

Abstract
Cytoglobin (Cygb) has been identified as the major nitric oxide (NO) metabolizing protein in vascular smooth muscle cells (VSMCs) and is crucial for the regulation of vascular tone. In the presence of its requisite cytochrome B5a (B5)/ B5 reductase-isoform-3 (B5R) reducing system, Cygb controls NO metabolism through the oxygen-dependent process of NO dioxygenation. Tobacco cigarette smoking (TCS) induces vascular dysfunction; however, the role of Cygb in the pathophysiology of TCS-induced cardiovascular disease has not been previously investigated. While TCS impairs NO biosynthesis, its effect on NO metabolism remains unclear. Therefore, we performed studies in aortic VSMCs with tobacco smoke extract (TSE) exposure to investigate the effects of cigarette smoke constituents on the rates of NO decay, with focus on the alterations that occur in the process of Cygb-mediated NO metabolism. TSE greatly enhanced the rates of NO metabolism by VSMCs. An initial increase in superoxide-mediated NO degradation was seen at 4 hours of exposure. This was followed by much larger progressive increases at 24 and 48 hours, accompanied by parallel increases in the expression of Cygb and B5/B5R. siRNA-mediated Cygb knockdown greatly decreased these TSE-induced elevations in NO decay rates. Therefore, upregulation of the levels of Cygb and its reducing system accounted for the large increase in NO metabolism rate seen after 24 hours of TSE exposure. Thus, increased Cygb-mediated NO degradation would contribute to TCS-induced vascular dysfunction and partial inhibition of Cygb expression or its NO dioxygenase function could be a promising therapeutic target to prevent secondary cardiovascular disease.
Keywords: globins, nitric oxide metabolism, superoxide, tobacco cigarette smoking, cardiovascular disease, cytochrome B5 / cytochrome B5 reductase
1. Introduction
Despite cessation efforts, tobacco cigarette smoking (TCS) continues to be a major health risk and contributor to cardiovascular disease (CVD) [1]. TCS induces vascular dysfunction that in turn leads to CVD [2; 3; 4]. Decreased nitric oxide (NO) bioavailability serves as a central hallmark of this critical pathophysiological process [4; 5]. Originally identified as endothelium-derived relaxing factor, NO, is a key regulator of vascular tone and blood flow [6; 7]. NO binding to soluble guanylate cyclase (sGC) in the smooth muscle of vessels induces cyclic guanosine monophosphate (cGMP)-mediated vascular relaxation [8; 9]. In addition to its vasodilator effect, NO is also a key regulator of vascular homeostasis via inhibition of smooth muscle cell proliferation, platelet aggregation, leukocyte adhesion, and expression of pro-inflammatory cytokines [10; 11; 12].
Vascular NO levels are controlled by the balance between the rates of NO synthesis and NO metabolism [13; 14]. Under normal physiological conditions, NO is synthesized in the endothelium by endothelial NO synthase (eNOS) which utilizes the amino acid L-arginine and molecular oxygen to generate NO and L-citrulline. Several co-factors, such as tetrahydrobiopterin (BH4) and Ca2+/calmodulin, regulate the production of NO from eNOS [15; 16]. Whilst the process of NO synthesis has been well-defined and extensively studied for decades, the precise process of NO metabolism in the vascular wall had remained elusive.
Early studies observed O2-dependent NO metabolism in the vascular wall and suggested that there is a NO dioxygenase present in vascular smooth muscle [17; 18]. Most recently, it has been demonstrated that vascular NOD activity is primarily mediated by the novel globin, cytoglobin (Cygb), which in the presence of its requisite cytochrome b5a/cytochrome b5 reductase isoform 3 (B5/B5R) reducing system, uniquely exhibits high O2-dependent NO dioxygenase activity [13; 19; 20; 21; 22]. Cygb belongs to the globin family, which includes hemoglobin, myoglobin, and neuroglobin [19; 23]. While all globins can metabolize NO to nitrate via NO dioxygenation, Cygb has much higher NOD activity than the other globins [24; 25; 26]. In addition, Cygb is also expressed at much higher levels in vascular smooth muscle than other globins. Cygb has been shown to be the main NO degradation pathway in the blood vessel wall serving to regulate vascular tone and blood pressure [24]. This has been confirmed with the observation of abrogated NO consumption with increased vascular relaxation, and low systemic blood pressure in Cygb−/− mice [13; 24].
While Cygb is now established as a key regulator of vascular tone and is inducible under some pathological conditions [27; 28], there is a lack of knowledge regarding the alterations in Cygb expression and function that occur in response to CVD risk factors including TCS. While TCS can trigger eNOS dysfunction and uncoupling with resultant loss of NO synthesis and increase in superoxide, O2•−, generation [5; 29; 30], the alterations in the process of cellular NO metabolism remain unclear. The levels of Cygb expressed in vascular smooth muscle would be expected to modulate the rate of NO degradation and vascular NO bioavailability, in turn affecting subsequent onset of TCS-induced CVD. Thus, it is of great importance to determine how TCS exposure impacts the expression of Cygb, its NOD activity, and overall NO metabolism.
Therefore, in the present work, studies are performed in vascular smooth muscle cells (VSMCs) characterizing the effects of tobacco smoke extract (TSE) exposure on Cygb expression and the process of cellular NO decay. The expression of Cygb and its requisite reducing system, B5/B5R, greatly increased 24 hours post TSE exposure, with further increases at 48 hours. The rate of Cygb-mediated NO decay paralleled the increase in Cygb expression, while with Cygb knockdown this increased NO degradation was abrogated. Thus, TSE-exposure triggers enhanced Cygb-mediated NO metabolism leading to decreased NO bioavailability.
2. Materials and Methods
2.1. Materials:
Chemicals, reagents, and other materials were obtained from Millipore Sigma-Aldrich (St. Louis, MO) unless noted otherwise.
2.2. Cell culture
Mouse aortic VSMCs were purchased from American Type Culture Collection (CRL-2797, ATCC, Manassas, VA) were maintained in RPMI 1640 medium (A10491–01, Gibco LTC, Waltham, MA) supplemented with 10% (v/v) fetal bovine serum (FBS; Invitrogen, Waltham, MA), penicillin (100 U/ml), VSMCs Growth supplement (Lonza Group Ltd, Switzerland), and streptomycin (100 μg/ml), at 37 °C with a 5% CO2 atmosphere in a humidified incubator. Culture medium was changed every other day and 70% confluent cells were used for experiments.
2.3. Preparation of tobacco cigarette smoking extract
Tobacco smoking extract was prepared from conditioned 3R4F Kentucky research grade cigarettes (Center for Tobacco Reference Products, University of Kentucky, USA) according to the Massachusetts Intense Regimen [29; 31]. Four cigarettes were sequentially bubbled into 15 ml of phosphate buffered saline (PBS). The TSE pH was corrected to 7.4 and a 1:10 (v/v) diluted aliquot was used to measure the absorbance at 320 nm, using a UV-VIS spectrophotometer (Molecular Devices, CA). TSE preparations with an absorbance of 0.5 in a 1:10 (v/v) diluted aliquot were considered as 100% stocks and then diluted with low serum medium as needed. TSE was freshly prepared and used within 15 minutes.
2.4. Cell viability assay
XTT (2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxanilide) (NB12, VWR Life Science, Radnor, PA) was used to measure cellular metabolic activity, which reflects the number of viable cells, and thus provides information on cytotoxicity and the inhibition of cell growth. This colorimetric assay is based on the reduction of the yellow tetrazolium salt to an orange formazan dye by metabolically active cells. Cells were grown in 96-well plates, incubated in a humidified atmosphere at 37 °C for 24 hours to reach ~ 70 % confluency and then exposed for 4, 24 or 48 hours to serial dilutions from TSE (0.0625, 0.125, 0.25, 0.5, 1, 2, 4 and 8%). At the end of the treatment period, medium was removed, cells were incubated with 200 μl/well of 0.25% XTT solution containing 1% 10 mM phenazine methosulfate (PMS) for 2 hours. Absorbance was measured at 450 nm using a SpectraMaxplus 384 Microplate Reader (Molecular Devices, Hampton, NH). The cell viability was expressed relative to that of untreated control cells.
2.5. Preparation of NO stock solution
NO solution was prepared from NO gas as described previously [24; 25; 32]. Firstly, NO gas was scrubbed by passage through a U-shaped tube containing NaOH pellets and then through 1 M deaerated (bubbled with 100% argon) NaOH solution in a custom-designed apparatus connected using only glass or stainless-steel tubing and fittings. Secondly, the purified gas was collected by saturating a deaerated buffer solution (0.2 M potassium phosphate, pH 7.4) contained in a glass sampling flask (Kimble/Kontes, Vineland, NJ) fitted with a septum. A gas-tight Hamilton syringe (Hamilton Robotics, Reno, NV) was used for anaerobic extraction of NO solution.
2.6. Measurements of NO metabolism rate
Measurements were performed in a four-port water-jacketed electrochemical chamber (NOCHM-4, WPI, Sarasota, FL) at 37 °C in air-equilibrated buffer as described previously [24; 33]. Briefly, two Clark-type NO electrodes (ISO-NOP, WPI, Sarasota, FL) were inserted through two ports on the side wall of the chamber for measuring the rate of NO metabolism. An O2 electrode (ISO-OXY-2, WPI, Sarasota, FL) was placed through another port on the side wall of the chamber for continuously monitoring O2 level, which was maintained at ~ 200 μM through passing purified air over the head space of the chamber. Measurements were done at 37 °C in air-equilibrated buffer continuously stirred with a magnetic stir bar. The rate of NO decay in the buffer solution was measured before adding the VSMCs into the solution following injection of NO to achieve an initial concentration of 1.0 μM using a 10 μl gas-tight Hamilton syringe. The NO electrode reads a given current that is proportional to the concentration in solution. This is calibrated before the experiment with known concentrations of NO. The rate of NO decay is determined from the derivative of the observed current over time with the initial decay rate measured after the initial peak. The delay in observing the peak after bolus addition of NO is due to the time required for mixing and the response time of the platinum electrode. Time 0 of the electrode reading [NO]0 is defined at the time of this initial peak with a value of 1 assigned and decay from this value measured over time [22; 33]. The rate of NO decay was detected in non-exposed and exposed cells. Normal VSMCs untreated or treated with TSE 1.0% for 4, 24 and 48 hours were assayed for NO decay in the absence or presence of the superoxide dismutase mimetic (SODm), MnTBAP [Mn(III)tetrakis(4-benzoic acid)porphyrin], 100 μM (sc-221954, Santa Cruz Biotechnology Inc., Santa Cruz, CA) [29]. Untreated and 1% TSE treated for 48 hours, scrambled siRNA (Scr-RNA) and Cygb siRNA treated VSMCs were also assayed for NO metabolism. Cell suspension concentrations in the chamber were fixed at 3.5 × 106 cells per ml [22]. In order to obtain the NO decay rate secondary to the VSMCs, the baseline rate of NO decay in the buffer solution was subtracted from the total measured decay rate as reported [21; 29; 34].
2.7. Knockdown of Cygb in VSMCs
Aortic VSMCs were transfected with Cygb siRNA (sc-45548, Santa Cruz Biotechnology Inc., Santa Cruz, CA) using Lipofectamine RNAiMAX (Invitrogen, Waltham, MA) according to the manufacturer’s recommendations. Briefly, Lipofectamine RNAiMAX was mixed gently with antibiotic-free Opti-MEM medium (Invitrogen, Waltham, MA) in a ratio of 6 μl:100 μl for each 1 ml growth medium followed by incubation at room temperature for 5 minutes. An aliquot of Cygb siRNA was mixed with the Lipofectamine RNAiMAX/Opti-MEM mixture to reach a final concentration of 100 nM. Following incubation at room temperature for 30 minutes, 1 ml of mixture was added to a separate 150 × 25 mm culture plate containing VSMCs exponentially growing in 9 ml of antibiotic-free Opti-MEM medium. Cygb siRNA and the corresponding Scr-RNA-transfected cells were incubated at 37 °C in a 5% CO2-humidified incubator. Seven hours later, Opti-MEM medium was changed to RPMI 1640 complete medium to provide essential nutrients and growth factors for optimal growth and cell survival. Forty-eight hours post transfection, the cells were collected or treated with TSE 1.0% for further studies, and protein expression was evaluated by Western blotting.
2.8. Western blotting
Exponentially growing cells were exposed to 1.0% TSE for 4, 24 or 48 hours. Whole-cell protein extracts from harvested cells were prepared using ice-cold lysis RIPA buffer containing 20 mM Tris-HCl (pH 6.8), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP40, 1% Na-deoxycholate, and protease/phosphatase inhibitor cocktail (Roche, Mannheim, Germany). Protein concentration was determined using a Bio-Rad DC protein assay kit (Bio-Rad Laboratories, Hercules, CA). Equal protein amounts of cell lysate were separated on a gradient 4–20% SDS- polyacrylamide gel and electro-blotted on PVDF membranes (Bio-Rad Laboratories Inc., Hercules, CA). The membranes were blocked with 5% non-fat dry milk in Tris buffered saline-tween 20 (TBST) (Bio-Rad Laboratories Inc., Hercules, CA) for 1 hour at 37 °C and then incubated overnight at 4°C with primary antibodies. The following primary antibodies were used: rabbit polyclonal anti-Cygb (diluted 1:500) (PA5–75671, Invitrogen, Waltham, MA), rabbit polyclonal anti-B5R (1:1500) (10894-AP, Proteintech Inc., Rosemont, IL), rabbit polyclonal anti-B5 (1:200) (sc-33174, Santa Cruz Biotechnology Inc., Santa Cruz, CA), and rabbit monoclonal anti-B-actin (1:1000) (8457S, Cell Signaling Technology Inc., Danvers, MA). All antibodies were validated for specificity using dual color molecular weight markers (Bio-Rad Laboratories Inc., Hercules, CA). Membranes were then washed three times in TBST and incubated with horseradish peroxidase (HRP)-linked anti-rabbit secondary antibody (1:2000) (7074P2, Cell signaling technology Inc., Danvers, MA) in 1% milk/TBST for one hour at room temperature followed by washing with TBST. The Bio-Rad ECL western blotting detection reagent (Clarity MaxTM Western ECL, Hercules, CA) was used to develop the membranes. Protein expression was captured and analyzed by a high-resolution KwikQuant Imager Molecular Imager and KwikQuant Image manager software, respectively (Kindle Biosciences LLC., Greenwich, CT).
2.9. Measurement of O2•− generation
VSMCs were cultured on sterile cover slips in a 6-well plate at a density of 2 × 105 cells/well and incubated in a humidified atmosphere at 37 °C and 5% CO2 for 4, 24 or 48 hours. Cells were unexposed or exposed to 1.0% TSE, washed with PBS and incubated with the probe dihydroethidium (DHE; 10 μM) and the nuclear stain 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; 1 μM) in the dark, for 30 min at 37 °C. DHE-derived fluorescence was used to determine O2•− generation. Preincubation with or without the SODm (MnTBAP, 100 μM) for 15 minutes before DHE, was performed to confirm that the observed fluorescence was derived from O2•− [31]. Stained cells on cover slips were rinsed extensively with PBS, mounted in antifade mounting medium Fluoromount-G (Southern Biotechnology Associates, Birmingham, AL), and coverslipped. Different fields were captured at 40x using an Olympus FV3000 spectral confocal microscope (Tokyo, Japan). The fluorescence intensity of the images was analyzed using the software provided by the manufacturer.
2.10. Statistical analysis
Data are presented as means ± SE. Statistical analysis was performed using one-way analysis of variance (ANOVA) followed by a Tukey-Kramer test. GraphPad Prism software, version 8 (GraphPad, San Diego, USA), was used to compute statistical data. Statistical differences were considered significant from control at p < 0.05.
3. Results
3.1. Dose-dependence of TSE–induced cell injury and death.
Cell viability following TSE exposure was performed by XTT assay to determine the optimal TSE concentrations and exposure times to be used for further studies. The viability of the VSMCs decreased in a TSE concentration and time dependent manner (Fig. 1). Significant reduction in cell viability was first observed with 2.0% TSE after 24 and 48 hours of exposure with values of 80 ± 3 % and 64 ± 4 %, respectively. With 4.0 % and 8.0% TSE, further decreases in cell viability were seen at 24 hours (65 ± 3 % and 54 ± 3%) and 48 hours (53 ± 3 % and 45 ± 3 %), respectively. However, only 8.0% TSE showed a significant decrease in cell viability after four hours treatment. Based on these cell viability studies, 1.0% TSE was selected for use in the subsequent studies, in order to minimize induction of cell death.
Figure 1: Effect of TSE exposure on cell viability:
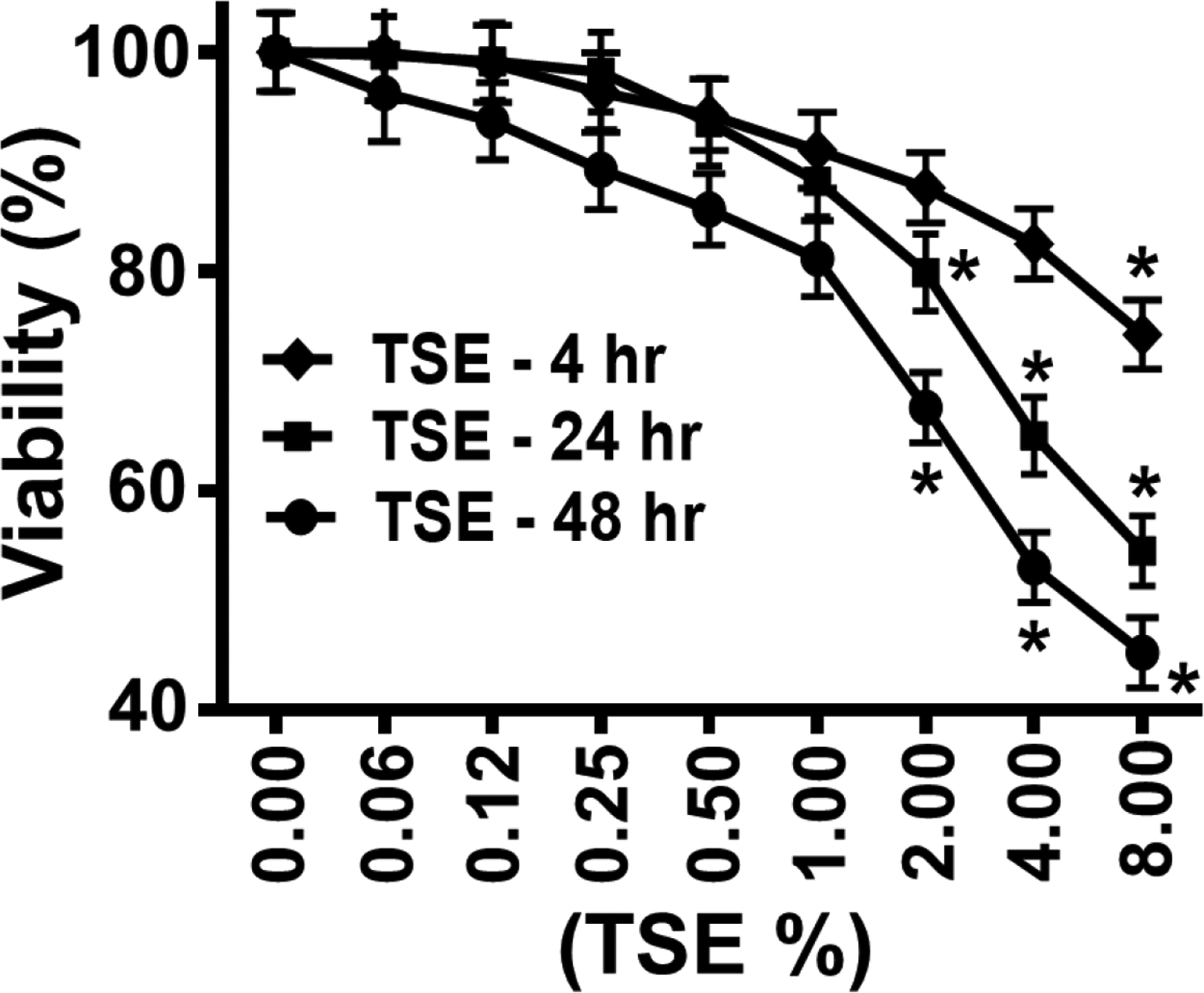
VSMCs were grown to 70% confluency, exposed for 4, 24 and 48 hr to different concentrations of Tobacco smoke extract (TSE) and then cell viability was assayed, compared to corresponding control untreated cells, using the XTT assay. Each value represents the mean of four independent experiments ± SEM. *: Denotes significant difference from untreated control at p < 0.05.
3.2. TSE increases the rates of NO metabolism by VSMCs.
In order to assess the effect of TSE exposure on NO metabolism, we measured the rate of NO decay in TSE-treated VSMCs. As seen in Figure 2A and D, treatment of VSMCs with 1.0 % TSE for 4 hours resulted in a significant increase in NO consumption rate from 11.6 ± 0.6 nMs−1 in control untreated VSMCs to 15.4 ± 0.6 nMs−1 (p < 0.02). After 24 hours of exposure, TSE-treated VSMCs showed further marked increase in NO decay rate to 19.2 nMs−1, while the untreated cells exhibited a rate of 11.4 nMs−1 (p < 0.001) (Fig. 2B, E). Further increase was seen following 48 hours of TSE-treatment with NO decay rate of 27.9 nMs−1 compared to 10.8 nMs−1 in the untreated cells (p < 0.001) (Fig. 2C, F). Thus, TSE exposure triggered an increase in the rate of NO metabolism that increased with the time of exposure.
Figure 2: Effects of TSE on NO metabolism by VSMCs.
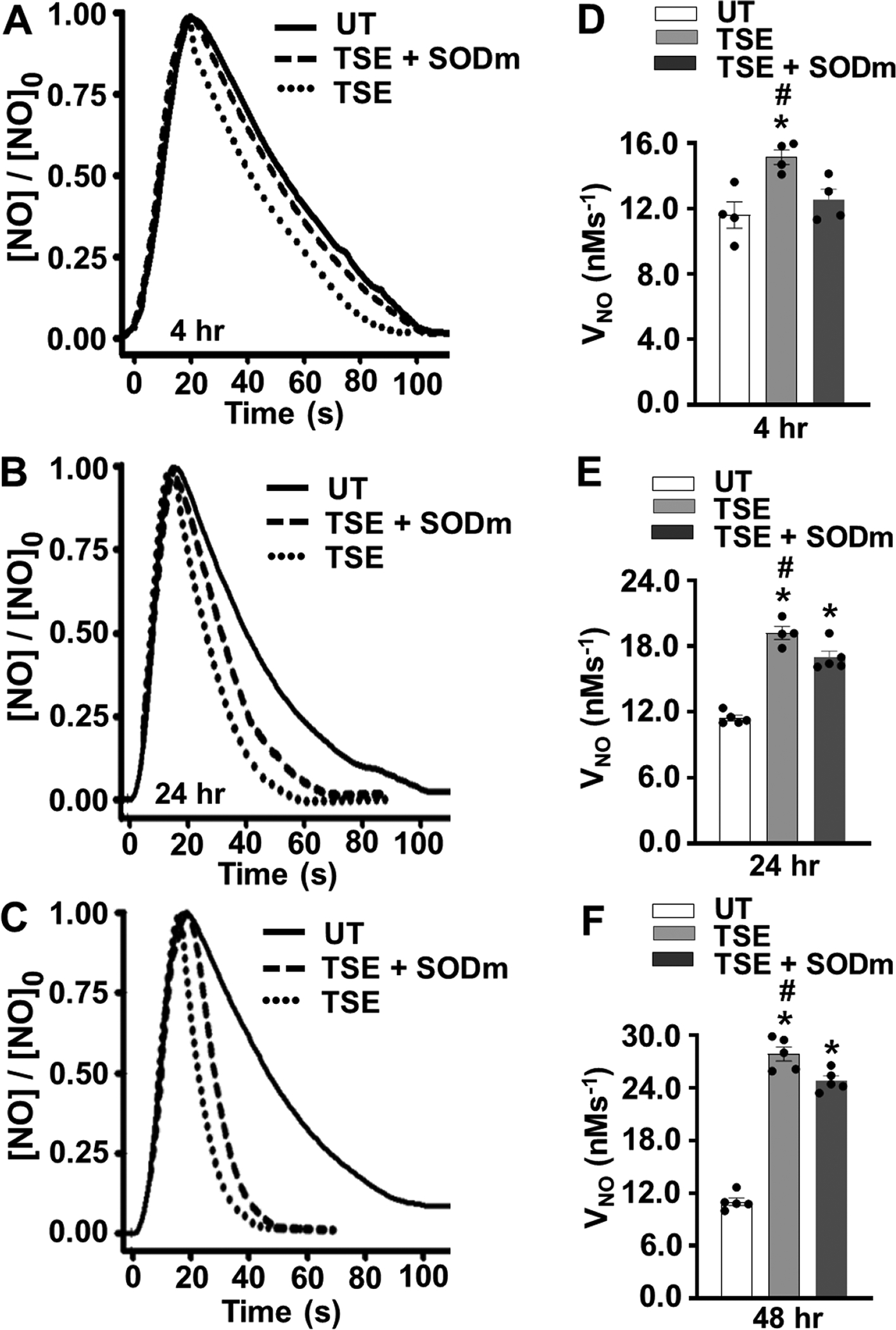
VSMCs cells were either untreated (UT) or treated with 1% tobacco smoke extract (TSE) for 4, 24 and 48 hr and then harvested for measurement of nitric oxide (NO) metabolism, with or without pre-incubation with SOD mimetic (SODm). A, B, and C, show the time course of NO decay after VSMCs treatment with TSE for 4, 24 and 48 hr, respectively. Values are shown as the ratio of the [NO] measured over time to the initial peak value, [NO]0. A cell density of 3.5 × 106 VSMCs per ml was used in all groups. NO was injected into the solution to achieve an initial concentration of 1 μM. D, E and F, show the mean ± SEM of the NO metabolism rate (VNO) measured from 4–5 independent experiments. VNO was detected in VSMCs after 4, 24 and 48 hr of exposure, respectively. *: Denotes significant difference from untreated control at p < 0.05. #: Denotes significant difference from TSE-treated group in presence of SODm p < 0.05.
Additional experiments were performed in the presence of a SODm in order to determine the role of O2•− in the TSE-induced increase in NO decay rate. In the presence of the SODm, the increase in the rate of NO decay after 4 hours of exposure was largely, 76%, reversed (Fig. 2 A, D). However, with the much larger increases in NO metabolism rates seen after 24 or 48 hours of TSE treatment, only modest 25% or 18%, respective decreases were seen with the SODm (Fig. 2 B,C, E, F). Thus, with only 4 hours of exposure the reaction of NO with O2•− accounted for most of the TSE-induced increase in NO metabolism rate, while with longer exposures of 24 to 48 hours most of NO metabolism was not O2•− -mediated but due to induction of another process.
3.3. TSE exposure increases cellular superoxide generation in VSMCs.
Generation of O2•− was measured in freshly collected VSMCs from TSE-treated and untreated VSMCs using the fluorescence probe DHE (Fig. 3A, B). DHE reacts with O2•− to form 2-hydroxyethidium with characteristic red fluorescence [35]. After four hours exposure, TSE-treated VSMCs clearly exhibited higher O2•− generation with fluorescence intensity 2.7-fold of that in untreated cells. After 24 hours of treatment, O2•−-derived fluorescence further increased to values 3.6 -fold those in matched untreated cells and with treatment for 48 hours a larger 5.8 -fold increase was seen (Fig. 3B). The specificity of the observed fluorescence was confirmed by measurements with pre-incubation with SODm, which largely abolished the red fluorescence (Fig. 3A, right panel).
Figure 3: Effect of TSE on superoxide generation in VSMCs.
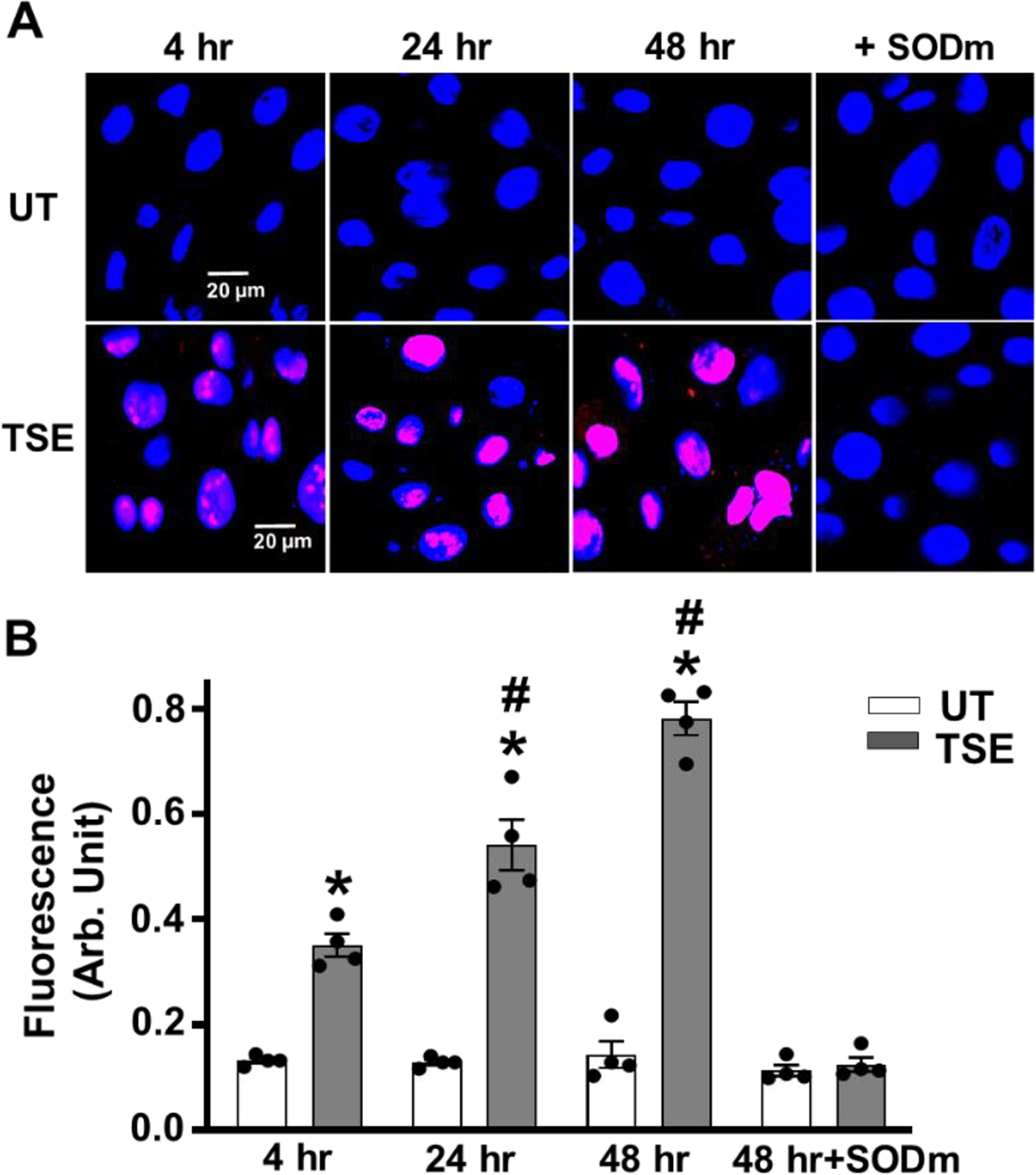
Confluent cells were tobacco smoke extract (TSE)-untreated (UT) or treated with 1% of TSE for 4, 24 and 48 hr, with or without pre-incubation with SOD mimetic (SODm) and then incubated with DHE and DAPI and imaged by confocal fluorescence microscopy. A, representative images of VSMCs for each group, following 4, 24 and 48 hr of exposure. TSE + SODm is shown for 48 hr exposure. B, quantitation of fluorescence intensity in VSMCs from repeat experiments performed as in A. Data are means ± SEM of 4–5 independent experiments. *: Denotes significant difference from exposure time matched untreated control at p < 0.001. #: Denotes significant difference from TSE treated value at 4 hr at p < 0.01.
3.4. TSE increases Cygb expression in VSMCs.
As was seen in figure 2 above, with 4 hours of TSE-treatment the process of accelerated NO decay was reversed by addition of the SODm suggesting a role of O2•−, while after 24 or 48 hours the rate of NO decay further increased and became independent of SODm treatment. This suggests that another NO metabolizing process is increased such as that through NO dioxygenation. Therefore, we investigated whether Cygb expression changes following TSE-treatment. As shown in Fig. 4A and B, no significant alteration in Cygb expression occurred with 4 hours TSE-treatment, compared to untreated cells. However, after 24 hours treatment a clear increase in the cellular Cygb expression to 210 ± 6 % of basal levels was seen, p <0.001, with further increase to 340 ± 7 % of basal levels after 48 hours, p <0.001, (Fig. 4 A, B). Thus, after 24 hours of TSE-treatment, the higher Cygb levels would be expected to contribute to the increased NO metabolism rates observed and even more to the increase seen after 48 hours.
Figure 4: Effects of TSE on Cygb expression in VSMCs.
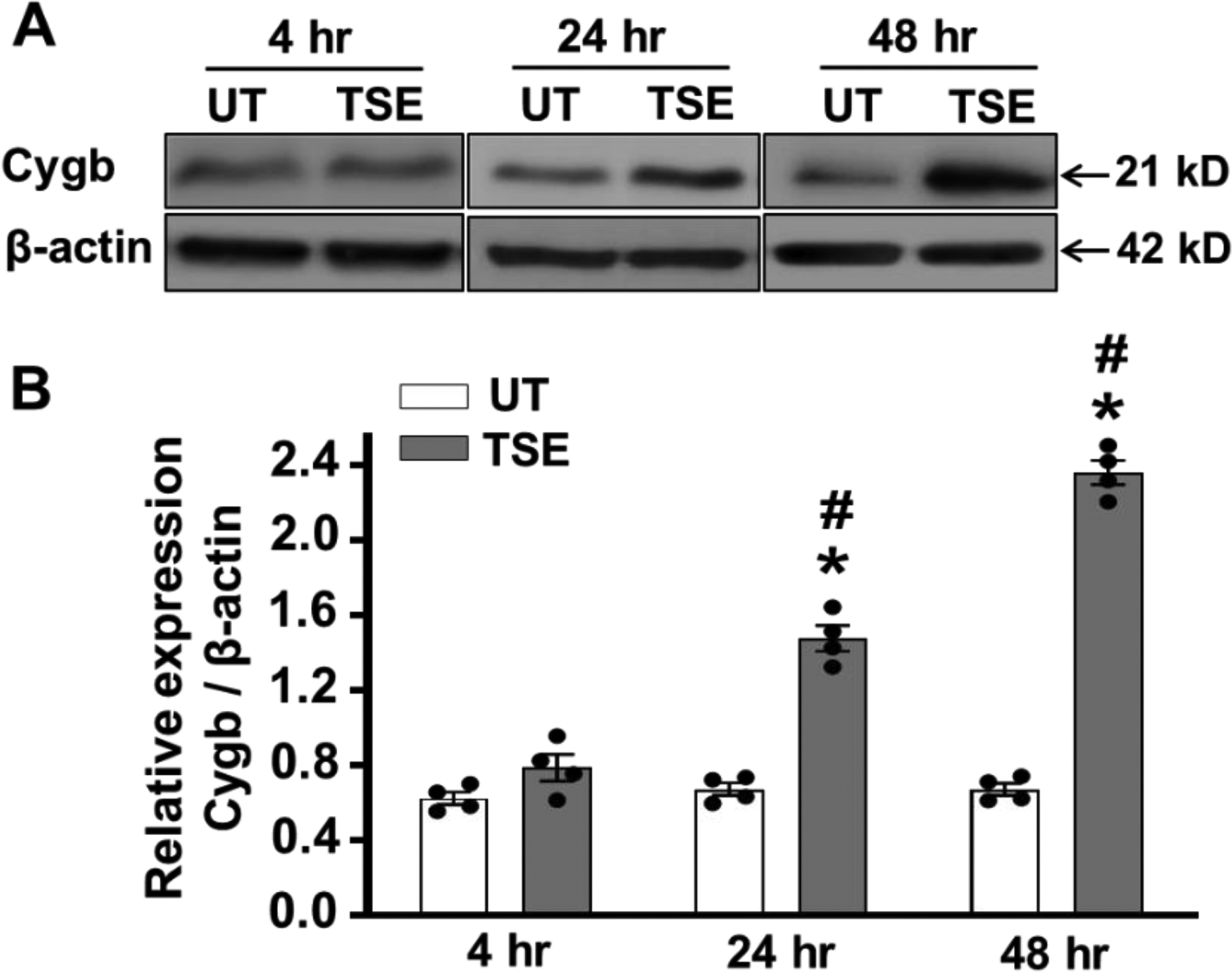
Cells were treated with 1% tobacco smoke extract (TSE) for 4, 24 and 48 hr and then whole cell lysate was blotted against Cytoglobin (Cygb) and β-actin antibodies. A, western blots for Cygb and β-actin in VSMCs, after treatment with TSE for 4, 24 and 48 hr. B, quantitation of Cygb expression from densitometry of the observed bands normalized for the β-actin loading control. Data shown are means ± SEM of 4 independent experiments. *: Denotes significant difference from untreated control at p < 0.001; #: Denotes significant difference from the TSE treated value at 4 hr at p < 0.001.
3.5. TSE increases expression of B5R and B5 in VSMCs.
Since the NOD activity of Cygb requires the presence of its reducing system, we assessed if the cellular expression of B5R and B5, which have been recently identified as the major reducing system for Cygb in VSMCs, were also increased [22; 36]. As shown in Fig. 5A and B, treatment with TSE for 24 hours increased the expression of B5R to 230 ± 5 % of that in untreated control VSMCs, p <0.005. Additionally, with extending TSE treatment duration to 48 hours a further increase to 350 ± 9 % of untreated control was seen, p <0.001, compared to the respective untreated cells.
Figure 5: Effects of TSE on B5R and B5 expressions in VSMCs.
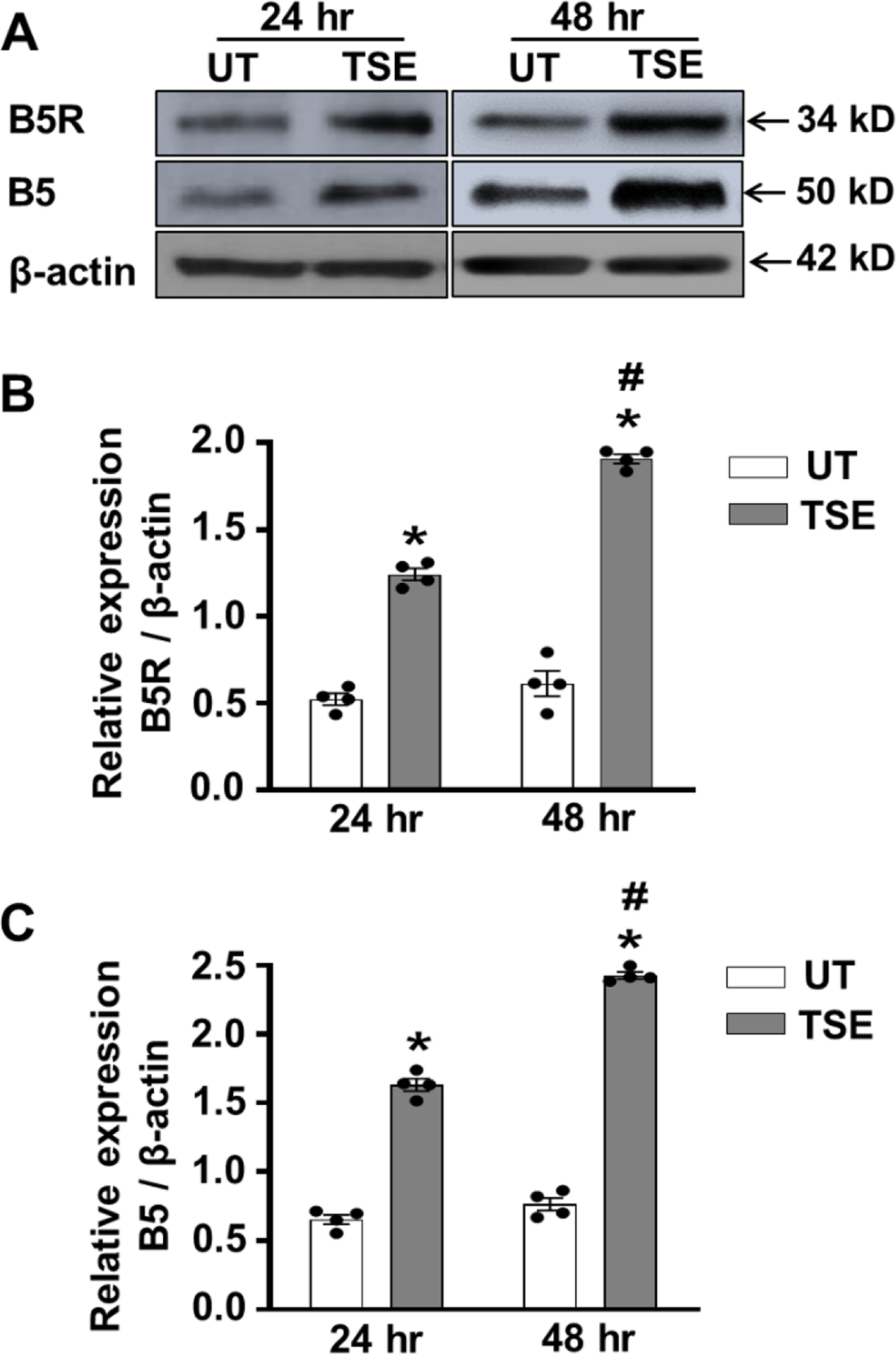
Confluent cells were treated with 1% tobacco smoke extract (TSE) for 24 and 48 hr and then whole cell lysate was blotted against cytochrome b5 reductase 3 (B5R), cytochrome b5 (B5) and β-actin antibodies. A, western blots for B5R, B5 and β-actin expressions in VSMCs, after treatment with TSE for 24 and 48 hr. B, quantitation of B5R expression from densitometry of the bands in VSMCs, after treatment with TSE for 24 and 48 hr. C, quantitation of B5 expression from densitometry of the bands in VSMCs, after treatment with TSE for 24 and 48 hr. The band intensities were normalized for β-actin levels. Data shown are means ± SEM of at least 4 independent experiments. *: Denotes significant difference from TSE-untreated control (UT) at p < 0.005; #: Denotes significant difference from 24 hr TSE treatment at p < 0.01.
For B5, western blotting similarly exhibited increased expression to 240 ± 6 % of basal levels after 24 hours treatment with TSE, p <0.005, and further increased to 321 ± 8 % of basal levels after 48 hours, p <0.001, (Fig. 5A, C). Thus, we observe similar upregulation of Cygb expression along with its B5R/B5 reducing system and together this would account for the large increase in Cygb-mediated NO metabolism seen following 24 or 48 hours of TSE-exposure.
3.6. Cygb knockdown largely reverses the increased rate of NO metabolism induced by TSE.
Since TSE treatment increased the expression levels of Cygb and this was associated with an increase in the rate of NO metabolism, we performed further experiments to test the effect of genetic knockdown (KD) of Cygb expression using siRNA. First experiments were performed in control VSMCs that were either maintained untreated or transfected with either Cygb siRNA (Cygb-KD) or non-specific scrambled RNA (Scr-RNA).
We evaluated the effectiveness of Cygb knockdown by immunoblotting. As shown in Fig. 6A and B, Cygb siRNA-treated cells showed a marked, over 90%, reduction in Cygb expression compared to scrRNA treated cells. In addition, Cygb knockdown greatly decreased the rate of NO metabolism in siRNA-treated VSMCs by ~80%, compared to that measured in corresponding scrRNA VSMCs or control non-transfected cells (Fig. 6C, D). Thus, in the basal state Cygb profoundly influences the rate of NO decay.
Figure 6: Effect of Cygb-knockdown on NO metabolism by VSMCs.
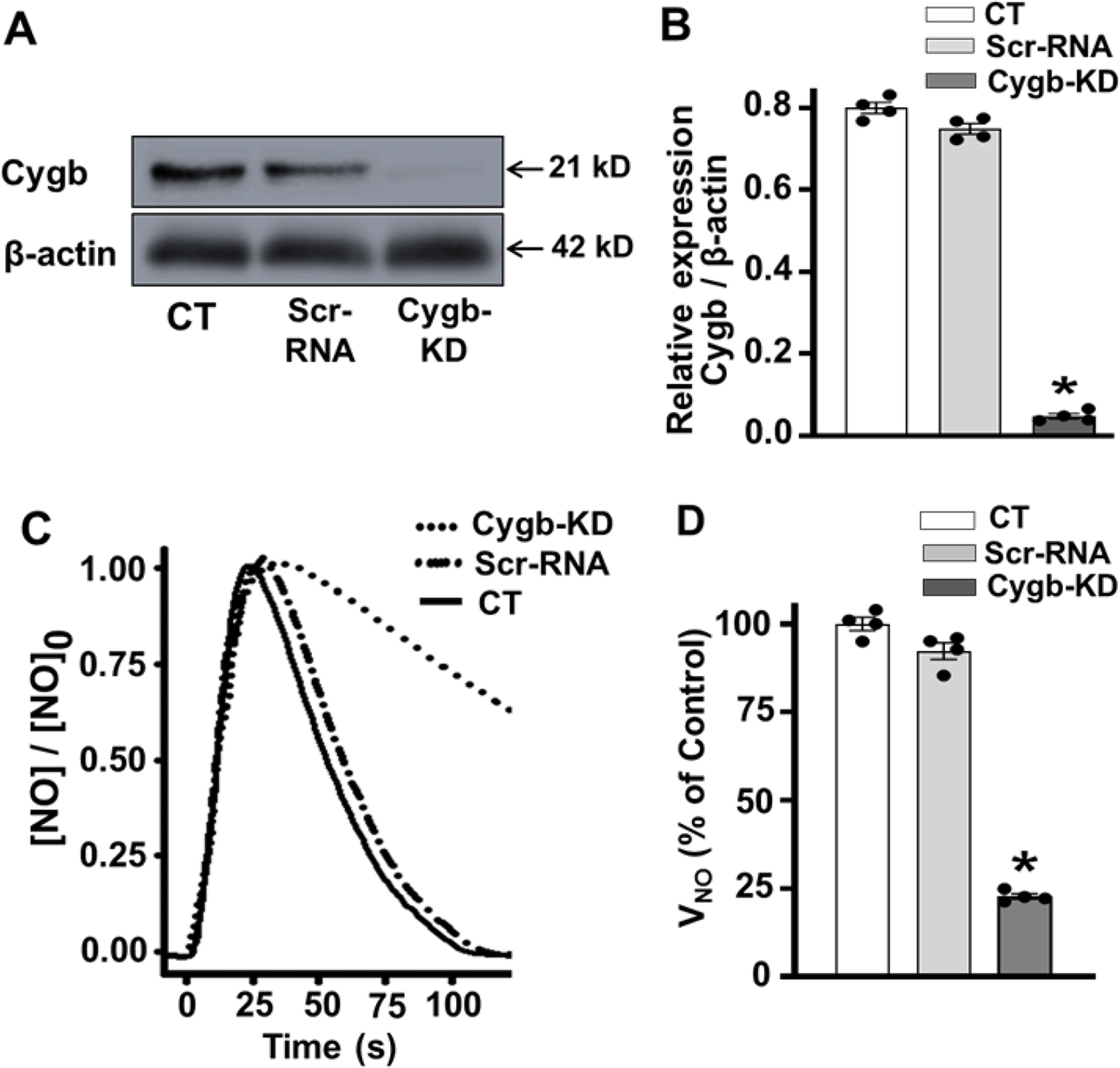
Cells were treated with Cytoglobin (Cygb) siRNA or scrambled RNA (ScrRNA) and then harvested for immunoblotting and measurement of nitric oxide (NO) metabolism. A, B, western blots and quantitation for Cygb and β-actin in control (CT), ScrRNA and Cygb-siRNA treated (Cygb-KD) VSMCs, respectively. C, the measured NO concentration over time after addition of NO in control, Scr-RNA and Cygb-KD treated VSMCs. D, shows the mean ± standard error of the NO metabolism rate (VNO), detected in control, Scr-RNA and Cygb-KD treated VSMCs, represented as % of the value in control cells. Each value represents the mean of 4–5 independent experiments ± SEM. A cell density of 3.5 × 106 VSMCs per ml was used in all groups. NO was injected as in figure 2. *: Denotes significant difference from control at p < 0.001.
Then, we performed experiments to determine the effect of siRNA-mediated Cygb knockdown on the increased rate of NO decay induced by TSE treatment. In cells not treated with TSE, Cygb knockdown decreased the rate of NO metabolism by ~ 75 – 80% compared to the corresponding ScrRNA VSMCs, p < 0.001 (Fig. 7 A, B). Furthermore in TSE-treated cells, where the rate of NO decay was increased to 2.8-fold that in the control or ScrRNA-treated cells, Cygb knockdown reduced the NO decay rate by ~4-fold, p < 0.001, to a value below that observed in control or ScrRNA cells that were not TSE-treated (Fig. 7 A, B). In addition, knockdown of Cygb expression provided an even larger decrease in the NO metabolism rate in TSE-treated cells than that in TSE-untreated cells consistent with the higher expression of Cygb observed in the TSE-treated cells (Fig. 7B). Thus, these results confirm that upregulation of Cygb expression accounts for the large increase in the NO decay rate observed after 48 hours of TSE exposure with downregulation of Cygb expression largely reversing the increased rate of NO decay induced by TSE exposure.
Figure 7: Effect of Cygb knockdown on NO metabolism by TSE-exposed VSMCs.
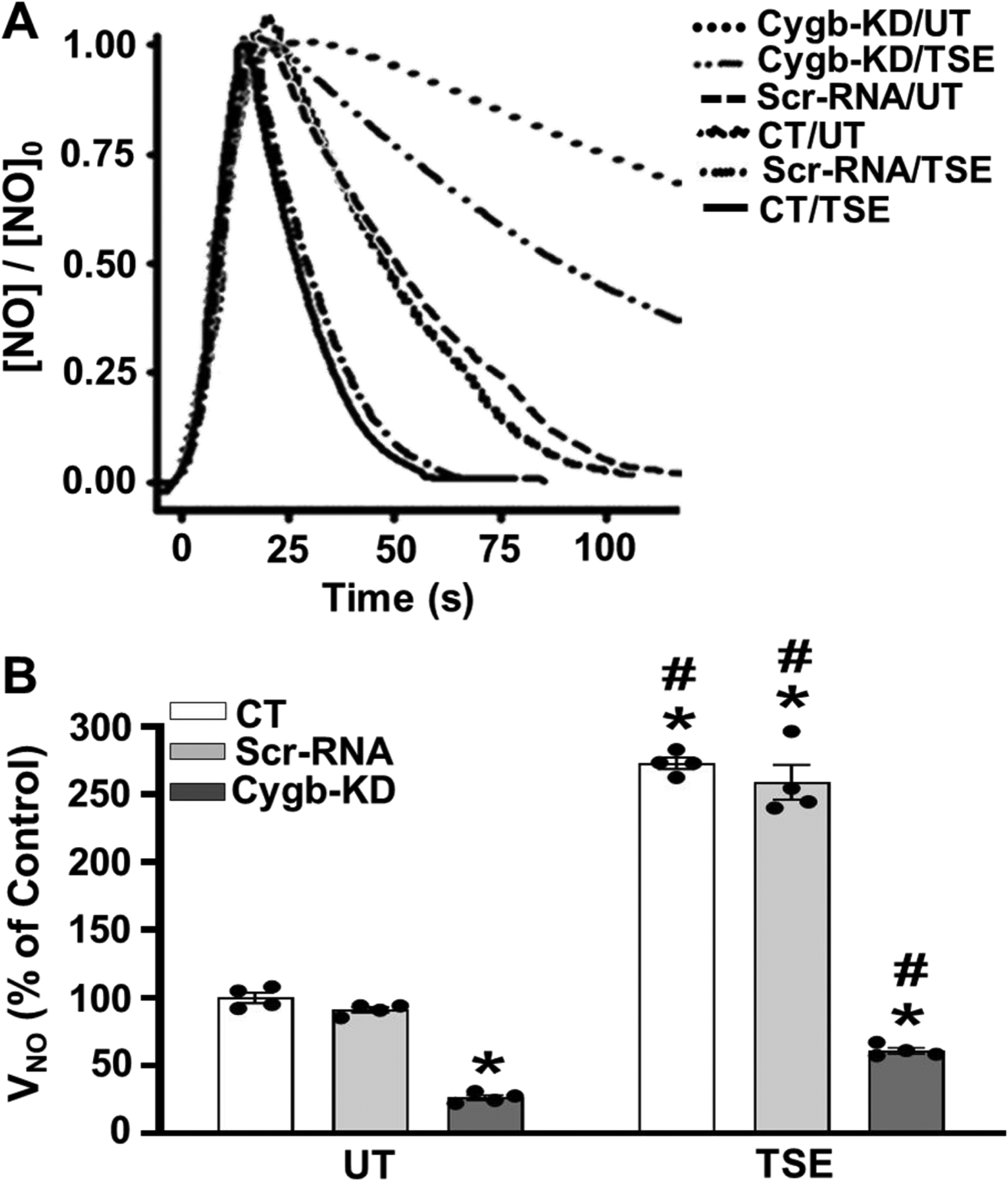
Control non-transfected (CT), Scr-RNA pretreated and Cygb-siRNA pretreated (Cygb-KD) cells were untreated (UT) or treated with 1% tobacco smoke extract (TSE) for 48 hr and then harvested for measurement of nitric oxide (NO) metabolism. A, NO metabolism was measured as in Figure 2. Cell density of 3.5 × 106 VSMCs per ml was used in all groups. B, the mean ± standard error of the NO metabolism rate, VNO, from repeat experiments performed as in A. Values are shown for UT and TSE treated CT, Scr-RNA and Cygb-KD VSMCs with values depicted as % of untreated CT. Similar to figure 2, VNO in untreated cells was 10.9 ± 0.5 nMs−1. Each value represents the mean of 4–5 independent experiments ± SEM. *: Denotes significant difference from the UT control cells at p < 0.01. #: Denotes significant difference of each type of TSE-treated cells with respect to the corresponding untreated cells at p < 0.01.
3.7. Role of superoxide in the TSE-induced increase in NO decay rate in Cgb depleted VSMCs.
With Cygb knockdown the rate of NO decay greatly decreased; however, it still remained 2.4-fold higher in TSE exposed than in untreated cells, p < 0.01, suggesting that in addition to Cygb-mediated NO dioxygenation, other processes such as increased O2•− generation also contribute to increase the NO decay rate (Fig. 8). In order to further investigate the role of O2•− in the TSE exposure-induced increase in NO decay rate seen in Cygb knockdown cells, experiments were performed in the presence of the SODm. With the SODm, the NO metabolism rate in these cells exposed to TSE for 48 hours was decreased ~2.4-fold to values similar to those observed in untreated cells (Fig. 8). The effect of the SODm was much more prominent in the TSE exposed Cygb knockdown cells than that observed in the similar experiments in control cells of figure 2, due to the lower overall rate of NO decay as well as the larger decrease in this rate with the SODm. With Cygb knockdown, SODm treatment decreased the rate of NO decay by 5.2 nMs−1, almost twice the change seen in similarly exposed control cells (Fig. 2). Thus, this data suggests that while Cygb depletion greatly decreased the overall NO decay rate, it may increase the levels of O2•− present in TSE-exposed VSMCs.
Figure 8: Effect of SODm on the TSE-induced increase in NO metabolism rate in Cygb-KD VSMCs.
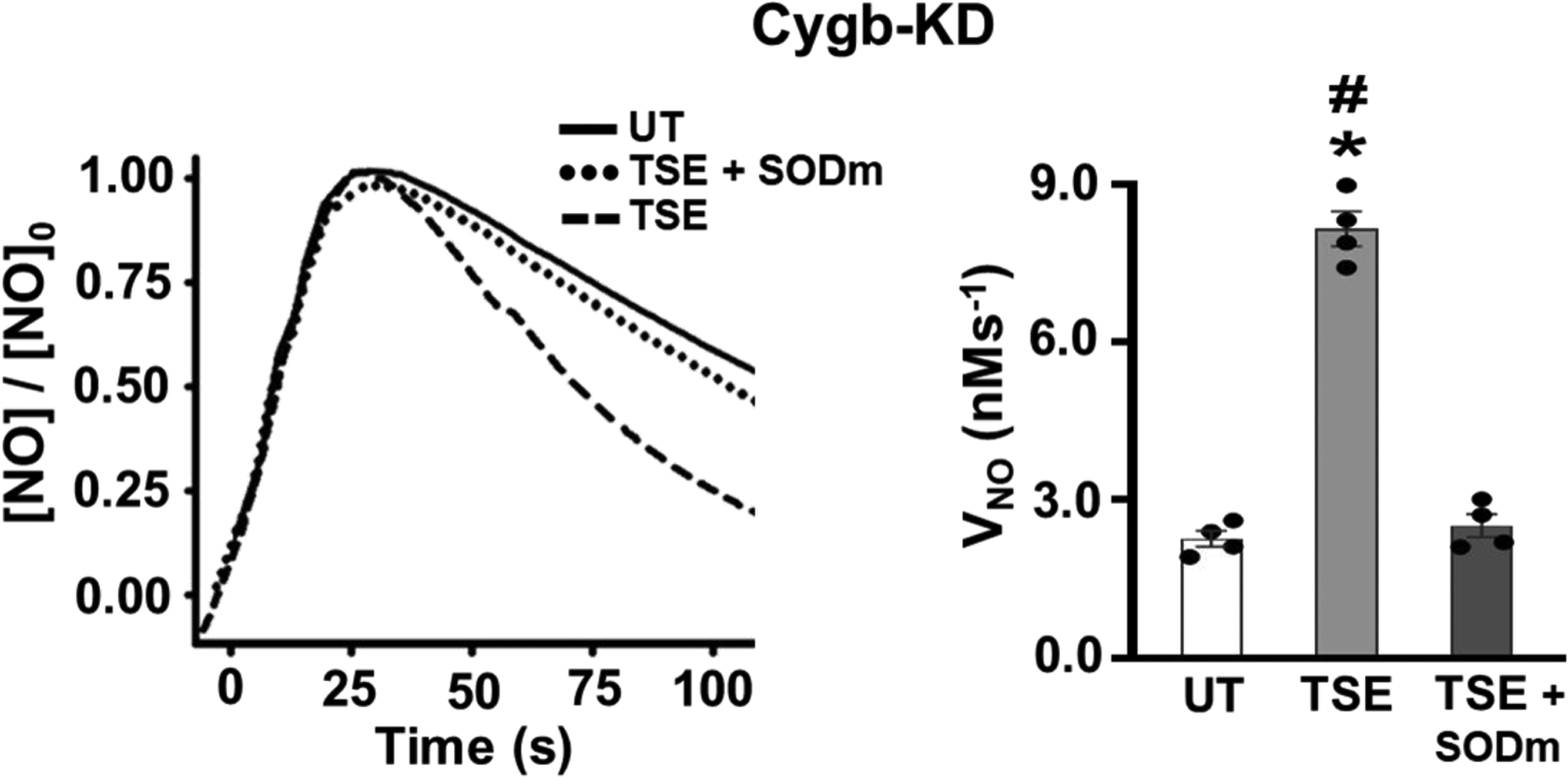
Cygb-siRNA pretreated (Cygb-KD) cells were either untreated (UT) or treated with 1% tobacco smoke extract (TSE), in the presence or absence of the SODm, for 48 hr and then harvested for measurement of NO metabolism, VNO, measured as in Figure 2. Cell density of 3.5 × 106 VSMCs per ml was used in all groups. Each value represents the mean of 4 independent experiments ± SEM. *: Denotes significant difference from the UT control cells at p < 0.001. #: Denotes significant difference from TSE-treated cells pre-incubated with SODm at p < 0.001.
In order to determine the effects of Cygb-KD on O2•− production in TSE-treated cells, Cygb-KD and scrRNA treated VSMCs were exposed to TSE for 48 hours, with or without pre-incubation with SODm and then incubated with DHE. While untreated cells exhibited only minimal DHE-derived fluorescence, with TSE-exposure higher levels of O2•− production were seen in the Cygb knockdown cells than in identically treated scrRNA cells with ~2-fold higher fluorescence intensity (Fig. 9 A,B). The specificity of the observed fluorescence was confirmed by pre-incubation with SODm, which abolished the observed fluorescence.
Figure 9: Effect of Cygb-KD on O2•− production in VSMCs.
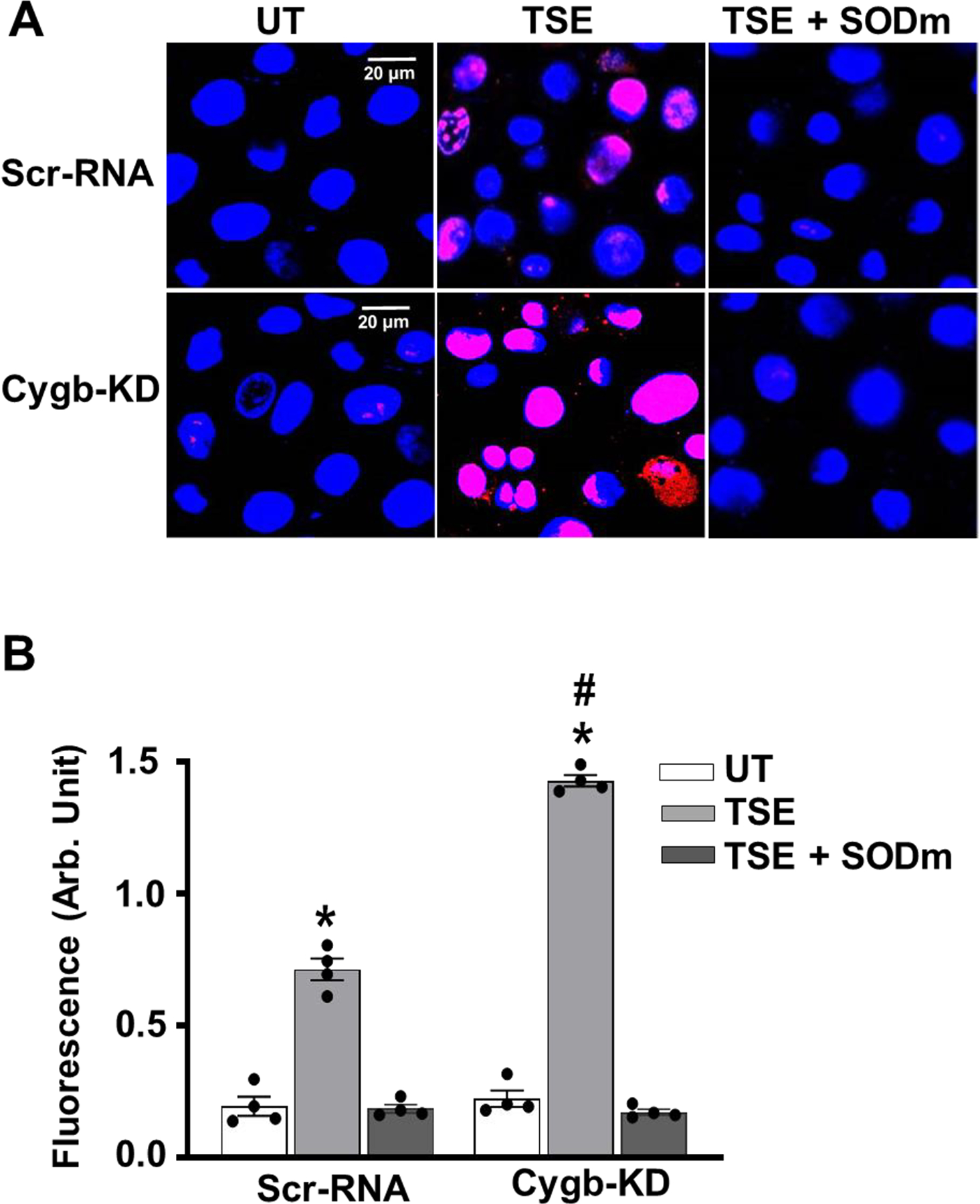
Confluent cells were treated with 1% of TSE for 48 hours, with or without pre-incubation with SODm and then incubated with DHE and DAPI and visualized by confocal fluorescence microscope. A, representative images of VSMCs from untreated (UT) and TSE treated Cygb knock down (Cygb-KD) or control scr-RNA treated VSMCs. B, quantitation of fluorescence intensity in VSMCs from a series of repeat experiments. Data shown are means ± SEM of 4 independent experiments. *: Denotes significant difference from corresponding untreated VSMCs at p < 0.001. #: Denotes significant difference of TSE-treated cells with respect to the corresponding Scr-RNA-treated cells at p < 0.001.
4. Discussion
We have previously shown that Cygb is the major NO metabolizing protein in VSMCs and plays a crucial role in regulating vascular tone and blood pressure through its control of NO metabolism [13; 24]. Cygb is a potent NO dioxygenase exerting oxygen-dependent regulation of NO levels in the vascular wall [13; 21; 24]. While this critical function was identified under normal physiological conditions, the role of Cygb in different settings of vascular pathology remains unclear. Although TCS is a major cardiovascular disease risk factor, its effect on NO metabolism in vascular smooth muscle was not previously characterized. The current study examines how Cygb in VSMCs responds to TSE exposure and provides the first evidence for a critical role of Cygb in the elevated rates of NO decay observed following TSE. We observe that following exposure to tobacco cigarette constituents, over 3-fold up-regulation of Cygb expression occurs with parallel upregulation of its major reducing system, B5/B5R. These findings provide the first identification of the major role of this novel globin in smoking induced smooth muscle cell dysfunction.
Tobacco cigarette smoking is well known to cause potent oxidative stress and secondary inflammation triggering the pathogenesis of CVD [30; 31; 37]. This in turn causes the depletion of NO levels within the vasculature, thereby impairing vasodilatory responses, inducing vascular dysfunction leading to CVD. TCS has been shown to decrease the expression and activation of eNOS, diminishing de novo synthesis of NO [29; 30; 38]. Moreover, TCS-generated ROS depletes BH4, an essential cofactor for eNOS function, resulting in eNOS uncoupling, which further depletes NO by generating O2•− [5; 29]. Given the major role of Cygb in regulating intravascular levels of NO [13; 24], the large increase in Cygb expression in VSMCs following TSE exposure could also serve an important role in the development of TCS-induced vascular dysfunction.
In our studies, we utilized an in vitro model for TSE exposure in VSMCs in which we observed that TSE enhances the cellular rate of NO metabolism. This was accompanied by increases in the expression of both Cygb and its B5/B5R reducing system that were first detected after 24 hours of TSE exposure, with a further increase seen with 48 hours of exposure, to levels ~3.5-fold higher than those in untreated cells. This observation is consistent with a prior report that exposure to side-stream TCS increases Cygb gene and protein expression in rat heart, brain, and lung tissue [39]. Alterations in Cygb expression have also been previously detected in response to different pathological insults. For example, Cygb in neuroblastoma cells was upregulated following exposure to hydrogen peroxide, where it conferred protection against oxidative stress [40]. Moreover, in a vascular injury model, Cygb levels gradually increased exerting protective effects via regulating neointima formation and inhibiting apoptosis during vascular injury [34; 36]. In these pathological models, Cygb exerted an adaptive response to the external insult. While Cygb acts as the main NOD in the vascular wall, the effects of TSE on Cygb and its NO metabolism in VSMCs were not previously characterized.
The Cygb-mediated process of NO dioxygenation occurs with reduction of CygbFe3+ to CygbFe2+ by a cellular reducing system, followed by O2 binding to form CygbFe2+-O2, which rapidly oxidizes NO to nitrate, with its conversion back to CygbFe3+ [21; 25; 41]. Notably, sustained Cygb NOD activity depends on an efficient Cygb reducing system, to recycle ferric-Cygb to the ferrous state [13; 22; 24], and in turn activate O2-dependent NO metabolism within the vascular wall. Although several cellular reducing systems, including ascorbate, cytochrome P450 reductase, and B5/B5R have been proposed to support Cygb reduction, the latter has been recently demonstrated to function as the main Cygb reducing system within the vasculature [22].
In the current study, we observed that the increased expression of Cygb was accompanied by comparable upregulation in the expression of B5 and B5R, indicating their availability to support the higher rates of Cygb reduction and NO dioxygenation. Our data are in line with multiple recent studies that have reported the importance of the B5/B5R reducing system to maintain the NOD function of Cygb, and its role as the major pathway controlling vascular NO metabolism [13; 24; 36]. The B5/B5R system has several roles in cellular physiology as it catalyzes the redox reactions between NADH and a variety of acceptor molecules [42]. In red blood cells, it serves to reduce hemoglobin, maintaining it in the ferrous state allowing it to bind oxygen [42; 43]. Similarly in skeletal and cardiac muscle it maintains the ferrous state of myoglobin [44; 45]. Several studies have reported that B5R is induced by oxidative stress to maintain the antioxidant activity of several endogenous antioxidants, including coenzyme Q, α-tocopherol, and ascorbate [46; 47]. Since TCS is a major inducer of oxidative stress, the marked increase in B5 and B5R levels in TSE treated cells might be triggered by TCS-induced oxidative stress.
With the major increases in the expression of Cygb and its reducing system observed following TSE exposure, one would expect that the rate of cellular NO dioxygenation in the vascular smooth muscle would also be increased. We observed that the rate of NO metabolism by VSMCs was increased in a TSE treatment duration-dependent manner (Fig. 10). Interestingly, we initially observed that with short TSE-exposure periods of 4 hours, administration of an SODm, largely reversed the accelerated NO decay indicating the primary role of O2•− in the initial increase in NO decay rate. However, after 24 or 48 hours of TSE-exposure, the rate of NO decay further increased and this increase in rate was only modestly decreased by SODm treatment. This suggests that another NO metabolizing process increased such as enzyme-mediated NO dioxygenation. Consistent with this, following 4 hours of TSE exposure Cygb expression was not significantly altered, while after 24 hours exposure Cygb expression was clearly increased, with further increase to 3.5-fold of basal levels following 48 hours of exposure. This increase in Cygb expression paralleled the increases observed in NO metabolism rates (Fig. 10). Furthermore, knockdown of Cygb markedly blunted the increase in NO decay rate in response to TSE treatment with ~ 80% reduction, confirming the role of Cygb in mediating the TSE-induced increase in NO consumption by VSMCs. In line with this, we previously showed that down-regulation of Cygb prevented angiotensin II-induced vascular dysfunction and systemic hypertension in mice [24]. Thus, the current study is consistent with prior work demonstrating that Cygb has a pivotal role in regulating intravascular levels of NO [13; 24]. With the major increase in Cygb expression with TSE exposure in VSMCs, increased rates of NO metabolism occur which would be expected to contribute to TSE-induced vascular dysfunction [30; 37].
Figure 10. Exposure time-dependent mechanisms by which tobacco smoke constituents increase NO metabolism rate.
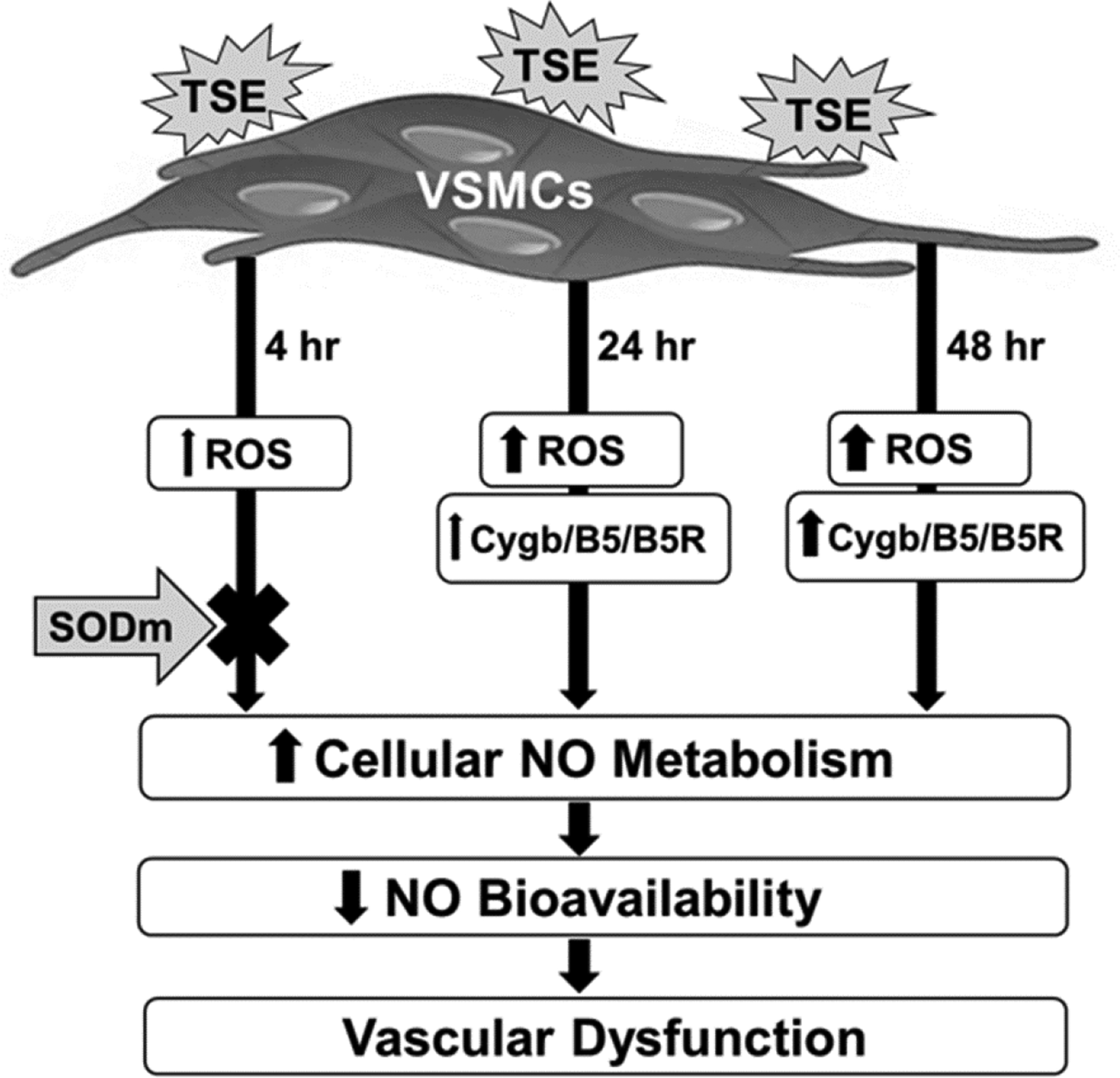
TSE (tobacco smoke extract) stimulates VSMCs (vascular smooth muscle cells) to increase ROS (reactive oxygen species) after 4 hours of exposure. SODm, pre-incubation largely blocks this increased NO metabolism with 4 hours of exposure but not after 24 hours. Longer durations of exposure of 24 or 48 hours progressively increase Cygb and also B5/B5R expression. The increased cellular rate of NO metabolism decreases NO bioavailability leading to vascular dysfunction.
With TSE exposure times of 24 or 48 hours, while increased O2•− generation was observed, the SODm had only a small effect on lowering the NO decay rate. This may be attributed to the large increase in Cygb expression with these exposure times, along with its antioxidant activity that together would intrinsically provide increased ability to metabolize O2•−. It has been recently discovered that Cygb has potent SOD activity [48]. Therefore, its increased expression in VSMCs would enhance O2•− dismutation so that addition of the SODm would provide limited additional benefit. While Cygb would contribute to O2•− degradation in the VSMCs, elevated production of O2•− compared to the control untreated cells would still be observed if there is only partial scavenging due to the relative concentrations of Cygb and the detection probe, as well as the effect of compartmentalized O2•− production at sites were Cygb is not present. As Cygb expression is largely confined to the cytosol [20; 21; 24], while O2•− generation can occur at a variety of membrane and organelle sites, only partial quenching may occur. Consistent with a role of Cygb in partial quenching of O2•−, in Cygb knockdown cells exposed to TSE for 48 hours, higher O2•− production was observed than in matched Scr-RNA treated cells. With Cygb knockdown, SODm treatment had almost a two-fold larger effect on decreasing the rate of NO decay and it abolished the TSE-induced increase. This suggests that in addition to NO dioxygenation by Cygb, increased O2•− generation also contributes to increase the rate of NO decay. Thus, with longer exposure times Cygb-mediated NO dioxygenation is the major process of NO degradation; however, O2•− -mediated NO degradation also occurs and is more prominent with abrogation of Cygb expression.
The upregulation of Cygb expression together with its reducing system, which occurred following TSE exposure, might be a compensatory response to TSE-induced oxidative stress. There is increasing evidence that Cygb has important antioxidant properties [40; 48; 49; 50; 51]. This is in line with results of the current study that, TSE induces exposure duration-dependent oxidative stress reflected by a significant increase in O2•− generation in TSE-exposed VSMCs, confirming previous findings from our group and others [29; 30; 31; 52]. Nevertheless, while Cygb can exert antioxidant function in a variety of cells and tissues, its NOD function in vascular smooth muscle uniquely contributes to vascular dysfunction with loss of NO bioavailability [53; 54]. Thus, in considering therapeutic approaches to reverse tobacco smoking-induced or other CVD, selective targeting inhibition of the NOD function of Cygb may be more beneficial than abrogation of its expression.
Over the last decade there have been major therapeutic efforts aimed at increasing endothelial-derived NO synthesis by enhancing eNOS activity via antioxidants/antioxidant enzymes, cofactor supplements, or direct eNOS activators; however, these failed to show clinical benefit [55; 56]. Increasing the synthesis of NO in face of greatly increased processes of NO degradation may be insufficient to prevent disease. Therefore, in view of the observations of the current work and related prior studies, increased focus should be directed at enhancing NO bioavailability in the vascular smooth muscle through inhibition of the processes of NO decay. Indeed, interventions to modulate Cygb expression and NOD function offer a promising new approach to modulate vascular tone, reverse vascular dysfunction and prevent CVD.
In conclusion, we provide the first demonstration that TSE-treatment triggers a major increase in Cygb-mediated NO metabolism in vascular smooth muscle cells. This was due to upregulation in the expression of Cygb and its B5/B5R reducing system. Knockdown of Cygb reversed this TSE-mediated increase in NO metabolism. Therefore, selective modulation of Cygb expression or its NOD function represents a promising target for treatment of tobacco smoking-induced and other cardiovascular disease.
Highlights.
Cytoglobin controls nitric oxide (NO) metabolism through NO dioxygenation.
Tobacco smoking exposure induces loss of vascular NO; however, the cause was unknown.
Vascular smooth muscle cell exposure first triggered increased superoxide generation.
This was followed by induction of cytoglobin and the B5/B5R reducing system.
Together, this greatly increased the rate of NO decay in vascular smooth muscle cells.
Acknowledgements
This work was supported by National Institutes of Health/National Heart, Lung, and Blood Institute grants R01HL131941 and R01HL135648 to JLZ. EMM was supported in part by a graduate fellowship from the Egyptian Ministery of Education. We thank Dr. Tamer Abdelghany for his assistance with EMM’s initial training at The Ohio State University, and thank Dr. Gouda K. Helal and Dr. Adel R. Abd-Allah, professors at the Department of Pharmacology & Toxicology, Al-Azhar University, Egypt for their role in facilitating EMM’s fellowship.
List of abbreviations:
- TCS
tobacco cigarette smoking
- TSE
tobacco smoke extract
- CVD
cardiovascular disease
- O2•−
superoxide
- NO
nitric oxide
- NOD
nitric oxide dioxygenase
- Cygb
cytoglobin
- sGC
soluble guanylate cyclase
- cGMP
cyclic guanosine monophosphate
- eNOS
endothelial NO synthase
- BH4
tetrahydrobiopterin
- B5/B5R
cytochrome b5a/cytochrome b5 reductase isoform 3
- VSMCs
vascular smooth muscle cells
- TSE
tobacco cigarette smoking extract
- PBS
phosphate buffered saline
- XTT
2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxanilide
- PMS
phenazine methosulfate
- SODm
superoxide dismutase mimetic
- MnTBAP
Mn(III)tetrakis(4-benzoic acid)porphyrin
- SEM
standard error of the mean
- ANOVA
Analysis of variance
- Scr-RNA
scrambled siRNA
- TBST
Tris buffered saline-tween 20
- HRP
horseradish peroxidase
- DHE
dihydroethidium
- DAPI
4′,6-diamidino-2-phenylindole dihydrochloride
- UT
TSE-untreated
- CT
control
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Bhatnagar A, Environmental cardiology: studying mechanistic links between pollution and heart disease, Circ Res 99 (2006) 692–705. [DOI] [PubMed] [Google Scholar]
- [2].Talukder MH, Johnson WM, Varadharaj S, Lian J, Kearns PN, El-Mahdy MA, Liu X, Zweier JL, Chronic cigarette smoking causes hypertension, increased oxidative stress, impaired NO bioavailability, endothelial dysfunction, and cardiac remodeling in mice, American Journal of Physiology-Heart and Circulatory Physiology 300 (2011) H388–H396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Puranik R, Celermajer DS, Smoking and endothelial function, Progress in cardiovascular diseases 45 (2003) 443–458. [DOI] [PubMed] [Google Scholar]
- [4].Flammer AJ, Anderson T, Celermajer DS, Creager MA, Deanfield J, Ganz P, Hamburg NM, Lüscher TF, Shechter M, Taddei S, Vita JA, Lerman A, The assessment of endothelial function: from research into clinical practice, Circulation 126 (2012) 753–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Golbidi S, Edvinsson L, Laher I, Smoking and endothelial dysfunction, Current vascular pharmacology 18 (2020) 1–11. [DOI] [PubMed] [Google Scholar]
- [6].Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G, Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide, Proceedings of the National Academy of Sciences 84 (1987) 9265–9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tejero J, Shiva S, Gladwin MT, Sources of vascular nitric oxide and reactive oxygen species and their regulation, Physiological reviews 99 (2019) 311–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Arnold WP, Mittal CK, Katsuki S, Murad F, Nitric oxide activates guanylate cyclase and increases guanosine 3′: 5′-cyclic monophosphate levels in various tissue preparations, Proceedings of the National Academy of Sciences 74 (1977) 3203–3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Karow DS, Pan D, Davis JH, Behrends S, Mathies RA, Marletta MA, Characterization of functional heme domains from soluble guanylate cyclase, Biochemistry 44 (2005) 16266–16274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Moncada S, Nitric oxide: physiology, pathophysiology and pharmacology, Pharmacol rev 43 (1991) 109–142. [PubMed] [Google Scholar]
- [11].Moncada S, Radomski MW, Palmer RM, Endothelium-derived relaxing factor: identification as nitric oxide and role in the control of vascular tone and platelet function, Biochemical pharmacology 37 (1988) 2495–2501. [DOI] [PubMed] [Google Scholar]
- [12].Delledonne M, Polverari A, Murgia I, The functions of nitric oxide-mediated signaling and changes in gene expression during the hypersensitive response, Antioxidants and Redox Signaling 5 (2003) 33–41. [DOI] [PubMed] [Google Scholar]
- [13].Zweier JL, Ilangovan G, Regulation of Nitric Oxide Metabolism and Vascular Tone by Cytoglobin, Antioxidants & Redox Signaling (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tousoulis D, Kampoli A-M, Tentolouris Nikolaos Papageorgiou C, Stefanadis C, The role of nitric oxide on endothelial function, Current vascular pharmacology 10 (2012) 4–18. [DOI] [PubMed] [Google Scholar]
- [15].Alderton WK, Cooper CE, Knowles RG, Nitric oxide synthases: structure, function and inhibition, Biochemical journal 357 (2001) 593–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Stuehr DJ, Mammalian nitric oxide synthases, Biochimica et Biophysica Acta (BBA)-Bioenergetics 1411 (1999) 217–230. [DOI] [PubMed] [Google Scholar]
- [17].Liu X, Cheng C, Zorko N, Cronin S, Chen YR, Zweier JL, Biphasic modulation of vascular nitric oxide catabolism by oxygen, Am J Physiol Heart Circ Physiol 287 (2004) H2421–6. [DOI] [PubMed] [Google Scholar]
- [18].Liu X, Srinivasan P, Collard E, Grajdeanu P, Lok K, Boyle SE, Friedman A, Zweier JL, Oxygen regulates the effective diffusion distance of nitric oxide in the aortic wall, Free Radic Biol Med 48 (2010) 554–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kawada N, Kristensen DB, Asahina K, Nakatani K, Minamiyama Y, Seki S, Yoshizato K, Characterization of a stellate cell activation-associated protein (STAP) with peroxidase activity found in rat hepatic stellate cells, Journal of Biological Chemistry 276 (2001) 25318–25323. [DOI] [PubMed] [Google Scholar]
- [20].Halligan KE, Jourd’heuil FL, Jourd’heuil D, Cytoglobin is expressed in the vasculature and regulates cell respiration and proliferation via nitric oxide dioxygenation, J Biol Chem 284 (2009) 8539–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu X, Follmer D, Zweier JR, Huang X, Hemann C, Liu K, Druhan LJ, Zweier JL, Characterization of the function of cytoglobin as an oxygen-dependent regulator of nitric oxide concentration, Biochemistry 51 (2012) 5072–5082. [DOI] [PubMed] [Google Scholar]
- [22].Ilangovan G, Khaleel SA, Kundu T, Hemann C, El-Mahdy MA, Zweier JL, Defining the reducing system of the NO dioxygenase cytoglobin in vascular smooth muscle cells and its critical role in regulating cellular NO decay, Journal of Biological Chemistry 296 (2021) 100196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Trent JT 3rd, Hargrove MS, A ubiquitously expressed human hexacoordinate hemoglobin, J Biol Chem 277 (2002) 19538–45. [DOI] [PubMed] [Google Scholar]
- [24].Liu X, El-Mahdy MA, Boslett J, Varadharaj S, Hemann C, Abdelghany TM, Ismail RS, Little SC, Zhou D, Thuy LT, Kawada N, Zweier JL, Cytoglobin regulates blood pressure and vascular tone through nitric oxide metabolism in the vascular wall, Nat Commun 8 (2017) 14807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu X, Tong J, Zweier JR, Follmer D, Hemann C, Ismail RS, Zweier JL, Differences in oxygen-dependent nitric oxide metabolism by cytoglobin and myoglobin account for their differing functional roles, The FEBS journal 280 (2013) 3621–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gardner AM, Cook MR, Gardner PR, Nitric-oxide dioxygenase function of human cytoglobin with cellular reductants and in rat hepatocytes, J Biol Chem 285 (2010) 23850–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jusman SWA, Sari DH, Ningsih SS, Hardiany NS, Sadikin M, Role of Hypoxia Inducible Factor-1 Alpha (HIF-1α) in Cytoglobin Expression and Fibroblast Proliferation of Keloids, Kobe J Med Sci 65 (2019) E10–e18. [PMC free article] [PubMed] [Google Scholar]
- [28].Shaw RJ, Omar MM, Rokadiya S, Kogera FA, Lowe D, Hall GL, Woolgar JA, Homer J, Liloglou T, Field JK, Risk JM, Cytoglobin is upregulated by tumour hypoxia and silenced by promoter hypermethylation in head and neck cancer, Br J Cancer 101 (2009) 139–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Abdelghany TM, Ismail RS, Mansoor FA, Zweier JR, Lowe F, Zweier JL, Cigarette smoke constituents cause endothelial nitric oxide synthase dysfunction and uncoupling due to depletion of tetrahydrobiopterin with degradation of GTP cyclohydrolase, Nitric Oxide 76 (2018) 113–121. [DOI] [PubMed] [Google Scholar]
- [30].El-Mahdy MA, Abdelghany TM, Hemann C, Ewees MG, Mahgoup EM, Eid MS, Shalaan MT, Alzarie YA, Zweier JL, Chronic cigarette smoke exposure triggers a vicious cycle of leukocyte and endothelial-mediated oxidant stress that results in vascular dysfunction, Am J Physiol Heart Circ Physiol 319 (2020) H51h65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].El-Mahdy MA, Mahgoup EM, Ewees MG, Eid MS, Abdelghany TM, Zweier JL, Long-Term Electronic Cigarette Exposure Induces Cardiovascular Dysfunction Similar to Tobacco Cigarettes: Role of Nicotine and Exposure Duration, Am J Physiol Heart Circ Physiol (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Liu X, Zweier JL, Application of electrode methods in studies of nitric oxide metabolism and diffusion kinetics, Journal of Electroanalytical Chemistry 688 (2013) 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Liu X, Liu Q, Gupta E, Zorko N, Brownlee E, Zweier JL, Quantitative measurements of NO reaction kinetics with a Clark-type electrode, Nitric Oxide 13 (2005) 68–77. [DOI] [PubMed] [Google Scholar]
- [34].Jourd’heuil FL, Xu H, Reilly T, McKellar K, El Alaoui C, Steppich J, Liu YF, Zhao W, Ginnan R, Conti D, The hemoglobin homolog cytoglobin in smooth muscle inhibits apoptosis and regulates vascular remodeling, Arteriosclerosis, thrombosis, and vascular biology 37 (2017) 1944–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Benov L, Sztejnberg L, Fridovich I, Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical, Free Radical Biology and Medicine 25 (1998) 826–831. [DOI] [PubMed] [Google Scholar]
- [36].Amdahl MB, Sparacino-Watkins CE, Corti P, Gladwin MT, Tejero J, Efficient Reduction of Vertebrate Cytoglobins by the Cytochrome b 5/Cytochrome b 5 Reductase/NADH System, Biochemistry 56 (2017) 3993–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Dikalov S, Itani H, Richmond B, Arslanbaeva L, Vergeade A, Rahman SJ, Boutaud O, Blackwell T, Massion PP, Harrison DG, Tobacco smoking induces cardiovascular mitochondrial oxidative stress, promotes endothelial dysfunction, and enhances hypertension, American Journal of Physiology-Heart and Circulatory Physiology 316 (2019) H639–H646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].DiGiacomo SI, Jazayeri M-A, Barua RS, Ambrose JA, Environmental tobacco smoke and cardiovascular disease, International journal of environmental research and public health 16 (2019) 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tae B, Oliveira KC, Conceicao RR, Valenti VE, de Souza JS, Laureano-Melo R, Sato MA, Maciel RM, Giannocco G, Evaluation of globins expression in brain, heart, and lung in rats exposed to side stream cigarette smoke, Environ Toxicol 32 (2017) 1252–1261. [DOI] [PubMed] [Google Scholar]
- [40].Li D, Chen XQ, Li WJ, Yang YH, Wang JZ, Yu AC, Cytoglobin up-regulated by hydrogen peroxide plays a protective role in oxidative stress, Neurochem Res 32 (2007) 1375–80. [DOI] [PubMed] [Google Scholar]
- [41].Gödecke A, On the impact of NO–globin interactions in the cardiovascular system, Cardiovascular research 69 (2006) 309–317. [DOI] [PubMed] [Google Scholar]
- [42].Amdahl MB, Petersen EE, Bocian K, Kaliszuk SJ, DeMartino AW, Tiwari S, Sparacino-Watkins CE, Corti P, Rose JJ, Gladwin MT, The zebrafish cytochrome b 5/cytochrome b 5 reductase/NADH system efficiently reduces cytoglobins 1 and 2: conserved activity of cytochrome b 5/cytochrome b 5 reductases during vertebrate evolution, Biochemistry 58 (2019) 3212–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Saleh M, McConkey S, NADH-dependent cytochrome b5 reductase and NADPH methemoglobin reductase activity in the erythrocytes of Oncorhynchus mykiss, Fish physiology and biochemistry 38 (2012) 1807–1813. [DOI] [PubMed] [Google Scholar]
- [44].Carew NT, Altmann HM, Galley JC, Hahn S, Miller MP, Shiva S, McNamara D, Straub AC, Cytochrome b5 Reductase 3 Regulates Myoglobin Redox State and Controls Cardiac Function, Circulation Research 123 (2018) A103–A103. [Google Scholar]
- [45].Arihara K, Cassens RG, Greaser ML, Luchansky JB, Mozdziak PE, Localization of metmyoglobin-reducing enzyme (NADH-cytochrome b5 reductase) system components in bovine skeletal muscle, Meat Science 39 (1995) 205–213. [DOI] [PubMed] [Google Scholar]
- [46].Siendones E, SantaCruz-Calvo S, Martín-Montalvo A, Cascajo MV, Ariza J, López-Lluch G, Villalba JM, Acquaviva-Bourdain C, Roze E, Bernier M, de Cabo R, Navas P, Membrane-bound CYB5R3 is a common effector of nutritional and oxidative stress response through FOXO3a and Nrf2, Antioxid Redox Signal 21 (2014) 1708–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Fan J, Du W, Kim-Muller JY, Son J, Kuo T, Larrea D, Garcia C, Kitamoto T, Kraakman MJ, Owusu-Ansah E, Cyb5r3 links FoxO1-dependent mitochondrial dysfunction with β-cell failure, Molecular metabolism 34 (2020) 97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zweier JL, Hemann C, Kundu T, Ewees MG, Khaleel SA, Samouilov A, Ilangovan G, El-Mahdy MA, Cytoglobin Has Potent Superoxide Dismutase Function, PNAS (In press) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Le Thi Thanh Thuy HH, Kawada N, Role of cytoglobin, a novel radical scavenger, in stellate cell activation and hepatic fibrosis, Clinical and Molecular Hepatology 26 (2020) 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Fang J, Ma I, Allalunis-Turner J, Knockdown of cytoglobin expression sensitizes human glioma cells to radiation and oxidative stress, Radiation research 176 (2011) 198–207. [DOI] [PubMed] [Google Scholar]
- [51].Mathai C, Jourd’heuil FL, Lopez-Soler RI, Jourd’heuil D, Emerging perspectives on cytoglobin, beyond NO dioxygenase and peroxidase, Redox Biol 32 (2020) 101468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Messner B, Bernhard D, Smoking and cardiovascular disease: mechanisms of endothelial dysfunction and early atherogenesis, Arteriosclerosis, thrombosis, and vascular biology 34 (2014) 509–515. [DOI] [PubMed] [Google Scholar]
- [53].Romero-Puertas MC, Sandalio LM, Nitric oxide level is self-regulating and also regulates its ROS partners, Frontiers in plant science 7 (2016) 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Di Meo S, Reed TT, Venditti P, Victor VM, Role of ROS and RNS sources in physiological and pathological conditions, Oxidative medicine and cellular longevity 2016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Pechanova O, Simko F, Chronic antioxidant therapy fails to ameliorate hypertension: potential mechanisms behind, Journal of Hypertension 27 (2009) S32–S36. [DOI] [PubMed] [Google Scholar]
- [56].Davies AM, Holt AG, Why antioxidant therapies have failed in clinical trials, Journal of Theoretical Biology 457 (2018) 1–5. [DOI] [PubMed] [Google Scholar]