

Abstract
Aims:
To gain insights into FKBP regulation of cardiac ryanodine receptor (RyR2) and Ca2+ signaling, we introduced the point mutation (N771D-RyR2) corresponding to skeletal muscle mutation (N760D-RyR1) associated with central core disease (CCD) via CRISPR/Cas9 gene-editing in the RyR2 FKBP binding site expressed in human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs). Patients inflicted with CCD and other hereditary skeletal muscle diseases often show higher incidence of atrial or ventricular arrhythmias.
Methods and Results:
Ca2+ imaging of voltage-clamped N771D-RyR2 mutant compared to WT hiPSC-CMs showed: (1) ~30% suppressed ICa with no significant changes in the gating kinetics of ICa; (2) 29% lower SR Ca2+ content and 33% lower RyR2 Ca2+ leak; (3) higher CICR gain and 30–35% increased efficiency of ICa-triggered Ca2+ release; (4) higher incidence of aberrant SR Ca2+ releases, DADs, and Ca2+ sparks; (5) no change in fractional Ca2+-release, action potential morphology, sensitivity to isoproterenol, and sarcomeric FKBP-binding pattern.
Conclusions:
The more frequent spontaneous Ca2+ releases and longer Ca2+ sparks underlie the increased incidence of DADs and cellular arrhythmogenesis of N771D-RyR2 mutant. The smaller RyR2 Ca2+ leak and SR content result from suppressed ICa that is compensated by higher CICR gain.
Keywords: FKBP binding site, Ryanodine Receptor mutation, CRISPR/Cas9, Skeletal Myopathy, human induced Pluripotent Stem Cells, CICR gain
Graphical Abstract

1. INTRODUCTION
Ryanodine receptors are Ca2+ release channels of sarcoplasmic reticulum that mediate the skeletal and cardiac muscle excitation-contraction (EC) coupling. In cardiac myocytes, influx of Ca2+ through voltage-gated L-type Ca2+ channels trigger large releases of Ca2+ (Ca2+ induced Ca2+ release, CICR) from type-2 ryanodine receptors [1–5], that underlies development of Ca2+ sparks and Ca2+ stripes at sarcomeric z-lines of cardiomyocytes [6]. Ca2+ sparks also activate spontaneously from random openings of dyadic RyR2, under resting conditions, without activation of Ca2+ channels [7]. The frequency of spontaneous Ca2+ sparks appears to be enhanced when mutations in RyR2 [8] or calsequestrin [9] render them leaky to Ca2+, triggering often life-threatening arrhythmias [10, 11].
RyR2 is a homotetramer protein of ~5000 amino acids bounded by a number of accessory proteins (FKBP12.6, calmodulin, protein kinases and phosphatases) and small molecules (Ca2+, Mg2+) that affect its gating [12, 13]. The effects of FKBPs on regulation of RyR2 gating are controversial as some reports show that FKBP12.6 associates with RyR2 with high affinity, promoting RyR2 closed channel state and stabilizing its function [14–16]. Supporting this idea, number of studies show that unbinding of FKBPs leads to leaky RyRs in pathologies of muscular dystrophy [17], sarcopenia [18], cardiac arrhythmias and heart failure [14, 19–21]. In this respect, overexpression of FKBP12.6, appears to counter arrhythmogenesis by reducing diastolic RyR2 Ca2+ leak and terminating spontaneous Ca2+ release [22–25]. Other reports, however, suggest that although FKBP12 stabilizes RyR1, neither FKBP12.6 nor FKBP12 affect RyR2 function [26, 27]. There are also reports that FKBP binding destabilizes RyR function [28, 29]. Despite these apparently contradictory findings, pharmacological agents such as 1,4-benzothiazepine derivatives JTV519 and S107, that stabilize FKBP–RyR interaction improve RyR function [15, 20, 30, 31], and may have therapeutic potential.
In this study, we introduced a human skeletal muscle myopathy-associated mutation (N771D-RyR2, corresponding to skeletal muscle N760D-RyR1, (Yuchi et al., 2015) in the RyR2-FKBP binding site using CRISPR/Cas9 gene editing of hiPSCs [32] and characterized the Ca2+-signaling phenotype of mutant cardiomyocytes as compared to WT K3-line hiPSC-CMs. Although we found higher frequencies of aberrant SR Ca2+ release events and enhanced CICR gain in N771D mutants, the mutation did not increase diastolic RyR2 Ca2+ leak or the frequency of spontaneous Ca2+ sparks, most likely because of significant suppression of ICa, resulting in lower SR Ca2+ content.
2. MATERIAL AND METHODS
2.1. Cell culture of human pluripotent stem cells and cardiac differentiation
Human pluripotent stem cells (hiPSC-K3) were obtained from Stephen Duncan at Medical University of South Carolina [33]. hiPSC-K3 were routinely cultured in StemFlex medium (GIBCO) on Vitronectin (GIBCO) coated tissue culture plates with daily media change at 37 °C with 5% (vol/vol) CO2. Differentiation was performed following the protocols of Xiaojun Lian et al [34]. Briefly, dissociated hiPSCs were plated in 12 well plates with matrigel and then treated with 12 μM CHIR 99021, a GSK3β inhibitor for 24 h in RPMI/B-27 without insulin. 72 h after CHIR99021 treatment, 5 μM IWR-1, a wnt processing inhibitor, was added to culture with the same media for 48 h. After 48 h of continued culture in RPMI/B-27 without insulin, the cells were maintained in RPMI/B-27 medium with insulin for the rest of the time.
2.2. Genome editing in hiPSCs
To introduce the human disease-associated N771D mutation located at the FKBP binding site of RyR2 in the hiPSC genome, we used CRISPR/Cas9 gene-editing technique according to the established protocol [32, 35]. Briefly, the target sequence (5’-AGCATCTCGTTCCGAATTAA-3’) adjacent to Pam sequence in RYR2 gene exon21 (Fig. 1A) was cloned into pX459 plasmid vector (Addgene). The plasmid was then transfected into WT hiPSC to express Cas9 exonuclease, which is guided by the target sequence and thereby digest the target site in the genome. N771D-RyR2 point mutation was introduced during homology-directed repair of the digested genome by co-transfection of single stranded oligo donor nucleotide (ssODN: 5’TTTAGATCTGAGTGCCCCAAGCATCTCGTTCCGGATTGATGGACAACCTGTTCAAG GAATGTTTGAGAATTTCAACATCGATGGCCTCTTCTTTCCAGTCGTTAGTTTCTCTGC AGGAATAAAGTT -3’), carrying two mutations (underline); one is for N771D mutation and the other one is silent mutation (no amino acid change) but creates a new restriction enzyme site (HpaII). Mutated locus in each single isolated cell colony was amplified by PCR, followed by the restriction digestion by HpaII to identified gene-edited colonies (Fig. 1B). Correct gene-editing was verified by sequencing of PCR products from genomic DNA of mutated hiPSCs (Fig. 1C) and RT-PCR products from mRNA in the cardiac differentiated clones (not shown). We have established two N771D-RyR2 hiPSC clones, named #49–1 and #4–1. Both clones carry homozygous N771D mutation (Fig. 1C), while the clone #49–1 has homozygous HpaII site and the clone #4–1 is heterozygote (Fig. 1B and C). Quantitative RT-PCR analysis of the differentiated hiPSC-CMs showed that RyR2 gene expression levels in three lines (WT, N771D #49–1, and N771D #4–1) were not significantly different (Fig. 1D). Unless otherwise mentioned, electrophysiology and calcium signaling data were obtained from mutant clone #49–1.
Fig. 1.-. Panel A, schematic for CRISPR/Cas9 gene-editing of RYR2 exon 21 in hiPSC.

RYR2 gene is digested by Cas9 exonuclease at the target sequence. During homology directed repair two mutations are introduced; a mutation introducing N771D amino acid substitution and a mutation creating HpaII restriction site but not causing amino acid change. Panel B, the targeted locus of the genome was amplified by PCR, followed by restriction digestion by HpaII. The PCR product is 387 bp, and PCR product with mutations is digested into two smaller fragments (276bp and 111bp) by HpaII. Panel C, sequencing of PCR products amplified from the targeted RYR2 gene locus showed two mutations, verifying correct gene-editing by CRISPR/Cas9. Both clones carry homozygous N771D mutation. The clone #49–1 has homozygous HpaII site, while the clone #4–1 shows heterozygous restriction site, which is consistent with the restriction digestion pattern in panel B. Panel D, quantification of RyR2 transcription levels in wild-type (WT) and two N771D hiPSC-CMs (n=3–4) by quantitative RT-PCR. Data are mean ± SEM. RyR2 transcription levels are not significantly different among three lines by one-way ANOVA.
2.3. Quantitative RT-PCR
Total RNAs of hiPSC-CMs were extracted with TRIzol LS reagent (ambion, life technology) at day 40 after cell beating and reverse transcribed into cDNA. The cDNAs were synthesized from total RNAs by reverse transcription with Verso cDNA Synthesis Kit (Thermo Scientific). Quantitative PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems, Thermo Fisher Scientific) in the CFX96 TOUCH qPCR instrument (Bio-Rad). The thermal profile for qPCR was 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The RyR2 expressions were normalized to those obtained for the 18S expression levels in the same samples. Each cell line was run in 3–4 different groups of cardiomyocytes, and each group is the average of 3–6 hiPSC-CM samples, which were differentiated from the same passage of hiPSC but in individually separated culture dishes. Primers for 18S are 5’-TAGAGGGACAAGTGGCGTTC-3′ (forward) and 5′-CGCTGAGCCAGTCAGTGT-3′ (reverse). Primers for RyR2 are 5’-AAGGATGTGGGCTTCTTTCA-3’ (forward) and 5’-AGTTGCAGGAATCGGAAGAG-3’ (reverse).
2.4. hiPSC-CMs dissociation
The hiPSC-CM cell lines were grown in culture for 30–40 days before dissociating and re-plating for electrophysiological and Ca2+ imaging experiments. The mechanical dissociation of hiPSC-CM clusters into single cardiomyocytes was made according to following protocol: 1) Visualize spontaneously beating EBs under the microscope, and mechanically dissect them from the gelatin coated wells with a curved 23G needle. 2) Aspirate the dissected EBs with their original medium and transfer them to a 50ml test tube. 3) Centrifuge the EBs suspension within the test tube at a rate of 1000 rpm for 5 min. 4) Resuspend the EBs in 10 ml of fresh medium and transfer them to a 15 ml test tube. 5) Centrifuge at a rate of 900 rpm for 2 min. 6) Wash the cells 3×with PBS (centrifuge between washings at a rate of 900 rpm for 2 min). 7) Add 5 ml of 1 mg/ml collagenase type IV solution (5mg collagenase IV-low Ca2+ solution) to the centrifuged washed cells. 8) Incubate the solution with EBs in 37°C bath for 5 min. 9) Incubate with rotator for additional 45 min in 37°C. 10) Centrifuge at a rate of 1000 rpm for 5 min. 11) Resuspend cells in 3ml of resuspension solution. Incubate in 37°C for additional 15 min. 12) Centrifuge at a rate of 1000 rpm for 5 min. Single hiPSC-CMs were then placed on fibronectin (2.5 μg/ml)-coated glass coverslips of 6-well plates after collagenase B treatment, and then incubated for 36 to72 h before their use in electrophysiological or Ca2+ imaging experiments.
2.5. Electrophysiological recordings
Whole-cell Ca2+ currents (ICa), action potentials, and caffeine induced INCX were recorded using the perforated-patch mode of the patch-clamp technique [36, 37], induced by amphotericin B (1mg/ml) [38, 39]. After waiting approximately 5–10 minutes, series resistance fell below 20 MΩ and tight seals (>1 GΩ) were achieved using an intracellular solution composed (in mM): 145 Glutamic Acid, 9 NaCl, 1 MgCl2 and 10 HEPES, pH 7.2~7.3, adjust with CsOH. Cells were bathed in a Tyrode’s solution composed of (in mM): 137 NaCl, 1 MgCl2, 2 CaCl2, 5.3 KCl, 10 glucose, and 10 HEPES.
In all experiments ICa was recorded at room temperature (22–25 °C) using a Dagan voltage-clamp amplifier controlled by pClamp-9 software running on a personal computer. All other experiments that integrate the different sections of results were also done at similar controlled room temperature ~22–25 °C except the TIRF measurements of Ca2+ sparks that were carried out at warmer temperature of ~25–30 °C. Borosilicate patch pipettes with 3–5 MΩ resistance were prepared using a horizontal pipette puller (Model P-87, Sutter Instruments, CA). The series resistance of patched cell was monitored until it decreased to < 20 MΩ, after which the experiments were began. The liquid junction potential was corrected before seal formation.
In the ICa recordings, the holding potential was set at −40 mV in order to inactivate Na+ channels. ICa was activated by 100-ms depolarizing voltage pulses to 0 mV measured at 5 s intervals. To measure fractional release, ICa was first activated using 100-ms depolarizing pulses to 0 mV followed by application of 5 or 20mM Caffeine during 1 second and then the generated calcium release transients and accompanying INCX were measured. The measured currents were filtered at 1 or 10 kHz, digitized at 10 or 100 kHz, and plotted and analyzed in terms of magnitude and time constants of their inactivation, using Graph Prism (GraphPad Corp., San Diego, CA, USA) and pCLAMP 9.0 software.
To measure membrane potential (Em) oscillations the current-clamp configuration of the patch-clamp technique was used. The holding potential was first set at −50 mV in voltage clamp mode, before the amplifier was shifted to the current-clamp mode, where zero current injection was indicated. Only cells with a tight seal and leak currents of < 5 pA were used for experimental analysis [40]. Cell size was estimated from membrane capacitance measurements. This approach is particularly useful in hiPSC-CMs which have few membrane invaginations or t-tubular system at these stages of development [41, 42]. Note that earlier studies have shown a positive linear correlations between membrane capacitance and cell volume in cardiomyocytes of several species [43].
2.6. Fluorometric Ca2+ measurements in voltage-clamped cells
For short-term experiments (figures 2 to 5 and S4–S5), intracellular global Ca2+ signals triggered by ICa or caffeine were measured in single isolated beating hiPSC-CMs incubated for 25 minutes with Ca2+- indicator dye Fluo-4AM (1 μM, Invitrogen) at 37 °C and 5% CO2. SR Ca2+ Leak and load were calculated according to protocols already established [44]. SR Ca2+ leak and load values in WT and N771D mutant hiPSC-CMs were obtained from changes of Ca2+-dependent fluorescence signal on rapid exposure to 1 mM tetracaine (Tet) followed by rapid application of caffeine. The SR was kept Ca2+ loaded by pacing at ~0.5–1.0 Hz in Tyrode’s solution, and then the solution was rapidly switched to 1 mM tetracaine (Tet) in zero Na+ and Ca2+ Tyrode’s solution, followed by 1s long application of 5 mM caffeine (Caff) in zero Na+ and Ca2+ Tyrode’s solution without tetracaine. For long-term experiments, (figures 6–8 and S1 to S3, S6 and S8), cytosolic Ca2+ signals were measured in WT and N771D mutant hiPSC-CMs, using the genetically engineered virally introduced biosensor GCaMP6 (Kd=200 nM, λex=488 nm). In figure 6, S6 and S8 additionally, RyR2 Ca2+ μ-domains were monitored using the genetically engineered virally introduced biosensor GCaMP6-FKBP targeted to FKBP-12.6 (calstabin-2) binding site of RyR2 (Kd=250 nM, λex=488 nm). The GCaMP6 probes and Fluo-4M were excited at 460 nm using a LED-based illuminator (Prismatix, Modiin Ilite, Israel) and Ca2+-dependent emitted light (>500nm) was detected with a photomultiplier tube using a Zeiss Axiovert 100 TV inverted microscope. For both probes and dye the parameters quantified were the basal fluorescence (F0) and the peak of the Ca2+ transient (ΔF).
Fig. 2.-. Voltage-dependent inward current study carried by 2mM Ca2+ through L-type calcium channel in WT and N771D mutant hiPSC-CMs.

Panels A and B show in the left and right depolarization-activated whole-cell currents from WT and N771D hiPSC-CMs cell lines respectively obtained by 10 mV step depolarizations of 100 ms duration from −50 mV to +60 mV with a holding potential of −50 mV. Panel C include in the bottom I-V relationship curves for 2 mM extracellular Ca2+, plotted with the average values of the ICa (mean) and the SEM for each voltage, in black or green colors assigned to the WT and the N771D mutant; the corresponding calcium transients evoked by the voltage depolarizations are shown at the top. Panel D plotted the CICR gain voltage-dependent relationship curve estimated how the relative increase in fluorescence elicited by the voltage depolarization pulses and normalized with respect to cell size and SR Ca2+ content values described in the manuscript. The number of cells (n) and the number of different cultures conducted to obtain these data (N) are indicated in parentheses as (n, N). Data were presented as the mean ± SEM in this figure. * p<0.05; n.s., not significant by unpaired two-tailed Student’s t-test in this figure.
Fig. 5.-. SR Ca2+ leak and load in WT and N771D mutant hiPSC-CMs.

Panels A and B show representative time courses of normalized Ca2+-dependent fluorescence changes after rapid exposure to 1 mM tetracaine (Tet) followed by application of 5 mM caffeine (Caff) in zero Na+ and Ca2+ Tyrode’s solution. Panel D exhibit quantified SR Ca2+ load levels at zero Na+ and Ca2+ conditions. Panel C and E show quantified SR Ca2+ leak levels and normalized to the SR store size respectively for the WT and the N771D mutant hiPSC-CMs. The number of cells (n) and the number of different cultures conducted to obtain these data (N) are indicated in parentheses as (n, N) in panel C. Data were presented as the mean ± SEM in this figure. n.s., not significant; *P <0.05; compared to WT by unpaired two-tailed Student’s t-test in this figure.
Fig. 6.-. Spontaneous SR Ca2+ release modulation by FKBP12.6.
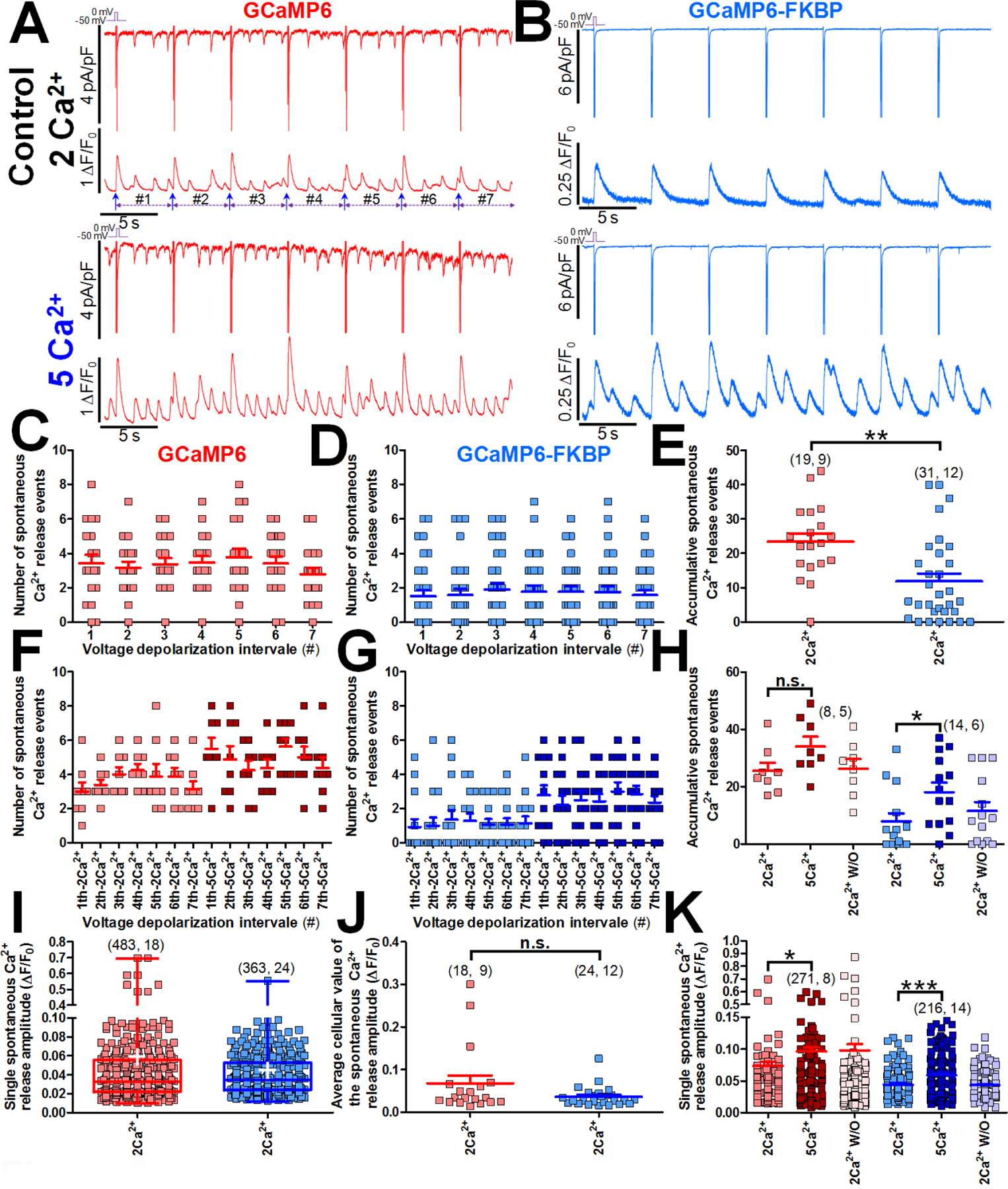
Panels A and B show two cell examples (treated with 2 and 5 mM extracellular Ca2+) of voltage clamp pulses from N771D hiPSC-CMs that were infected with the global GCaMP6 and local GCaMP6-FKBP probes respectively to measure the cytosolic and RyR2 μ-domains calcium concentration respectively, Original traces show ICa induced by voltage-depolarizing pulse trains forming 7 intervals between the voltage depolarization of 5 s each (above, 0.2 Hz) and its corresponding calcium signals (below). Throughout each of the 7 voltage depolarization intervals, between the calcium transients induced by ICa we found frequent spontaneous SR Ca2+ release events of small magnitude which can activate the Na+/Ca2+ exchanger. Panel C and D show in scatter plots the number of spontaneous Ca2+ release events found from each cell studied in each of the 7 intervals. The red or blue colors refer to cells treated with GCaMP6 or GCaMP6-FKBP respectively in all cases, with light or dark colors for 2 or 5 mM extracellular Ca2+. Panel E plotted the total number of spontaneous Ca2+ release events found over the 35s of full recording for every single cell studied. In panels C-E we show the frequency of events found only in cells subjected to extracellular 2 mM Ca2+ control condition, while panels F-H show the results of the group of cells where we studied the effect of extracellular 5 mM Ca2+ in the spontaneous Ca2+ release events. Panel I plotted the amplitude of every single spontaneous Ca2+ release event from all cells studied with at least 3 events in total while in the panel J each plotted point represents the mean value of all the events from each cells studied. Panel K show the effect of 5 mM extracellular Ca2+ in the amplitude of single spontaneous Ca2+ release events. Data are mean ± SEM. The number of cells (n) and the number of different cultures (N) are indicated as (n, N) with the exception of panels I and K which represent the total number of individual spontaneous Ca2+ release events and the number of cells from which they were obtained. n.s., not significant; *P <0.05; **P<0.01; ***P<0.001, compared to 5 mM extracellular Ca2+ condition or GCaMP6 FKBP by unpaired two-tailed Student’s t-test in this figure.
Fig. 8.-. Spontaneous Ca2+ release from SR in WT and N771D mutant hiPSC-CMs.

Panels A and B present original records of consecutive ICa induced by voltage-depolarizing pulse trains (above, 0.2 Hz) and its corresponding cytosolic calcium transients (below) for the WT (black traces) and N771D mutant (green traces) hiPSC-CMs; between the calcium transients induced by ICa we found very frequent spontaneous SR Ca2+ release events of small magnitude (red arrows) which can activate the Na+/Ca2+ exchanger (blue arrows). Panel C plotted the frequency of spontaneous SR Ca2+ release events for the WT and N771D mutant hiPSC-CMs with the ratio of cells that present these events indicated in red brackets. Panels D and E show time courses of spontaneous Ca2+ sparks recorded in two single cells of WT and N771D mutant hiPSC-CMs. Note the higher number of these events in N771D mutant hiPSC-CMs (panel D). The time course of normalized Ca2+ sparks recorded at different color code regions (pane E). Panel F and G show the frequency of spontaneous Ca2+ sparks and a frequency histogram of the spark duration at its half maximum respectively for WT and the two different clones of N771D mutant hiPSC-CMs studied. ROI: Color coded regions of interest corresponding to locations of Ca2+ sparks. TIRF images of Ca2+ sparks correspond to the Ca2+ fluorescence changes in the panels. Ca2+ sparks were recorded at 60–70 Hz. The number of cells that present spontaneous SR Ca2+ release events (n) and the number of total cells studied (N) are indicated with the red parentheses as (n, N) in panel C. The number of spontaneous Ca2+ sparks analyzed is indicated as (n) in panel G for WT and the two clones of N771D mutant hiPSC-CMs. Data were presented as the mean ± SEM in this figure. **P<0.01, compared to WT by unpaired two-tailed Student’s t-test in this figure.
2.7. Cell staining and confocal microscopy imaging
Two weeks post cardiac differentiation, beating hiPSC-CMs were treated with fatty acids cocktail (BSA-bound fatty acids: 52.5 μM palmitate, 40.5 μM oleate, 22.5 μM linoleate, and 120 μM L-carnitine) for 2–3 weeks, drives hiPSC-CMs towards functionally and morphologically more mature state with a more developed sarcomeric pattern [45]. Cardiomyocytes were infected during 9 hours with adenovirus carrying the targeted genes (FKBP12.6-calstabin- linked to GCaMP6) and cell-staining for confocal imaging was performed after 48 hours in culture. hiPSC-CMs were fixed with 4% paraformaldehyde and mounted with ProLong™ Gold antifade reagent with DAPI. The cell-staining was visualized using Leica SP8 confocal microscope at 488 nm excitation (GCaMP6-FKBP) and 500–550 nm emission.
2.8. Chemical products
Chemical as well as isoprenaline hydrochloride were purchased from Sigma (Sigma-Aldrich, St Louise, MO, USA). Amphotericin B was acquired from Fisher Scientific (Pittsburgh, PA, USA). Stock solution of isoprenaline hydrochloride was prepared daily in deionized water and amphotericin B in DMSO. The inhibitor CHIR99021 was acquired from Selleckchem (Houston, TX, USA) and IWR1 from TOCRIS (Minneapolis, MN, USA), StemFlex, RPMI and B-27 culture mediums were purchased from ThermoFisher Scientific/GIBCO (Grand Island, NY, USA).
2.9. Statistical analysis
Data are presented mainly as columns scatter plot, showing the individual data value of each cell together with the mean (large horizontal line) ± standard error of the mean (SEM, small horizontal line), presented in black or green color for the WT or N771D mutant hiPSC-CMs respectively. The number of cells and cultures are indicated in parentheses as (n, N). Paired or unpaired two-tailed Student’s t test was used to compare means according to the dependent or independent between the data set to compare. A P value equal or smaller than 0.05 was selected as the limit of significance. Significance levels were indicated with an increasing number of asterisks (*P < 0.05, **P < 0.01, ***P < 0.001) and when not being significant by (n.s., P > 0.05). Data sets were tested for normality (Kolmogorov-Smirnov normality test), an assumption for the application of the Student’s t-test. To analyze ICa decay or tau inactivation of the ICa (τi) and the tau of clearance of the calcium transients (τCl), single exponential fits were applied to the decaying part of individual ICa or Ca2+ transients traces using a simplex optimization algorithm as follows: y = y0 + {1 − [Ai exp(− t / τi)]} where Ai represent the amplitudes of the ICa or the Ca2+ transient and τi represent the time constants of inactivation or clearance respectively. All statistical analysis was performed using GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA) and MS Excel (Microsoft, Redmond, WA).
3. RESULTS
3.1. Calcium current and Ca2+ transients in N771D mutants
Calcium currents were measured in 3 to 5 day hiPSC-CM cellular cultures (day zero corresponds to mechanical dissociation into single cells). N771D mutant hiPSC-CMs had the typical bell-shaped voltage dependence of ICa and Ca-transients (Fig. 2C) as the WT cells, with no significant differences in the activation threshold (~−40 to −30 mV), reversal potential (~ +50 mV), voltage at which ICa peaks (~ +0 mV) or the magnitude of calcium transients evoked by ICa (Fig. 2A, B and C). The gain of CICR (corrected for SR Ca2+ content) was, however, significantly enhanced in N771D mutants compared to WT cells Fig.2D. There was no significant difference in the inactivation kinetics of ICa: τi = 25.96 ± 3.75ms, n=34 cells in WT vs. τi = 28.18 ± 3.47 ms, n=41 cells in N771D mutant hiPSC-CMs (Fig. 3A, B and E). Cell size, estimated from membrane capacitance, was not significantly larger, in the N771D mutant (45.29 ± 4.06 pF vs. 37.08 ± 2.04 pF) than WT cells, Fig. 3C. However, normalizing ICa for cell size, revealed a smaller Ca2+ current density in N771D mutants (−5.13 ± 0.51 pA/pF vs. −7.068 ± 0.61 pA/pF, n=41 vs. n=34 cells; Fig. 3A, B and D). Surprisingly, the magnitude of Ca-transients (0.45 ± 0.04 vs. 0.58 ± 0.06, n=41 vs. n=34 cells; Fig. 3A, B and F), its time-to-peak (169.7 ± 16.3 ms vs. 152.1 ± 12.56 ms, n=41 vs. n=34 cells; Fig. 3A, B and G) and its relaxation time (442.9 ± 31.3 ms vs. 487.2 ± 33.1 ms, n=41 vs. n=34 cells; Fig. 3A, B and H), activated by ~30% smaller ICa, were not significantly different than the WT cells.
Fig. 3.-. Calcium current and SR Ca2+ release comparison induced by voltage depolarization pulses in WT and N771D mutant hiPSC-CMs.

Panels A and B show examples of original traces of calcium currents (above) and cytosolic calcium transients (below) obtained from two different WT and N771D mutant hiPSC-CMs respectively, activated by 100 ms depolarization from −40 mV to 0 mV, exhibiting the clearance rates of the calcium transient in red color (τCl). Panel C plotted the cell size values, estimated from membrane capacitance measurements, and panels D and E ICa parameters as density of the ICa peak and inactivation tau constant of the ICa (τi) obtained from WT (black circles) and N771D mutant (green squares) hiPSC-CMs. Panels F-H exhibit in scatter plots the parameters of calcium release from SR such as the magnitude (panel F), activation time (panel G) and clearance (panel H) of the cytosolic calcium transients. Cytosolic calcium transient clearance values are a measure extrapolated to SR SERCA2a activity. The number of cells (n) and the number of different cultures conducted to obtain these data (N) are indicated in parentheses as (n, N). Data were presented as the mean ± SEM in this figure. *P < 0.05; n.s., not significant; by unpaired two-tailed Student’s t-test in this figure.
Using depolarizing pulses to ~0mV followed by 1s-long puffs of 5mM caffeine, allowed quantification of SR Ca2+ content as compared to ICa-gated Ca2+ release, Fig. 4A & B. The integral of caffeine fluorescence signal, was 36.09 % smaller in N771D mutant compared to WT cells (1149 ± 134.2 vs. 1798 ± 210.4 V × ms, n=37 vs. n=34 cells; Fig. 4C). Consistent with the fluorescence measurements, the integral of INCX activated by caffeine-triggered Ca2+ release was also reduced by ~30% in N771D mutants compared to WT cells (30033 ± 2188 vs. 42857 ± 2637 pA × ms, n=37 vs. n=34 cells; Fig. 4D), suggesting significantly smaller SR Ca2+ store in the mutant cells. Since N771D mutants show the same proportionate reduction in both SR Ca2+ release and SR Ca2+ content compared to WT cells, the fractional Ca2+ release (measured as the ratio of I - and caffeine-induced Ca2+ Ca release), remained about the same in both cell types (38.81 ± 2.57% vs. 44.62 ± 3.51 %, n=37 vs. n=34 cells; Fig. 4G). Considering the calcium current values were significantly lower in the mutants (~7pA vs. 5pA, Fig.4F) and Ca2+ content was smaller by ~30% (Fig.4C), the CICR gain or efficiency of calcium release appears to be significantly higher in mutant cells (10.01 ± 1.04 vs. 6.47 ± 0.52, n=37 vs. n=34 cells; Fig. 4H), suggesting that SR Ca2+ release was more effectively triggered by ICa in the N771D mutant cells.
Fig. 4.-. Magnitude and efficiency of the calcium-induced calcium release (CICR) from SR in WT and N771D mutant of hiPSC-CMs.

Panels A and B show representative original records of ICa, INCX (above) and Ca2+ signals triggered by ICa or 5mM Caffeine (below) in WT and N771D mutant of hiPSC-CMs respectively. Panel C show the SR Ca2+ content expressed as the integral of the fluorescence area under the calcium transient curve evoked by a saturating concentration of 5 mM caffeine. Panel D plotted the activity of the sodium-calcium exchanger to clearance the cytosolic calcium released by the caffeine pulse, measured as the integral of electric charge under the INCX curve, an another way to measure indirectly the SR Ca2+ content. In panel E we presented the average values of diastolic Ca2+ measured how the basal fluorescence (F0). Efficiency (fractional release, panel G) was calculated as the ratio of the ICa-triggered Ca2+ transient [ΔF/F0 (ICa)], between the amplitude of the Ca2+ transient generated by application of 1 second 5mM Caffeine [ΔF/F0 (Caffeine)]. Panel F plotted the average values of density of ICa peak and note how this parameter is significantly smaller in the N771D mutant, consequently the parameter which represent the magnitude of Gain (panel H), normalized respect to the density of the ICa, are significantly higher in the N771D mutant hiPSC-CMs. The number of cells (n) and the number of different cultures conducted to obtain these data (N) are indicated in parentheses as (n, N) in panel C. Data were presented as the mean ± SEM in this figure. *P < 0.05; **p<0.01; n.s., not significant, compared to WT by unpaired two-tailed Student’s t-test in this figure.
Similar results were found with another clone (#4_1) of N771D mutant cells that also expressed significantly reduced ICa current density (−4.45 ± 0.44 pA/pF vs. −7.56 ± 0.80 pA/pF in WT cells, n=23 vs. n=19 cells; Fig. S4A, B and F); smaller ~49% SR Ca2+ content (1756 ± 189 vs. 2625 ± 390 V × ms, n=23 vs. n=19 cells; Fig. S4C & D) and higher CICR gain (10.60 ± 0.96 vs. 4.63 ± 0.55, n=23 vs. n=19 cells; Fig. S4H).
3.2. RyR2 Ca2+ leak and SR Ca2+ load in WT and N771D mutant cells
To measure SR leak from RyR2, cells were super-perfused with 1mM tetracaine in zero Na+/zero Ca2+ solutions and the decrease in diastolic Ca2+ fluorescence levels was quantified as the SR Ca2+ leak (Fig. 5A, B). The Ca2+ content of SR, quantified from magnitude of Ca2+ transients triggered by 5mM caffeine, was not significantly different in N771D mutants compared to WT hiPSC-CMs (1.93 ± 0.12 vs. 2.02 ± 0.17, n=32 vs. n=24 cells; Fig. 5A, B and D). Unexpectedly, however, the magnitude of RyR2 Ca2+ leak was smaller in the N771D mutants (0.27 ± 0.02 vs. 0.35 ± 0.02, n=32 vs. n=24 cells; Fig. 5A, B and C), especially when corrected for SR Ca2+ load (16.27 ± 1.39% vs. 21.76 ± 2.68%, n=32 vs. n=24 cells; Fig. 5E). Similarly, the second N771D mutant line (#4_1), also showed smaller RyR2 Ca2+ leak compared to WT cells (0.20 ± 0.01 vs. 0.32 ± 0.01, n=50 vs. n=47 cells; Fig. S5).
Since the equivalent N760D-RyR1 mutation in skeletal muscle was reported to reduce FKBP binding to RyR1 by ~70% [49], we examined whether the N771D mutation in RyR2 would similarly destabilize the FKBP binding to RyR2. RyR2 targeted GCaMP6-FKBP probe over-expression approach was used to address this issue, because previous studies had shown that such an approach resulted both in lower diastolic Ca2+ leak and reduced frequency of spontaneous SR Ca2+ releases in WT adult cardiomyocytes [22, 50, 51]. Figure 6 A–E, shows that the frequency of spontaneous SR Ca2+ release events were reduced by 49% in the N771D mutant hiPSC-CMs that were infected with RyR2 targeted GCaMP6-FKBP probe compared to cells infected with untargeted GCaMP6 probe (18 of 31 cells infected with the GCaMP6-FKBP probe were silent or had event frequency < 10 as compared to only 1 of 19 cells infected with the untargeted GCaMP6 probe), Fig. 6E. These findings are consistent with previous studies suggesting that overexpression of FKBP via the GCaMP6-FKBP approach stabilizes RyR2 activity. Interestingly, increasing extracellular calcium to 5mM enhanced the frequency of spontaneous Ca2+ release events more effectively in N771D mutant cells expressing targeted GCaMP6-FKBP probe than in cells infected with the untargeted GCaMP6 probe (~ 130% vs. 33%, n=14 vs. n=8 cells; Fig. 6F–H), suggesting that FKBP12.6 continues to bind to N771D-RyR2 mutant and to lower the frequency of aberrant Ca2+ releases. In WT hiPSC-CMs, FKBP overexpression also reduced the frequency of spontaneous SR Ca2+ releases by 47% (Fig. S6, n=43 cells). Consistent with this finding, the frequency of aberrant spontaneous Ca2+ releases were higher in another N771D mutant clone (Fig. S8, see also Fig. 6 and Fig. 8 vs. Fig. S6, n=161 cells). Note that even though the frequency of aberrant releases was higher in mutant cells, their amplitude was unaffected in either mutant or wild type cells by FKBP (Fig. S8H, n=138 cells). The reduction in the aberrant spontaneous Ca2+ releases by FKBP in the mutant cells were supported by confocal structural studies showing that the characteristic striated pattern of RyR2 binding to FKBP12.6 remained mostly unaltered in N771D compared to WT hiPSC-CMs, Figure S7.
3.3. β-adrenergic modulation of ICa, spontaneous action potential and Ca-transients
We have already reported that submicromolar concentrations of isoproterenol (ISO) increase ICa significantly in WT hiPSC-CMs [46]. Fig. S1 shows that even though the density of ICa is significantly smaller in the N771D mutants compared to WT cells (−3.75 ± 0.60 pA/pF vs. −6.08 ± 0.84 pA/pF, n=6 vs. n=6 cells; panels A, B and C), ISO enhanced ICa by ~ 56 % in N771D mutants vs. ~ 37% in the WT hiPSC-CMs, (from −3.75 ± 0.60 pA/pF to −5.92 ± 0.82 pA/pF, Fig. S1B and C, vs from −6.08 ± 0.84 pA/pF to −7.96 ± 1.54 pA/pF, Fig. S1A and C). Nevertheless, ISO appeared to be less effective in enhancing the calcium transients in N771D mutants, ~ 21% from 0.37 ± 0.07 ΔF/F0 to 0.45 ± 0.06 ΔF/F0, as compared to ~47% in WT cells from 0.47 ± 0.07 ΔF/F0 to 0.64 ± 0.12 ΔF/F0, n=6 vs. n=6 cells; Fig. S1 C&D. These results suggest that although N771D mutants are more efficient in releasing calcium from SR compared to WT cells under control conditions (Fig. 4H), ISO-treatment failed to enhance further the efficiency of Ca2+ release in N771D mutants compared to WT cells (Fig. S1D).
Detailed analysis of the action potentials parameters (AP amplitude, duration, spike rise-time, spike decay-time, after hyperpolarization amplitude, Fig 7) did not show significant differences between the action potential parameters of the two cell types, either in control or ISO-treated cells (Fig. 7C and data not shown). N771D mutants expressed, however, APs with lower spike decay times (Fig. 7D) and more hyperpolarized resting membrane potentials (−52.68 ± 1.92 mV vs. −61.38 ± 2.45 mV, n=6 vs. n=11 cells; Fig. 7E). There were also no significant differences in the calcium transients triggered by APs in the two cell types or in ISO-treated cells (Fig. 7C and data not shown). N771D mutant cells showed a higher frequency of occurrence of delayed afterdepolarizations (DADs) (7 of 11 cells vs. only 1 of 6 in the WT cells). The aberrancies in the membrane potential (Fig.7A and B, red arrows) mostly appeared in phase 4 of the APs (Fig.7A and B, blue arrows), as noted in arrhythmogenesis of patients with heart disease [47, 48]. Acute 20 second applications of 500nM ISO increased the frequency of spontaneous beating that ranged between 5–1.0 Hz in control solutions, by ~ 20–30 % in both WT (Fig. S2A, C and D) and N771D mutants (Fig. S2B, C and D). The diastolic and systolic Ca2+ levels remained similar and were not significantly changed by ISO (Fig. S2A, B, E and F).
Fig. 7.-. β-adrenergic regulation and measurement of the action potential parameters from WT and N771D mutant hiPSC-CMs.

Panels A and B show representative original traces of spontaneous APs (below) simultaneously recorded with the induced cytosolic Ca2+ fluctuations (above) in the WT and N771D mutant hiPSC-CMs respectively. APs were recorded under the current-clamp mode of the perforated patch-clamp technique with a holding current of zero, allowing the amplifier to act as a voltage follower and measure the actual membrane potential. Isoprenaline 500 nM was perfused during 20 seconds between the initial 20 seconds of 2 mM Ca2+ Tyrode control condition and another 20 seconds of ISO washout. Note how during simultaneous recording under control, ISO and ISO washout conditions, delayed after depolarizations (DADs) were occasionally observed (red arrows) along with their corresponding cytosolic Ca2+ transients (blue arrows). Panel C exhibit individual original examples of APs and spontaneous Ca2+ release events with the ISO and control condition superimposed. In order to make the figure less complex panels D to E display only the two scatter plots of eleven APs parameters studied that show significant differences (spike decay time and resting membrane potential) with WT and N771D mutant hiPSC-CMs represented by circles or squares respectively (gray circles and light green squares show the effect of ISO). The number of action potentials (n) and the number of cells conducted to obtain these data (N) are indicated in parentheses as (n, N) in panel D. Data were presented as the mean ± SEM in this figure. n.s., not significant; *P <0.05; compared to WT by unpaired two-tailed Student’s t-test in this figure.
3.4. Aberrant spontaneous Ca2+ releases in WT and N771D mutant hiPSC-CMs.
Long-duration voltage clamp pulses (35s at −50mVs) were used to record the frequency of spontaneously occurring calcium release events. The cells were first subjected to trains of depolarizing pulses that activated ICa and triggered Ca2+ release transients (Fig. 8A and B). Interspersed between the regularly triggered calcium transients, smaller spontaneous SR Ca2+ release events (Fig. 8B, red arrows) activating INCX (Fig. 8B, blue arrows) were often recorded. The frequency of these spontaneous Ca2+ release events and the number of cells expressing them were significantly higher in the N771D mutant hiPSC-CMs, Fig 8C, consistent with enhanced proclivity to arrhythmogenesis of cells expressing N771D mutation. Note, that although the number of spontaneously occurring Ca2+ sparks were similar (14.65 ± 2.00 vs. 19.09 ± 2.82 or 16.86 ± 1.43 sparks/s, n=13 vs. n=17 or n=11 cells; Fig. 8D and F) in both clones of N771D mutants and WT cells, the spark durations were consistently longer by ~46% in mutant myocytes, (54.73 ± 2.43 vs. 80.21 ± 2.75 ms in the N771D, n=13 vs. n=17 cells; Fig. 8G), consistent with the higher frequency of DADs (Fig.7) and aberrant Ca2+ releases (Fig.8 A, B and C; red arrows).
4. DISCUSSION
4.1. Ca2+ signaling consequences of FKBP-RyR2 binding site mutation in hiPSC-CMs
The major finding of this report is that introduction of N771D-RyR2 mutation in hPSC-CMs increased CICR gain, the frequency of DADs, and spontaneous SR calcium releases, and proclivity to arrhythmogenesis, while decreasing unexpectedly ICa, the diastolic RyR2 Ca2+ leak, and SR Ca2+ content. It is likely that the suppressed ICa was responsible for both the lower SR Ca2+ content and decreased SR leak. The suppressed ICa in the face of increased CICR gain in human FKBP mutant cardiomyocytes may have resulted from a cellular calcium compensatory mechanism that regulates the expression of calcium handling proteins to maintain the cellular Ca2+ homeostasis (67, 68), somewhat similar to suppression of ICa reported in NCX deleted mice (67). The lower SR Ca2+ content of mutant cells resulting from ~30% lower ICa density (3D, & 4F), could also have obscured or masked the enhanced SR Ca2+ leak and been responsible for lower diastolic RyR2 Ca2+ leak [3, 52, 53]. It is, therefore, reasonable to conclude that mutant cells express an emptier SR because of suppressed Ca2+ influx through L-type calcium channels (Fig. 4D). The suppression of aberrant Ca2+ releases in FKBP overexpressed mutant cells (Fig. 6 and Fig. S8), also suggests that FKBP12.6 must still bind to RyR2 in mutant cells, as reported in adult cardiomyocytes [22, 50, 51]. Lack of significant distortion in sarcomeric striation patterns of targeted FKBP-GCaMP6 confocal images of N771D mutant compared to WT hiPSC-CMs (Fig. S7), also supports the idea that unlike the mutation in skeletal muscle (N760D-RyR1), the equivalent mutation in RyR2 does not greatly distort FKBP binding to RyR2. Since the corresponding skeletal muscle mutation (residue N760D-RyR1) had been shown to alter RyR1 folding and FKBP binding to RyR1 [49], it was reasonable to have expected that corresponding RyR2-N771D mutation would similarly cause conformational changes in the residues that bind FKBP, altering its binding to RyR2 and destabilizing its activity [14–16, 26–29], and functionally increasing RyR2 Ca2+ leak. It appears, however, that cellular compensatory mechanism activated by a destabilized FKBP binding to RyR2 counteracted this process, most likely by activating transcription factors that downregulate the expression of calcium channels that result in lower SR Ca2+load and mask the higher leakiness of RyR2.
Alternatively, the observed higher CICR gain, increased DAD and aberrant Ca2+ release frequency (Figs. 4, 6, 7, 8 & S8) in RyR2-N771D mutant cells could have resulted from sensitization of RyR2 to luminal Ca2+ [54–56] or even enhanced mitochondrial contribution to cytosolic Ca2+ [57]. The faster relaxation of Ca2+ transients in the N771D mutants compared to WT cells expressing GCaMP6 (Fig. S6F and S8F) or GCaMP6-FKBP (Fig. S6F), as well as confocal images of asymmetric distribution of mitochondria in cytosolic spaces in the N771D mutants as compared to diffuse distribution of mitochondria in WT hiPSC-CMs (Fig. S9), might also reflect differential role of mitochondrial calcium transporters in the two sets of cells [58, 59].
4.2. Suppressed ICa but enhanced CICR gain in N771D mutant
The enhanced EC coupling gain in mutant cells, despite their ~30% suppressed calcium channel currents (Figs. 3, S4) may have resulted from decreased redundancy of calcium channels to trigger SR Ca2+ release, in a manner similar to activation of GTPase “Rad” regulating the trafficking of L-type β subunit to the cell membrane [65]. The enhanced EC-coupling gain in N771D-RyR2 mutant are also consistent with findings in myo-tubes from patients with dominant CCD RYR1 mutations showing increased excitation-coupled Ca2+ entry, providing further support for the possibility that pathological dysregulation of Ca2+ homeostasis is not only caused by aberrancies of RyR1 Ca2+ release, but also by altered Ca2+ influx through L-type Ca2+ channels [66]. We speculate that enhanced RyR2 Ca2+sensitivity to release Ca2+ and resultant CICR gain activate cellular transcription factors that regulate Ca2+ homeostasis to compensate for FKBP RyR2 destabilization by down regulating the L-type calcium channels, which results in observed lower SR Ca2+ content and SR Ca2+ leak. Similar compensatory mechanisms of increased CICR gain and reduced ICa were reported for NCX deleted myocytes to maintain Ca2+ homeostasis [67]. It is likely that activation of CaMKII and Ca-calmodulin-dependent phosphatase calcineurin pathways that regulate the expression of membrane Ca2+ channels and transporters mediates this process [68, 69].
4.3. β-adrenergic regulation of CICR and SR Ca2+ store in N771D mutant cells
The enhancement in ICa-triggered Ca2+-transients in presence of ISO were modest in N771D mutants, and not significantly different from those of WT cells (~ 36% in WT vs. ~ 21% in the N771D mutant; Fig. S1D). Similarly, the chronotropic effects of ISO were negligible as evaluated by measurements of rate of spontaneous beating or altered AP morphology (Fig. 7C, S2C). We also checked whether the SR Ca2+ load was differentially enhanced by ISO in the two cell types, but found only modest (~16%) enhancement of the SR Ca2+ load in N771D mutants as compared to WT cells (Fig. S3A, C). This modest but directional effect on the SR Ca2+ content in the N771D mutant cells maybe be due to the observed emptier SR Ca2+ content in basal conditions [52, 60].
4.4. Pathophysiological consequences of N771D mutation
We created N771D-RyR2 mutation to investigate the role of FKBP in cardiac calcium signaling because the parallel N760D-RyR1 mutation that associates with CCD in patients [61], reduces FKBP binding to RyR1-SPRY1 domain by~75% [49]. Athough there are no reports of cardiac pathology associated with this mutation, it is not inconceivable that future studies could identify a low-incidence arrhythmia as reported recently for I784F-RyR2 variant, in the same RyR2-SPRY1 domain near the N771D-RyR2 mutation, that produces lethal ventricular tachycardia [62]. Interestingly, somewhat similar to our findings, cellular calcium imaging of this mutation also show reduced SR Ca2+ content with higher propensity for spontaneous Ca2+ oscillations at rest. Even though we confirmed the higher frequencies of DADs and aberrant Ca2+ releases in both N771D-RyR2 mutant lines (#49_1, #4_1), we did not find a higher RyR2 Ca2+ leak that generally associates with arrhythmogenic calcium release conditions, most likely because the low SR Ca2+ content of N771D mutants may have “obscured” the higher SR Ca2+ leak, it can be extrapolated from model of Shannon et al. [44]. Consistent with the stabilizing role of FKBP12.6, we confirmed that its over-expression significantly suppressed the aberrant Ca2+ releases, suggesting that FKBP is still regulating RyR2 in N771D mutant (Fig. 6 and S8). The lower room temperatures used in our study may alternatively explain these divergent results, as calcium homeostasis in excitable cells is highly temperature dependent [63, 64].
4.5. Conclusion
The effects of FKBPs on RyR2 gating and its role in pathologies of muscular dystrophy, sarcopenia, cardiac arrhythmias and heart failure regulation are somewhat controversial. Our data showing higher frequency of aberrant calcium releases, longer spontaneous Ca2+ sparks, and greater efficiency of EC-coupling in N771D-RyR2 mutant cardiomyocytes support the observed higher proclivity to arrhythmogenesis, despite the suppressed ICa and smaller SR Ca2+ leak. The failure to observe the higher SR Ca2+ leakiness, predicted by earlier studies showing destabilization of FKBP binding, most likely are caused by novel effect of this mutation on suppressing ICa that results in lower SR Ca2+ content and masks the enhanced SR Ca2+ leakiness.
Supplementary Material
Highlights.
CRISPR/Cas9 gene-editing at the RyR2 FKBP binding site (N771D-RyR2) in hiPSC-CMs.
The parallel skeletal mutation (N760D-RyR1) is related to CCD and arrhythmogenesis.
The N771D-RyR2 mutation induced cellular arrhythmogenesis.
Calcium signaling from N771D-RyR2 show suppressed ICa, SR Ca2+ content and leak.
ACKNOWLEDGMENTS:
The authors thank Dr. Naohiro Yamaguchi for his continued support in the design of the mutant plasmids and editing of this manuscript. We thank Ms. Caroline Everett for preparation of hiPSC-CMs and confocal imaging experiments.
Funding Statement:
This work was supported by the National Institute of Health grants to M.M.: (1) R56HL147054 and Ro1HL153504.
LIST OF ABBREVIATIONS
- APs
action potentials
- [Ca2+]i
intracellular calcium-concentration
- CCD
central core disease
- CICR
calcium-induced calcium release
- DADs
delayed afterdepolarizations
- EADs
early afterdepolarizations
- GCaMP6
cytosolic-targeted Ca2+ biosensor
- hiPSC-CMs
human induced pluripotent stem cell-derived cardiomyocytes
- ICa
calcium current
- ISO
Isoproterenol
- MH
malignant hyperthermia
- RYR
ryanodine receptor
- SR
sarcoplasmic reticulum
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CREDIT AUTHOR STATEMENT
Designed the experiments and wrote the final version of the MS: M. Morad.
Conducted experiments and analysis and wrote the first versions of the MS: Jose-Carlos Fernandez-Morales, Xiao-Hua Zhang, Yanli Xia.
Creation of N771D mutant cardiomyocytes and their maintenance in culture: Taylor J. Rienzo.
Editing the manuscript: Jose-Carlos Fernandez-Morales, Xiao-Hua Zhang, Yanli Xia and M. Morad.
CONFLICT OF INTEREST STATEMENT
No conflicts of interest, financial or otherwise, are declared by the author(s).
DATA AVAILABILITY STATEMENT
All relevant data that support the findings of this study are available from the authors upon reasonable request.
REFERENCES
- [1].Santana LF, Cheng H, Gomez AM, Cannell MB, Lederer WJ, Relation between the sarcolemmal Ca2+ current and Ca2+ sparks and local control theories for cardiac excitation-contraction coupling, Circ Res, 78 (1996) 166–171. [DOI] [PubMed] [Google Scholar]
- [2].Stern MD, Song LS, Cheng H, Sham JS, Yang HT, Boheler KR, Rios E, Local control models of cardiac excitation-contraction coupling. A possible role for allosteric interactions between ryanodine receptors, J Gen Physiol, 113 (1999) 469–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nabauer M, Callewaert G, Cleemann L, Morad M, Regulation of calcium release is gated by calcium current, not gating charge, in cardiac myocytes, Science, 244 (1989) 800–803. [DOI] [PubMed] [Google Scholar]
- [4].Wehrens XH, Lehnart SE, Marks AR, Intracellular calcium release and cardiac disease, Annu Rev Physiol, 67 (2005) 69–98. [DOI] [PubMed] [Google Scholar]
- [5].Sham JS, Cleemann L, Morad M, Functional coupling of Ca2+ channels and ryanodine receptors in cardiac myocytes, Proc Natl Acad Sci U S A, 92 (1995) 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cleemann L, Wang W, Morad M, Two-dimensional confocal images of organization, density, and gating of focal Ca2+ release sites in rat cardiac myocytes, Proc Natl Acad Sci U S A, 95 (1998) 10984–10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lopez-Lopez JR, Shacklock PS, Balke CW, Wier WG, Local, stochastic release of Ca2+ in voltage-clamped rat heart cells: visualization with confocal microscopy, J Physiol, 480 (Pt 1) (1994) 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhang XH, Wei H, Xia Y, Morad M, Calcium signaling consequences of RyR2 mutations associated with CPVT1 introduced via CRISPR/Cas9 gene editing in human-induced pluripotent stem cell-derived cardiomyocytes: Comparison of RyR2-R420Q, F2483I, and Q4201R, Heart Rhythm, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wang W, Cleemann L, Jones LR, Morad M, Modulation of focal and global Ca2+ release in calsequestrin-overexpressing mouse cardiomyocytes, J Physiol, 524 Pt 2 (2000) 399–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Priori SG, Chen SR, Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis, Circ Res, 108 (2011) 871–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Marks AR, Priori S, Memmi M, Kontula K, Laitinen PJ, Involvement of the cardiac ryanodine receptor/calcium release channel in catecholaminergic polymorphic ventricular tachycardia, J Cell Physiol, 190 (2002) 1–6. [DOI] [PubMed] [Google Scholar]
- [12].Meissner G, Molecular regulation of cardiac ryanodine receptor ion channel, Cell Calcium, 35 (2004) 621–628. [DOI] [PubMed] [Google Scholar]
- [13].Yamaguchi N, Xu L, Evans KE, Pasek DA, Meissner G, Different regions in skeletal and cardiac muscle ryanodine receptors are involved in transducing the functional effects of calmodulin, J Biol Chem, 279 (2004) 36433–36439. [DOI] [PubMed] [Google Scholar]
- [14].Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR, PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts, Cell, 101 (2000) 365–376. [DOI] [PubMed] [Google Scholar]
- [15].Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N, D’Armiento JM, Napolitano C, Memmi M, Priori SG, Lederer WJ, Marks AR, FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death, Cell, 113 (2003) 829–840. [DOI] [PubMed] [Google Scholar]
- [16].Brillantes AB, Ondrias K, Scott A, Kobrinsky E, Ondriasova E, Moschella MC, Jayaraman T, Landers M, Ehrlich BE, Marks AR, Stabilization of calcium release channel (ryanodine receptor) function by FK506-binding protein, Cell, 77 (1994) 513–523. [DOI] [PubMed] [Google Scholar]
- [17].Fauconnier J, Thireau J, Reiken S, Cassan C, Richard S, Matecki S, Marks AR, Lacampagne A, Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy, Proc Natl Acad Sci U S A, 107 (2010) 1559–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Andersson DC, Betzenhauser MJ, Reiken S, Meli AC, Umanskaya A, Xie W, Shiomi T, Zalk R, Lacampagne A, Marks AR, Ryanodine receptor oxidation causes intracellular calcium leak and muscle weakness in aging, Cell Metab, 14 (2011) 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wehrens XH, Lehnart SE, Reiken SR, Deng SX, Vest JA, Cervantes D, Coromilas J, Landry DW, Marks AR, Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2, Science, 304 (2004) 292–296. [DOI] [PubMed] [Google Scholar]
- [20].Wehrens XH, Lehnart SE, Reiken S, van der Nagel R, Morales R, Sun J, Cheng Z, Deng SX, de Windt LJ, Landry DW, Marks AR, Enhancing calstabin binding to ryanodine receptors improves cardiac and skeletal muscle function in heart failure, Proc Natl Acad Sci U S A, 102 (2005) 9607–9612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Marks AR, Ryanodine receptors, FKBP12, and heart failure, Front Biosci, 7 (2002) d970–977. [DOI] [PubMed] [Google Scholar]
- [22].Prestle J, Janssen PM, Janssen AP, Zeitz O, Lehnart SE, Bruce L, Smith GL, Hasenfuss G, Overexpression of FK506-binding protein FKBP12.6 in cardiomyocytes reduces ryanodine receptor-mediated Ca(2+) leak from the sarcoplasmic reticulum and increases contractility, Circ Res, 88 (2001) 188–194. [DOI] [PubMed] [Google Scholar]
- [23].Gomez AM, Schuster I, Fauconnier J, Prestle J, Hasenfuss G, Richard S, FKBP12.6 overexpression decreases Ca2+ spark amplitude but enhances [Ca2+]i transient in rat cardiac myocytes, Am J Physiol Heart Circ Physiol, 287 (2004) H1987–1993. [DOI] [PubMed] [Google Scholar]
- [24].Guo T, Cornea RL, Huke S, Camors E, Yang Y, Picht E, Fruen BR, Bers DM, Kinetics of FKBP12.6 binding to ryanodine receptors in permeabilized cardiac myocytes and effects on Ca sparks, Circ Res, 106 (2010) 1743–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhang JZ, Waddell HM, Wu E, Dholakia J, Okolo CA, McLay JC, Jones PP, FKBPs facilitate the termination of spontaneous Ca2+ release in wild-type RyR2 but not CPVT mutant RyR2, Biochem J, 473 (2016) 2049–2060. [DOI] [PubMed] [Google Scholar]
- [26].Timerman AP, Onoue H, Xin HB, Barg S, Copello J, Wiederrecht G, Fleischer S, Selective binding of FKBP12.6 by the cardiac ryanodine receptor, J Biol Chem, 271 (1996) 20385–20391. [DOI] [PubMed] [Google Scholar]
- [27].Barg S, Copello JA, Fleischer S, Different interactions of cardiac and skeletal muscle ryanodine receptors with FK-506 binding protein isoforms, Am J Physiol, 272 (1997) C1726–1733. [DOI] [PubMed] [Google Scholar]
- [28].Venturi E, Galfre E, O’Brien F, Pitt SJ, Bellamy S, Sessions RB, Sitsapesan R, FKBP12.6 activates RyR1: investigating the amino acid residues critical for channel modulation, Biophys J, 106 (2014) 824–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Galfre E, Pitt SJ, Venturi E, Sitsapesan M, Zaccai NR, Tsaneva-Atanasova K, O’Neill S, Sitsapesan R, FKBP12 activates the cardiac ryanodine receptor Ca2+-release channel and is antagonised by FKBP12.6, PLoS One, 7 (2012) e31956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yano M, Kobayashi S, Kohno M, Doi M, Tokuhisa T, Okuda S, Suetsugu M, Hisaoka T, Obayashi M, Ohkusa T, Kohno M, Matsuzaki M, FKBP12.6-mediated stabilization of calcium-release channel (ryanodine receptor) as a novel therapeutic strategy against heart failure, Circulation, 107 (2003) 477–484. [DOI] [PubMed] [Google Scholar]
- [31].Andersson DC, Marks AR, Fixing ryanodine receptor Ca leak - a novel therapeutic strategy for contractile failure in heart and skeletal muscle, Drug Discov Today Dis Mech, 7 (2010) e151–e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wei H, Zhang XH, Clift C, Yamaguchi N, Morad M, CRISPR/Cas9 Gene editing of RyR2 in human stem cell-derived cardiomyocytes provides a novel approach in investigating dysfunctional Ca(2+) signaling, Cell Calcium, 73 (2018) 104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Si-Tayeb K, Noto FK, Sepac A, Sedlic F, Bosnjak ZJ, Lough JW, Duncan SA, Generation of human induced pluripotent stem cells by simple transient transfection of plasmid DNA encoding reprogramming factors, BMC Dev Biol, 10 (2010) 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lian X, Zhang J, Azarin SM, Zhu K, Hazeltine LB, Bao X, Hsiao C, Kamp TJ, Palecek SP, Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions, Nat Protoc, 8 (2013) 162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F, Genome engineering using the CRISPR-Cas9 system, Nat Protoc, 8 (2013) 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lindau M, Fernandez JM, A patch-clamp study of histamine-secreting cells, J Gen Physiol, 88 (1986) 349–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Horn R, Marty A, Muscarinic activation of ionic currents measured by a new whole-cell recording method, J Gen Physiol, 92 (1988) 145–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Aggett PJ, Fenwick PK, Kirk H, The effect of amphotericin B on the permeability of lipid bilayers to divalent trace metals, Biochim Biophys Acta, 684 (1982) 291–294. [DOI] [PubMed] [Google Scholar]
- [39].Rae J, Cooper K, Gates P, Watsky M, Low access resistance perforated patch recordings using amphotericin B, J Neurosci Methods, 37 (1991) 15–26. [DOI] [PubMed] [Google Scholar]
- [40].Perkins KL, Cell-attached voltage-clamp and current-clamp recording and stimulation techniques in brain slices, J Neurosci Methods, 154 (2006) 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lieu DK, Liu J, Siu CW, McNerney GP, Tse HF, Abu-Khalil A, Huser T, Li RA, Absence of transverse tubules contributes to non-uniform Ca(2+) wavefronts in mouse and human embryonic stem cell-derived cardiomyocytes, Stem Cells Dev, 18 (2009) 1493–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Satin J, Itzhaki I, Rapoport S, Schroder EA, Izu L, Arbel G, Beyar R, Balke CW, Schiller J, Gepstein L, Calcium handling in human embryonic stem cell-derived cardiomyocytes, Stem Cells, 26 (2008) 1961–1972. [DOI] [PubMed] [Google Scholar]
- [43].Satoh H, Delbridge LM, Blatter LA, Bers DM, Surface:volume relationship in cardiac myocytes studied with confocal microscopy and membrane capacitance measurements: species-dependence and developmental effects, Biophys J, 70 (1996) 1494–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Shannon TR, Ginsburg KS, Bers DM, Quantitative assessment of the SR Ca2+ leak-load relationship, Circ Res, 91 (2002) 594–600. [DOI] [PubMed] [Google Scholar]
- [45].Yang X, Rodriguez ML, Leonard A, Sun L, Fischer KA, Wang Y, Ritterhoff J, Zhao L, Kolwicz SC Jr., Pabon L, Reinecke H, Sniadecki NJ, Tian R, Ruohola-Baker H, Xu H, Murry CE, Fatty Acids Enhance the Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells, Stem Cell Reports, 13 (2019) 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Fernandez-Morales JC, Hua W, Yao Y, Morad M, Regulation of Ca(2+) signaling by acute hypoxia and acidosis in cardiomyocytes derived from human induced pluripotent stem cells, Cell Calcium, 78 (2019) 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Verkerk AO, Veldkamp MW, Baartscheer A, Schumacher CA, Klopping C, van Ginneken AC, Ravesloot JH, Ionic mechanism of delayed afterdepolarizations in ventricular cells isolated from human end-stage failing hearts, Circulation, 104 (2001) 2728–2733. [DOI] [PubMed] [Google Scholar]
- [48].Venetucci LA, Trafford AW, O’Neill SC, Eisner DA, The sarcoplasmic reticulum and arrhythmogenic calcium release, Cardiovasc Res, 77 (2008) 285–292. [DOI] [PubMed] [Google Scholar]
- [49].Yuchi Z, Yuen SM, Lau K, Underhill AQ, Cornea RL, Fessenden JD, Van Petegem F, Crystal structures of ryanodine receptor SPRY1 and tandem-repeat domains reveal a critical FKBP12 binding determinant, Nat Commun, 6 (2015) 7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gellen B, Fernandez-Velasco M, Briec F, Vinet L, LeQuang K, Rouet-Benzineb P, Benitah JP, Pezet M, Palais G, Pellegrin N, Zhang A, Perrier R, Escoubet B, Marniquet X, Richard S, Jaisser F, Gomez AM, Charpentier F, Mercadier JJ, Conditional FKBP12.6 overexpression in mouse cardiac myocytes prevents triggered ventricular tachycardia through specific alterations in excitation-contraction coupling, Circulation, 117 (2008) 1778–1786. [DOI] [PubMed] [Google Scholar]
- [51].Pahlavan S, Morad M, Total internal reflectance fluorescence imaging of genetically engineered ryanodine receptor-targeted Ca(2+) probes in rat ventricular myocytes, Cell Calcium, 66 (2017) 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bassani JW, Yuan W, Bers DM, Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes, Am J Physiol, 268 (1995) C1313–1319. [DOI] [PubMed] [Google Scholar]
- [53].Bers DM, Eisner DA, Valdivia HH, Sarcoplasmic reticulum Ca2+ and heart failure: roles of diastolic leak and Ca2+ transport, Circ Res, 93 (2003) 487–490. [DOI] [PubMed] [Google Scholar]
- [54].Gyorke S, Gyorke I, Lukyanenko V, Terentyev D, Viatchenko-Karpinski S, Wiesner TF, Regulation of sarcoplasmic reticulum calcium release by luminal calcium in cardiac muscle, Front Biosci, 7 (2002) d1454–1463. [DOI] [PubMed] [Google Scholar]
- [55].Laver DR, Ca2+ stores regulate ryanodine receptor Ca2+ release channels via luminal and cytosolic Ca2+ sites, Clin Exp Pharmacol Physiol, 34 (2007) 889–896. [DOI] [PubMed] [Google Scholar]
- [56].Jiang D, Chen W, Wang R, Zhang L, Chen SR, Loss of luminal Ca2+ activation in the cardiac ryanodine receptor is associated with ventricular fibrillation and sudden death, Proc Natl Acad Sci U S A, 104 (2007) 18309–18314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zhang XH, Wei H, Saric T, Hescheler J, Cleemann L, Morad M, Regionally diverse mitochondrial calcium signaling regulates spontaneous pacing in developing cardiomyocytes, Cell Calcium, 57 (2015) 321–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Drago I, De Stefani D, Rizzuto R, Pozzan T, Mitochondrial Ca2+ uptake contributes to buffering cytoplasmic Ca2+ peaks in cardiomyocytes, Proc Natl Acad Sci U S A, 109 (2012) 12986–12991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Weber CR, Ginsburg KS, Philipson KD, Shannon TR, Bers DM, Allosteric regulation of Na/Ca exchange current by cytosolic Ca in intact cardiac myocytes, J Gen Physiol, 117 (2001) 119–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zhang XQ, Ng YC, Moore RL, Musch TI, Cheung JY, In situ SR function in postinfarction myocytes, J Appl Physiol (1985), 87 (1999) 2143–2150. [DOI] [PubMed] [Google Scholar]
- [61].Bharucha-Goebel DX, Santi M, Medne L, Zukosky K, Dastgir J, Shieh PB, Winder T, Tennekoon G, Finkel RS, Dowling JJ, Monnier N, Bonnemann CG, Severe congenital RYR1-associated myopathy: the expanding clinicopathologic and genetic spectrum, Neurology, 80 (2013) 1584–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Touat-Hamici Z, Blancard M, Ma R, Lin L, Iddir Y, Denjoy I, Leenhardt A, Yuchi Z, Guicheney P, A SPRY1 domain cardiac ryanodine receptor variant associated with short-coupled torsade de pointes, Sci Rep, 11 (2021) 5243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Herve JC, Yamaoka K, Twist VW, Powell T, Ellory JC, Wang LC, Temperature dependence of electrophysiological properties of guinea pig and ground squirrel myocytes, Am J Physiol, 263 (1992) R177–184. [DOI] [PubMed] [Google Scholar]
- [64].Padin JF, Fernandez-Morales JC, Olivares R, Vestring S, Arranz-Tagarro JA, Calvo-Gallardo E, de Pascual R, Gandia L, Garcia AG, Plasmalemmal sodium-calcium exchanger shapes the calcium and exocytotic signals of chromaffin cells at physiological temperature, Am J Physiol Cell Physiol, 305 (2013) C160–172. [DOI] [PubMed] [Google Scholar]
- [65].Wang G, Zhu X, Xie W, Han P, Li K, Sun Z, Wang Y, Chen C, Song R, Cao C, Zhang J, Wu C, Liu J, Cheng H, Rad as a novel regulator of excitation-contraction coupling and beta-adrenergic signaling in heart, Circ Res, 106 (2010) 317–327. [DOI] [PubMed] [Google Scholar]
- [66].Treves S, Vukcevic M, Jeannet PY, Levano S, Girard T, Urwyler A, Fischer D, Voit T, Jungbluth H, Lillis S, Muntoni F, Quinlivan R, Sarkozy A, Bushby K, Zorzato F, Enhanced excitation-coupled Ca(2+) entry induces nuclear translocation of NFAT and contributes to IL-6 release from myotubes from patients with central core disease, Hum Mol Genet, 20 (2011) 589–600. [DOI] [PubMed] [Google Scholar]
- [67].Pott C, Philipson KD, Goldhaber JI, Excitation-contraction coupling in Na+-Ca2+ exchanger knockout mice: reduced transsarcolemmal Ca2+ flux, Circ Res, 97 (2005) 1288–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wu X, Zhang T, Bossuyt J, Li X, McKinsey TA, Dedman JR, Olson EN, Chen J, Brown JH, Bers DM, Local InsP3-dependent perinuclear Ca2+ signaling in cardiac myocyte excitation-transcription coupling, J Clin Invest, 116 (2006) 675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bers DM, Ca(2)(+)-calmodulin-dependent protein kinase II regulation of cardiac excitation-transcription coupling, Heart Rhythm, 8 (2011) 1101–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data that support the findings of this study are available from the authors upon reasonable request.