients from the WC and local files (protein identifications, functions, molecular weights, locations, concentrations, UniProt codes and structural models). The CSV files were imported into Mesoscope where we visually inspect every structure, selecting appropriate protein chains and biological assemblies, and curating associated ingredient properties. Proper membrane orientations were assigned to membrane proteins, either manually or by automatically retrieving data from the Orientations of Proteins in Membranes (OPM) database [80]. Mesoscope was employed to generate and export color palettes into files readable by CellPACK-gpu, which allowed easier data interpretation and navigation of the whole 3D-WC-MG model. Ingredient properties currently visualized through color palettes include structure confidence, structure quality, data source, function and copy number. Finally, mesh geometry and coarse-grain bead representations were assigned to each ingredient based on their size. Two levels of coarse-graining were used: a very coarse model with a minimum number of beads per molecule, and a more detailed model with an automatically assigned number of uniformly-sized (17Å radius) beads. Both approaches use the k-means algorithm to calculate the bead positions. For the two largest ingredients (PDH and ribosome) we use an alternative approach to avoid internal holes in the representation, creating beads on a regular grid that fills the volume of the structure.
At this stage, interactions between ingredients were compiled. Protein ingredients that interact with fibrous ingredients such as DNA or RNA were manually ‘tagged’ and binding regions on the protein ingredients were specified using beads with a fixed radius of 17Å. Visual feedback is provided within Mesoscope by coloring the interacting beads to highlight them. In the future we plan to develop automatic assignment of the interacting beads when the structure of a complex exists and interactive assignment with a point-and-click interface for the remaining complexes. Recipe files were finally exported in JSON format readable from CellPACKgpu.
Lattice Nucleoid Model
The nucleoid was generated using the LatticeNucleoids software [37], with several new enhancements to allow modeling of time points from the WC simulations. LatticeNucleoids builds procedural coarse-grain models of nucleoids and associated macromolecules in several steps. The genome is modeled as a supercoiled circular loop, composed of a collection of unsupercoiled segments that are closed to form a circle, punctuated with supercoiled plectonemes. This simple model is designed to recapitulate results from fluorescence microscopy and HiC [81].
The unsupercoiled segments were originally generated using a subdivision method. However, this method was unstable and did not consistently fill the available space. In the new version, points for all segments are equally spaced on a simple curve similar to a Lissajous curve. This starts with a circle and applies sine displacements radially and perpendicular to the plane of the circle, with a magnitude of roughly half the radius of the circle and periods randomly chosen between 4 and 7 repeats around the circle. These curves, which typically have very tightly spaced points, are then relaxed within the space of the cell to create a curve with proper spacing between beads.
This curve is then embedded in a lattice filling the cell, and beads corresponding to the root of plectonemes are assigned to the nearest lattice point. Plectonemes are then added as in previous work with a self-avoiding random walk within the lattice, followed by doubling of the random-walk trace to form a superhelix with the desired supercoiling density. Nascent and free mRNA strands are added with the same algorithm, by seeding the random walk at the DNA bead corresponding to the site of transcription or a random point, performing the random walk with a reduced persistence length, and omitting the doubling/supercoiling step.
Finally, the lattice model is relaxed and optimized, including specific constraints for nucleoid-associated molecules. Proteins and ribosomes are treated as hard spheres. The relaxation method provides very simple constraints: a single collider may be applied to a DNA bead, or a distance constraint may be applied between pairs of beads, and a spherical collider may be placed at the center of the distance constraint or two spherical colliders may be placed at positions 1/3 and 2/3 along the distance constraint. Parameters in this study were assigned as follows, with distance constraint (d) and hard-sphere radius (r) in lattice units==3.4nm:
a single constraint at the origin of replication closes the circular genome (d=1.0, r=1.0);
RNA polymerase is modeled with a constraint between the DNA bead at the site of transcription and the mRNA (d=1.0, r=2.2);
SMC protein dimers, which form loops in the DNA, are treated with two spheres (d=1.324, r=0.662);
all other molecules are treated as spheres centered on DNA positions (d=0.0) with radii: ribosome, 2.94; DNA gyrase, 1.47; MG_101_MONOMER, 0.88; MG_205_DIMER, 1.18; MG_236_MONOMER, 0.88; MG_428_DIMER, 0.88; MG_469_1MER_ATP, MG_469_2, MG_469_6MER_ATP, MG_469_7MER_ATP, 1.03.
The relaxation method has been described previously [37].
CellPACKgpu Molecule Distribution and Flex Optimization
The overall workflow for model generation and optimization is summarized in Figure 10. 3D-WC-MG models were defined in Mesoscope [9] and instances were built using CellPACKgpu [82]. CellPACKgpu builds on the method used in CellPACK [45], where the available space is voxelized, defining acceptable locations for placement of ingredients. The GPU implementation allows interactive generation of model instances including stochastic distribution of randomly-oriented rigid molecules within a given space, rigid molecules placed and oriented within membranes, and a few simple approaches to generating fibrous structures such as nucleic acids and cytoskeletal filaments. Other tools are incorporated for specialized higher-order structures, including import of entire nucleoid structures from LatticeNucleoids [37] and lipid bilayer generation using the cookie-cutter approach of LipidWrapper [46]. As described below, the current version of CellPACKgpu developed as part of this project includes additional methods for optimization, visualization, and export of model instances.
Figure 10.
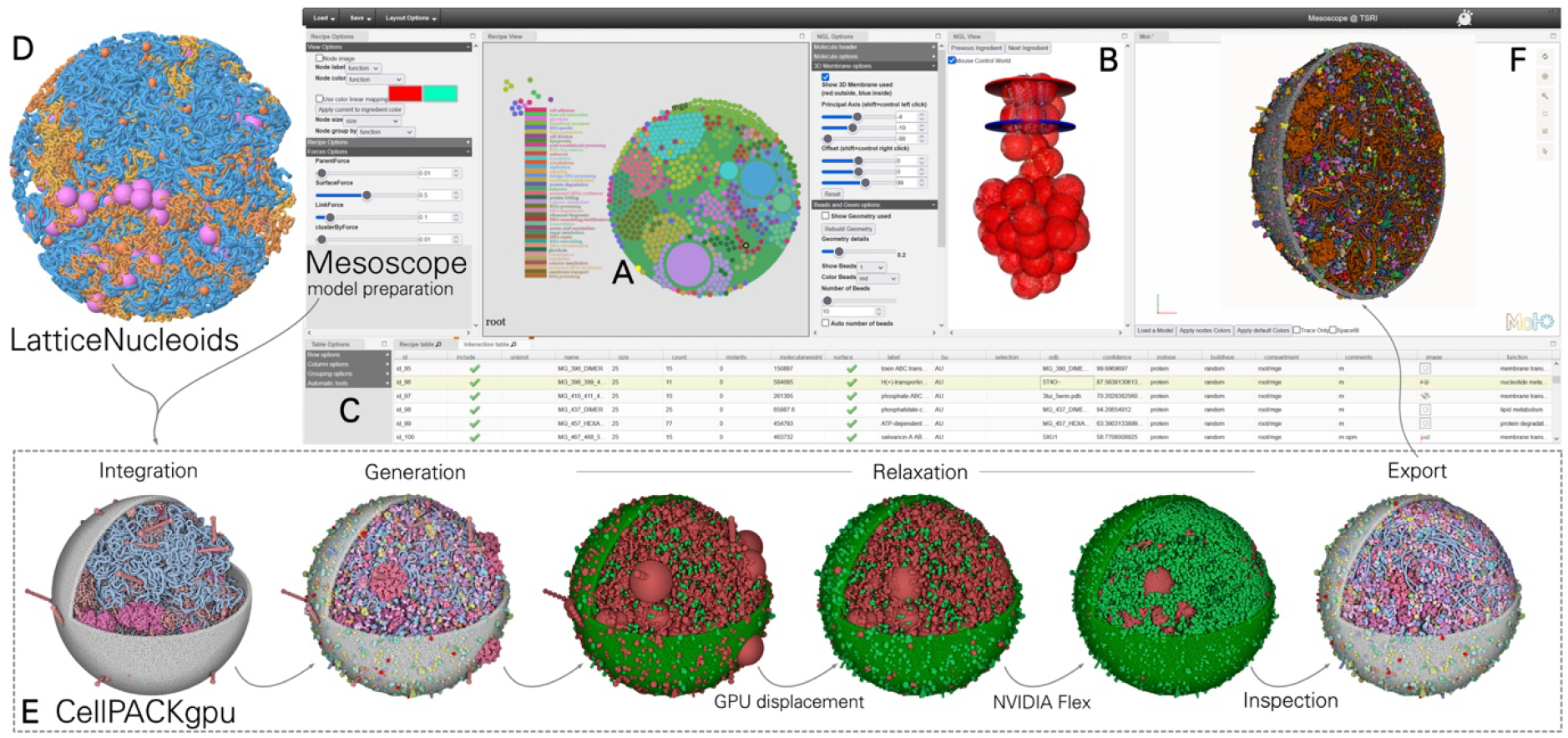
Modeling workflow for generating 3D-WC-MG model instances. A coarse-grain model of the DNA, mRNA, ribosomes and associated proteins is generated with LatticeNucleoids. The recipe is assembled and curated in Mesoscope, assigning abundance, localization, bead representations, and membrane localization for each protein and the cell size. For example, this snapshot shows the curated Frame 149 s recipe, and ATP synthase is being selected in the Mesoscope graphical browser (A, small yellow dot), launching an interactive NGL molecular viewer that allows specification of the membrane location and level of detail (B). Other parameters such as abundance and localization are included in the Mesoscope spreadsheet (C). The LatticeNucleoid model (D) and the Mesoscope recipe are integrated in CellPACKgpu (E), and several steps of model generation and relaxation produce a final model instance for inspection. The model instance may be exported in mmCIF and other formats, and then visualized using Mol* within Mesoscope (F) or used in other applications. In the MG structural models, DNA is in blue, RNA in orange, proteins in magenta, and in the three relaxation figures, molecules with steric clashes are in red.
3D-WC-MG models are typically generated from the recipe interactively in several steps (Figure 10, bottom). In CellPACKgpu, the nucleoid is embedded within a container representing the cell membrane. Nucleoid-associated molecules, represented as spheres in the LatticeNucleoid model, are then replaced with appropriately-oriented bead models. Soluble and membrane-bound ingredients are distributed randomly in the remaining soluble space and the membrane. This results in a model with all ingredients, but often showing many small overlaps between neighboring molecules. Finally, positions and orientations of molecules are optimized with the NVIDIA Flex library (developer.nvidia.com/flex) to minimize these clashes, and the model is exported and/or visualized.
LatticeNucleoid integration.
LatticeNucleoid treats proteins as spheres, so coarse-grain models of nucleoid-associated proteins need to be aligned for use in CellPACKgpu. Proteins were centered on LatticeNucleoid beads and reoriented according to a principal axis manually defined in Mesoscope for each protein type, using DNA and RNA bead coordinates as control points. LatticeNucleoid models are currently loaded into CellPACKgpu through a simple menu, and the remaining molecules are distributed around the nucleoid in numbers as defined in the recipe. The lattice models also contain information on constraints required to define interactions between nucleoid-associated molecules, DNA, and RNA, initially extracted from WC-MG data for each time step, which will be used in the relaxation.
Lipid bilayer.
Lipid bilayers are generated using a GPU implementation of the LipidWrapper algorithm [46]. LipidWrapper uses a cookie-cutter approach, by randomly placing pre-equilibrated lipid triangular patches onto the triangular mesh that represents the cell. Currently, our implementation exports models for the macromolecular components and the lipids separately and does not explicitly resolve clashes at triangle edges or with macromolecules. However, during visualization, we resolve lipid-protein overlaps on-the-fly using the bead-level representation of proteins to remove lipids within proteins.
Currently, our implementation exports models for the macromolecular components and the lipids separately and does not explicitly resolve clashes at triangle edges as in the original implementation of LipidWrapper. However, we currently use a shortcut to resolve clashes during construction and visualization of models that include lipids. Using the underlying voxelization that stores the identifier for protein instances within each voxel, we can test for overlap between every lipid residue head group and the protein beads present in the same voxel. This may be used to interactively discard lipids with clashes during the distribution of lipids using a compute shader or at render time using the vertex shader.
Optimization of models.
While the distribution of molecules by CellPACKgpu is interactive, the parallel nature of the algorithm results in overlap between neighboring molecules. To resolve these clashes in real-time we use the following approach. In the first step, we use a custom GPU implementation of a simple displacement method to relax soluble ingredients that overlap with the membrane, to move the molecules inside the compartment. This step uses the coarser level of beads for every protein. In this step we can individually select and relax exceptionally-large ingredients such as PDH. In the second step we switch to the finer level of beads and use the NVIDIA Flex library (developer.nvidia.com/flex) to relax the entire model while the surface proteins and an optional selection of proteins (e.g. PDH) are fixed in place. Proteins are defined as rigid bodies and nucleic acids as bead-spring chain models. The chains of beads are attached by springs between consecutive beads and additional springs are added to control the chain flexibility. Springs in Flex are specified as pairs of particle indices, a rest length, and a stiffness coefficient, however, they are not a spring in the classical sense, but rather a distance constraint with a user-specified stiffness. For the current work, we used the default stiffness coefficient of 1.0 provided in Flex. We specified a rest-length equal to two times the particle radius for all direct connections, and we used the distance between the particles in a fully extended state for constraining the DNA flexibility. In the current work, we used 10 springs (bead n to beads n+2 to n+11) to obtain a persistence length of around 35nm. Nucleic-acid-binding protein constraints use similar springs between the local beads of the rigid body and beads along the nucleic acid chain.
As some molecules present interior holes in their coarse-grain representation, other proteins or nucleic acids can get interlocked and never resolve overlaps. We resolve locked molecules by repeatedly increasing and decreasing the main radius of collision during the simulation. This breathing mechanism allows locked beads to escape their trap. Since the system is under stress due to the DNA and nucleic-acid-binding proteins, we add a final step and relax the entire system using a slightly expanded radius for all beads (Flex uses a unique radius for all beads). The Standard Protocol with 17Å beads yielded a model with ~7% of the proteins showing steric overlaps with neighbors. We tested expanded radii in the range of 18Å to 27Å (see Figure 5 inset graph) and found optimal reduction of contacts with a radius of 21Å, which resulted in a model with only 2.2 % of proteins with steric contacts for all models.
Typically, we monitor the current level of clashes during the interactive optimization by visually inspecting the model with a cutting plane and coloring the molecules that overlap. Overlaps are estimated using the list of closest pairs of protein-protein or protein-fiber control points retrieved in the underlying packing grid. The overlap is then computed using distances between beads representing the two molecules. Two proteins are considered overlapping when any of their beads are at a distance lesser than the sum of their radii minus a constant threshold (typically in the range of 0.0 to one bead radius, or 17Å). Finally, the number of overlapping proteins is reported to the user.
Model export.
Final models are exported in several file formats, including a binary format specific to CellPACK, a custom PDB file holding the coarse-grain bead coordinates, and an mmCIF file holding the coarse-grain or the fully atomistic model. We heavily rely on the entity biological unit matrices definition of the mmCIF dictionary to reduce the final size of the files. Lipids can also be exported using the same matrix definition, however they retain overlap at triangle edges and protein/lipid positions.
Visualization and Interactivity
As mentioned above, CellPACKgpu is built on top of the cellVIEW rendering paradigm [51], providing advanced features such as dynamic coloring and clipping geometry. We further developed additional rendering capabilities to address several challenges encountered with the 3D-WC-MG model. Most of the rendering development was focused on the DNA and the RNA. The dynamic level-of-details approach used for adaptive display of proteins has been extended to nucleic acids and is also now capable of using a sequence. Namely, we can tailor the units that are repeated along the control points that define the fibers to store the mapping for atom, segment, gene and transcription unit and retrieve it at run time, allowing selection and coloration of the DNA by base pair or by gene. The gene id is also stored for the protein monomer, thus we are able to specify the color of a given mRNA, which will in turn color all the protein products of that particular gene that are present in the model.
To improve usability, we exposed options through the user interface to manually choose the level-of-detail, change the coloring methods (automatic, using loaded color palette, using specific information such as gene ID), load and save color palette files (user provided color per ingredient) and a widget to change color palettes on the fly, toggle the shadows, enable a spinning model, enable visibility of a ghost (e.g. contours of objects hidden by cutting primitives), visual jitter, and coloring based on overlapping beads. We added two specific panels, one for the relaxation option and one for the MG modeling recipes and time stamp. The relaxation panel is used to switch between relaxation methods: GPU, embedded Flex, external Flex. For each method, options are available to change the bead display and the DNA persistence length constraints. The MG-specific panel is used to switch between the two different recipes as well as the different time steps.
Supplementary Material
Supplementary Table S1: MG Proteins List of all protein ingredients, monomers, and complexes included in our 3D-WC-MG recipe. The table reports the type of each ingredient and its molecular weight, monomer length, DNA interaction information, complex biosynthesis, functional annotations, protein localization (compartment), and details on the structural representation selected for each ingredient, including data source, templates for homology models, PDB ID and chain ID for experimental structures, and quality measures.
Supplementary Table S2: MG Genes This table lists the genes captured by the 3D-WC-MG model, including the genomic location of each MG gene; the type of each gene and codon and amino acid for tRNA genes; start coordinate, length, direction; the transcription unit into which each gene is organized; the essentiality of each gene; and a functional annotation where available. Additionally, for each protein-coding gene, the table reports the UniProt ID and the name of the protein monomer.
Supplementary Table S3: Assignment of Membrane Proteins Protein monomers listed in this table were assigned to different cell compartments in the WC-MG and CYT-MG models. Data from both models are reported on this table, with computational predictions of protein localization and signal peptides (BUSCA, SignalP-5.0, SOSUI, Phobius, Psortb), and the consensus localization that is used in the current study.
HIGHLIGHTS.
3D whole cell modeling requires new bioinformatics and computational methods
Information for generating 3D cell models is gathered and curated with Mesoscope
A multi-step workflow generates structural models of an entire proteome
Entire bacterial cells are interactively modeled and visualized with CellPACKgpu
This work demonstrates the feasibility of building 3D models of an entire cell
ACKNOWLEDGEMENTS
We thank Michel Sanner, Stefano Forli, and Andrew Jewett for helpful discussions and Jerome Eberhardt for coding support. We thank the SWISS-MODEL team and Prof. McGuffin for running modeling predictions on their local servers. This work was supported by NIH R01 GM120604 (DSG), R35 GM119771 (JK), and an Allen Discovery Center award from the Paul G. Allen Family Foundation (MWC). This is manuscript 30120 from the Scripps Research Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- 1.Tomita M (2001) Whole-cell simulation: a grand challenge of the 21st century. Trends Biotechnol. 19, 205–210 [DOI] [PubMed] [Google Scholar]
- 2.Goldberg AP, Szigeti B, Chew YH, Sekar JA, Roth YD, and Karr JR (2018) Emerging whole-cell modeling principles and methods. Curr. Opin. Biotechnol 51, 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rust MJ, Bates M, and Zhuang X (2006) Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3, 793–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng Y (2015) Single-particle cryo-EM at crystallographic resolution. Cell. 161, 450–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feig M, and Sugita Y (2019) Whole-cell models and simulations in molecular detail. Annu. Rev. Cell Dev. Biol 35, 191–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goodsell DS, Olson AJ, and Forli S (2020) Art and science of the cellular mesoscale. Trends Biochem. Sci 45, 472–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Im W, Liang J, Olson A, Zhou H-X, Vajda S, and Vakser IA (2016) Challenges in structural approaches to cell modeling. J. Mol. Biol 428, 2943–2964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singla J, McClary KM, White KL, Alber F, Sali A, and Stevens RC (2018) Opportunities and challenges in building a spatiotemporal multi-scale model of the human pancreatic β cell. Cell. 173, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Autin L, Maritan M, Barbaro BA, Gardner A, Olson AJ, Sanner M, and Goodsell DS (2020) Mesoscope: A web-based tool for mesoscale data integration and curation. Workshop Mol. Graph. Vis. Anal. Mol. Data Eurographics Assoc 10.2312/MOLVA.20201098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goodsell DS, Franzen MA, and Herman T (2018) From atoms to cells: Using mesoscale landscapes to construct visual narratives. J. Mol. Biol 430, 3954–3968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu H, Li F, Sánchez BJ, Zhu Z, Li G, Domenzain I, Marcišauskas S, Anton PM, Lappa D, Lieven C, Beber ME, Sonnenschein N, Kerkhoven EJ, and Nielsen J (2019) A consensus S. cerevisiae metabolic model Yeast8 and its ecosystem for comprehensively probing cellular metabolism. Nat. Commun 10, 3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Modi V, and Dunbrack RL (2016) Assessment of refinement of template-based models in CASP11: Template-Based Models in CASP11. Proteins Struct. Funct. Bioinforma 84, 260–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, Lepore R, and Schwede T (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, and Hassabis D (2021) Highly accurate protein structure prediction with AlphaFold. Nature. 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Evans R, O’Neill M, Pritzel A, Antropova N, Senior A, Green T, Žídek A, Bates R, Blackwell S, Yim J, Ronneberger O, Bodenstein S, Zielinski M, Bridgland A, Potapenko A, Cowie A, Tunyasuvunakool K, Jain R, Clancy E, Kohli P, Jumper J, and Hassabis D (2021) Protein complex prediction with AlphaFold-Multimer, Bioinformatics, 10.1101/2021.10.04.463034 [DOI] [Google Scholar]
- 16.Baek M, DiMaio F, Anishchenko I, Dauparas J, Ovchinnikov S, Lee GR, Wang J, Cong Q, Kinch LN, Schaeffer RD, Millán C, Park H, Adams C, Glassman CR, DeGiovanni A, Pereira JH, Rodrigues AV, van Dijk AA, Ebrecht AC, Opperman DJ, Sagmeister T, Buhlheller C, Pavkov-Keller T, Rathinaswamy MK, Dalwadi U, Yip CK, Burke JE, Garcia KC, Grishin NV, Adams PD, Read RJ, and Baker D (2021) Accurate prediction of protein structures and interactions using a three-track neural network. Science. 10.1126/science.abj8754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rigden DJ, and Fernández XM (2021) The 2021 Nucleic Acids Research database issue and the online molecular biology database collection. Nucleic Acids Res. 49, D1–D9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szigeti B, Roth YD, Sekar JAP, Goldberg AP, Pochiraju SC, and Karr JR (2018) A blueprint for human whole-cell modeling. Curr. Opin. Syst. Biol 7, 8–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.wwPDB Consortium, Burley SK, Berman HM, Bhikadiya C, Bi C, Chen L, Costanzo LD, Christie C, Duarte JM, Dutta S, Feng Z, Ghosh S, Goodsell DS, Green RK, Guranovic V, Guzenko D, Hudson BP, Liang Y, Lowe R, Peisach E, Periskova I, Randle C, Rose A, Sekharan M, Shao C, Tao Y-P, Valasatava Y, Voigt M, Westbrook J, Young J, Zardecki C, Zhuravleva M, Kurisu G, Nakamura H, Kengaku Y, Cho H, Sato J, Kim JY, Ikegawa Y, Nakagawa A, Yamashita R, Kudou T, Bekker G-J, Suzuki H, Iwata T, Yokochi M, Kobayashi N, Fujiwara T, Velankar S, Kleywegt GJ, Anyango S, Armstrong DR, Berrisford JM, Conroy MJ, Dana JM, Deshpande M, Gane P, Gáborová R, Gupta D, Gutmanas A, Koča J, Mak L, Mir S, Mukhopadhyay A, Nadzirin N, Nair S, Patwardhan A, Paysan-Lafosse T, Pravda L, Salih O, Sehnal D, Varadi M, Vařeková R, Markley JL, Hoch JC, Romero PR, Baskaran K, Maziuk D, Ulrich EL, Wedell JR, Yao H, Livny M, and Ioannidis YE (2019) Protein Data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res. 47, D520–D528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.UniProt Consortium (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 47, D506–D515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perez-Riverol Y, Bai M, da Veiga Leprevost F, Squizzato S, Park YM, Haug K, Carroll AJ, Spalding D, Paschall J, Wang M, del-Toro N, Ternent T, Zhang P, Buso N, Bandeira N, Deutsch EW, Campbell DS, Beavis RC, Salek RM, Sarkans U, Petryszak R, Keays M, Fahy E, Sud M, Subramaniam S, Barbera A, Jiménez RC, Nesvizhskii AI, Sansone S-A, Steinbeck C, Lopez R, Vizcaíno JA, Ping P, and Hermjakob H (2017) Discovering and linking public omics data sets using the Omics Discovery Index. Nat. Biotechnol 35, 406–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roth YD, Lian Z, Pochiraju S, Shaikh B, and Karr JR (2021) Datanator: an integrated database of molecular data for quantitatively modeling cellular behavior. Nucleic Acids Res. 49, D516–D522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keseler IM, Mackie A, Santos-Zavaleta A, Billington R, Bonavides-Martínez C, Caspi R, Fulcher C, Gama-Castro S, Kothari A, Krummenacker M, Latendresse M, Muñiz-Rascado L, Ong Q, Paley S, Peralta-Gil M, Subhraveti P, Velázquez-Ramírez DA, Weaver D, Collado-Vides J, Paulsen I, and Karp PD (2017) The EcoCyc database: reflecting new knowledge about Escherichia coli K-12. Nucleic Acids Res. 45, D543–D550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kadir SR, Lilja A, Gunn N, Strong C, Hughes RT, Bailey BJ, Rae J, Parton RG, and McGhee J (2021) Nanoscape, a data-driven 3D real-time interactive virtual cell environment. eLife. 10, e64047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson GT, Goodsell DS, Autin L, Forli S, Sanner MF, and Olson AJ (2014) 3D molecular models of whole HIV-1 virions generated with cellPACK. Faraday Discuss. 169, 23–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amaro RE, and Mulholland AJ (2018) Multiscale methods in drug design bridge chemical and biological complexity in the search for cures. Nat. Rev. Chem 2, 0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karr JR, Sanghvi JC, Macklin DN, Gutschow MV, Jacobs JM, Bolival B, Assad-Garcia N, Glass JI, and Covert MW (2012) A whole-cell computational model predicts phenotype from genotype. Cell. 150, 389–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tully JG, Taylor-Robinson D, Rose DL, Cole RM, and Bove JM (1983) Mycoplasma genitalium, a new species from the human urogenital tract. Int. J. Syst. Bacteriol 33, 387–396 [Google Scholar]
- 29.Ando T, and Skolnick J (2010) Crowding and hydrodynamic interactions likely dominate in vivo macromolecular motion. Proc. Natl. Acad. Sci 107, 18457–18462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGuffee SR, and Elcock AH (2010) Diffusion, crowding & protein stability in a dynamic molecular model of the bacterial cytoplasm. PLoS Comput. Biol 6, e1000694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cossins BP, Jacobson MP, and Guallar V (2011) A new view of the bacterial cytosol environment. PLoS Comput. Biol 7, e1002066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frembgen-Kesner T, and Elcock AH (2013) Computer simulations of the bacterial cytoplasm. Biophys. Rev 5, 109–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oliveira Bortot L, Bashardanesh Z, and van der Spoel D (2020) Making soup: Preparing and validating models of the bacterial cytoplasm for molecular simulation. J. Chem. Inf. Model 60, 322–331 [DOI] [PubMed] [Google Scholar]
- 34.Feig M, Harada R, Mori T, Yu I, Takahashi K, and Sugita Y (2015) Complete atomistic model of a bacterial cytoplasm for integrating physics, biochemistry, and systems biology. J. Mol. Graph. Model 58, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu I, Mori T, Ando T, Harada R, Jung J, Sugita Y, and Feig M (2016) Biomolecular interactions modulate macromolecular structure and dynamics in atomistic model of a bacterial cytoplasm. eLife. 5, e19274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hacker WC, Li S, and Elcock AH (2017) Features of genomic organization in a nucleotide-resolution molecular model of the Escherichia coli chromosome. Nucleic Acids Res. 45, 7541–7554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goodsell DS, Autin L, and Olson AJ (2018) Lattice models of bacterial nucleoids. J. Phys. Chem. B 122, 5441–5447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yildirim A, and Feig M (2018) High-resolution 3D models of Caulobacter crescentus chromosome reveal genome structural variability and organization. Nucleic Acids Res. 46, 3937–3952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gilbert BR, Thornburg ZR, Lam V, Rashid F-ZM, Glass JI, Villa E, Dame RT, and Luthey-Schulten Z (2021) Generating Chromosome Geometries in a Minimal Cell From Cryo-Electron Tomograms and Chromosome Conformation Capture Maps. Front. Mol. Biosci 8, 644133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amaro RE, Ieong PU, Huber G, Dommer A, Steven AC, Bush RM, Durrant JD, and Votapka LW (2018) A computational assay that explores the hemagglutinin/neuraminidase functional balance reveals the neuraminidase secondary site as a novel anti-influenza target. ACS Cent. Sci 4, 1570–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Durrant JD, Kochanek SE, Casalino L, Ieong PU, Dommer AC, and Amaro RE (2020) Mesoscale All-atom influenza virus simulations suggest new substrate binding mechanism. ACS Cent. Sci 6, 189–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, Müller SA, Rammner B, Gräter F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmüller H, Heuser J, Wieland F, and Jahn R (2006) Molecular anatomy of a trafficking organelle. Cell. 127, 831–846 [DOI] [PubMed] [Google Scholar]
- 43.Wilhelm BG, Mandad S, Truckenbrodt S, Krohnert K, Schafer C, Rammner B, Koo SJ, Classen GA, Krauss M, Haucke V, Urlaub H, and Rizzoli SO (2014) Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science. 344, 1023–1028 [DOI] [PubMed] [Google Scholar]
- 44.Singharoy A, Maffeo C, Delgado-Magnero KH, Swainsbury DJK, Sener M, Kleinekathöfer U, Vant JW, Nguyen J, Hitchcock A, Isralewitz B, Teo I, Chandler DE, Stone JE, Phillips JC, Pogorelov TV, Mallus MI, Chipot C, Luthey-Schulten Z, Tieleman DP, Hunter CN, Tajkhorshid E, Aksimentiev A, and Schulten K (2019) Atoms to phenotypes: Molecular design principles of cellular energy metabolism. Cell. 179, 1098–1111.e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson GT, Autin L, Al-Alusi M, Goodsell DS, Sanner MF, and Olson AJ (2015) cellPACK: a virtual mesoscope to model and visualize structural systems biology. Nat. Methods 12, 85–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Durrant JD, and Amaro RE (2014) LipidWrapper: An algorithm for generating large-scale membrane models of arbitrary geometry. PLoS Comput. Biol 10, e1003720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karr JR, Sanghvi JC, Macklin DN, Arora A, and Covert MW (2012) WholeCellKB: model organism databases for comprehensive whole-cell models. Nucleic Acids Res. 41, D787–D792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karr JR, Phillips NC, and Covert MW (2014) WholeCellSimDB: a hybrid relational/HDF database for whole-cell model predictions. Database. 2014, bau095–bau095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee R, Karr JR, and Covert MW (2013) WholeCellViz: data visualization for whole-cell models. BMC Bioinformatics. 14, 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jewett AI, Zhuang Z, and Shea J-E (2013) Moltemplate: A coarse-Grained model assembly tool. Biophys. J 104, 169a [Google Scholar]
- 51.Muzic ML, Autin L, Parulek J, and Viola I (2015) cellVIEW: a tool for illustrative and multi-scale rendering of large biomolecular datasets. Eurographics Workshop Vis. Comput. Biol. Med 10.2312/VCBM.20151209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, and Steinegger M (2021) ColabFold - Making protein folding accessible to all, Bioinformatics, 10.1101/2021.08.15.456425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guzenko D, Lafita A, Monastyrskyy B, Kryshtafovych A, and Duarte JM (2019) Assessment of protein assembly prediction in CASP13. Proteins Struct. Funct. Bioinforma 87, 1190–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lafita A, Bliven S, Kryshtafovych A, Bertoni M, Monastyrskyy B, Duarte JM, Schwede T, and Capitani G (2018) Assessment of protein assembly prediction in CASP12. Proteins Struct. Funct. Bioinforma 86, 247–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lensink MF, Brysbaert G, Nadzirin N, Velankar S, Chaleil RAG, Gerguri T, Bates PA, Laine E, Carbone A, Grudinin S, Kong R, Liu R, Xu X, Shi H, Chang S, Eisenstein M, Karczynska A, Czaplewski C, Lubecka E, Lipska A, Krupa P, Mozolewska M, Golon Ł, Samsonov S, Liwo A, Crivelli S, Pagès G, Karasikov M, Kadukova M, Yan Y, Huang S, Rosell M, Rodríguez-Lumbreras LA, Romero-Durana M, Díaz-Bueno L, Fernandez-Recio J, Christoffer C, Terashi G, Shin W, Aderinwale T, Maddhuri Venkata Subraman SR, Kihara D, Kozakov D, Vajda S, Porter K, Padhorny D, Desta I, Beglov D, Ignatov M, Kotelnikov S, Moal IH, Ritchie DW, Chauvot de Beauchêne I, Maigret B, Devignes M, Ruiz Echartea ME, Barradas-Bautista D, Cao Z, Cavallo L, Oliva R, Cao Y, Shen Y, Baek M, Park T, Woo H, Seok C, Braitbard M, Bitton L, Scheidman-Duhovny D, Dapkūnas J, Olechnovič K, Venclovas Č, Kundrotas PJ, Belkin S, Chakravarty D, Badal VD, Vakser IA, Vreven T, Vangaveti S, Borrman T, Weng Z, Guest JD, Gowthaman R, Pierce BG, Xu X, Duan R, Qiu L, Hou J, Ryan Merideth B, Ma Z, Cheng J, Zou X, Koukos PI, Roel-Touris J, Ambrosetti F, Geng C, Schaarschmidt J, Trellet ME, Melquiond ASJ, Xue L, Jiménez-García B, Noort CW, Honorato RV, Bonvin AMJJ, and Wodak SJ (2019) Blind prediction of homo- and hetero-protein complexes: The CASP13-CAPRI experiment. Proteins Struct. Funct. Bioinforma 87, 1200–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O’Donoghue SI, Baldi BF, Clark SJ, Darling AE, Hogan JM, Kaur S, Maier-Hein L, McCarthy DJ, Moore WJ, Stenau E, Swedlow JR, Vuong J, and Procter JB (2018) Visualization of Biomedical Data. Annu. Rev. Biomed. Data Sci 1, 275–304 [Google Scholar]
- 57.Gardner A, Autin L, Fuentes D, Maritan M, Barad BA, Medina M, Olson AJ, Grotjahn DA, and Goodsell DS (2021) CellPAINT: Turnkey illustration of molecular cell biology. Front. Bioinforma 1, 660936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alva V, Nam S-Z, Söding J, and Lupas AN (2016) The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res. 44, W410–W415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zimmermann L, Stephens A, Nam S-Z, Rau D, Kübler J, Lozajic M, Gabler F, Söding J, Lupas AN, and Alva V (2018) A completely reimplemented MPI Bioinformatics Toolkit with a new HHpred server at its core. J. Mol. Biol 430, 2237–2243 [DOI] [PubMed] [Google Scholar]
- 60.Kelley LA, Mezulis S, Yates CM, Wass MN, and Sternberg MJE (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc 10, 845–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McGuffin LJ, Adiyaman R, Maghrabi AHA, Shuid AN, Brackenridge DA, Nealon JO, and Philomina LS (2019) IntFOLD: an integrated web resource for high performance protein structure and function prediction. Nucleic Acids Res. 47, W408–W413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Källberg M, Margaryan G, Wang S, Ma J, and Xu J (2014) RaptorX server: A resource for template-based protein structure modeling. in Protein Structure Prediction (Kihara D ed), pp. 17–27, Methods in Molecular Biology, Springer New York, New York, NY, 1137, 17–27 [DOI] [PubMed] [Google Scholar]
- 63.Yang J, Yan R, Roy A, Xu D, Poisson J, and Zhang Y (2015) The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baek M, Park T, Heo L, Park C, and Seok C (2017) GalaxyHomomer: a web server for protein homo-oligomer structure prediction from a monomer sequence or structure. Nucleic Acids Res. 45, W320–W324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kryshtafovych A, Schwede T, Topf M, Fidelis K, and Moult J (2019) Critical assessment of methods of protein structure prediction (CASP)—Round XIII. Proteins Struct. Funct. Bioinforma 87, 1011–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haas J, Barbato A, Behringer D, Studer G, Roth S, Bertoni M, Mostaguir K, Gumienny R, and Schwede T (2018) Continuous Automated Model EvaluatiOn (CAMEO) complementing the critical assessment of structure prediction in CASP12. Proteins Struct. Funct. Bioinforma 86, 387–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Haas J, Gumienny R, Barbato A, Ackermann F, Tauriello G, Bertoni M, Studer G, Smolinski A, and Schwede T (2019) Introducing “best single template” models as reference baseline for the Continuous Automated Model Evaluation (CAMEO). Proteins Struct. Funct. Bioinforma 87, 1378–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lewis JS, Spenkelink LM, Jergic S, Wood EA, Monachino E, Horan NP, Duderstadt KE, Cox MM, Robinson A, Dixon NE, and van Oijen AM (2017) Single-molecule visualization of fast polymerase turnover in the bacterial replisome. eLife. 6, e23932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Spenkelink LM, Lewis JS, Jergic S, Xu Z-Q, Robinson A, Dixon NE, and van Oijen AM (2019) Recycling of single-stranded DNA-binding protein by the bacterial replisome. Nucleic Acids Res. 47, 4111–4123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Duderstadt KE, Chuang K, and Berger JM (2011) DNA stretching by bacterial initiators promotes replication origin opening. Nature. 478, 209–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Milne JLS, Wu X, Borgnia MJ, Lengyel JS, Brooks BR, Shi D, Perham RN, and Subramaniam S (2006) Molecular structure of a 9-MDa icosahedral pyruvate dehydrogenase subcomplex containing the E2 and E3 enzymes using cryoelectron microscopy. J. Biol. Chem 281, 4364–4370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Milne JLS (2002) Molecular architecture and mechanism of an icosahedral pyruvate dehydrogenase complex: a multifunctional catalytic machine. EMBO J. 21, 5587–5598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Savojardo C, Martelli PL, Fariselli P, Profiti G, and Casadio R (2018) BUSCA: an integrative web server to predict subcellular localization of proteins. Nucleic Acids Res. 46, W459–W466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hirokawa T, Boon-Chieng S, and Mitaku S (1998) SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics. 14, 378–379 [DOI] [PubMed] [Google Scholar]
- 75.Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Sahinalp SC, Ester M, Foster LJ, and Brinkman FSL (2010) PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 26, 1608–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Almagro Armenteros JJ, Tsirigos KD, Sønderby CK, Petersen TN, Winther O, Brunak S, von Heijne G, and Nielsen H (2019) SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol 37, 420–423 [DOI] [PubMed] [Google Scholar]
- 77.Käll L, Krogh A, and Sonnhammer ELL (2004) A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol 338, 1027–1036 [DOI] [PubMed] [Google Scholar]
- 78.Maghrabi AHA, and McGuffin LJ (2017) ModFOLD6: an accurate web server for the global and local quality estimation of 3D protein models. Nucleic Acids Res. 45, W416–W421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Olechnovič K, and Venclovas Č (2019) VoroMQA web server for assessing three-dimensional structures of proteins and protein complexes. Nucleic Acids Res. 47, W437–W442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lomize MA, Pogozheva ID, Joo H, Mosberg HI, and Lomize AL (2012) OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–D376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Trussart M, Yus E, Martinez S, Baù D, Tahara YO, Pengo T, Widjaja M, Kretschmer S, Swoger J, Djordjevic S, Turnbull L, Whitchurch C, Miyata M, Marti-Renom MA, Lluch-Senar M, and Serrano L (2017) Defined chromosome structure in the genome-reduced bacterium Mycoplasma pneumoniae. Nat. Commun 8, 14665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Klein T, Autin L, Kozlikova B, Goodsell DS, Olson A, Groller ME, and Viola I (2018) Instant construction and visualization of crowded biological environments. IEEE Trans. Vis. Comput. Graph 24, 862–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Diebold-Durand M-L, Lee H, Ruiz Avila LB, Noh H, Shin H-C, Im H, Bock FP, Bürmann F, Durand A, Basfeld A, Ham S, Basquin J, Oh B-H, and Gruber S (2017) Structure of full-length SMC and rearrangements required for chromosome organization. Mol. Cell 67, 334–347.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tomasello G, Armenia I, and Molla G (2020) The Protein Imager: a full-featured online molecular viewer interface with server-side HQ-rendering capabilities. Bioinformatics. 36, 2909–2911 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1: MG Proteins List of all protein ingredients, monomers, and complexes included in our 3D-WC-MG recipe. The table reports the type of each ingredient and its molecular weight, monomer length, DNA interaction information, complex biosynthesis, functional annotations, protein localization (compartment), and details on the structural representation selected for each ingredient, including data source, templates for homology models, PDB ID and chain ID for experimental structures, and quality measures.
Supplementary Table S2: MG Genes This table lists the genes captured by the 3D-WC-MG model, including the genomic location of each MG gene; the type of each gene and codon and amino acid for tRNA genes; start coordinate, length, direction; the transcription unit into which each gene is organized; the essentiality of each gene; and a functional annotation where available. Additionally, for each protein-coding gene, the table reports the UniProt ID and the name of the protein monomer.
Supplementary Table S3: Assignment of Membrane Proteins Protein monomers listed in this table were assigned to different cell compartments in the WC-MG and CYT-MG models. Data from both models are reported on this table, with computational predictions of protein localization and signal peptides (BUSCA, SignalP-5.0, SOSUI, Phobius, Psortb), and the consensus localization that is used in the current study.
Data Availability Statement
The CellPACK binaries dedicated to the MG modeling, the source code and scripts for generating LatticeNucleoid MG nucleoid models, representative models (in different file formats including PDB, CIF, binary), ingredient modeling notes and scripts for the modeling workflow, and tables and structure files for the 3D-WC-MG recipes are freely available on GitHub at: https://github.com/ccsb-scripps/MycoplasmaGenitalium. Mesoscope is available online at mesoscope.scripps.edu/beta, and examples of the Frame 149 s curated and auto models are available in the Load→New Recipe→From Examples menu. Note that these examples do not include lipids, which seriously impact performance of the Mol* visualization.