

Abstract
Cells activate distinctive regulatory pathways that prevent excessive initiation of DNA replication to achieve timely and accurate genome duplication. Excess DNA synthesis is constrained by protein-DNA interactions that inhibit initiation at dormant origins. In parallel, specific modifications of pre-replication complexes prohibit post-replicative origin re-licensing. Replication stress ensues when the controls that prevent excess replication are missing in cancer cells, which often harbor extrachromosomal DNA that can be further amplified by recombination-mediated processes to generate chromosomal translocations. The genomic instability that accompanies excess replication origin activation can provide a promising target for therapeutic intervention. Here we review molecular pathways that modulate replication origin dormancy, prevent excess origin activation, and detect, encapsulate and eliminate persistent excess DNA.
Keywords: Replication origins, dormant origins, re-replication, genomic instability, DNA damage, over-replication, extrachromosomal DNA, extrachromosomal circular DNA
Flexible use of replication origins
To achieve complete and timely chromosome duplication, large metazoan genomes initiate replication from thousands of sites (replication origins) within a consistent time frame during the S-phase of the cell cycle. Typically, genome duplication under unperturbed conditions requires initiation from ~50–70k origins. Each of these origins replicates a defined region, often referred to as a replicon. Replicons are organized into larger, synchronously duplicated chromatin segments known as ‘replication domains’. DNA replication proceeds under a finely orchestrated spatio-temporal program to achieve a consistent replication order (i.e., ‘replication timing’), eventually duplicating the entire genome once prior to each cell division. The failure to properly regulate origin selection and activation may result in catastrophic genomic instability and potentially tumorigenesis.
In budding yeast, replication starts at distinct replication origins that share a consensus sequence [1–4]. The sequence-specificity of yeast replication origins can be recapitulated in vitro, as replicative helicases can assemble with low affinity on non-origin DNA sequences, but with high affinity on canonical origin sequences [5]. In metazoans, replication origins do not share similar consensus sequences, but they do share critical features such as purine/pyrimidine asymmetry [1, 4]. Yet, replication initiation processes in yeast and metazoans exhibit similar fundamental principles in that they involve two distinct steps: first, the assembly of pre-replication complexes (‘pre-RCs’) on potential origins (‘licensing’) during the G1 phase of the cell cycle, and second, the activation of the pre-RC complexes by S-phase-specific kinases to facilitate helicase activity. In parallel with the second step, S-phase kinases phosphorylate pre-RC components to prevent further pre-RC assembly, ensuring that licensing of additional origins is blocked until the next cell division [6–10].
In metazoans, pre-RCs are assembled in excess at potential origins. Most origins are activated intermittently so that only about a fraction of these origins actually initiate replication during each cell cycle. Excess pre-RCs serve to ensure a sufficient abundance of backup origins that are activated in cases of replication fork failure [11, 12]. Because only a fraction of the potential origins initiate replication [1, 2, 4, 13], the remaining dormant licensed origins are passively replicated by replication forks initiated from adjacent origins (Figure 1). The intermittent activation of replication origins is evidenced experimentally by the results of single molecule analyses of DNA replication, in which origins seem to be separated by 120–200 kb, whereas population-based sequencing analyses have detected much shorter inter-origin distances [1, 3, 4, 12, 14].
Figure 1. Replication origin flexibility.
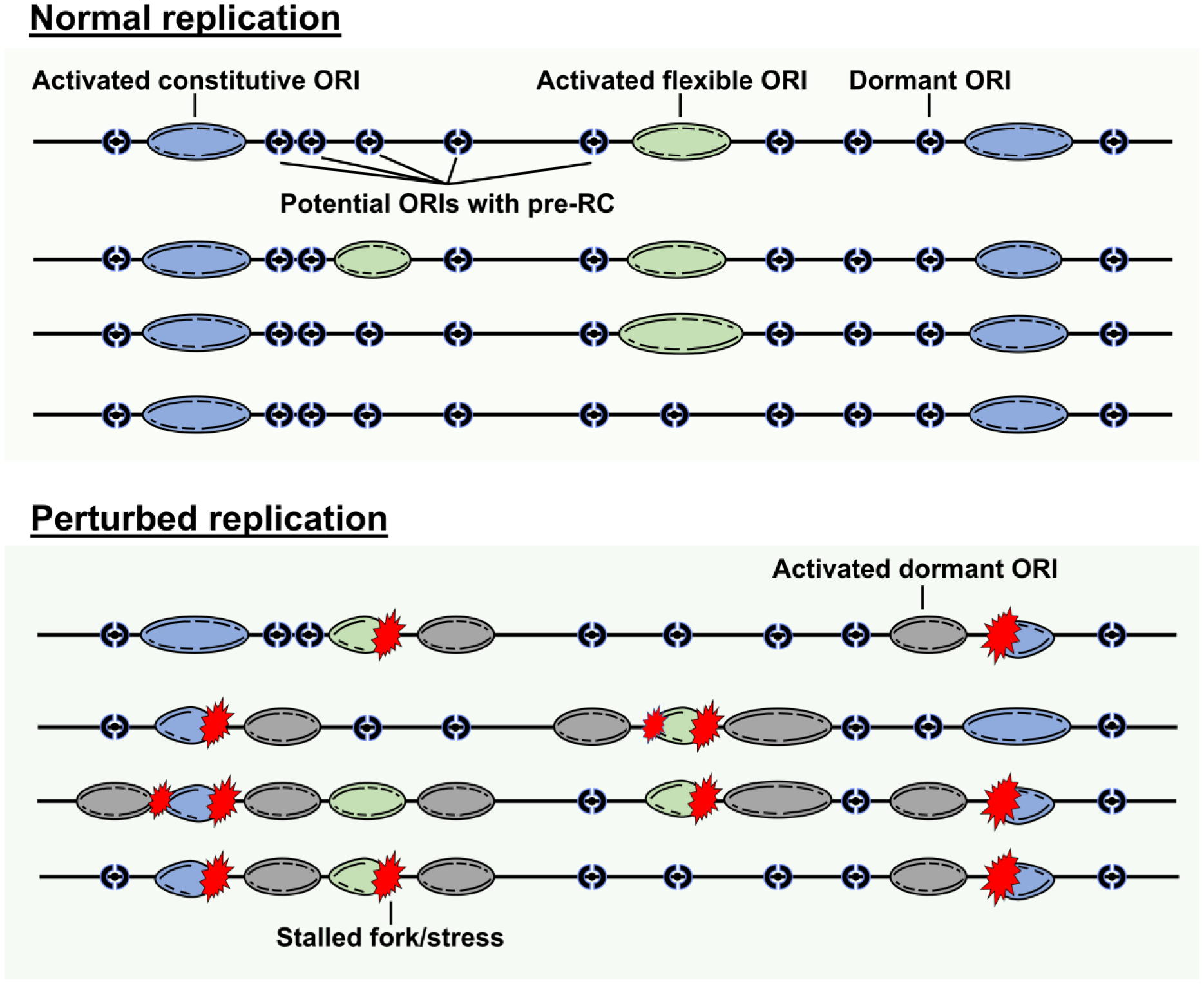
Replication origins can be designated either as constitutive (origins that initiate replication in all cells), flexible (origins that initiate replication irregularly with initiation occurring in some cells but not others) or dormant (origins that nucleate pre-RC assembly but never or rarely initiate replication when DNA synthesis is unperturbed). Some dormant origins are activated during perturbed replication; those origins are typically in the vicinity of stalled or damaged replication forks (bottom panel) and they serve to complete DNA replication in the loci where replication stress occurred.
The choice of active replication origins is intermittent and flexible, but it is not random. Metazoan cells contain a group of origins that are constantly active in a broad range of cell and tissue types, and referred to as constitutive origins, as well as a group of origins that could initiate replication intermittently or in a cell-type-specific manner, hence referred to as flexible origins. Cells also contain a group of potential origin sequences that nucleate pre-RCs but are not activated during routine cell growth. These potential origins, however, can be activated in the presence of local replication fork stalling or in response to global stress agents. Here we broadly refer to the origins (both constitutive and flexible) that are utilized during normal, unperturbed growth as ‘baseline origins’ and to the origins that do not initiate replication during unperturbed growth as ‘dormant origins’.
Although not necessary for routine genome duplication, dormant origins are key determinants of genomic stability, facilitating faithful completion of genome duplication under perturbed conditions. They also play a critical role in prevention of mutations and other genotoxic lesions that increase with the distance travelled by replication forks. The role of dormant replication origins in ensuring genome stability during DNA replication has been extensively reviewed previously [11, 13, 15, 16]. Below we discuss current advances in understanding the selective activation of replication origins, the mechanisms underlying excess origin activation along with their consequences, and the cellular pathways that prevent excessive initiation of DNA replication or eliminate the products of such excess replication.
Signaling pathways constraining replication origin activation.
Distribution of pre-replication complexes
Pre-RCs are nucleated in both yeast and metazoans by an Origin Recognition Complex (ORC), which facilitates the sequential recruitment of CDC6, CDT1 adaptor proteins, and the MCM (Mini Chromosome Maintenance 2–7) complex. The S-phase kinases DDK (DBF4 Dependent Kinase) and CDK (Cyclin Dependent Kinases) activate the MCM complex and facilitate recruitment of other components, CDC45 and GINS, to form the active helicase (CMG: CDC45-MCM-GINS) that catalyzes DNA unwinding, facilitating DNA synthesis at replication forks (reviewed earlier [1, 2, 4]). Concomitant with the establishment of replication forks, S-phase kinases prohibit further pre-RC assembly, limiting the licensing of replication origins to the G1 phase prior to the onset of DNA synthesis. The separation of the genome duplication process into two distinct phases, origin licensing in G1 and replication initiation in S phase, promote faithful replication by preventing relicensing and re-activation of origins that have already initiated replication [1, 6, 7, 9, 10]. Disruption of the balance between licensing and initiation can result in untimely or improper replication initiation. When licensing frequency is reduced (for example, in cells harboring mutations in MCM proteins), cells can proceed into the S-phase with reduced MCM loading but they exhibit increased susceptibility to fork stalling, replicative stress, and genome instability even in the absence of exogenous DNA damaging agents [6–8, 17, 18]. On the other hand, increased or untimely activation of licensed origins can result in excess replication, either by re-replication on the same origins (for example, by inhibition of ubiquitin ligases that target pre-RC components [6–8, 19, 20]) or by the activation of dormant origins (for example, when cells encounter replication or metabolic stress [16, 21–23]). Both re-replication and dormant origin activation lead to DNA damage and genomic instability.
Computational models depict replication initiation as a time-stochastic process in which the initiation of replication in some origins induces subsequent initiation of adjacent replicons and concomitantly inhibits initiation at adjacent origins within the same replicon [24]. In concordance, single-fiber analyzes demonstrate that at the onset of S-phase, replication starts in a heterogenous and stochastic manner, with no discrete regions of high-efficiency initiation within zones of accessible chromatin. Such replication initiation zones are distributed in a cell-type specific manner and colocalize with distinct chromatin marks [25]. These patterns are consistent with the premise that all licensed replication origins associate with inactive pre-RC complexes and that only a fraction of those complexes, which are residing in chromatin regions associated with specific chromatin marks, are activated [1, 11, 26–28]. Following pre-RC activation, when ongoing replication forks encounter adjacent, inactive Pre-RCs, the unused MCM2-7 complexes are removed from the DNA and the inactive origin region is passively replicated. Because passive replication of dormant origins is delayed when adjacent replication forks slow or stall, the probability of initiation from such dormant origins increases, facilitating the completion of DNA synthesis in the vicinity of the stalled replication fork [11].
In yeast, earlier-activated origins initiate replication in a tighter temporal window than later-activated origins, and the density of MCM complexes on earlier-replicating regions is higher than in later-replicating regions [28, 29]. These observations raise the hypothesis that the abundance of pre-replication complexes on origins regulates the frequency of origin activation [28]. Origin activity in yeast is also affected by other factors including chromatin modifications, variations in nucleosome occupancy, the activities of histone deacetylases Rpd3 and Sir2 and the Fkh1/w transcription factors, which modulate the interaction of replication origins with the origin recognition complex [30–34]. In humans, experimental evidence supporting the MCM density model was observed in HeLa cells, where MCM7-binding sites that associate with replication initiation were more prevalent in open chromatin than in heterochromatin, whereas MCM7 binding sites that did not associate with initiation sites were uniformly distributed throughout the genome [35]. Notably however, although ORC/MCM are more abundant in early than in late replication-timing domains [36], they exhibit a widespread genome distribution and do not delineate replication initiation regions [29], suggesting that pre-RC density does not solely regulate the probability of replication initiation [37].
Chromatin modifiers
Chromatin features and transcriptional activity underlie the order of origin activation (the replication timing program) [1, 24, 38]. In yeast, replication initiation events reflect both chromatin organization (nucleosome occupancy) as well as replication timing. For example, the histone deacetylase Rpd3 [39] and the transcription factors Fkh1/2 modulate genome-wide replication timing [31, 34] while nucleosome occupancy also plays a role in defining origins [40].
Nucleosome positioning and histone H3 occupancy also restrict origin activity when quiescent yeast cells re-enter the cell cycle, attenuating the interaction between replication origins and the MCM subunit MCM4 and necessitating a prolonged G1 period prior to the onset of DNA replication [41].
In mammalian cells, euchromatic histone post-translational modifications (e.g. H3K4me3 and H3K9ac), as well as unmethylated CpGs, often associate with early replication whereas transcriptionally repressed or silent heterochromatic regions are replicated later [1, 2, 4, 14]. In agreement, open chromatin (DNase-hypersensitivity) was the best predictor of replication order in mathematical models [38]. Experimental data concur with this interpretation, as origins that are constitutively active and shared across various cell types are primarily associated with euchromatic regions, whereas origins within heterochromatic regions are often active only in a particular cell type or lineage [3, 14]. Microscopy-based analyses in HeLa cells also show that replication origins that initiate DNA synthesis at a high frequency relocate to the Topologically Associated Domains (TAD) periphery prior to the onset of S-phase in a transcription and CTCF-dependent manner [42]. Transcriptionally active and euchromatic regions often associate with demethylation of H3K79(me2). Depletion of DOT1L, the sole enzyme responsible for methylation of H3K79, results in partial over-replication, indicating that H3K79me2 possibly marks replicated chromatin during S-phase to prevent re-replication in these regions [43]. The H4K20 methyltransferases SET8 (PR-SET7) and SUV420H1/2 and the H4 acetyltransferase HBO1/KAT7, although dispensable for replication initiation, modulate the extent of replication licensing [36, 44, 45].
Post-translational modifications of non-histone proteins can also play a role in replication origin activation. For example, in yeast, the interaction of the histone acetyl transferase Esa1 with the replication initiation factor Sld3 plays a role in regulating the timing of replication origin activation at the HML alpha locus [46]. In mammalian cells, HBO1 is a coactivator of the licensing factor and pre-RC component CDT1 [36], and mutants deficient in HBO1 activity are unable to load the MCM complex.
The mammalian NAD+ dependent deacetylase SIRT1, an ortholog of the yeast deacetylase Sir2, catalyzes the deacetylation of several histone residues including H3K9ac and H4K16ac. The yeast Sir2 and mammalian SIRT1 also both deacetylate MCM proteins and play essential roles in replication fork protection [30, 47–49]. In yeast, although Sir2 and the nucleosome binding protein Sir3 primarily reside in heterochromatin, these proteins attenuate MCM complex loading on euchromatic replication origins by depleting H4K16ac in origin-adjacent nucleosomes [30, 47]. In humans, a phosphorylated form of SIRT1 at the threonine 530 residue associates with both baseline and dormant replication origins. This interaction facilitates replication fork progression while inhibiting dormant origin activation and is essential to prevent DNA breakage upon replication stress [21]. It is possible that the acetylation of H4K16, which is catalyzed by HBO1, recruits SIRT1 to replication origins for maintenance of origin dormancy.
Kinase activity during S-phase
Phosphorylation and dephosphorylation of pre-RC components often drive proliferation decisions during the metazoan cell cycle [50, 51]. During S-phase, CDK2 mediated phosphorylation limits the loading of CDC45 on MCM2-7 on chromatin and consequently prevents origin over-activation, thus regulating initiation frequency and replication fork speed [52, 53]. When cells sustain replication stress, the ATR (Ataxia telangiectasia and Rad3-related) - CHK1 (Checkpoint kinase 1) kinase pathway is activated. This pathway inhibits DNA synthesis at chromatin domains that had not yet initiated replication while facilitating initiation from adjacent dormant origins at chromatin domains that contain stalled replication forks [11, 52, 53]. Replication inhibition is achieved by ATR-mediated phosphorylation of CHK1 at serines 217 and 345, leading to CHK1-mediated phosphorylation and inactivation of the CDK activator CDC25, triggering CDK inhibition followed by cell cycle arrest [54].
DNA topoisomerase II-binding protein 1 (TOPBP1), which binds the MCM2-7 helicase and facilitates replication initiation, is an activator of ATR kinase required for suppression of local dormant origin activation [55, 56]. TOPBP1 is also a SIRT1 substrate, suggesting that TopBP1 acetylation provides a possible link between SIRT1-mediated replication origin dormancy and the ATR-CHK1 pathway that modulates origin activity [49, 57]. The TOPBP1 binding protein Treslin can be phosphorylated by CHK1, in turn preventing CDC45 loading and inhibiting the activation of CMG helicase at dormant origins [58].
Inhibitory interactions by S-phase kinases are in place not only to prevent the initiation of DNA synthesis at dormant origins but also to prevent premature initiation at late-replicating origins [37, 59–61]. Notably, the activity of the CDC7 subunit of DDK, which facilitates replication initiation through the activation of the CMG helicase, is modulated by RIF1 (RAP1-interacting-factor-1), a protein localized to mid-S-phase replication domains [60, 61]. RIF1 plays several crucial roles in genomic stability, acting in DNA replication origin regulation, telomere homeostasis and DNA double-strand break repair pathway choice [62–64]. RIF1 forms a complex with protein phosphatase 1 (PP1), which counteracts DDK-mediated phosphorylation and activation of CMG in mid-S-phase replicating regions. The RIF1-PP1 axis, therefore, prevents replication origins programmed to initiate replication in mid-S-phase from initiating replication earlier in S-phase [60, 61]. The RIF1-PP1 interaction is also modulated by ATR-CHK1 signaling during normal unperturbed genome duplication by affecting CDK1-mediated phosphorylation of RIF1 on S2205, which in turn can disrupt RIF1-PP1 association [37]. Unlike during replication stress, when high levels of ATR and CHK1 globally inhibit initiation of DNA replication, low levels of ATR and CHK1 in normally replicating cells selectively inhibit CDK1-mediated RIF1 phosphorylation, further stabilizing RIF1-PP1 association and constraining excess replication origin activity [37]. In parallel, the RIF1-PP1 complex directly stimulates origin licensing by preventing hyperphosphorylation of ORC1 and MCM proteins, thus protecting ORC1 from proteasome-mediated degradation [59].
Pathways circumventing origin re-licensing
Cells activate several mechanisms to regulate licensing factors and cell cycle related proteins to prevent re-replication and limit genome duplication to only once per cell cycle [8]. These pathways often involve phosphorylation, ubiquitination and acetylation of licensing proteins to promote their degradation and prevent untimely pre-RC assembly. A crucial process that prevents re-replication is the proteasomal targeting and degradation of both CDC6 and CDT1, two key regulatory licensing factors. A second mechanism for CDT1 inhibition that does not involve protein degradation evolved in metazoans. This mechanism involves CDT1 sequestration in late S phase and G2 by Geminin to prevent MCM recruitment to chromatin [65].
Proteasomal targeting is mediated through ubiquitination catalyzed by redundant E3 ubiquitin ligase pathways involving the Cullin Ring Ligases (CRLs) and the anaphase promoting (APC/C) complexes. CDC6 is targeted for degradation during mitosis by the APC/CCDH1 complex so that it is virtually absent in mid-G1 and is also a CRL4 substrate, preventing its accumulation at the G1/S boundary [66]. Chromatin-associated CRL4 catalyzes the ubiquitination of CDT1 and CDC6 through its substrate receptor CDT2 [6, 17, 67–69]. CDT1 and CDC6 are also targeted for degradation during the S/G2 phases by the ubiquitin ligase complexes SCFSKP2 / SCFFBXO31 and SCFCYCLIN F [8, 20, 67]. Inhibition of CRL-mediated ubiquitination triggers re-replication [6–8, 68, 70–72].
The CRL4 CDT2 ubiquitin ligase complex can be recruited to chromatin prior to the onset of S-phase by the replication initiation determinant protein RepID, which binds a subset of replication origins [70, 73]. SKP2, a component of the alternative ubiquitin ligase complex SCF, associates with replication origins that do not bind RepID [70], thus recruiting SCFSKP2 and CRL4CDT2 to distinct, non-overlapping groups of replication origins for efficient CDT1 degradation [20, 70, 73]. In agreement, inhibition of SCFSKP2 in RepID-deficient cells results in massive re-replication [19, 70].
At least two disparate pathways prevent excess initiation
Although excess initiation is induced in various instances of replication stress, the identities of the additional replication origins that are activated under specific challenges differ. As shown in simulations [74] and experimental studies [19], when the regulatory pathways that prevent re-replication fail, re-replication is mediated by the same baseline origins that are normally activated during normal mitotic growth, with a marked preference for replication origins located at early-replicating domains. Therefore, dormant origins seem to play an insignificant role in re-replication induced by inhibition of ubiquitin ligase complexes or over-expression of pre-RC components [19]. Conversely, induction of potentially genotoxic replication stress, either by exposure to the DNA polymerase inhibitor aphidicolin [19] or by inhibition of the ATR kinase [37], induces initiation of DNA replication from dormant origins that are not utilized during normal mitotic proliferation. Inactivation of the SIRT1 histone deacetylase also activates dormant origins that do not colocalize with baseline origins [21] suggesting that SIRT1 plays a unique role in suppressing dormant origins that is distinct from the inhibition of re-replication by ubiquitin ligase complexes.
Combined, these observations suggest that cells possess at least two pathways to prevent excess replication. One pathway directly targets pre-RC components to prevent re-initiation from baseline origins whereas the other suppresses a distinct group of dormant origins, which are maintained as dormant origins by a mechanism that involves SIRT1 and the ATR-CHK1 axis (Figure 2, Key figure). Further studies will be required to elucidate how dormant origins are maintained, how these potential origins respond to diverse challenges, and how the pathways that regulate initiation from these origins can be exploited as therapeutic targets.
Figure 2, Key figure. Replication origins and excess replication.
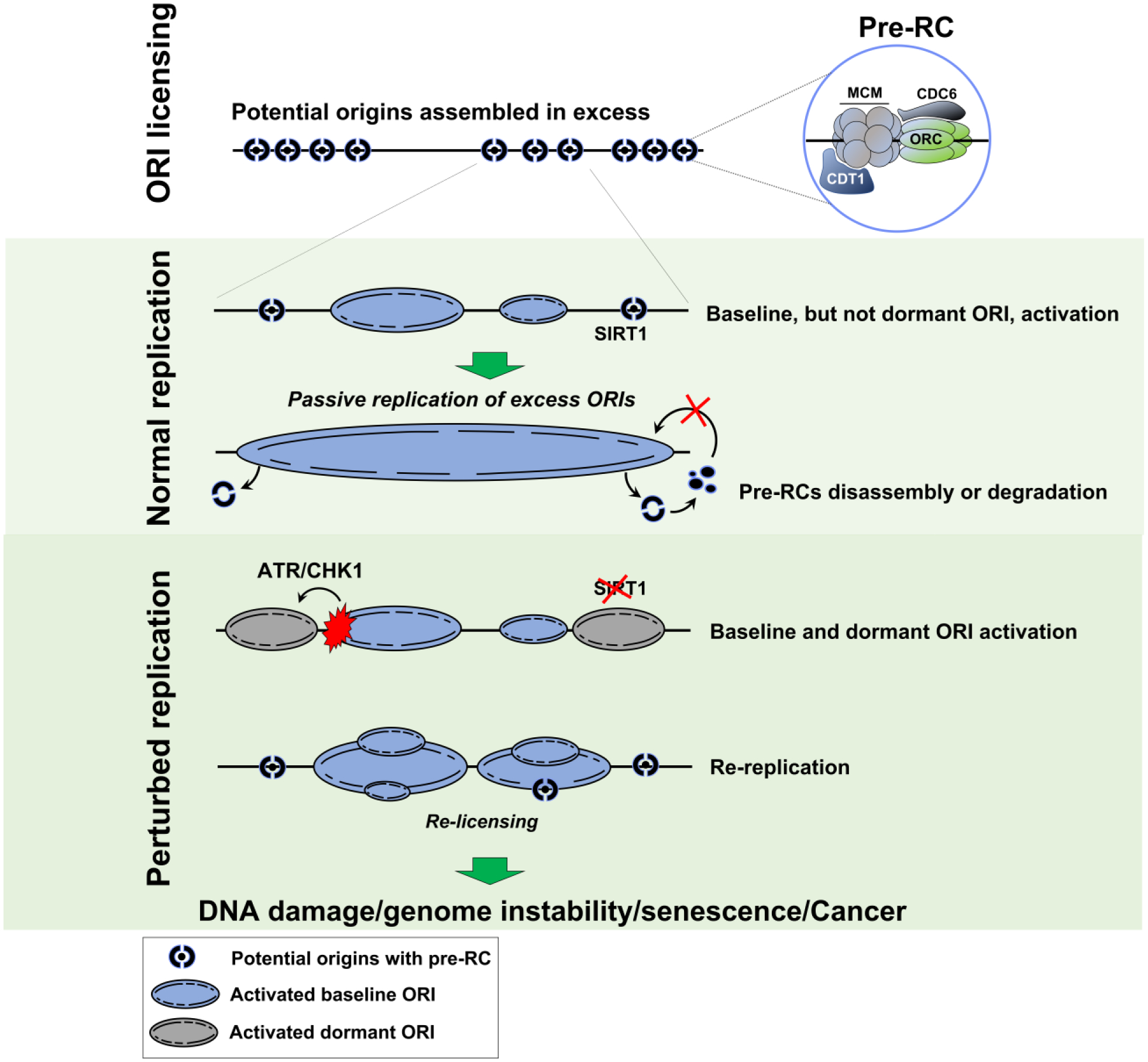
Top panel: Potential replication origins are licensed in excess. The distribution of licensed origins throughout the genome varies based on chromatin marks, gene density, replication timing and other restrictions on nuclear organization. Pre-RCs, which are a prerequisite for DNA replication, are first nucleated by the assembly of six ORC proteins (ORC1-6) onto chromatin. CDC6 then binds the ORC complex to form a ring-shaped complex that will recruit, together with CDT1, several heterohexamers of the six MCM proteins (MCM2-7) (top panel). Middle panel: During normal unperturbed replication, only a small fraction of licensed origins is activated with most licensed origins remaining dormant. These dormant origins are then passively replicated with pre-RCs subunits being dismantled, degraded or recycled to avoid re-licensing during S phase. Inhibition of dormant origin activation may be facilitated by SIRT1. Bottom panel: Two different pathways can result in over-replication. The first pathway, dormant origin activation, can occur during replication stress through ATR/CHK1 mediated phosphorylation or due to the loss of a protein SIRT1, which is involved in dormant origin silencing. The second pathway, DNA re-replication, occurs when origins are re-licensed during S phase before DNA replication is completed and can be triggered by inhibition of pre-RC component degradation, chromatin decondensation or oncogene over-expression. Both pathways could lead to DNA damage and genomic instability, leading to senescence or tumor progression.
Special cases: repeated sequences
The stability of repeat sequences has a major impact on human aging and diseases [75–77]. Repetitive sequences are especially vulnerable to mismatches, error-prone repair leading to copy number alterations or structural chromosomal aberrations. These include two important genomic regions: ribosomal DNA (rDNA) repeats and telomeric sequence.
The genetic loci for ribosomal DNA (rDNA) constitute large regions of repetitive DNA and code for ribosomal RNA. Due to its repetitive nature (~300–600 copies), rDNA is susceptible to recombination, deletions and insertions, which makes rDNA one of the largest fragile sites of the genome [78]. Unlike telomeric repeats, the repetitive rDNA loci are actively transcribed in the nucleolar region, increasing the frequency of collisions between transcription and replication. To evade this, budding yeast rDNA loci contain multiple replication fork barrier (RFB) sites that recruits the Fob1 protein to induce replication fork arrest [79]. In mammalian cells, an interaction between TIMELESS and TTF1 at transcription termination site act as RFB [80]. In addition to these replication fork barriers, other mechanisms have evolved to circumvent collisions between transcription and replication, which would lead to chromosome breaks, recombination and ultimately, to genome instability. Some copies of active rDNA are replicated in early S phase at the nucleolar periphery prior to relocating to the nucleolar interior to resume transcription. In contrast, transcriptionally silent rDNA copies replicate inside nucleoli during middle and late S phase [81]. However, excess replication and recombination can still occur in rDNA loci, as is evidenced by the extra chromosomal circular DNA (eccDNA) detected in aging yeast [78, 82, 83] and human cells [84, 85].
In yeast, excess replication in both rDNA and telomeric sequences is modulated by Sir2, which affects the distribution of licensing complexes on both rDNA [86] and telomeric [47] loci. Specifically, Sir2 represses replication initiation at the rDNA loci and preventing over-accumulation of MCM complexes in telomeric regions. The formation of rDNA eccDNA is repressed by the histone deacetylase Sir2, which blocks the recombination between inter-rDNA repeats [87]. The human Sir2 ortholog, SIRT1, also suppresses eccDNA and gene amplification [21, 88], suggesting a role for sirtuin-mediated suppression in preventing excess replication in repetitive sequences.
Telomeres are short, repetitive non-coding TG-rich sequences present at the end of chromosomes [89]. Single-molecule DNA-fiber analyses have revealed that human telomeres do not display a common origin activation pattern, with each chromosome carrying its own replication program [90]. Telomeres are subject to gradual shortening since semiconservative DNA replication leaves an incomplete lagging strand during each cell cycle, and shortening is often reversed in developing tissues and in cancer cells by the telomerase complex [91] or by a recombination-mediated alternative lengthening of telomeres (ALT) that can form extrachromosomal telomeric repeats [92, 93]. In cancer cells, ALT-generated extrachromosomal telomeric DNA fragments are prevalent [92, 94]. Such repeats can be detected by the cGAS-STING pathway, triggering an innate immune response [94]. Indeed, many ALT+ cancers were found to express high levels of Tousled-Like Kinases 1 and 2 (TLK1/2), which attenuate the formation of extrachromosomal telomeric repeats by maintaining telomeres in a heterochromatic chromatin conformation, along with low cGAS-STING levels, possibly facilitating DNA damage tolerance in such cells [94].
The high abundance of extrachromosomal telomeric DNA in cancer cells suggests that repetitive nature of telomeric sequences and the high prevalence of G-quadruplex repeats in telomeres hinder faithful DNA replication, posing a challenge to genomic integrity [95]. Specialized enzymes have evolved to address telomere-specific obstacles to DNA synthesis. For example, Tousled-Like Kinases and ORCA/LRWD1 maintain telomeres as heterochromatin [94, 96] and the TRF2 and TAZ1 members of the shelterin complex recruit the ORC complex to telomeric repeats via an interaction with TERRA long non-coding RNA [97] and mediate chromatin reorganization at telomeric and subtelomeric regions in response to replication stress [98]. In addition, the BLM helicase facilitates telomeric leading strand replication [93] and prevents replication stress at telomeres by interacting with the FANCM associated complex and BRCA1 [99] as well as TOP3 and RMI [100] to prevent TERRA-mediated telomeric R-loops [101, 102].
Dynamics of excess replicated DNA in cancer
Despite the prevalence of regulatory pathways that inhibit excess replication, excess DNA synthesis can occur and extrachromosomal DNA (ecDNA) is often detected in cancer cells. Inappropriate replication is implicated in the etiology of cancer, as oncogenes such as RAS, mutated CYCLIN D1, MOS and CDC6 can induce over-replication [18, 23, 71, 103, 104]. Hyper-replication can also be induced by aberrant activation of the RB-E2F pathway, either by HPV E6/E7 or by overexpression of CYCLIN E [22]. If such hyper-replication is combined with suppression of the p53 pathway and/or the DNA damage response, oncogene-induced proliferation can underlie the etiology of early events in tumorigenesis [22, 23, 103, 105]. Excess replication can also play a role in tumor progression because ecDNA can be transcribed to confer selective growth advantages to cancer cells (for example, ecDNA can contain oncogenes or enzymes that confer drug resistance) [105, 106].
Another manifestation of excess replication in cancer cells is the prevalence of induced, transient and locus-specific increases in copy numbers of genomic regions that frequently harbor pro-survival genes and oncogenes. These copy number changes can be induced by epigenetic modifications such as overexpression and stabilization of histone 3 lysine 9/36 (H3K9/36) tri-demethylase KDM4A. Such changes can amplify specific drug resistance-associated loci without causing genome-wide chromosomal instability, with amplification occurring during a single S phase, likely originating from re-replication. Such localized gene amplification can originate from activation of excess replication origins via a mechanism that involved a collapsed replication fork meeting a converging replication fork from a downstream origin, generating a single-stranded nick that can be ligated and continue replicating using the previously replicated DNA as template [107]
The excess DNA fragments are subsequently lost during the late S/early G2 phases [108, 109]. Multiple parallel signaling pathways have evolved to counter the deleterious effects of excess replication by detecting, encapsulating and eliminating extrachromosomal DNA (ecDNA). These pathways often cross-interact with the inflammatory response machinery that protects organisms from deleterious consequences of viral infection. For example, excess extrachromosomal DNA can be eliminated via autophagy, which is activated by cyclic GMP-AMP synthase (cGAS) binding to cytoplasmic DNA, thus producing cGAMP that facilitates the activation of the innate immune response via the Stimulator of Interferon Genes (STING) pathway [110]. Although primarily detecting cytoplasmic DNA, cGAS can also associate with nuclear replicating DNA and possibly affect the rate of DNA synthesis [111]. When activated, cGAS can suppress nuclear DNA repair and prevents homologous recombination and is associated with cancer progression, invasion and metastasis [112].
Another route to elimination of excess DNA is micronucleation, a process that is often detected in cancer. Cells harboring acentric chromatin fragments, which can be generated by multiple mechanisms including excess replication and/or DNA breakage, can encapsulate such fragments in small, membrane-bounded cellular compartments termed micronuclei [113]. Micronuclei are spatially separated from the primary nucleus by intact nuclear membranes and can support transcription, some repair processes, and recombination. Following unequal segregation during mitosis, transcription of amplified oncogenes and enzymes involved in drug resistance could provide a selective advantage for genes entrapped in micronuclei [113, 114]. Micronuclei with encapsulated ecDNAs can be eliminated from cells as intact structures [115, 116] and can also be subject to nuclear envelope rupture. Nuclear rupture releases chromatin fragments that are potential substrates for exonucleases (for example, TREX1 is a DNA 3’ repair exonuclease I), which play an important role in the clearance of cytosolic DNA [117, 118]). Exonuclease activity on ecDNA can generate a spectrum of genomic rearrangements, including the formation of shattered chromosomes known as chromothripsis [113, 114].
As cancer cells tolerate high levels of replication stress, including excess DNA synthesis that might result in unstable and consistently dynamic genomes, this property that can be exploited as a therapeutic strategy. For example, excess DNA released into the circulation can be utilized in diagnostic ‘liquid biopsies’ [82]. In contrast to non-cancerous cells, which induce senescence in response to oncogene-induced over-replication [23, 103], limited over-replication is tolerated in cancer cells and does not result in senescence. Cancer cells that undergo massive over-replication can exhibit high levels of genomic instability and eventually activate an apoptosis pathway and can be therefore targeted by employing ubiquitin ligase and DNA repair inhibitors [71, 119] and inhibitors of checkpoint kinases such as ATR [120].
Concluding Remarks
Replication origin flexibility, derived from the excess of pre-RCs, is essential for maintenance of genomic stability in two ways. First, for baseline origins, it may maintain replication order and help adapt to the changing transcriptional environment, therefore preventing collisions with the transcription machinery. Second, for dormant origins, it maintains dormancy but allows those origins to be used as backups if needed, and therefore allows cells to overcome minor DNA damage without activating apoptotic or senescence pathways. On the other hand, excess origin activation, regardless of mechanism, is deleterious due to the potential to enhance transcription-replication collisions, partial genome duplication and DNA breakages, leading in turn to chromosome aberrations, mutations, chromothripsis and oncogene activation. Recent mechanistic studies imply that there are at least two distinct pathways cells use to repress over-replication, suggesting that replication origins serve as endpoints for several parallel pathways that can be employed in therapeutic strategies to target cells that tolerate replication stress.
Our current understanding of replication origin flexibility and the cellular pathways that regulate excess replication raise several open questions (see Outstanding Questions). For example, it is unclear if dormant origins respond to specific signaling cascades and if so, it would be important to learn if such signals can be targeted in cancer cells to trigger replication catastrophe. It is unclear if the pathways that maintain replication order (e.g. the RIF1-PP1 pathway) also play a direct role in preventing re-replication, and if so, under which conditions. In addition, the replication origins that respond to oncogenes have not yet been identified, and it is unclear if oncogenes induce excess replication in all baseline origins or in dormant origins are prone to over-replication. Understanding the specific protein-DNA interactions that induce excess replication at origins could identify the specific roles over-replication might play in carcinogenesis. For example, the locations and associations between dormant origins and transcribed regions should be further explored, as it is evident for baseline origins that are often associated with promoters. Finally, the details of the activation of the cGAS-STING pathway need to be elucidated. Addressing some of these questions could lead to a better understanding of the mechanism(s) that suppress, detect, and eliminate the products of excess replication during normal growth, as well as the role(s) excess replication plays in the etiology of cancer and the pathways that allow cancer cells to tolerate excess replication, leading to potential interventions.
Outstanding questions:
Can we identify new chromatin markers for origin licensing and/or origin activation?
Can we identify a chromatin signature (i.e., histones or pre-RC) that would associate with dormant origin activation?
Can we identify specific protein-DNA interactions that induce excess replication at origins?
Is there an association (positive or negative) between the locations of dormant origins and transcriptional regulatory elements?
Are cellular signaling pathways that maintain replication order (e.g. the RIF1-PP1 pathway) also play a direct role in preventing re-replication and maintaining replication origin dormancy?
Which group of origins (baseline origins or dormant origins) are prone to oncogene-induced over-replication?
How is the cGAS-STING pathway activated in response to excess replication?
Can origin dormancy be modulated to trigger a replication catastrophe in cancer cells?
Highlights:
Cells have evolved specialized mechanisms to avoid the deleterious effects of excess initiation from replication origins.
During unperturbed proliferation, most potential replication origins are dormant and do not initiate replication but are prepared for activation to facilitate complete genome duplication if cells encounter replication stress.
Cells prevent initiation at dormant origins by adjusting chromatin structure to constrain the assembly and activation of pre-replication complexes, targeting components of the replication machinery for degradation or modulating specific protein-DNA interactions at origins.
Cancer cells often lack the controls that prevent excess activation of replication origins, resulting in the amplification of extrachromosomal DNA that can persist if they contain genes that can confer a selective growth advantage.
Cells activate metabolic and inflammatory responses to sense, encapsulate and eliminate excess DNA.
Because excess replication origin activation can directly or indirectly lead to genomic instability, targeting the pathways that prevent excess replication or eliminate excess DNA is a promising avenue for therapeutic intervention.
Acknowledgements
Studies in the authors’ lab are supported by the Intramural Research Program of the NIH, Center for Cancer Research, National Cancer Institute (project ZIA BC010411 to MIA). We thank Drs. Haiqing Fu and Robin Sebastian for critical reading of insightful comments and Ms. Iris Helen Indig for the Baseline Origin suggestion. We would like to apologize to colleagues whose primary work could not be cited directly due to space limitations.
Glossary
- Baseline origins
Replication origins active during an unperturbed S-phase of the cell cycle
- Constitutive origins
Replication origins that are constantly active in a broad range of cell and tissue types
- Dormant origins
Replication origins that do not initiate replication during unperturbed S-phases but are activated during perturbed S-phases
- Extrachromosomal circular DNA (eccDNA)
Extra chromosomal, circular and self-replicating DNA located mostly in metazoan or plant cell nuclei
- Flexible origins
Replication origins that can initiate replication intermittently or in a cell-type-specific manner
- Origin licensing
Assembly of pre-replication complexes on chromatin. Pre-replication complexes are assembled in an inactive form, and are activated by cyclin dependent kinases or DBF4-dependent kinase leading to the initiation of DNA replication
- Pre-replication complex
An inactive replication initiation complex composed of ORCs, CDT1, MCM2-7 and CDC6 that is assembled on potential origin sites during the G1-phase of cell cycle
- Re-replication
Over-replication leading to increased total DNA content, leading to a situation where a genome can be replicated more than once per cell cycle
- Replication domain or replicon
A chromosomal region that is replicated from a single replication origin
- Replication fork barrier (RFB)
Chromosomal sites present in ribosomal DNA repeats which recruit replication arrest proteins (i.e., Fob1, TIMELESS and TTF1) to avoid collisions between replication and transcription
- Replication order/timing
Timely order in which segments of a chromosome are duplicated through the S-phase of the cell cycle
- Replication origins (ORI)
Chromosomal sites which initiate DNA replication
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aladjem MI and Redon CE (2017) Order from clutter: selective interactions at mammalian replication origins. Nat Rev Genet 18 (2), 101–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prioleau MN and MacAlpine DM (2016) DNA replication origins-where do we begin? Genes Dev 30 (15), 1683–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marks AB et al. (2016) Replication origins: determinants or consequences of nuclear organization? Curr Opin Genet Dev 37, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fragkos M et al. (2015) DNA replication origin activation in space and time. Nat Rev Mol Cell Biol 16 (6), 360–74. [DOI] [PubMed] [Google Scholar]
- 5.Gros J et al. (2014) Origin plasticity during budding yeast DNA replication in vitro. EMBO J 33 (6), 621–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou Y et al. (2020) Distinct and sequential re-replication barriers ensure precise genome duplication. PLoS Genet 16 (8), e1008988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Limas JC and Cook JG (2019) Preparation for DNA replication: the key to a successful S phase. FEBS Lett 593 (20), 2853–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blow JJ and Dutta A (2005) Preventing re-replication of chromosomal DNA. Nat Rev Mol Cell Biol 6 (6), 476–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Machida YJ et al. (2005) Right place, right time, and only once: replication initiation in metazoans. Cell 123 (1), 13–24. [DOI] [PubMed] [Google Scholar]
- 10.Remus D and Diffley JF (2009) Eukaryotic DNA replication control: lock and load, then fire. Curr Opin Cell Biol 21 (6), 771–7. [DOI] [PubMed] [Google Scholar]
- 11.Alver RC et al. (2014) The contribution of dormant origins to genome stability: from cell biology to human genetics. DNA Repair (Amst) 19, 182–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hyrien O (2015) Peaks cloaked in the mist: the landscape of mammalian replication origins. J Cell Biol 208 (2), 147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shima N and Pederson KD (2017) Dormant origins as a built-in safeguard in eukaryotic DNA replication against genome instability and disease development. DNA Repair (Amst) 56, 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith OK et al. (2016) Distinct epigenetic features of differentiation-regulated replication origins. Epigenetics Chromatin 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brambati A et al. (2018) Dormant origins and fork protection mechanisms rescue sister forks arrested by transcription. Nucleic Acids Res 46 (3), 1227–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yekezare M et al. (2013) Controlling DNA replication origins in response to DNA damage - inhibit globally, activate locally. J Cell Sci 126 (Pt 6), 1297–306. [DOI] [PubMed] [Google Scholar]
- 17.Petropoulos M et al. (2019) Replication Licensing Aberrations, Replication Stress, and Genomic Instability. Trends Biochem Sci 44 (9), 752–764. [DOI] [PubMed] [Google Scholar]
- 18.Blow JJ and Gillespie PJ (2008) Replication licensing and cancer--a fatal entanglement? Nat Rev Cancer 8 (10), 799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fu H et al. (2021) Dynamics of replication origin over-activation. Nat Commun 12 (1), 3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nishitani H et al. (2006) Two E3 ubiquitin ligases, SCF-Skp2 and DDB1-Cul4, target human Cdt1 for proteolysis. EMBO J 25 (5), 1126–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Utani K et al. (2017) Phosphorylated SIRT1 associates with replication origins to prevent excess replication initiation and preserve genomic stability. Nucleic Acids Res 45 (13), 7807–7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bester AC et al. (2011) Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145 (3), 435–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bartkova J et al. (2006) Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444 (7119), 633–7. [DOI] [PubMed] [Google Scholar]
- 24.Lob D et al. (2016) 3D replicon distributions arise from stochastic initiation and domino-like DNA replication progression. Nat Commun 7, 11207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang W et al. (2021) Genome-wide mapping of human DNA replication by optical replication mapping supports a stochastic model of eukaryotic replication. Mol Cell 81 (14), 2975–2988 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ding Q and Koren A (2020) Positive and Negative Regulation of DNA Replication Initiation. Trends Genet 36 (11), 868–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DePamphilis ML (1993) Origins of DNA replication in metazoan chromosomes. J Biol Chem 268 (1), 1–4. [PubMed] [Google Scholar]
- 28.Das SP et al. (2015) Replication timing is regulated by the number of MCMs loaded at origins. Genome Res 25 (12), 1886–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirstein N et al. (2021) Human ORC/MCM density is low in active genes and correlates with replication time but does not delimit initiation zones. Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoggard TA et al. (2018) Yeast heterochromatin regulators Sir2 and Sir3 act directly at euchromatic DNA replication origins. PLoS Genet 14 (5), e1007418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ostrow AZ et al. (2014) Fkh1 and Fkh2 bind multiple chromosomal elements in the S. cerevisiae genome with distinct specificities and cell cycle dynamics. PLoS One 9 (2), e87647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fox CA and Weinreich M (2008) Beyond heterochromatin: SIR2 inhibits the initiation of DNA replication. Cell Cycle 7 (21), 3330–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodriguez J et al. (2017) Nucleosome occupancy as a novel chromatin parameter for replication origin functions. Genome Res 27 (2), 269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoggard T et al. (2021) The Fkh1 Forkhead associated domain promotes ORC binding to a subset of DNA replication origins in budding yeast. Nucleic Acids Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sugimoto N et al. (2018) Genome-wide analysis of the spatiotemporal regulation of firing and dormant replication origins in human cells. Nucleic Acids Res 46 (13), 6683–6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miotto B et al. (2016) Selectivity of ORC binding sites and the relation to replication timing, fragile sites, and deletions in cancers. Proc Natl Acad Sci U S A 113 (33), E4810–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moiseeva TN et al. (2019) An ATR and CHK1 kinase signaling mechanism that limits origin firing during unperturbed DNA replication. Proc Natl Acad Sci U S A 116 (27), 13374–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gindin Y et al. (2014) A chromatin structure-based model accurately predicts DNA replication timing in human cells. Mol Syst Biol 10, 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aparicio JG et al. (2004) The Rpd3-Sin3 histone deacetylase regulates replication timing and enables intra-S origin control in Saccharomyces cerevisiae. Mol Cell Biol 24 (11), 4769–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eaton ML et al. (2010) Conserved nucleosome positioning defines replication origins. Genes Dev 24 (8), 748–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee PH and Osley MA (2021) Chromatin structure restricts origin utilization when quiescent cells re-enter the cell cycle. Nucleic Acids Res 49 (2), 864–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Y et al. (2021) Transcription-coupled structural dynamics of topologically associating domains regulate replication origin efficiency. Genome Biol 22 (1), 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fu H et al. (2013) Methylation of histone H3 on lysine 79 associates with a group of replication origins and helps limit DNA replication once per cell cycle. PLoS Genet 9 (6), e1003542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tardat M et al. (2010) The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat Cell Biol 12 (11), 1086–93. [DOI] [PubMed] [Google Scholar]
- 45.Kuo AJ et al. (2012) The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature 484 (7392), 115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tanaka S (2021) Interaction of replication factor Sld3 and histone acetyl transferase Esa1 alleviates gene silencing and promotes the activation of late and dormant replication origins. Genetics 217 (1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoggard T et al. (2020) Sir2 mitigates an intrinsic imbalance in origin licensing efficiency between early- and late-replicating euchromatin. Proc Natl Acad Sci U S A 117 (25), 14314–14321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fatoba ST et al. (2013) Human SIRT1 regulates DNA binding and stability of the Mcm10 DNA replication factor via deacetylation. Nucleic Acids Res 41 (7), 4065–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang RH et al. (2014) SIRT1 deacetylates TopBP1 and modulates intra-S-phase checkpoint and DNA replication origin firing. Int J Biol Sci 10 (10), 1193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Depamphilis ML et al. (2012) “The Octet”: Eight Protein Kinases that Control Mammalian DNA Replication. Front Physiol 3, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Novak B et al. (2010) Regulated protein kinases and phosphatases in cell cycle decisions. Curr Opin Cell Biol 22 (6), 801–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Petermann E and Helleday T (2010) Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol 11 (10), 683–7. [DOI] [PubMed] [Google Scholar]
- 53.Shechter D et al. (2004) ATR and ATM regulate the timing of DNA replication origin firing. Nat Cell Biol 6 (7), 648–55. [DOI] [PubMed] [Google Scholar]
- 54.Saldivar JC et al. (2017) The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat Rev Mol Cell Biol 18 (10), 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sokka M et al. (2018) The ATR-Activation Domain of TopBP1 Is Required for the Suppression of Origin Firing during the S Phase. Int J Mol Sci 19 (8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kumagai A et al. (2010) Treslin collaborates with TopBP1 in triggering the initiation of DNA replication. Cell 140 (3), 349–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu T et al. (2014) A divergent role of the SIRT1-TopBP1 axis in regulating metabolic checkpoint and DNA damage checkpoint. Mol Cell 56 (5), 681–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo C et al. (2015) Interaction of Chk1 with Treslin negatively regulates the initiation of chromosomal DNA replication. Mol Cell 57 (3), 492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hiraga SI et al. (2017) Human RIF1 and protein phosphatase 1 stimulate DNA replication origin licensing but suppress origin activation. EMBO Rep 18 (3), 403–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hiraga S et al. (2014) Rif1 controls DNA replication by directing Protein Phosphatase 1 to reverse Cdc7-mediated phosphorylation of the MCM complex. Genes Dev 28 (4), 372–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yamazaki S et al. (2012) Rif1 regulates the replication timing domains on the human genome. EMBO J 31 (18), 3667–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alavi S et al. (2021) G-quadruplex binding protein Rif1, a key regulator of replication timing. J Biochem 169 (1), 1–14. [DOI] [PubMed] [Google Scholar]
- 63.Gnan S et al. (2021) Nuclear organisation and replication timing are coupled through RIF1-PP1 interaction. Nat Commun 12 (1), 2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Garzon J et al. (2019) Human RIF1-Protein Phosphatase 1 Prevents Degradation and Breakage of Nascent DNA on Replication Stalling. Cell Rep 27 (9), 2558–2566 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wohlschlegel JA et al. (2000) Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science 290 (5500), 2309–12. [DOI] [PubMed] [Google Scholar]
- 66.Clijsters L and Wolthuis R (2014) PIP-box-mediated degradation prohibits re-accumulation of Cdc6 during S phase. J Cell Sci 127 (Pt 6), 1336–45. [DOI] [PubMed] [Google Scholar]
- 67.Pozo PN and Cook JG (2016) Regulation and Function of Cdt1; A Key Factor in Cell Proliferation and Genome Stability. Genes (Basel) 8 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Panagopoulos A et al. (2020) CRL4(Cdt2): Coupling Genome Stability to Ubiquitination. Trends Cell Biol 30 (4), 290–302. [DOI] [PubMed] [Google Scholar]
- 69.Gilberto S and Peter M (2017) Dynamic ubiquitin signaling in cell cycle regulation. J Cell Biol 216 (8), 2259–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jang SM et al. (2018) The replication initiation determinant protein (RepID) modulates replication by recruiting CUL4 to chromatin. Nat Commun 9 (1), 2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vaziri C et al. (2003) A p53-dependent checkpoint pathway prevents rereplication. Mol Cell 11 (4), 997–1008. [DOI] [PubMed] [Google Scholar]
- 72.Liontos M et al. (2007) Deregulated overexpression of hCdt1 and hCdc6 promotes malignant behavior. Cancer Res 67 (22), 10899–909. [DOI] [PubMed] [Google Scholar]
- 73.Zhang Y et al. (2016) A replicator-specific binding protein essential for site-specific initiation of DNA replication in mammalian cells. Nat Commun 7, 11748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Menzel J et al. (2020) Isolation and analysis of rereplicated DNA by Rerep-Seq. Nucleic Acids Res 48 (10), e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roake CM and Artandi SE (2020) Regulation of human telomerase in homeostasis and disease. Nat Rev Mol Cell Biol 21 (7), 384–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Warmerdam DO and Wolthuis RMF (2019) Keeping ribosomal DNA intact: a repeating challenge. Chromosome Res 27 (1–2), 57–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hannan AJ (2018) Tandem repeats mediating genetic plasticity in health and disease. Nat Rev Genet 19 (5), 286–298. [DOI] [PubMed] [Google Scholar]
- 78.Salim D and Gerton JL (2019) Ribosomal DNA instability and genome adaptability. Chromosome Res 27 (1–2), 73–87. [DOI] [PubMed] [Google Scholar]
- 79.Castan A et al. (2017) The abundance of Fob1 modulates the efficiency of rRFBs to stall replication forks. Nucleic Acids Res 45 (17), 10089–10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Akamatsu Y and Kobayashi T (2015) The Human RNA Polymerase I Transcription Terminator Complex Acts as a Replication Fork Barrier That Coordinates the Progress of Replication with rRNA Transcription Activity. Mol Cell Biol 35 (10), 1871–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dimitrova DS (2011) DNA replication initiation patterns and spatial dynamics of the human ribosomal RNA gene loci. J Cell Sci 124 (Pt 16), 2743–52. [DOI] [PubMed] [Google Scholar]
- 82.Paulsen T et al. (2018) Discoveries of Extrachromosomal Circles of DNA in Normal and Tumor Cells. Trends Genet 34 (4), 270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Potapova TA and Gerton JL (2019) Ribosomal DNA and the nucleolus in the context of genome organization. Chromosome Res 27 (1–2), 109–127. [DOI] [PubMed] [Google Scholar]
- 84.Cohen S et al. (2010) Extrachromosomal circles of satellite repeats and 5S ribosomal DNA in human cells. Mob DNA 1 (1), 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moller HD et al. (2018) Circular DNA elements of chromosomal origin are common in healthy human somatic tissue. Nat Commun 9 (1), 1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yoshida K et al. (2014) The histone deacetylases sir2 and rpd3 act on ribosomal DNA to control the replication program in budding yeast. Mol Cell 54 (4), 691–7. [DOI] [PubMed] [Google Scholar]
- 87.Kobayashi T et al. (2004) SIR2 regulates recombination between different rDNA repeats, but not recombination within individual rRNA genes in yeast. Cell 117 (4), 441–53. [DOI] [PubMed] [Google Scholar]
- 88.Taniguchi R et al. (2021) SIRT1 stabilizes extrachromosomal gene amplification and contributes to repeat-induced gene silencing. J Biol Chem, 100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhao Y et al. (2008) Quantitative telomeric overhang determination using a double-strand specific nuclease. Nucleic Acids Res 36 (3), e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Drosopoulos WC et al. (2012) Human telomeres replicate using chromosome-specific, rather than universal, replication programs. J Cell Biol 197 (2), 253–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Maciejowski J and de Lange T (2017) Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Biol 18 (3), 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen YA et al. (2017) Extrachromosomal telomere repeat DNA is linked to ALT development via cGAS-STING DNA sensing pathway. Nat Struct Mol Biol 24 (12), 1124–1131. [DOI] [PubMed] [Google Scholar]
- 93.Drosopoulos WC et al. (2015) BLM helicase facilitates telomere replication during leading strand synthesis of telomeres. J Cell Biol 210 (2), 191–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Segura-Bayona S et al. (2020) Tousled-Like Kinases Suppress Innate Immune Signaling Triggered by Alternative Lengthening of Telomeres. Cell Rep 32 (5), 107983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Higa M et al. (2017) DNA Replication Origins and Fork Progression at Mammalian Telomeres. Genes (Basel) 8 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hsu RYC et al. (2020) ORCA/LRWD1 Regulates Homologous Recombination at ALT-Telomeres by Modulating Heterochromatin Organization. iScience 23 (5), 101038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Deng Z et al. (2009) TERRA RNA binding to TRF2 facilitates heterochromatin formation and ORC recruitment at telomeres. Mol Cell 35 (4), 403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mizuguchi T et al. (2017) Shelterin components mediate genome reorganization in response to replication stress. Proc Natl Acad Sci U S A 114 (21), 5479–5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pan X et al. (2017) FANCM, BRCA1, and BLM cooperatively resolve the replication stress at the ALT telomeres. Proc Natl Acad Sci U S A 114 (29), E5940–E5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lu R et al. (2019) The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT). Nat Commun 10 (1), 2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Silva B et al. (2019) FANCM limits ALT activity by restricting telomeric replication stress induced by deregulated BLM and R-loops. Nat Commun 10 (1), 2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pan X et al. (2019) FANCM suppresses DNA replication stress at ALT telomeres by disrupting TERRA R-loops. Sci Rep 9 (1), 19110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Di Micco R et al. (2006) Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444 (7119), 638–42. [DOI] [PubMed] [Google Scholar]
- 104.Aggarwal P et al. (2007) Nuclear accumulation of cyclin D1 during S phase inhibits Cul4-dependent Cdt1 proteolysis and triggers p53-dependent DNA rereplication. Genes Dev 21 (22), 2908–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Turner KM et al. (2017) Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543 (7643), 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nikolaev S et al. (2014) Extrachromosomal driver mutations in glioblastoma and low-grade glioma. Nat Commun 5, 5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Johansson E and Diffley JFX (2021) Unchecked nick ligation can promote localized genome re-replication. Curr Biol 31 (11), R710–R711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Black JC et al. (2013) KDM4A lysine demethylase induces site-specific copy gain and rereplication of regions amplified in tumors. Cell 154 (3), 541–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mishra S et al. (2018) Cross-talk between Lysine-Modifying Enzymes Controls Site-Specific DNA Amplifications. Cell 175 (6), 1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nassour J et al. (2019) Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 565 (7741), 659–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen H et al. (2020) cGAS suppresses genomic instability as a decelerator of replication forks. Sci Adv 6 (42). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu H et al. (2018) Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 563 (7729), 131–136. [DOI] [PubMed] [Google Scholar]
- 113.Krupina K et al. (2021) Causes and consequences of micronuclei. Curr Opin Cell Biol 70, 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang CZ et al. (2015) Chromothripsis from DNA damage in micronuclei. Nature 522 (7555), 179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shimizu N et al. (2005) Tracking of microinjected DNA in live cells reveals the intracellular behavior and elimination of extrachromosomal genetic material. Nucleic Acids Res 33 (19), 6296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kumar P et al. (2017) Normal and Cancerous Tissues Release Extrachromosomal Circular DNA (eccDNA) into the Circulation. Mol Cancer Res 15 (9), 1197–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kustanovich A et al. (2019) Life and death of circulating cell-free DNA. Cancer Biol Ther 20 (8), 1057–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Stetson DB et al. (2008) Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134 (4), 587–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Helleday T et al. (2008) DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 8 (3), 193–204. [DOI] [PubMed] [Google Scholar]
- 120.Thomas A et al. (2021) Therapeutic targeting of ATR yields durable regressions in small cell lung cancers with high replication stress. Cancer Cell 39 (4), 566–579 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]