

Summary
Functional precision medicine is a strategy in which live patient tumor cells are directly perturbed with drugs to provide immediately translatable, personalized information to guide patient therapy. The heterogeneity of human cancer has led to the realization that personalized approaches are needed to improve treatment outcomes. Precision oncology has traditionally used static features of the tumor to dictate which therapies should be used. Static features can include expression of key targets or genomic analysis of mutations to identify therapeutically-targetable “drivers.” While a surprisingly small proportion of patients receive clinical benefit from the static approach, functional precision medicine can provide additional information on tumor vulnerabilities. We discuss emerging technologies for functional precision medicine, as well as limitations and challenges to using these assays in the clinical trials that will be necessary to determine if functional precision medicine can improve patient outcomes and eventually become a standard tool in clinical oncology.
Introduction
Targeted cancer therapies are capable of inhibiting specific oncogenic proteins. Many of these targeted therapies are administered following a positive companion diagnostic predicting therapy outcomes. For example, the presence of HER2 amplification in breast cancer predicts response to HER2-targeted therapy, and mutation of EGFR in lung cancer predicts response to EGFR-targeted therapeutics. The success of several targeted therapies, along with a comprehensive understanding of molecular diversity in cancer gave rise to the tantalizing idea that cancer therapy could be personalized, leading to the concept of precision oncology. Precision oncology is the approach in which properties specific to an individual patient’s tumor are used to direct effective therapy. Indeed, precision oncology has been conducted for decades in some types of cancer (e.g. hormone therapy in estrogen receptor-positive breast cancer and imatinib in Philadelphia chromosome-positive leukemia). The field has vastly expanded since several large cancer genome projects (Hudson et al., 2010) revealed common molecular features of tumors that could be targeted. This Perspective describes how precision oncology can be augmented by functional approaches to help realize the promise of personalized cancer treatment.
We contrast diagnostic approaches that are “functional” (measuring response to perturbations of living cells) with those that are “static” (measuring a fixed property). Current approaches to precision oncology have been dominated by genomics, a static diagnostic. Notable successes in identification of genomic variants that portend effective treatment with targeted therapies, in addition to those already mentioned, include targeting EGFR or ALK in lung adenocarcinoma; c-KIT in gastrointestinal stromal tumors; BRAF in melanoma and non-small-cell lung cancers; NTRK in a variety of rare tumors, PD-1 in microsatellite instability-high tumors, and PIK3CA in breast cancers. These successes, along with knowledge gleaned from TCGA data, launched the largest precision oncology trial to date: the National Cancer Institute’s Molecular Analysis for Therapy Choice (NCI-MATCH) trial. This trial features nearly 40 treatment arms, with at least 35 patients per arm, with tumors harboring a specific genetic alteration. A recent report on NCI-MATCH demonstrated the feasibility of rapid large-scale patient enrollment, DNA sequencing, and matching patients to trial arms (Flaherty et al., 2020); however, the clinical benefit was underwhelming. 38% of patients were matched to a targeted therapy, and 18% had access to the relevant therapy through one of the treatment arms. Unfortunately, a minority of the >6,000 participants experienced clinical benefit: overall response rates in individual arms ranged from 0–38%, with most on the low end of the spectrum (Flaherty et al., 2020; Commentary, 2021). Thus, the data from this large national trial essentially revealed that the current “upper limit” for patients with targetable genetic alterations is 38%, but that many tumors with predicted genetic vulnerabilities failed to respond to the matched targeted therapy. If all patients who received genetic analysis on NCI-MATCH are considered the denominator, the response rate in an intention-to-treat analysis would be well below 5%. Several other precision oncology trials, comprising more than 7,000 additional patients, have come to similar conclusions (Chen et al., 2021; Le Tourneau et al., 2015; Massard et al., 2017; Middleton et al., 2020; Sicklick et al., 2019; van Tilburg et al., 2021).
Given the broad use of genomics as a precision medicine tool it is worth considering why genomics has not had a greater impact in providing cancer patients with effective therapies. We think that there has been underestimation of the degree to which cancer phenotypes are driven by non-genetic mechanisms. If one were to perform whole genome sequencing on the fingernails, retinas, neutrophils, and hepatocytes of an individual, the same result would be obtained for each tissue, yet all would surely agree that there are physiologically important differences between the tissues. To put it another way, the reason why lymphoma patients can often be cured by the RCHOP drug combination regimen, and pancreatic cancer patients cannot, is not mainly because of mutations distinct for one of the tumor types, but rather due to non-genetic mechanisms determining cell type and cellular response to drug treatment. There are many discrete, at least metastable, states of cancer cells that are determined non-genetically; in many cases epigenetically (Baylin and Jones, 2016). In fact, there is a growing body of literature across many treatments and many tumor types that resistance can often be attributed to the establishment of an epigenetically determined “persister” state (Vallette et al., 2019). Thus, it is clear that many cancer phenotypes, including those that influence response to therapy, are determined by non-genetic mechanisms, in addition to the mutation-driven mechanisms that are commonly considered.
An additional challenge for genomic-only approaches is that it is rare for a genomic strategy to identify an active single agent, and even rarer for it to identify more than one. In a pattern repeated many times across many cancers, single agents, with only rare exceptions, are not capable of causing a sustained response. An excellent example is pediatric ALL. When early anti-metabolites were applied as single agents, there was patient benefit, but it was transient. It was only after several active drugs were combined that sustained responses and cures were obtained. Combinations were what made a previously incurable disease over 90% curable today. To assemble the type of novel combinations needed to make a substantial difference in cancer care, alternative precision methods are needed.
Functional precision medicine in oncology
Functional precision medicine (FPM)
Functional precision medicine (FPM) is an approach based on direct exposure of patient-derived tissues to drugs (Letai, 2017). This approach generates dynamic, functional data that may encompass key vulnerabilities including those conveyed by altered epigenetic states and/or altered signaling pathways not necessarily driven by distinct genomic aberrancies. A growing appreciation of the heterogeneity of human cancers, along with new enabling technologies, and a rapidly expanding set of active drugs has renewed the push for patient-derived models in research. Use of pre-clinical models, such as patient-derived xenografts (PDX) and organoids (PDO), representing tumors from patients may refine drug discovery and improve success of new therapies in the clinic. Improved feasibility of making patient-derived models on a large scale for research has also made these models accessible for personalized therapy. For example, realization that engraftment of breast and other tumors as PDX predicted metastatic relapse (DeRose et al., 2011; du Manoir et al., 2014; Garrido-Laguna et al., 2011; Kleine, 1986; Nemati et al., 2010) spawned the idea that the models could be used to personalize therapy upon recurrence, which led to the Functional Precision Oncology for Metastatic Breast Cancer study (FORESEE; NCT04450706).
FPM is not a new concept. In vitro culture or growth of cancer cells in mouse models for chemosensitivity testing began more than half a century ago using proliferation, viability, or clonogenic assays as readouts for drug efficacy. It is worth noting that nearly every curative cytotoxic regimen used today in oncology was derived based on information originating from these crude, but informative, assays (DeVita and Chu, 2008). The question arose whether these assays could be used to personalize therapy. However, two editorials in the New England Journal of Medicine in 1983 (Selby et al., 1983; Von Hoff, 1983) and later position papers of the American Society for Clinical Oncology (Burstein et al., 2011; Schrag et al., 2004) cast doubt on the utility of such assays to personalize cancer care. Problems included lack of tumor cells surviving in ex vivo cultures, unsophisticated culture conditions, and uninformative assays. Moreover, there were simply not many drugs to choose from so that the utility of the information, even when accurate, was limited. In addition, there was a lack of prospective data demonstrating clinical utility.
The past few decades have yielded dramatic increases in biological knowledge, available technology, and the number of drugs available, and re-kindled interest in ex vivo analysis of patient tumor cells to guide therapy. Advances in three-dimensional culture technology have accelerated work with patient tumors grown ex vivo as organoids for feasible drug screening (Liu et al., 2021; Tuveson and Clevers, 2019; Vlachogiannis et al., 2018); this was reviewed in (Veninga and Voest, 2021). The advent of new technologies allows comprehensive assessment of drug sensitivity or resistance on patient tumor samples, not only for evaluation of proliferation, survival, and metabolic activity, but also by incorporating single cell analysis to evaluate bulk or sub-clonal phenotypic effects. These “next generation” functional tests (Friedman et al., 2015) hold great promise to integrate with genomic evaluation for an improved approach to FPM, whereby drugs could be prioritized based on how well they work functionally and whether there is a genomic explanation for the effect. FPM should also be useful to identify which genomic abnormalities actually provide good targets. An example for this is the case for the previously mysterious lack of efficacy of RAF inhibitors in BRAF V600E-mutated colorectal cancer, despite proven efficacy in melanomas with the same mutation. It was not until functional studies were performed that it was revealed that in these colorectal cancers RAF inhibition causes feedback activation of EGFR, which supports tumor growth (Prahallad et al., 2012).
A key aspect to successful FPM is development and validation of short-term or long-term models that retain fidelity to the human tumors from which they arise. A recent large analysis of >500 PDX models from various cancer types showed that they recapitulate human tumors with relatively high fidelity (Woo et al., 2021). PDX also exhibit treatment responses that are concordant with those observed in the patients from which they are derived (Byrne et al., 2017; Zhang et al., 2013). Ex vivo cultures of patient tumors, such as PDOs, are less time-consuming and less expensive than PDX models, and data are accumulating to show organoids also have high fidelity to human tumor drug responses. Pancreatic and colorectal tumors, in particular, have been extensively modeled as PDOs. They show strong genotypic and phenotypic concordance with patient samples (Gendoo et al., 2019; Larsen et al., 2021) and are being used to predict therapeutic responses (Narasimhan et al., 2020; Tiriac et al., 2018). Validation of each model prior to performing FPM assays is paramount, though, as aggressive stromal cells or normal cells can sometimes overgrow the cultures depending on the conditions used (Dijkstra et al., 2020; Gao et al., 2017; Yu et al., 2017).
Emerging models and technologies for FPM
The development of new technologies at the intersection of engineering and biology can help meet some of the practical challenges of FPM (Figure 1). Notable advances have been made in technologies that aim to represent the tumor microenvironment, including extracellular matrices. Additionally, organ chips (also referred to as tissue chips or microphysiological systems) are being used to model cancer within its tumor microenvironment (Sontheimer-Phelps et al., 2019). These microfluidic devices, typically comprised of tissue specific cell types, extracellular matrix gels, and cancer cells, attempt to recapitulate the molecular and mechanical cues in human tissues(Ayuso et al., 2020; Choi et al., 2015; Hassell et al., 2017). Potential advantages of organ chips include the ability to model drug delivery, pharmacokinetics, pharmacodynamics, and drug toxicity (Barrile et al., 2018; Herland et al., 2020). Some obstacles to widespread use of organ chips in FPM are the low throughput of drug testing, and the requirement of special expertise to manufacture and operate these devices (Ayuso et al., 2021).
Figure 1. Models and readouts in functional precision medicine.
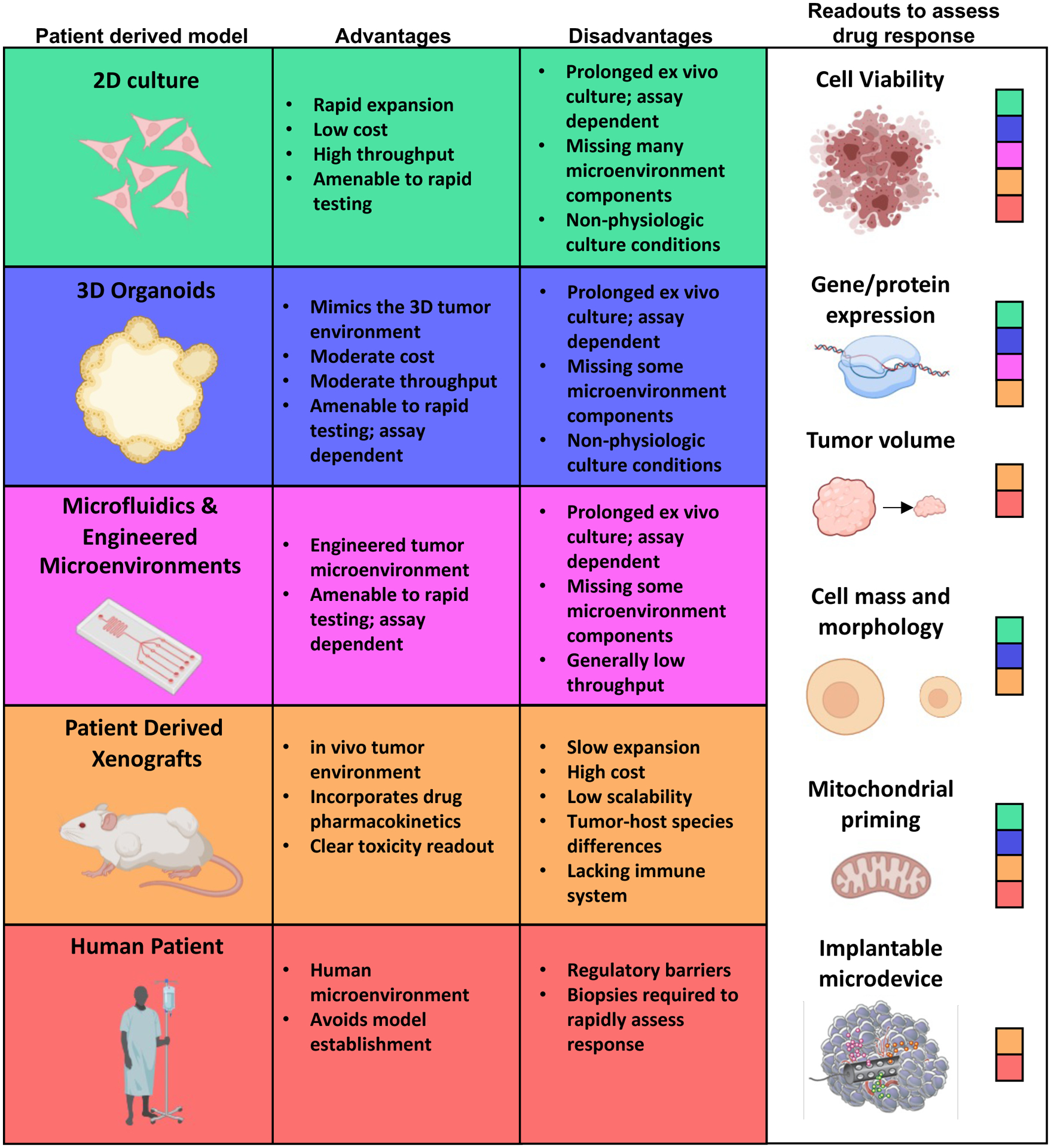
Functional precision medicine directly exposes patient derived cancer cells (left panel) to therapeutics and measures drug response using various readouts (right panel). Patient derived material amenable to specific readouts are indicated by color codes in the right panel. The use of specific patient derived models in functional precision medicine has strengths and weaknesses outlined in the center panels
Another approach to mimic the environment of tumors is to leave cells in their native environment during drug treatment. This includes drug treatment on tumor slices or core needle biopsies from patients where immunofluorescent markers of cell death can be used as a read out of drug sensitivity (Horowitz et al., 2020; Majumder et al., 2015; Naipal et al., 2016). Another compelling approach for FPM is the use of drug-containing microdevices that can be implanted directly into tumors in patients (Jonas et al., 2015; Klinghoffer et al., 2015). Drugs in these microdevices can act on the surrounding tissues and drug responses evaluated in the tumors upon resection. While implantable microdevices may face higher regulatory and clinical barriers than ex vivo approaches, they may enable drug testing within the patient tumor in situ.
Challenges of FPM
While there are both academic and commercial efforts using FPM to assign drugs to cancer patients (Table 1), these are not yet in the mainstream of patient diagnostics. Next, we will discuss the challenges to the routine use of FPM in oncology.
Table 1.
Companies conducting FPM services
| Company | Location | FPM assay(s) |
|---|---|---|
| 2CureX | Copenhagen, Denmark | patient tumor ex vivo assays |
| Allcyte | Vienna, Austria | patient tumor ex vivo assays |
| Celcuity | Minnesota, USA | patient tumor ex vivo assays |
| Cellesce | Cardiff, Wales | patient tumor ex vivo assays |
| Certis | California, USA | PDX |
| Champions | New Jersey, USA | PDX |
| Crown Bio | California, USA | patient tumor ex vivo assays |
| Imprimed | California, USA | veterinary ex vivo assays |
| Kibur Medical | Massachusetts, USA | implantable drug delivery microdevice |
| Knownmed | Utah, USA | patient tumor ex vivo assays |
| Nagourney Cancer Institute | California, USA | patient tumor ex vivo assays |
| Notable Labs | California, USA | patient tumor ex vivo assays |
| Presage Bio | Washington, USA | implantable drug delivery microdevice |
| SageMedic | California, USA | patient tumor ex vivo assays |
| SEngine | Washington, USA | patient tumor ex vivo assays |
| SpeciCare | Georgia, USA | patient sample provider |
| Tempus | Chicago, IL | patient tumor ex vivo assays |
| Travera | Massachusetts, USA | suspended microchannel resonator |
| Vivia Biotech | Madrid, Spain | patient tumor ex vivo assays |
Tissue handling
FPM methods by definition require viable tissues. All resections and biopsies start out viable until they are killed by time, fixation, or flash freezing. However, current diagnostic practice in oncology nearly always requires the rapid fixation or destruction of tissues so that they can be subjected to static tests like histology or genomics without uncontrolled degradation. Clinicians apply established algorithms and experience to use this information to make treatment decisions. Therefore, in order for FPM to get a foothold, standard tissue handling must change. This is something of a Catch 22. To change standard tissue handling so that valuable tissue is shunted to functional studies, pathologists and clinicians must feel confident that the information obtained will change management in a beneficial way. Yet, to provide the evidence that FPM methods benefit patients requires viable tissue in the first place. Many therapeutic clinical trials mandate the storage of patient samples for the evaluation of biomarkers so that once clinical response is known, a comparison can be made between biomarkers and clinical response. However, in our experience it is nearly unheard of for samples to be stored in viable fashion. Thus, nearly all of the existing tumor sample sets linked to completed and ongoing clinical trials are useless for testing FPM methods. Since biological signaling can change as the tumor is kept in the operating room, in banking facilities, or shipped to the laboratory, optimizing and standardizing storage and shipping conditions will be important for performing FPM clinical trials across different hospitals.
Another challenge in applying FPM is obtaining sufficient tumor cell yields from the types of samples that are acquired from patients within current clinical practice. For many hematologic malignancies, bone marrow aspirates or blood draws can yield cancer cell quantities that enable chemical screens. For solid tumors, however, large resections are relatively rare in the setting of metastatic disease, which may be the most impactful application of FPM. For most metastatic solid tumors, tissue sources typically include core needle biopsy, fine needle aspirates, or circulating tumor cells, all of which produce low cell numbers. When there are low cell numbers, establishment of the model can be a challenge, and FPM strategies that expand tissues prior to chemical screens are advantageous.
Calibration and decision making
Information obtained from most FPM methods is parametric, in contrast with genetic information, which is usually categorical. Thus, for any drug applied to a tumor model, a number is obtained that might vary along a range, like 0–100. This is often a parameter like % dead cells, or % loss of viability. Two main questions arise. First, at what value from the ex vivo test for a given drug is the desired clinical response obtained? Second, when obtaining results from different drugs, how can values be compared among drugs to derive a ranking of drugs according to predicted efficacy?
When considering the first question, the gold standard is simply the empiric comparison of FPM results for an individual drug with clinical outcome after treatment with that drug. As one acquires more comparisons, the calibration can be continuously updated. As one acquires experience for multiple drugs in multiple tumors, it may be that patterns emerge that can accelerate this process. For instance, perhaps a value output from the diagnostic assay will emerge, common to many drugs, above which a response is predicted. For this approach there is no substitute for experience.
Other strategies provide useful information while experience is being obtained. For instance, the assay signal of a drug on a tumor model can be compared with the response of that same drug on other patient models. Then, one can rank the response according to percentage or standard deviations. One can then set a cutoff, initially arbitrary, above which a response is predicted. For instance, one might identify a particular tumor as being in the top 5% of ex vivo signals from an individual drug, and thus classify it as a “hit.” Again, experience and clinical concordance data will refine how the output is interpreted.
A key consideration in the interpretation of ex vivo FPM tests is the drug concentration and drug treatment duration used in such tests. Poor calibration of drug concentration in FPM tests may lead to selection of drugs that do not work or may miss effective drugs. Unfortunately, the drug concentration that a tumor experiences in a patient is not trivial to measure accurately, and varies over time. One approach to identify the optimal drug concentration is to make empirical comparisons of ex vivo drug response and identify ranges of drug concentration that correlate with patient response. While attractive, this approach requires an ongoing clinical trial, which potentially limits the application of FPM.
Several pre-clinical trial strategies to calibrate ex vivo drug testing have emerged. One strategy involves comparing drug responses in 2D or organoid models with responses in matched in vivo models, to determine if the in vitro response is relevant (Bhola et al., 2020; Guillen et al., 2021; Lalazar et al., 2021). Another strategy involves comparing FPM tests on cancer cells and on healthy cells. Finally, leveraging inter-patient heterogeneity of drug response, it is possible to collect data at multiple drug concentrations across a range of tumors and identify drug concentrations where there is maximal variation between in vitro model responses. These “discriminating” drug concentrations is where the most information would be obtained, and hence would be the most useful for ranking predicted drug sensitivities.
Speed of FPM results
The clinical utility of FPM depends on how quickly results are returned to the clinician. FPM assays have various times to generate drug responses (Figure 1). For example, FPM assays on cancers that do not require ex vivo expansion can typically be completed within several days. This is frequently found in hematologic cancers where tumor cell yields from the patient are typically high. In contrast, many solid tumors, especially those starting from core needle biopsies, typically require expansion as cell lines, organoids, or PDX models prior to drug testing. This can take several weeks or months to establish, expand to sufficient quantity, and perform testing. One strategy to deal with competing pressures between FPM speed and model expansion time is to collect samples prior to standard of care treatment, perform FPM, and then treat based on the FPM information after a recurrence or upon progression (Guillen et al., 2021).
Other limitations
The models and strategies used for FPM assays have other limitations. The “take rate” of some cancers as ex vivo or PDX models varies, so not every cancer can be modeled for FPM. Organoid models tend to be simplistic, excluding cells of the tumor microenvironment, which can be key mediators regulating drug responses (Wu and Dai, 2017). Tumors grown as PDX develop a more realistic tumor microenvironment, but interactions with human microenvironmental components, such as immune cells, is lacking. Given the growing presence of immunotherapy as a mainstay in treatment of many cancers, models for FPM incorporating a functional immune microenvironment are paramount. New strategies for tumor-immune cocultures (Boucherit et al., 2020) and new microfluidic engineering approaches (Shelton et al., 2021) are beginning to address this challenge (Shelton et al., 2021). Lastly, the cost of FPM assays is variable and greatest utility will come from strategies that allow cost-effective drug testing, perhaps through automation and miniaturization of assays.
Examples of clinical trials and functional precision oncology tools
Despite the challenges, several clinical trials are now incorporating FPM approaches. This is a challenge in itself: Nearly all clinical trial designs are constructed to evaluate the performance of a therapeutic. There are few clinical trial structures to evaluate the performance of diagnostics like FPM assays. In fact, despite its widespread adoption, there still exists little prospective clinical trial data to support the widespread use of genomics in advanced cancer patients.
Nevertheless, the utility of FPM approaches is being tested in clinical trials (Table 2). Allcyte, a company based in Vienna, uses an approach based on morphologic changes in drug-treated cancer cells, which they call “pharmacoscopy” (Snijder et al., 2017). In the EXALT trial, a collaboration with the Medical University of Vienna, they tested the ability of this approach to assign therapy to 56 previously treated patients with hematologic malignancies. Strikingly, they found that most patients so treated demonstrated a progression-free survival time at least 1.3-fold longer than that of their prior therapy. As pan-resistance almost universally tends to accrue with subsequent rounds of therapy in oncology, the ability to identify drugs that outperform prior therapies was reasonably interpreted as evidence of the utility of their function approach. It’s notable that 40% of responders experienced exceptional responses lasting at least three times longer than expected (Kornauth et al., 2021) (Wheeler et al., 2021). EXALT 2.0 is a follow up study that randomizes patients to one of three arms in which treatments are based on either functional assays, genomic analysis, or physician choice. In another study, Malani et al evaluated response in 29 relapsed or refractory AML patients treated according to results from an ex vivo functional diagnostic, with most patients receiving a combination therapy (Malani et al., 2021). They observed a 59% objective response rate, with 45% receiving a complete remission of the leukemia (with or without complete hematologic recovery). The FORESEE study, mentioned above, is using a similar approach with FPM drug response assays being used on patient tumor organoids to predict therapy response in metastatic breast cancer patients, also assessing the response ratio of the “informed” to the previous “uninformed” therapy. The TuPro study, being conducted at the University Hospital Zurich, utilizes a constellation of genomics, proteomics, single cell analysis, and ex vivo drug sensitivity screening to predict treatment responses using the integrated information (Irmisch et al., 2021). While data are still very limited to assess how well FPM performs in clinical trials, we are encouraged that there is an increasing number of clinical trials active with results yet to be reported.
Table 2.
FPM trials currently listed in clinialtrials.gov
| NCT number | Acronym | Title | Sponsor | Status | FPM platform | Target sample size |
|---|---|---|---|---|---|---|
| NCT03860376 | none | Ex Vivo Drug Sensitivity Testing and Mutation Profiling | Florida International University | Recruiting | ex vivo assays | 22 |
| NCT03133273 | ONCOGRAM | Study of the Therapeutic Response and Survival of Patients With Metastatic Colorectal Cancer (Stage IV) and Treated According to the Guidelines of a Chemosensitivity Test, Oncogramme® (ONCOGRAM) | University Hospital, Limoges | Recruiting | ex vivo assays | 256 |
| NCT04470947 | EXALT-2 | Comprehensive Genomic Profiling and Next Generation Functional Drug Screening for Patients With Aggressive Haematological Malignancies | Medical University of Vienna | Recruiting | ex vivo assays | 150 |
| NCT02927106 | none | Beat AML Core Study | University of Florida | Completed | ex vivo assays | 22 |
| NCT01190241 | TSAP | Targeted Therapy Selection Based on Tumor Tissue Kinase Activity Profiles for Patients With Advanced Solid Malignancies, an Exploratory Study | Vanderbilt University | Terminated | ex vivo assays | 45 |
| NCT04450706 | FORESEE | Functional Precision Oncology for Metastatic Breast Cancer | University of Utah | Recruiting | Organoids | 15 |
| NCT03890614 | none | Novel 3D Myeloma Organoid to Study Disease Biology and Chemosensitivity | Wake Forest | Recruiting | Organoids | 70 |
| NCT04561453 | none | Feasibility Study of Multi-Platform Profiling of Resected Biliary Tract Cancer | University of Washingon | Recruiting | Organoids | 20 |
| NCT03896958 | PIONEER | The PIONEER Initiative: PrecisionInsights On N-of-1 Ex Vivo Effectiveness Research Based on Individual Tumor Ownership (Precision Oncology) | SpeciCare | Recruiting | Organoids | 1000 |
| NCT04768270 | none | The Culture of Ovarian Cancer Organoids and Drug Screening | Chongqing University | Recruiting | Organoids | 30 |
| NCT04755907 | none | 3D Bioprinted Models for Predicting Chemotherapy Response in Colorectal Cancer With/Without Liver Metastases | Peking Union Medical College Hospital | Not yet recruiting | Organoids | 120 |
| NCT04842006 | SYNCOPE | Systemic Neoadjuvant and Adjuvant Control by Precision Medicine in Rectal Cancer | Helsinki University Central Hospital | Not yet recruiting | Organoids | 93 |
| NCT04826913 | TUMOVASC | High Throughput Screening Device Based on 3D Nano-matrices and 3D Tumors With Functional Vascularization | University Hospital, Strasbourg, France | Not yet recruiting | Organoids | 100 |
| NCT03336931 | PRISM | PRecISion Medicine for Children With Cancer | Sydney Children’s Hospitals Network | Recruiting | PDX | 400 |
| NCT03219047 | none | Patient-Derived Xenografts in Personalizing Treatment for Patients With Relapsed/Refractory Mantle Cell Lymphoma | MD Anderson Cancer Center | Recruiting | PDX | 50 |
| NCT04745975 | none | Guided Treatment Based on Mini-PDX in Metastatic Triple Negative Breast Cancer | Fudan University | Recruiting | PDX | 100 |
| NCT04373928 | none | Personalized Precision Diagnosis and Treatment of Pancreatic Cancer | Changhai Hospital | Recruiting | PDX | 100 |
| NCT04602702 | PDXovo | Hyper-Personalized Medicine Using Patient Derived Xenografts (PDXovo) for Renal Cell Carcinoma Patients | Sunnybrook Health Sciences Centre | Not yet recruiting | PDX | 50 |
| NCT03878524 | SMMART | Serial Measurements of Molecular and Architectural Responses to Therapy (SMMART) PRIME Trial | OHSU Knight Cancer Center | Recruiting | multiple | 40 |
FPM can also function as a companion diagnostic for individual therapies. A rapid assay of drug-induced apoptotic signaling at the mitochondrion called dynamic BH3 profiling (DBP) was performed in a study of lenalidomide plus mitoxantrone, etoposide and cytarabine as an induction regimen for acute myelogenous leukemia (Garcia et al., 2020). Pretreatment patient blasts were exposed to lenalidomide ex vivo as part of the assay, performed blinded to patient outcome. A post-hoc comparison of DBP results and clinical outcome showed that clinical response could be nearly perfectly predicted via this approach. DBP has also been shown to predict response to varied therapies in multiple murine models (Bhola et al., 2020; Montero et al., 2015; Townsend et al., 2016). Similar functional methods identified mitochondrial sensitivity to BCL-2 antagonism in CLL and AML (Del Gaizo Moore et al., 2007; Vo et al., 2012). This work led to clinical trial programs that yielded multiple regulatory approvals for clinical use of the BCL-2 antagonist venetoclax in both diseases.
Assembling combinations
One of the advantages of functional methods is that they frequently identify several active drugs simultaneously. While this bodes well for the use of FPM to design novel, personalized combinations, it also offers a significant regulatory challenge. Many of these combinations are likely to have never been tested in the clinic before. Conventionally, a novel combination would require a Phase 1 multi-drug dose escalation trial with a dozen patients, at least, to identify a recommended Phase 2 dose for each drug to be tested in combination. That approach would be impractical and inconsistent with the goals of identifying novel, individualized combination regimens. The time and resources to assign a single patient to a single novel combination regimen would be impossible to maintain. However, given the urgent need for effective combination regimens, especially in the advanced cancer setting, a solution must be found. This likely will arise in some form of intra-patient dose escalation, facilitated by a union of enthusiasm from patients and patient advocates to accept the increased risk of toxicity, as well as an acknowledgment from IRBs and the FDA that the potential benefit outweighs the risk.
If the goal is to assemble two-, three-, or even four-way combinations from FPM strategies, it will be challenging to test every possible combination ex vivo, as this would be possible for only a very small panel of drugs before the number of conditions became unwieldy. While there is much attention placed on identification of mechanistic synergy in the assembly of combinations, which requires simultaneous testing of more than one drug, it is not clear that synergy is required for highly effective combinations. For instance, perhaps no therapy has cured more cancer patients than the R-CHOP therapy used in B-cell lymphomas, which has likely cured at least hundreds of thousands of patients worldwide. The components of CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) do not exhibit clinical synergy. Rather, each drug has an independent chance to kill a cancer cell (Palmer et al., 2019), so a cancer cell must be resistant to all drugs to survive. The same absence of synergy can be observed in other combination regimens (Palmer and Sorger, 2017). In practice, it is likely that more than one screening practice will be used. In some contexts, testing a limited number of combinations ex vivo will be a priority, while in others testing a larger number of single agents will be a priority. FPM single agent results can be combined with extrinsic information, including histology and other molecular annotations, to prioritize drugs to be included in higher order combinations.
To do list for FPM:
FPM has tremendous potential to provide a broadly applicable predictive biomarker for assigning patient therapy and for discovery. Here are three items that we feel will speed its adoption as a standard.
Limit pre-analytical variability. The requirement in FPM for viable, unfixed tissue can introduce pre-analytical variability into the downstream assays. At every step including initial sample acquisition, sample storage, short term viable freezing, shipping temperature and duration, sample dissociation, ex vivo culture conditions and duration, there is an opportunity for variability to be introduced that will baffle reliable comparison among different approaches and with clinical outcome. Reducing variability will require improved reporting of standard operating details all the way from the biopsy to the performance of the assay. Improved sharing and communication among FPM practitioners will be necessary, perhaps under the aegis of a consortium or organization like the Society for Functional Precision Medicine.
Report on clinical correlation with FPM assays. While the utility of FPM is indubitably plausible, there is no substitute for rigorous comparisons of FPM results with clinical outcome. Fortunately, as recorded above, we have started to see such reports in the literature, and are aware of many more that are in various stages of planning and execution (Table 2). As confidence grows in the utility of these assays to match patients with drugs that provide benefit, a virtuous cycle of increased acquisition and banking of viable tumor samples followed by increased application of these samples and their assays to clinical trials is to be expected. Such a virtuous cycle should ultimately result in adoption of high-performing assays as standard clinical tools.
Increase uptake of FPM by pharmaceutical companies. To a practitioner of FPM, the relative slowness of pharma to embrace FPM can seem puzzling, as FPM seems to offer solutions to problems that chronically vex pharma. FPM has the potential to provide a companion diagnostic for nearly any cancer therapeutic. Thus, drugs without companion diagnostics that falter in clinical trials with borderline response rates could be rescued by FPM tools that might stratify patients and dramatically improve response rates. Moreover, for agents that successfully target a protein or pathway but lack a clinical context, FPM can be used to survey a spectrum of cancer samples and identify those where biological activity is greatest. A few successes in this arena may enhance enthusiasm and establish FPM as an important clinical and pre-clinical stratification tool.
Share the data. FPM data in models that represent real-world cancer cases is an invaluable resource for research and development of new, more effective therapies. The utility of FPM assays is limited by the number of drugs that are available for patient treatment, but many of the models generated for FPM assays can be grown indefinitely and utilized for new research to develop new therapeutic strategies. Of course, it is important to continue to document the ongoing fidelity of the long-term models for such an approach to be useful. We recommend that validated models, their genomic data, and their associated drug response data be made publicly available through resources like the NCI’s PDX Network and Patient-derived Models Repository. A specific “data commons” for FPM might be especially useful, allowing both data deposition and sharing of knowledge within the scientific community. Eventually, one could imagine utilizing a vast resource of genomics and FPM data to predict therapeutic responses for future patients without actually having to grow their tumor for functional tests (Welm et al., 2021).
Conclusion
The full promise of precision medicine in oncology is yet to be realized, as more patients may benefit from functional approaches. Genomics has low operational barriers (minimal tissue handling; sequencing is relatively cheap and ubiquitous) and high intellectual barriers (how to go from mutations to active drug is often unclear), whereas functional precision medicine has high operational barriers (requirement fresh tissue, survival of cells, credentialing of readouts), and seemingly low intellectual barriers (active drugs are directly identified). We predict that the next five years will see an increasing marriage of these two approaches in clinical trials whose completion will be necessary for the establishment of functional precision medicine as a standard tool in clinical oncology. This union will be necessary for the rational selection of active combinations regimens that will ultimately be necessary for precision medicine in oncology to provide its greatest benefit to patients.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
A.L.W. has received royalties from licenses of patient-derived xenograft or organoid models issued by the University of Utah. The University may issue new licenses in the future at its discretion, which may result in additional royalties. A.L. owns equity and serves on the scientific advisory boards of Zentalis Pharmaceuticals, Dialectic Therapeutics, Anji Onco, and Flash Therapeutics. A.L. has received royalties from licenses of BH3 profiling issued by Dana-Faber Cancer Institute. The Institute may issue new licenses in the future at its discretion, which may result in additional royalties.
References
- Ayuso JM, Gong MM, Skala MC, Harari PM, and Beebe DJ (2020). Human Tumor-Lymphatic Microfluidic Model Reveals Differential Conditioning of Lymphatic Vessels by Breast Cancer Cells. Adv Healthc Mater 9, e1900925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayuso JM, Park KY, Virumbrales-Munoz M, and Beebe DJ (2021). Toward improved in vitro models of human cancer. APL bioengineering 5, 010902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrile R, van der Meer AD, Park H, Fraser JP, Simic D, Teng F, Conegliano D, Nguyen J, Jain A, Zhou M, et al. (2018). Organ-on-Chip Recapitulates Thrombosis Induced by an anti-CD154 Monoclonal Antibody: Translational Potential of Advanced Microengineered Systems. Clin Pharmacol Ther 104, 1240–1248. [DOI] [PubMed] [Google Scholar]
- Baylin SB, and Jones PA (2016). Epigenetic Determinants of Cancer. Cold Spring Harb Perspect Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhola PD, Ahmed E, Guerriero JL, Sicinska E, Su E, Lavrova E, Ni J, Chipashvili O, Hagan T, Pioso MS, et al. (2020). High-throughput dynamic BH3 profiling may quickly and accurately predict effective therapies in solid tumors. Sci Signal 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucherit N, Gorvel L, and Olive D (2020). 3D Tumor Models and Their Use for the Testing of Immunotherapies. Front Immunol 11, 603640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein HJ, Mangu PB, Somerfield MR, Schrag D, Samson D, Holt L, Zelman D, Ajani JA, and American Society of Clinical, O. (2011). American Society of Clinical Oncology clinical practice guideline update on the use of chemotherapy sensitivity and resistance assays. J Clin Oncol 29, 3328–3330. [DOI] [PubMed] [Google Scholar]
- Byrne AT, Alferez DG, Amant F, Annibali D, Arribas J, Biankin AV, Bruna A, Budinska E, Caldas C, Chang DK, et al. (2017). Interrogating open issues in cancer medicine with patient-derived xenografts. Nat Rev Cancer 17, 632. [DOI] [PubMed] [Google Scholar]
- Chen AP, Kummar S, Moore N, Rubinstein LV, Zhao Y, Williams PM, Palmisano A, Sims D, O’Sullivan Coyne G, Rosenberger CL, et al. (2021). Molecular Profiling-Based Assignment of Cancer Therapy (NCI-MPACT): A Randomized Multicenter Phase II Trial. JCO Precis Oncol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Hyun E, Seo J, Blundell C, Kim HC, Lee E, Lee SH, Moon A, Moon WK, and Huh D (2015). A microengineered pathophysiological model of early-stage breast cancer. Lab Chip 15, 3350–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commentary (2021). NCI-MATCH Sets “Benchmark of Actionability”. Cancer discovery 11, 6–7. [DOI] [PubMed] [Google Scholar]
- Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, and Letai A (2007). Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest 117, 112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, Factor R, Matsen C, Milash BA, Nelson E, et al. (2011). Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med 17, 1514–1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVita VT Jr., and Chu E (2008). A history of cancer chemotherapy. Cancer research 68, 8643–8653. [DOI] [PubMed] [Google Scholar]
- Dijkstra KK, Monkhorst K, Schipper LJ, Hartemink KJ, Smit EF, Kaing S, de Groot R, Wolkers MC, Clevers H, Cuppen E, and Voest EE (2020). Challenges in Establishing Pure Lung Cancer Organoids Limit Their Utility for Personalized Medicine. Cell reports 31, 107588. [DOI] [PubMed] [Google Scholar]
- du Manoir S, Orsetti B, Bras-Goncalves R, Nguyen TT, Lasorsa L, Boissiere F, Massemin B, Colombo PE, Bibeau F, Jacot W, and Theillet C (2014). Breast tumor PDXs are genetically plastic and correspond to a subset of aggressive cancers prone to relapse. Molecular oncology 8, 431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Gray RJ, Chen AP, Li S, McShane LM, Patton D, Hamilton SR, Williams PM, Iafrate AJ, Sklar J, et al. (2020). Molecular Landscape and Actionable Alterations in a Genomically Guided Cancer Clinical Trial: National Cancer Institute Molecular Analysis for Therapy Choice (NCI-MATCH). J Clin Oncol 38, 3883–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman AA, Letai A, Fisher DE, and Flaherty KT (2015). Precision medicine for cancer with next-generation functional diagnostics. Nat Rev Cancer 15, 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Huang C, Kernstine K, Pelekanou V, Kluger Y, Jiang T, Peters-Hall JR, Coquelin M, Girard L, Zhang W, et al. (2017). Non-malignant respiratory epithelial cells preferentially proliferate from resected non-small cell lung cancer specimens cultured under conditionally reprogrammed conditions. Oncotarget 8, 11114–11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JS, Bhatt S, Fell G, Sperling AS, Burgess M, Keshishian H, Yilma B, Brunner A, Neuberg D, Carr SA, et al. (2020). Increased mitochondrial apoptotic priming with targeted therapy predicts clinical response to re-induction chemotherapy. Am J Hematol 95, 245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido-Laguna I, Uson M, Rajeshkumar NV, Tan AC, de Oliveira E, Karikari C, Villaroel MC, Salomon A, Taylor G, Sharma R, et al. (2011). Tumor engraftment in nude mice and enrichment in stroma- related gene pathways predict poor survival and resistance to gemcitabine in patients with pancreatic cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 17, 5793–5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendoo DMA, Denroche RE, Zhang A, Radulovich N, Jang GH, Lemire M, Fischer S, Chadwick D, Lungu IM, Ibrahimov E, et al. (2019). Whole genomes define concordance of matched primary, xenograft, and organoid models of pancreas cancer. PLoS Comput Biol 15, e1006596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillen KP, Fujita M, Butterfield AJ, Scherer SD, Bailey MH, Chu Z, DeRose YS, Zhao L, Cortes-Sanchez E, Yang C-H, et al. (2021). A breast cancer patient-derived xenograft and organoid platform for drug discovery and precision oncology. bioRxiv, 2021.2002.2028.433268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassell BA, Goyal G, Lee E, Sontheimer-Phelps A, Levy O, Chen CS, and Ingber DE (2017). Human Organ Chip Models Recapitulate Orthotopic Lung Cancer Growth, Therapeutic Responses, and Tumor Dormancy In Vitro. Cell reports 21, 508–516. [DOI] [PubMed] [Google Scholar]
- Herland A, Maoz BM, Das D, Somayaji MR, Prantil-Baun R, Novak R, Cronce M, Huffstater T, Jeanty SSF, Ingram M, et al. (2020). Quantitative prediction of human pharmacokinetic responses to drugs via fluidically coupled vascularized organ chips. Nat Biomed Eng 4, 421–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz LF, Rodriguez AD, Dereli-Korkut Z, Lin R, Castro K, Mikheev AM, Monnat RJ Jr., Folch A, and Rostomily RC (2020). Multiplexed drug testing of tumor slices using a microfluidic platform. NPJ Precis Oncol 4, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson TJ, Anderson W, Aretz A, Barker AD, Bell C, Bernabé RR, Bhan MK, Calvo F, Eerola I, Gerhard DS, et al. (2010). International network of cancer genome projects. Nature 464, 993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irmisch A, Bonilla X, Chevrier S, Lehmann KV, Singer F, Toussaint NC, Esposito C, Mena J, Milani ES, Casanova R, et al. (2021). The Tumor Profiler Study: integrated, multi-omic, functional tumor profiling for clinical decision support. Cancer Cell 39, 288–293. [DOI] [PubMed] [Google Scholar]
- Jonas O, Landry HM, Fuller JE, Santini JT Jr., Baselga J, Tepper RI, Cima MJ, and Langer R (2015). An implantable microdevice to perform high-throughput in vivo drug sensitivity testing in tumors. Science translational medicine 7, 284ra257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleine W (1986). Prognostic significance of growth characteristics of xenotransplanted ovarian carcinomas into nude mice. Gynecol Oncol 25, 65–72. [DOI] [PubMed] [Google Scholar]
- Klinghoffer RA, Bahrami SB, Hatton BA, Frazier JP, Moreno-Gonzalez A, Strand AD, Kerwin WS, Casalini JR, Thirstrup DJ, You S, et al. (2015). A technology platform to assess multiple cancer agents simultaneously within a patient’s tumor. Science translational medicine 7, 284ra258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornauth C, Pemovska T, Vladimer GI, Bayer G, Bergmann M, Eder S, Eichner R, Erl M, Esterbauer H, Exner R, et al. (2021). Functional Precision Medicine Provides Clinical Benefit in Advanced Aggressive Hematological Cancers and Identifies Exceptional Responders. Cancer discovery, candisc.0538.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalazar G, Requena D, Ramos-Espiritu L, Ng D, Bhola PD, de Jong YP, Wang R, Narayan NJC, Shebl B, Levin S, et al. (2021). Identification of Novel Therapeutic Targets for Fibrolamellar Carcinoma Using Patient Derived Xenografts and Direct from Patient Screening. Cancer discovery. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen BM, Kannan M, Langer LF, Leibowitz BD, Bentaieb A, Cancino A, Dolgalev I, Drummond BE, Dry JR, Ho CS, et al. (2021). A pan-cancer organoid platform for precision medicine. Cell reports 36, 109429. [DOI] [PubMed] [Google Scholar]
- Le Tourneau C, Delord JP, Goncalves A, Gavoille C, Dubot C, Isambert N, Campone M, Tredan O, Massiani MA, Mauborgne C, et al. (2015). Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): a multicentre, open-label, proof-of-concept, randomised, controlled phase 2 trial. Lancet Oncol 16, 1324–1334. [DOI] [PubMed] [Google Scholar]
- Letai A (2017). Functional precision cancer medicine-moving beyond pure genomics. Nat Med 23, 1028–1035. [DOI] [PubMed] [Google Scholar]
- Liu L, Yu L, Li Z, Li W, and Huang W (2021). Patient-derived organoid (PDO) platforms to facilitate clinical decision making. J Transl Med 19, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder B, Baraneedharan U, Thiyagarajan S, Radhakrishnan P, Narasimhan H, Dhandapani M, Brijwani N, Pinto DD, Prasath A, Shanthappa BU, et al. (2015). Predicting clinical response to anticancer drugs using an ex vivo platform that captures tumour heterogeneity. Nat Commun 6, 6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malani D, Kumar A, Bruck O, Kontro M, Yadav B, Hellesoy M, Kuusanmaki H, Dufva O, Kankainen M, Eldfors S, et al. (2021). Implementing a functional precision medicine tumor board for acute myeloid leukemia. Cancer discovery. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massard C, Michiels S, Ferte C, Le Deley MC, Lacroix L, Hollebecque A, Verlingue L, Ileana E, Rosellini S, Ammari S, et al. (2017). High-Throughput Genomics and Clinical Outcome in Hard-to-Treat Advanced Cancers: Results of the MOSCATO 01 Trial. Cancer discovery 7, 586–595. [DOI] [PubMed] [Google Scholar]
- Middleton G, Fletcher P, Popat S, Savage J, Summers Y, Greystoke A, Gilligan D, Cave J, O’Rourke N, Brewster A, et al. (2020). The National Lung Matrix Trial of personalized therapy in lung cancer. Nature 583, 807–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero J, Sarosiek KA, DeAngelo JD, Maertens O, Ryan J, Ercan D, Piao H, Horowitz NS, Berkowitz RS, Matulonis U, et al. (2015). Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell 160, 977–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naipal KA, Verkaik NS, Sanchez H, van Deurzen CH, den Bakker MA, Hoeijmakers JH, Kanaar R, Vreeswijk MP, Jager A, and van Gent DC (2016). Tumor slice culture system to assess drug response of primary breast cancer. BMC Cancer 16, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan V, Wright JA, Churchill M, Wang T, Rosati R, Lannagan TRM, Vrbanac L, Richardson AB, Kobayashi H, Price T, et al. (2020). Medium-throughput Drug Screening of Patient-derived Organoids from Colorectal Peritoneal Metastases to Direct Personalized Therapy. Clinical cancer research : an official journal of the American Association for Cancer Research 26, 3662–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemati F, Sastre-Garau X, Laurent C, Couturier J, Mariani P, Desjardins L, Piperno-Neumann S, Lantz O, Asselain B, Plancher C, et al. (2010). Establishment and characterization of a panel of human uveal melanoma xenografts derived from primary and/or metastatic tumors. Clinical cancer research : an official journal of the American Association for Cancer Research 16, 2352–2362. [DOI] [PubMed] [Google Scholar]
- Palmer AC, Chidley C, and Sorger PK (2019). A curative combination cancer therapy achieves high fractional cell killing through low cross-resistance and drug additivity. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AC, and Sorger PK (2017). Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell 171, 1678–1691 e1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, and Bernards R (2012). Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483, 100–103. [DOI] [PubMed] [Google Scholar]
- Schrag D, Garewal HS, Burstein HJ, Samson DJ, Von Hoff DD, Somerfield MR, Sensitivity, A. W. G. o. C., and Resistance, A. (2004). American Society of Clinical Oncology Technology Assessment: chemotherapy sensitivity and resistance assays. J Clin Oncol 22, 3631–3638. [DOI] [PubMed] [Google Scholar]
- Selby P, Buick RN, and Tannock I (1983). A critical appraisal of the “human tumor stem-cell assay”. N Engl J Med 308, 129–134. [DOI] [PubMed] [Google Scholar]
- Shelton SE, Nguyen HT, Barbie DA, and Kamm RD (2021). Engineering approaches for studying immune-tumor cell interactions and immunotherapy. iScience 24, 101985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicklick JK, Kato S, Okamura R, Schwaederle M, Hahn ME, Williams CB, De P, Krie A, Piccioni DE, Miller VA, et al. (2019). Molecular profiling of cancer patients enables personalized combination therapy: the I-PREDICT study. Nat Med 25, 744–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder B, Vladimer GI, Krall N, Miura K, Schmolke AS, Kornauth C, Lopez de la Fuente O, Choi HS, van der Kouwe E, Gultekin S, et al. (2017). Image-based ex-vivo drug screening for patients with aggressive haematological malignancies: interim results from a single-arm, open-label, pilot study. Lancet Haematol 4, e595–e606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontheimer-Phelps A, Hassell BA, and Ingber DE (2019). Modelling cancer in microfluidic human organs-on-chips. Nat Rev Cancer 19, 65–81. [DOI] [PubMed] [Google Scholar]
- Tiriac H, Belleau P, Engle DD, Plenker D, Deschenes A, Somerville TDD, Froeling FEM, Burkhart RA, Denroche RE, Jang GH, et al. (2018). Organoid Profiling Identifies Common Responders to Chemotherapy in Pancreatic Cancer. Cancer discovery 8, 1112–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend EC, Murakami MA, Christodoulou A, Christie AL, Koster J, DeSouza TA, Morgan EA, Kallgren SP, Liu H, Wu SC, et al. (2016). The Public Repository of Xenografts Enables Discovery and Randomized Phase II-like Trials in Mice. Cancer Cell 29, 574–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuveson D, and Clevers H (2019). Cancer modeling meets human organoid technology. Science 364, 952–955. [DOI] [PubMed] [Google Scholar]
- Vallette FM, Olivier C, Lezot F, Oliver L, Cochonneau D, Lalier L, Cartron PF, and Heymann D (2019). Dormant, quiescent, tolerant and persister cells: Four synonyms for the same target in cancer. Biochem Pharmacol 162, 169–176. [DOI] [PubMed] [Google Scholar]
- van Tilburg CM, Pfaff E, Pajtler KW, Langenberg KPS, Fiesel P, Jones BC, Balasubramanian GP, Stark S, Johann PD, Blattner-Johnson M, et al. (2021). The Pediatric Precision Oncology INFORM Registry: Clinical Outcome and Benefit for Patients with Very High-Evidence Targets. Cancer discovery 11, 2764–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veninga V, and Voest EE (2021). Tumor organoids: Opportunities and challenges to guide precision medicine. Cancer Cell 39, 1190–1201. [DOI] [PubMed] [Google Scholar]
- Vlachogiannis G, Hedayat S, Vatsiou A, Jamin Y, Fernandez-Mateos J, Khan K, Lampis A, Eason K, Huntingford I, Burke R, et al. (2018). Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 359, 920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo TT, Ryan J, Carrasco R, Neuberg D, Rossi DJ, Stone RM, Deangelo DJ, Frattini MG, and Letai A (2012). Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell 151, 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Hoff DD (1983). “Send this patient’s tumor for culture and sensitivity”. N Engl J Med 308, 154–155. [DOI] [PubMed] [Google Scholar]
- Welm BE, Vaklavas C, and Welm AL (2021). Toward improved models of human cancer. APL bioengineering 5, 010901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DA, Takebe N, Hinoue T, Hoadley KA, Cardenas MF, Hamilton AM, Laird PW, Wang L, Johnson A, Dewal N, et al. (2021). Molecular Features of Cancers Exhibiting Exceptional Responses to Treatment. Cancer Cell 39, 38–53 e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo XY, Giordano J, Srivastava A, Zhao ZM, Lloyd MW, de Bruijn R, Suh YS, Patidar R, Chen L, Scherer S, et al. (2021). Conservation of copy number profiles during engraftment and passaging of patient-derived cancer xenografts. Nat Genet 53, 86–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, and Dai Y (2017). Tumor microenvironment and therapeutic response. Cancer Lett 387, 61–68. [DOI] [PubMed] [Google Scholar]
- Yu F, Lu Y, Tao L, Jiang YY, Lin DC, Wang L, Petersson F, Yoshiyama H, Koeffler PH, Goh BC, and Loh KS (2017). Non-malignant epithelial cells preferentially proliferate from nasopharyngeal carcinoma biopsy cultured under conditionally reprogrammed conditions. Sci Rep 7, 17359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Claerhout S, Prat A, Dobrolecki LE, Petrovic I, Lai Q, Landis MD, Wiechmann L, Schiff R, Giuliano M, et al. (2013). A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer research 73, 4885–4897. [DOI] [PMC free article] [PubMed] [Google Scholar]