

Abstract
Purpose:
Cancer immunotherapy has markedly improved the prognosis of patients with a broad variety of malignancies. However, benefits are weighed against unique toxicities, with immune-related adverse events (irAEs) that are frequent and potentially life-threatening. The diagnosis and management of these events are challenging due to heterogeneity of timing onset, multiplicity of affected organs, and lack of non-invasive monitoring techniques. We demonstrate the use of a granzyme B targeted PET imaging agent (GZP) for irAE identification in a murine model.
Experimental Design:
We generated a model of immunotherapy-induced adverse events in Foxp3-DTR-GFP mice bearing MC38 tumors. GZP PET imaging was performed to evaluate organs noninvasively. We validated imaging with ex vivo analysis, correlating the establishment of these events with the presence of immune infiltrates and granzyme B upregulation in tissue. To demonstrate the clinical relevance of our findings, the presence of granzyme B was identified through immunofluorescence staining in tissue samples of patients with confirmed checkpoint-inhibitor associated adverse events.
Results:
GZP PET imaging revealed differential uptake in organs affected by irAEs, such as colon, spleen and kidney, that significantly diminished after administration of the immunosuppressor dexamethasone. The presence of GZB and immune infiltrates were confirmed histologically and correlated with significantly higher uptake in PET imaging. The presence of GZB was also confirmed in samples from patients that presented with clinical irAEs.
Conclusion:
We demonstrate an interconnection between the establishment of immune-related adverse events and granzyme B presence and, for the first time, the visualization of those events through PET imaging.
Keywords: Granzyme B, Granzyme B Peptide, Adverse Events, Immunotherapy, PET Imaging
INTRODUCTION
Significant advances in understanding the role that immune-checkpoints play in downregulating the immune response have led to a revolution in the field of immuno-oncology. The development of immune-checkpoint inhibitors that activate cytotoxic T-cells have demonstrated strikingly positive clinical outcomes across multiple tumor types (1,2). With the growing clinical indications for immunotherapy agents, the number of cancer patients eligible to receive checkpoint inhibitor drugs went from 1.54% in 2011 to nearly 44% in 2018 (3). Since there are more than 1,700,000 new cases of cancer diagnosed per year (4), nearly 760,000 new patients would be eligible to receive immunotherapy in the United States alone. As immune-checkpoints are partly responsible for immune homeostasis, disturbing these pathways can contribute to the loss of immunological self-tolerance in patients (5). These therapy-induced immune-related adverse events (irAEs) have hindered the safe administration of checkpoint inhibitors. irAEs are frequent, with astonishing occurrence that varies between 15% and up to 91% of patients with combination treatment (6,7). irAEs may present within a few days of immunotherapy initiation and up to a year after completion of therapy (8). irAEs can occur with different severity levels in any organ system with dermatitis, colitis, hepatitis, pneumonitis and nephritis being the most common (7,9). irAEs may also be severe and life-threatening, often requiring prompt patient management with appropriate therapeutic decisions (10). In fact, approximately a third of patients will need to eventually stop immunotherapy treatment due to irAEs onset (11–14), and patients may also need to initiate steroids or other immunomodulatory drugs such as infliximab.
Even though the underlying mechanisms of irAEs remain unknown, they have been linked to overactivation of T cells resulting in healthy tissue damage (15,16). It is known that cytotoxic CD8+ T lymphocytes are directly responsible for killing tumor cells upon recognition by releasing granzyme B and perforin (17–19). In fact, these lymphocytes are the main effector cells in clinically approved immunotherapy drugs, as well as many under development (20–22). The link between anti-cancer effects and adverse events is more evident with reports that patients who presented with irAEs also presented improved outcomes such as higher response rates and overall survival (7).
Early evaluation and diagnosis are critical to achieve safe resolution of irAE. However, the diagnosis of irAE can be challenging, and currently relies primarily on invasive biopsies with their attendant risks (23). Furthermore, treatment of severe irAEs not only involves the discontinuation of therapy, but also treatment with immunomodulators that not only treat the irAE but can also stop the anti-tumoral immune response. Considering the fact that the time course for developing irAEs is variable, can occur after one dose or after several months of therapy and can affect multiple organs (8), a non-invasive approach of detecting and monitoring irAEs is urgently needed. Our group has developed a PET imaging tracer that specifically targets the active extracellular form of Granzyme B and has previously demonstrated successful use of granzyme B PET imaging as a biomarker for predicting tumoral immunotherapy response (24–26). Herein, we hypothesized that granzyme B release could potentially be harnessed as a biomarker to allow non-invasive detection of irAEs. In this study, we demonstrate the use of a granzyme B targeted peptide (GZP), radiolabeled with a PET isotope (68Ga), as an agent for irAE identification in a mouse model of irAE. Furthermore, we validated the usefulness of granzyme B for the assessment of irAEs by staining tissue specimens of human subjects with pathology confirmed checkpoint-inhibitor associated irAEs. To the best of our knowledge, this is the first study reporting non-invasive imaging of irAEs.
MATERIALS AND METHODS
Cell Culture and Tumor Implantation
All experimental procedures and animal studies were approved by the Institutional Animal Care and Use Committee (IACUC). Murine MC38 cell line derived from C57BL6 murine colon adenocarcinoma cells were obtained from Kerafast (Massachusetts, USA). Cells were grown using DMEM medium supplemented with 10% FBS at 37 °C and 5% CO2. Mycoplasma testing was carried out by PCR screening on a monthly basis and cells were discarded after 15 passages. All cell-based experiments were done with cells acquired within 6 months in order to ensure fidelity of the cell line identity. Allograft tumors were implanted with the subcutaneous injection of MC38 cells (1 × 106) in a 1:1 (v:v) ratio in Matrigel (Fisher Scientific,) into the right shoulder of animals.
Adverse Events Murine Model Generation in Tumor-bearing mice
We addressed the challenge that mice treated with ICI usually do not developed irAEs by utilizing a previously reported mouse model in which clinical and pathologic equivalents of irAEs could be generated in animals by combining Treg depletion, which lowers self-tolerance, with checkpoint inhibitor therapy. In the Foxp3-GFP-DTR animal model, transient Treg depletion induced in these mice by administration of diphtheria toxin, followed by administration of monoclonal antibody targeting immunomodulatory CD137 receptor, induced severe irAEs (27).
All animal studies were conducted using 8- to 12-week-old male B6.129(Cg)-Foxp3tm3(DTR/GFP)Ayr (Foxp3-DTR-GFP) mice, purchased from the Jackson Laboratory (Maine, USA). Male mice were used to ensure uniform knockout of the X-linked Foxp3 allele. Mice were housed and maintained by the Center for Comparative Medicine at Massachusetts General Hospital. To induce Treg depletion in vivo, tumor-bearing animals were injected intraperitoneally with 250 ng of diphtheria toxin (DT) (Sigma-Aldrich, MO, USA) diluted in PBS. This procedure was carried out three times, three days apart from each injection, that is, on days 3, 6 and 9 following MC38 tumor inoculation. The animals where then separated into 4 groups (n=5). Each group received intraperitoneally three doses at three days apart (days 12, 15 and 19) of either PBS (DT only group), both 200 μg of anti-PD-1(clone RMP1–14) (Bioxcell, NH, USA) and 100 μg of anti-CTLA-4 (clone 9D9) (Bioxcell, NH, USA), 250 μg of anti-CD137 (clone 3H3) (Bioxcell, NH, USA). An additional group of five mice was treated with intraperitoneal injection of 250 μg of anti-CD137 (clone 3H3) followed by five daily doses of dexamethasone (intraperitoneally; 5mg/kg dissolved in saline) (Sigma-Aldrich, MO, USA). A control group of animals that did not receive DT and only received PBS was also used. The humane end point criterion was selected as tumor diameter > than 20mm (Mean = (d+D)/2; where d and D are the shortest and longest diameter in mm), a body weight decrease of more than 20%, or if general health was deemed to be too poor to continue. Animals were monitored every other day and measurements of tumor volume and body weight were collected.
Radiolabeling, PET Imaging and Biodistribution Studies
68Ga was eluted from a 68Ge/68Ga generator (Eckert & Ziegler, Germany) with 0.1M HCl. Radiolabeling of NOTA-GZP was performed as previously described (25). In brief, 100 μg of NOTA-GZP (in 100 μL of PBS) was mixed with 2M HEPES buffer until pH was 3.5–4.0. Then, approximately 370 MBq of eluted 68Ga was added to the mixture and the reaction proceeded for 10 min at room temperature. The reaction product was purified using a reverse-phase C18 Sep-Pak mini cartridge, eluted with 70% ethanol and diluted with saline prior to administration. Radiolabeling yield was calculated through instant thin-layer chromatography (iTLC) using two solvent systems as reported elsewhere (28,29).
PET imaging was performed 1 h post-injection (p.i.) of approximately 7.4 MBq of 68Ga-NOTA-GZP via tail-vein injection. Tail-vein injection was performed on awake mice. For imaging, mice were anesthetized (isoflurane inhalation) and PET/CT Images were acquired on a rodent Triumph PET/CT (Trident Healthcare, SC, USA). PET acquisition was performed in static mode for 15 minutes with a single bed position, followed by CT acquisition. Images were reconstructed using 3D-MLEM algorithm (4 iterations and 20 subsets) and corrected for scatter and randoms. Uptake values, presented as percent injected dose per gram (%ID/g) for each organ was calculated in a 3D region of interest manually drawn around the tumor using CT images. Images were postprocessed using VivoQuant software (InviCRO, MA, USA). After the CT scan, the animals were euthanized, and major organs were harvested and decay-corrected retained activity per organ weight was measured using a gamma counter detector (Wizard2, Perkin Elmer, MA, USA). In order to validate the specificity of our tracer, we have also performed ex vivo biodistribution studies with a non-specific peptide (68Ga-NSP) in animals with the generated irAEs model (DT + anti-CD137). In addition, we have performed PET imaging after administration of the specific (68Ga-GZP) and non-specific (68Ga-NSP) peptides in a local inflammation animal model. To induce local inflammation, a subcutaneous injection of lipopolysaccharide (LPS) (100μL of 1mg/mL LPS with 100μL Matrigel) was administered into the right posterior shoulder of each mouse and the same volume of PBS into the contralateral shoulder as a control. Radiotracers were injected 24 hours post administration of LPS and PET Imaging was performed at 1 hour post injection time.
Ex Vivo Analysis of Mouse Tissue
Flow Cytometry.
A separate cohort of animals paralleling the different groups above were used for flow cytometric analysis. Spleen, liver, kidney and lungs were excised, minced and strained through a 70 μm nylon mesh and incubated with RPMI with 1 mg/mL collagenase type IV for 35 min at 37 °C for the generation of single cell suspension. Cells were then stained with live/dead fixable violet dead cell stain kit (ThermoFisher, MA, USA) followed by staining against murine anti-CD3 (clone17A2; ThermoFisher, MA, USA), anti-CD4 (clone GK1.5; ThermoFisher, MA, USA), anti-CD8 (clone 53-6.7; ThermoFisher, MA, USA) and anti-CD107a (clone HA3; ThermoFisher, MA, USA) according to the manufacturer’s specifications. Flow cytometry was performed on a cytometry cell analyzer BD LSR II and gating established using FlowJo software (version 8.7).
Immunofluorescence and Histological Staining.
Ex vivo tissue staining was performed on animals from the different groups following Treg depletion and therapeutic protocols. Three days after the last therapeutic doses were given, animals were euthanized and liver, spleen, colon, kidney, lungs and skin were harvested, fixed in 10% neutral buffered formalin overnight and submitted to the Specialized Histopathology Services of the MGH Pathology Core for processing, sectioning and H&E staining. Prior to immunofluorescence staining against granzyme B, tissue was deparaffinized and rehydrated. Antigen retrieval was carried out using standard heat-based antigen retrieval techniques (30). Blocking was carried out for 1 h at room temperature in PBS containing 5% goat serum. Primary antibody incubation was performed overnight at 4 °C with the addition of rabbit anti-mouse granzyme B antibody (ab255598; Abcam, UK). On the following day, stained slides were washed and incubated with AlexaFluor 647 conjugated goat anti-rabbit IgG secondary antibody (A32733; ThermoFisher, MA, USA) at room temperature in a moist dark chamber for 1 h. A coverslip was applied to each slide using Vectashield mounting medium for fluorescence microscopy with DAPI (4′, 6-diamidino-2-phenylindole). Semi-quantitative data of immunofluorescence was carried out by measuring total fluorescence in the red channel (granzyme B) divided by total fluorescence in the blue channel (DAPI). All images were acquired using Biotek Cytation 5 Cell Imaging Multi-Mode Reader and analyzed through Biotek Gen5 software.
Analysis of Human Patient Tissue Samples
Samples were provided by the Severe Immunotherapy Complications Service of Massachusetts General Hospital Cancer Center. All specimens were acquired from patients under an institutional review board-approved clinical protocol. Colon samples were obtained from patients undergoing checkpoint inhibitor treatment with or without immune-related colitis. The samples of patients that did not developed colitis were assigned as control. Kidney samples were obtained from patients that presented with checkpoint-inhibitor associated nephritis. Healthy kidney samples were used as control. Immunofluorescence staining against granzyme B was performed on either formalin-fixed samples (colon samples) as described above or on frozen samples (kidney samples). For these experiments, rabbit anti-human granzyme B antibody (ab243879; Abcam, UK) was used as primary antibody. Frozen tissue slices were fixed with 4% paraformaldehyde for 10 min, rinsed with PBS, and blocked with 5% goat serum before addition of primary and secondary antibodies. Rabbit anti-human granzyme B (ab243879; Abcam) primary antibody was incubated overnight at 4 °C, then Alexa Fluor Plus 647 conjugated goat anti-rabbit IgG (A32733; ThermoFisher) secondary antibody was added. Cell nuclei were stained with DAPI. Fluorescence images were acquired with Cytation 5 Cell Imaging Multi-Mode Reader equipped with three excitation lasers (488 nm, 546 nm, and 633 nm).
Statistical Analysis
All quantitative data were analyzed using GraphPad Prism (version 8.4.2) and are presented as the mean ± standard deviation. Comparisons between groups were made using two-way ANOVA or unpaired t test, where p < 0.05 was considered statistically significant.
RESULTS
Granzyme B PET Imaging allows detection of immune-related adverse events
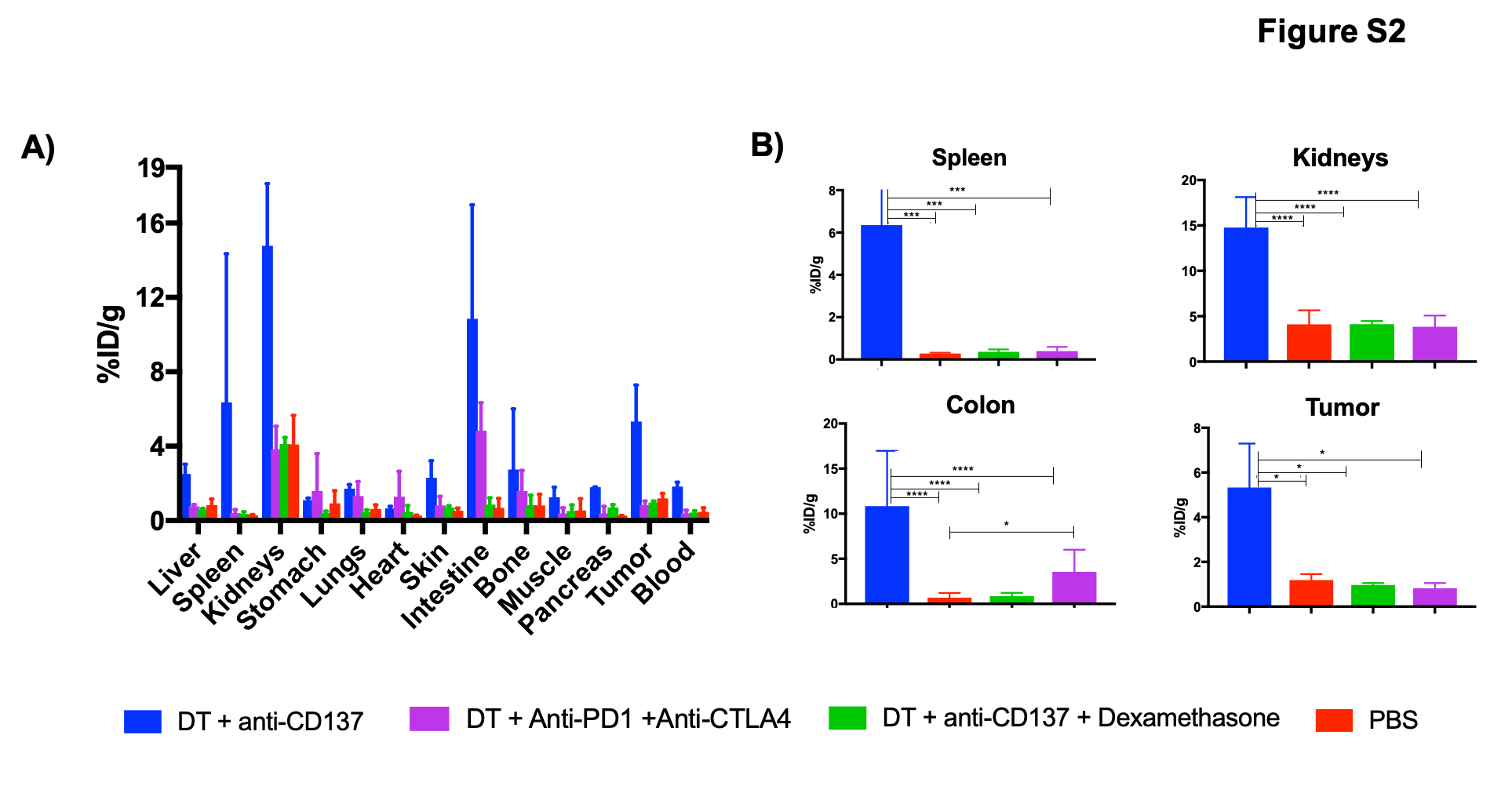
Schematics of animal studies workflow can be found in Figure S1 and Table S1. The peptide was radiolabeled with 68Ga with a radiolabeling yield of 74.2 ± 3.6 % and radiochemical purity of > 97% (data not shown), and an average calculated specific activity of 5180 ± 155 MBq/mg. PET/CT imaging of PBS and DT treatment control groups demonstrated low uptake in normal organs with tracer in the bladder reflecting its renal excretion; this is in line with our previous studies of the same probe in murine models (24). In contrast, in mice that received check point inhibitors, there was high uptake in the abdominal area localizing to liver, kidney, spleen and colon (Figure 1A). The group injected with DT + anti-CD137 had the highest uptake values for all organs when compared to all other groups, with percent injected dose per gram (%ID/g) values of 4.63 ± 0.77, 2.86 ± 1.69, 2.25 ± 1.33 and 1.58 ± 1.00 for colon, kidney, spleen and liver, respectively. Those values were statistically higher than the PBS injected group for spleen (p = 0.0034), colon (p < 0.0001), kidney (p = 0.0471) and tumor (p = 0.0063) and statistically higher than DT only injected group for spleen (p = 0.0140), colon (p < 0.0001), kidney (p = 0.0133) and tumor (p = 0.0322). %ID/g values for the group of animals injected with DT + anti-PD-1 + anti-CTLA-4 were 2.78 ± 1.98, 1.48 ± 1.03 and 1.17 ± 0.27 for colon, kidney and spleen, respectively. Colon was the organ with the highest %ID/g for animals injected with DT + anti-PD-1 + anti-CTLA-4, which was statistically higher than the colon uptake of animals injected with PBS (p = 0.0062) and DT only (p = 0.0018). Of note, the tumor in the group injected with DT + anti-CD137 showed markedly increased uptake with values of 2.14 ± 0.29, six times higher than the tumor uptake of PBS injected group, which likely indicates response to immunotherapy (25). Ex vivo biodistribution data, shown in Figure S2, were in agreement with findings from PET and showed that for the animals that received DT + anti-CD137, multiple organs including kidney, spleen, colon and tumor had significantly higher uptakes (p< 0.005) than the same organs in PBS-only injected mice. For the groups that received DT + anti-PD-1 + anti-CTLA-4, significantly (p = 0.0395) higher uptake in the colon was found when compared to PBS only treated group. The organ with the highest uptake among the groups that received immunotherapy was found to be the colon with values of 10.8 ± 6.1 and 4.8 ± 1.5 %ID/g for DT + anti-CD137 and DT + anti-PD-1 + anti-CTLA-4 groups, respectively; this was significantly higher (p < 0.05) than 0.7 ± 0.5 %ID/g found for the PBS group. Of note, uptake values for blood pool in PET images and biodistribution studies were not statistically different for any of the groups investigated. Secondary analysis of organ-to-blood pool ratios from ex vivo biodistribution data (Figure S3) was performed. Although the same trends were observed as Figure S2, kidney-to-blood ratios of animals that received anti-PD-1 + anti-CTLA-4 were higher than that of anti-CD137 group, suggesting the effects in the kidney might be more prominent in animals that received combinational therapy and further investigation is necessary.
Figure 1. In vivo PET/CT imaging of mice from different groups injected with 68Ga-NOTA-GZP reveal higher uptake in the abdominal area for animals injected with DT + anti-CD137 and DT + anti-PD-1 + anti-CTLA-4.
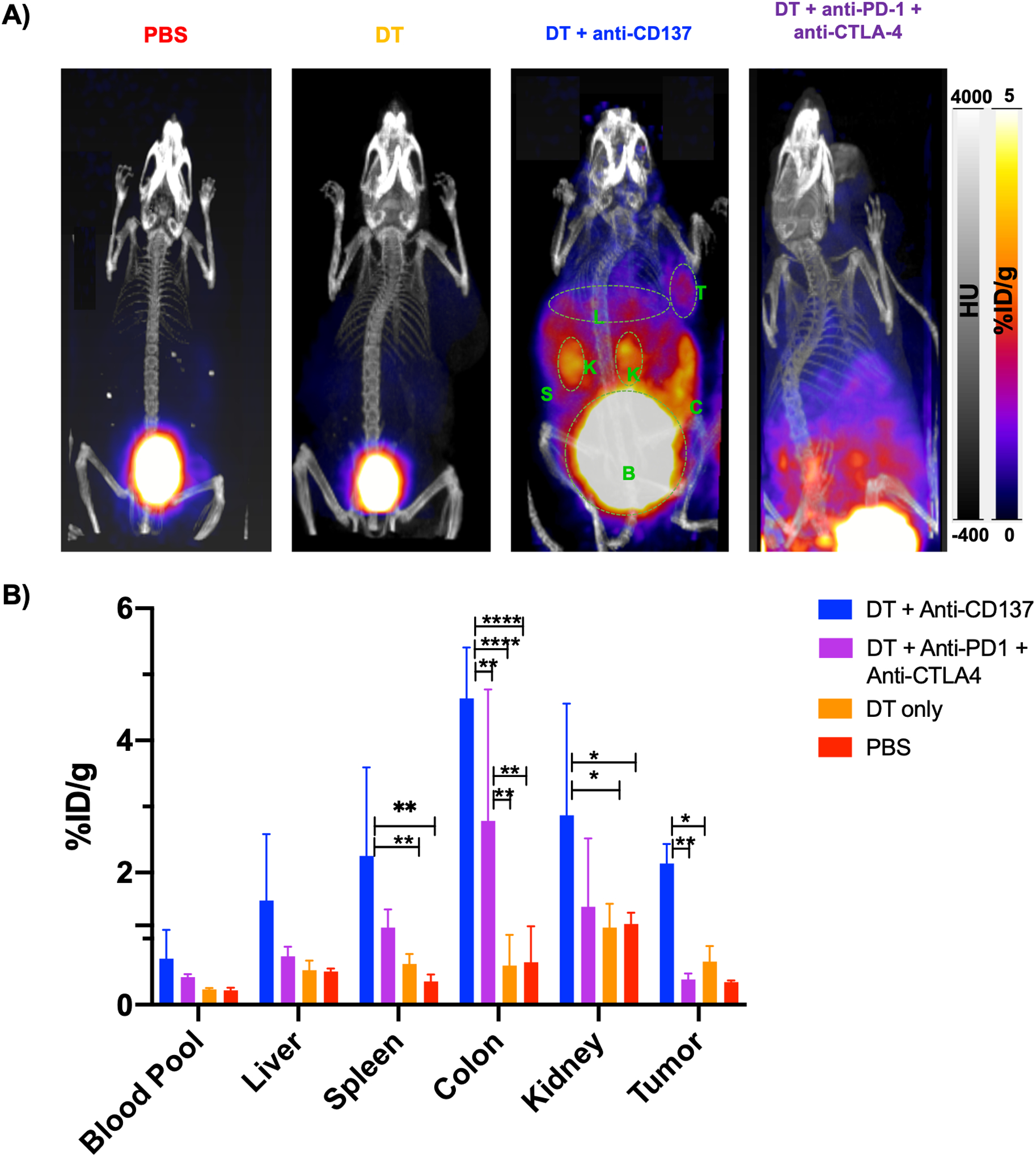
A) Representative PET/CT MIP images at 1 hour post-injection. Major organs are highlighted in green (K=Kidneys, B=Bladder, S=Spleen, T=Tumor, C=colon). B) Uptake values of blood pool and major organs, represented as %ID/g. Statistically significant *p<0.05, ** p<0.007, **** p<0.0001
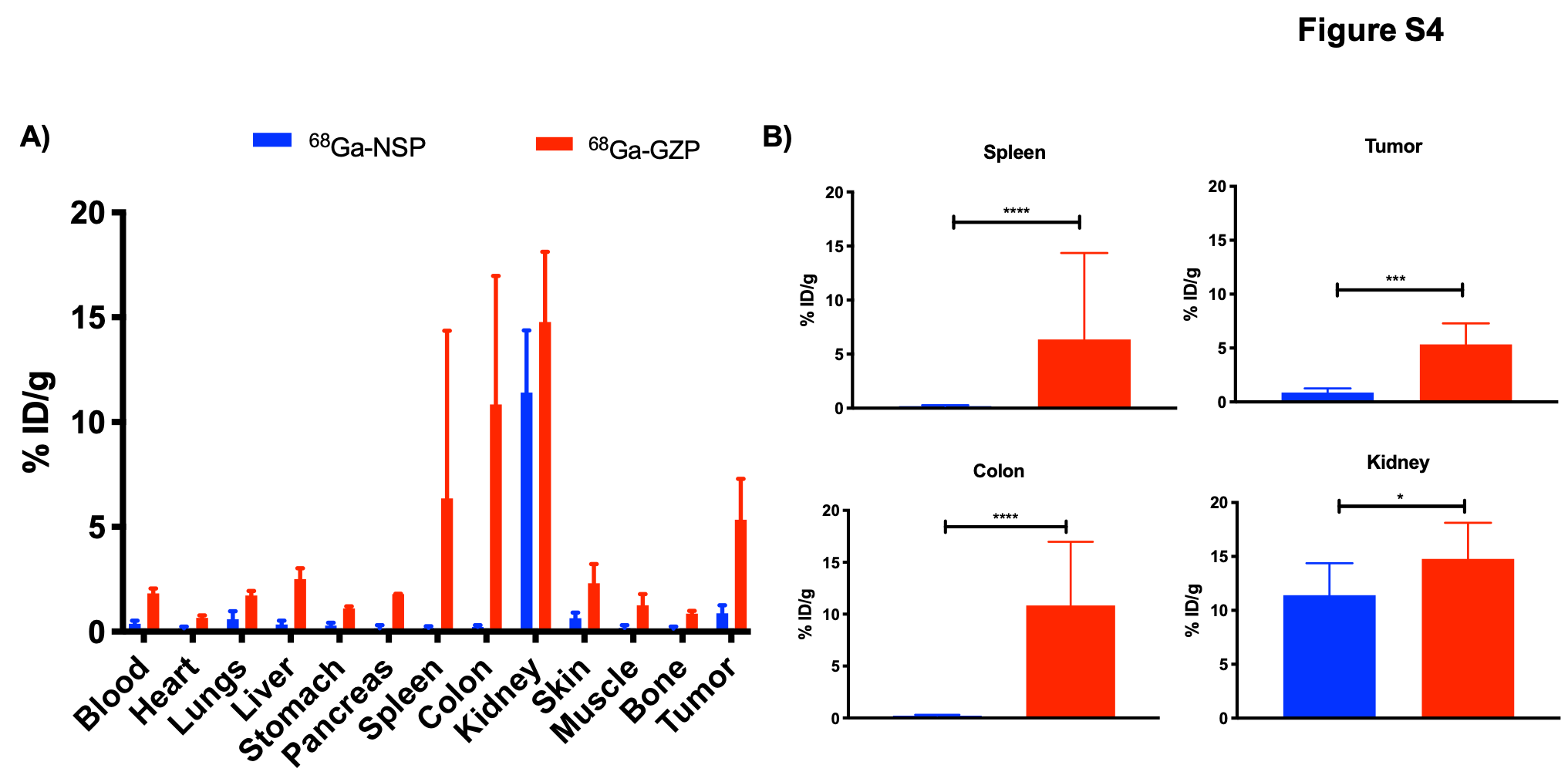
In order to validate the specificity of our tracer and rule out differences in tracer uptake due to perfusion and/or inflammation, we performed ex vivo biodistribution studies after administration of a non-specific DOTA-conjugated peptide radiolabeled with 68Ga (68Ga-NSP) in animals treated with DT + anti-CD137. Non-specific radiotracer uptake was negligible in major organs investigated (Figure S4A) except the kidneys, which is in agreement with the elimination pathway of the probe. Uptake in the kidneys, spleen, colon and tumors was significantly lower than uptake in the organs of animals injected with 68Ga-GZP (Figure S4B), indicating that inflammatory status was not the driver of radiotracer accumulation in those organs and that 68Ga-GZP uptake was indeed specific to GZB presence. Furthermore, as an additional investigation into the specificity of our tracer, we evaluated through PET imaging differences in uptake of our GZB specific (68Ga-GZP) and the non-specific peptide (68Ga-GZP) in local inflammation. Results (Figure S5) demonstrate specific and significantly higher (p < 0.001) 68Ga-GZP uptake in the region of LPS injection when compared with the PBS injected contralateral region, as seen in PET images (Figure S5A) and ROI analysis (Figure S5B). Imaging with 68Ga-NSP demonstrates negligible uptake in both LPS and PBS injected regions. More importantly, 68Ga-GZP uptake was significantly higher (p < 0.0001) than 68Ga-NSP uptake in the LPS injected region. This experiment demonstrates the specificity of our radiotracer in the context of granzyme B activity.
Treatment with steroids results in diminished granzyme B expression
Since corticosteroids are first-line treatment for the management of immune-related adverse events, we aimed to evaluate the impact of dexamethasone on granzyme B presence as well as on uptake of our imaging agent as a surrogate for whether resolution of irAE can also be non-invasively assessed with GZB PET imaging. For that, we selected mice treated with DT + anti-CD137 that had exhibited the most severe and abundant adverse events and administered 5 doses of dexamethasone (5mg/kg) on 5 consecutive days intraperitoneally three days after the last anti-CD137 treatment in five mice. We then performed PET imaging using the same protocol as specified before at 1 hour post-injection of 68Ga-NOTA-GZP. As observed in Figure 2A, overall PET signal in the abdominal area diminished considerably. Relatively high signal is still seen in bladder and kidney which is consistent with the elimination pathway of the probe. Values of ROI calculated from the PET images (Figure 2B) demonstrated that while heart and liver uptake did not significantly change in the group that receive dexamethasone, uptake values were significantly lower in spleen (p = 0.0015), colon (p < 0.0001), kidneys (p = 00085) and tumor (p = 001) when compared to the group that didńt receive dexamethasone. For the group that received DT + anti-CD137+ dexamethasone, %ID/g values were found to be 1.4 ± 1.01, 1.09 ± 0.59, 0.40 ± 0.17 and 0.43 ± 0.11 for colon, kidney, liver and spleen respectively. Of note, for the group injected with dexamethasone, colon had the highest uptake value among the organs investigated (except bladder) and uptake values were still higher than that found for PBS and DT only groups. Biodistribution data (Figure S2) validated findings from PET imaging.
Figure 2. In vivo PET imaging with diminished abdominal uptake in animals that received dexamethasone.
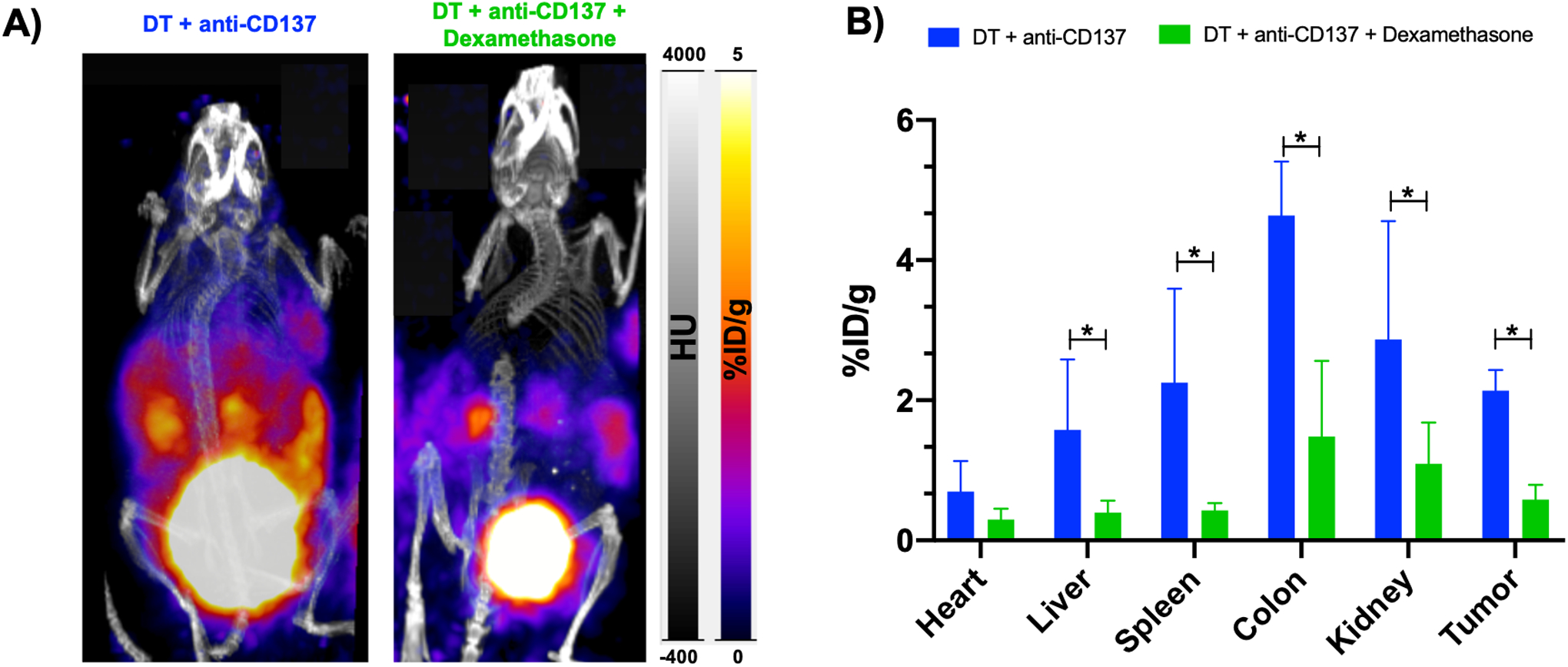
A) Representative PET/CT MIP images at 1 hour post-injection of 68Ga-NOTA-GZP of animals injected with immunotherapy and immunotherapy + dexamethasone. B) ROI analysis of blood pool and major organs, represented as %ID/g. *p = 0.01, ** p < 0.01, **** p < 0.0001
Immunofluorescence and histological staining reveal presence of immune infiltration and granzyme B upregulation in affected organs
After PET/CT imaging, major organs were dissected for ex vivo analysis. Immunofluorescence staining for granzyme B demonstrated that, when compared with animals from the PBS injected group, a higher granzyme B staining is seen for colon, liver, lung, kidney and skin samples of animals injected with DT + anti-CD137 or DT + anti-PD-1 + anti-CTLA-4, indicating that some extent of immunotherapy-associated inflammation occurred in those organs (Figure 3). The fluorescence staining was the strongest in specimens from mice injected with DT + anti-CD137, in concordance with findings from PET/CT scans. For the animals injected with anti-CD137, granzyme B staining was significantly higher in colon (p < 0.001), skin (p < 0.0001) and kidney (p < 0.05) when compared to PBS injected group (Figure S6). The group of animals that received DT + anti-PD-1 + anti-CTLA-4 had significantly higher staining in skin (p < 0.001) and colon samples (p < 0.05), when compared to PBS injected group.
Figure 3. Immunofluorescence staining against granzyme B (in red) of major organs of different treatment groups reveal granzyme B presence in the groups injected with DT + immune checkpoint inhibitors.
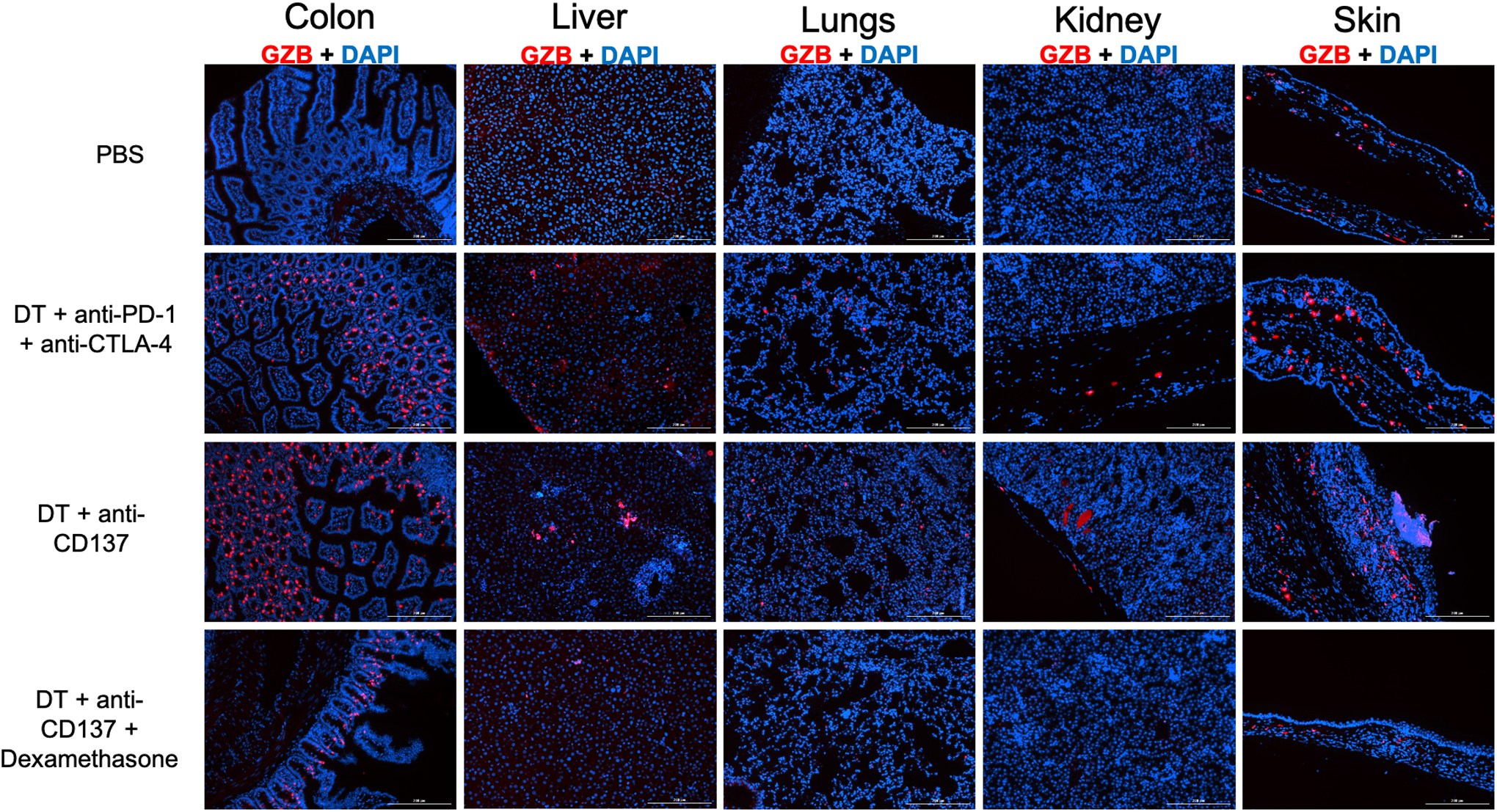
Cell nuclei is stained with DAPI (blue). Magnification 10X.
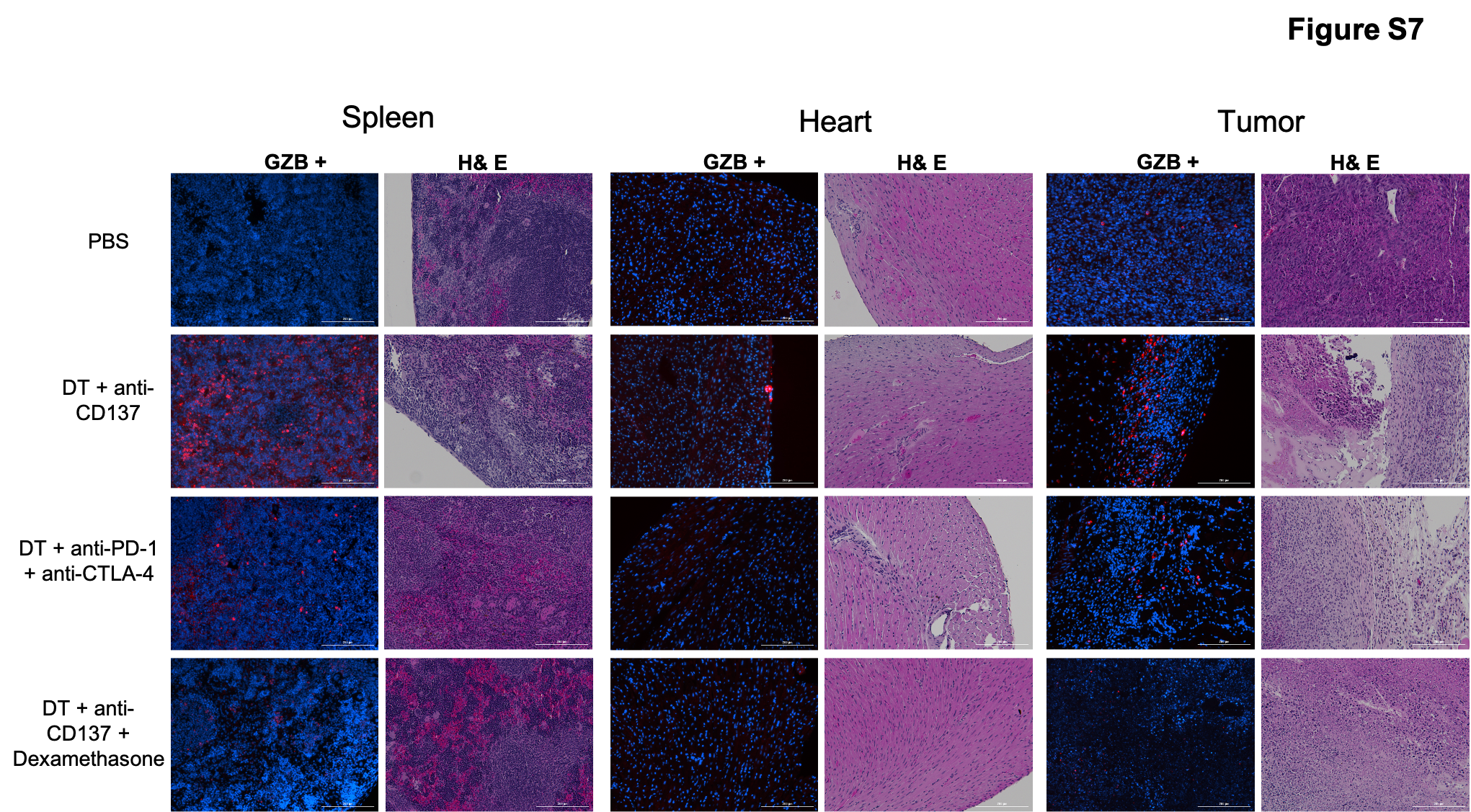
In addition, a visual evaluation of hematoxylin-eosin (H&E) stained samples was carried out to qualitatively assess the degree of inflammatory infiltration. As can be seen in Figure 4, immune infiltrates are present in colon, liver, lungs, kidney and skin for the animals injected with DT + anti-CD137 as well as in colon, liver, lungs and skin of the animals injected with DT + anti-PD-1 + anti-CTLA-4. Of the organs analyzed, colon and skin had a higher degree of immune infiltrate presence for the mice treated with DT + anti-CD137 while the lungs and skin had the higher degree of infiltration for those treated with DT + anti-PD-1 + anti-CTLA-4. The presence of immune infiltration is not visualized in the organs of animals injected with PBS only. Of note, immunofluorescence staining against granzyme B for spleen and tumor revealed high granzyme B signal in spleen and tumor samples of animals from both groups injected with DT + immunotherapy that diminished after administration of dexamethasone, while granzyme B signal was not observed in heart samples of any of the groups investigated (Figure S7).
Figure 4. Histological evaluation reveals presence of immune infiltrates in tissue samples of animals that received DT + immune checkpoint inhibitors.
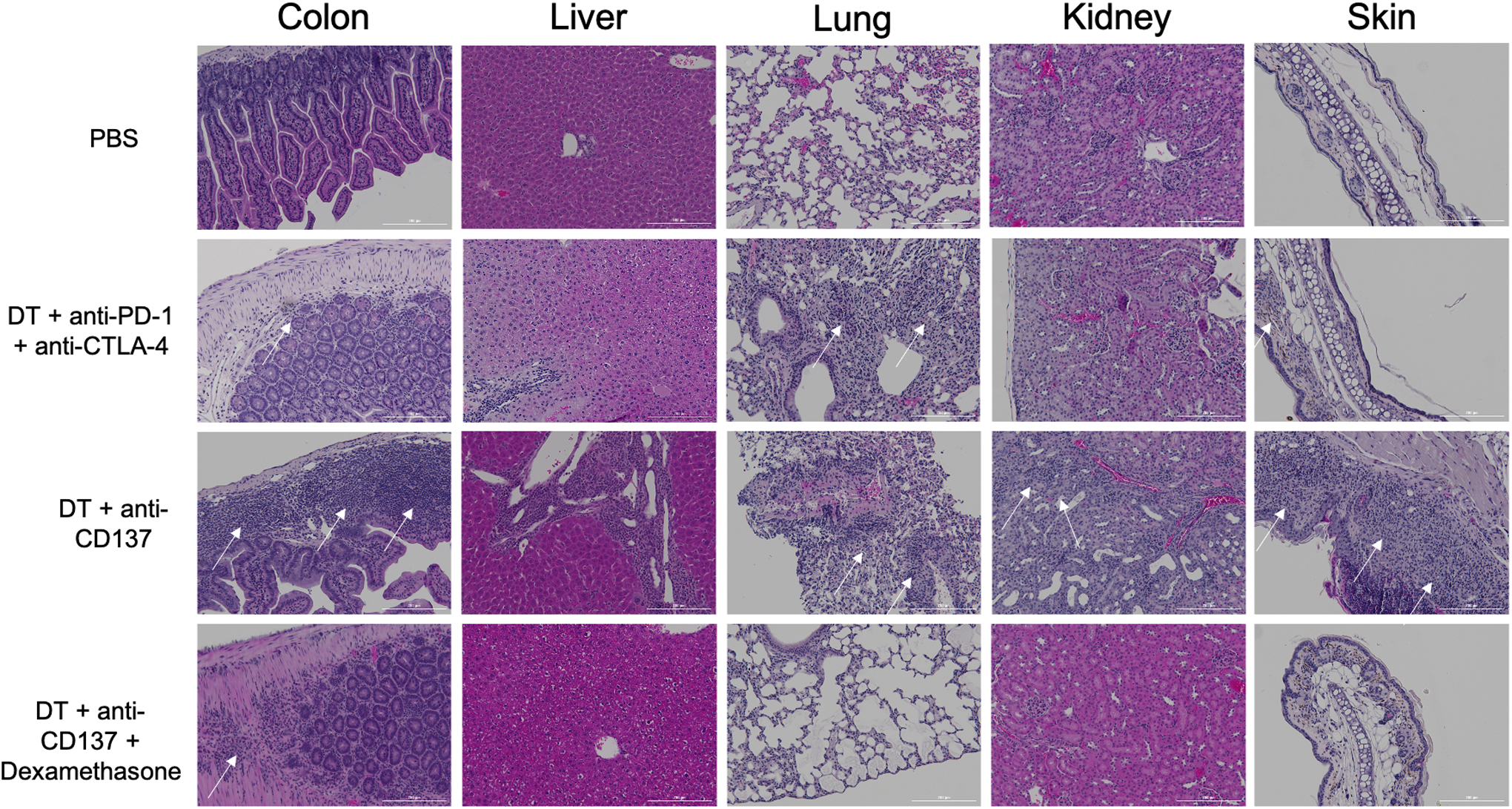
H&E staining of different tissues from different groups. White arrows point at immune infiltrates locations.
We proceeded to investigate effects of dexamethasone on histological evaluation of organ samples as well as on granzyme B expression within the tissues. It was possible to observe through immunofluorescence staining that granzyme B signal decreased in all organs investigated for animals injected with DT + anti-CD137 + dexamethasone when compared to those administered with DT + anti-CD137 only (Figure 3 and S6). In a similar manner, diminished presence of immune infiltrates is found on the dexamethasone administered group as confirmed by H&E staining (Figure 4).
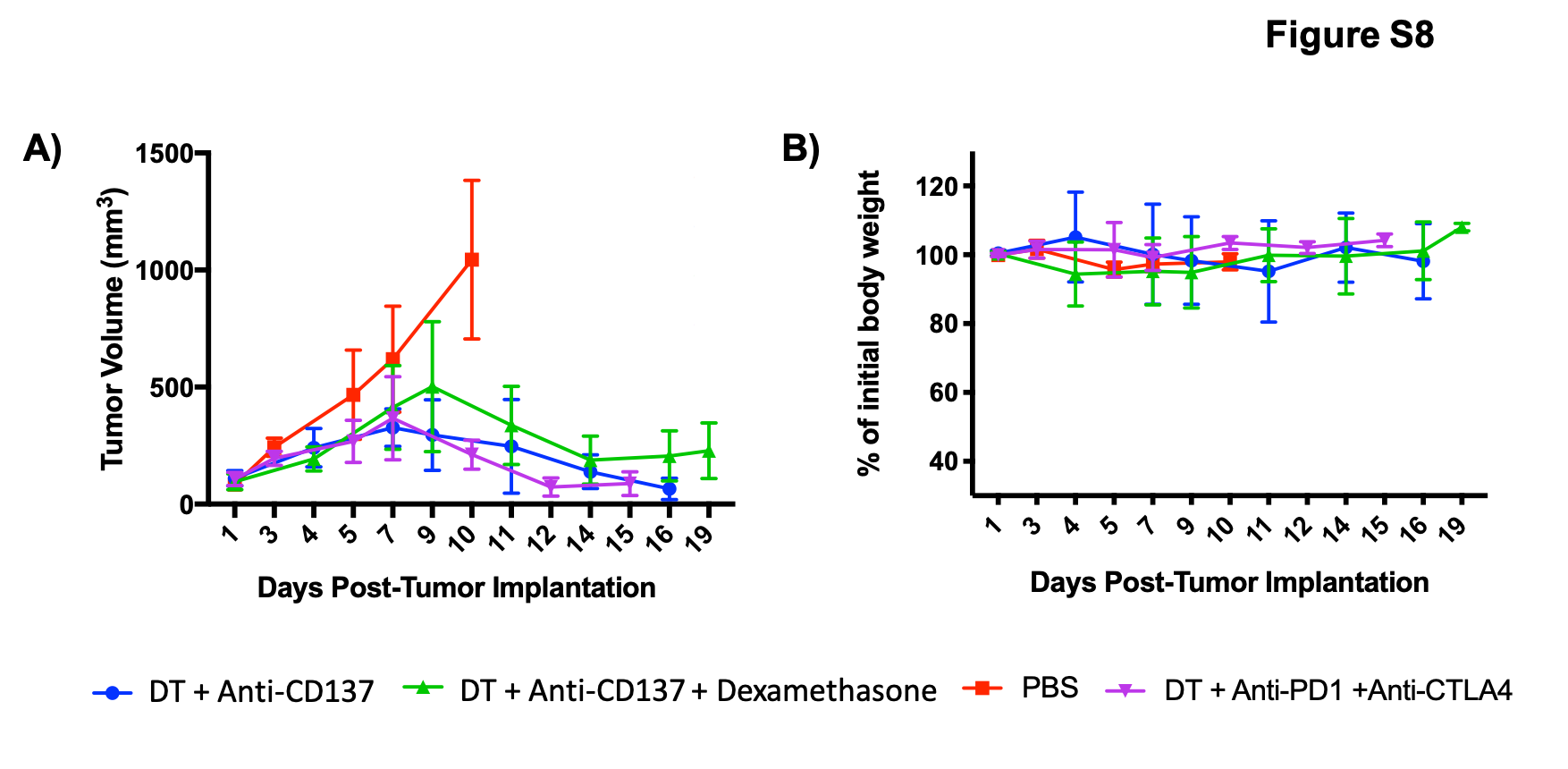
In addition, mice were monitored and body weight as well as tumor dimensions were measured throughout the course of the study (Figure S8). It is possible to observe that the tumor volumes decreased significantly in the groups that received immunotherapy when compared to the PBS-only treated group. Of note, after administration of dexamethasone, tumor volumes started to increase again, which can indicate that the immunosuppression affected immunotherapy efficacy. None of the animals from any of the groups investigated reached the humane end points criterion.
Flow Cytometry analysis demonstrate presence of immune-infiltrates and granzyme B release
Flow cytometry was performed in excised tissue samples of all groups studied to quantify levels of CD3, CD8 and CD107a positive cells as a separate means of analysis of cytotoxic T cell infiltrate and activity state of infiltrating cells. Results can be seen in Figures 5 and S9. CD3 positive cells were highest for the animals injected with DT + anti-CD137 with values of 14.35 ± 4.45 and 7.78 ± 1.94 % of total cells in kidney and liver samples respectively and highest in the animals injected with DT + anti-PD-1 + anti-CTLA-4 with values of 39.10 ± 9.47 and 27.95 ± 5.59 % of total cells in lung and spleen samples respectively. This reveals that CD3 positive cells were more prevalent in animals treated with DT + anti-CD137 or DT + anti-PD-1 + anti-CTLA-4 when compared to all other groups investigated. Levels of CD3 positive cells in animals treated with only PBS was found to be 2.55 ± 1.68, 3.01 ± 1.37, 1.83 ± 0.92 and 5.54 ± 0.14 % of total cells for kidney, liver, lungs and spleen respectively. Of note, when dexamethasone was administered, CD3 positive cells in kidney, liver and lungs diminished and reached values of 6.62 ± 0.17, 0.64 ± 0.08 and 25.7 ± 0.70 % of total cells respectively, similar to the levels of DT only group in all organs investigated. This same trend is seen for CD8 positive cells, with levels at least 4 times higher for animals injected with either DT + anti-CD137 or DT + anti-PD-1 + anti-CTLA-4 when compared to PBS only injected group. In kidney and liver samples, the highest values of CD8 positive cells were found for the group that received DT + anti-CD137 (5.69 ± 4.79 and 2.22 ± 1.36 % of total cells, respectively). The group injected with DT + anti-PD-1 + anti-CTLA-4 had the highest values of CD8 positive cells with 28.5 ± 1.41 % of total cells in the lungs. In addition, injection of dexamethasone decreased CD8 positive cells level in all organs investigated and reached values similar or lower than those found for the PBS injected animals in kidney and liver samples. Most importantly, in the animals that received DT + anti-CD137, levels of CD8 positive cells that also had the granzyme B release marker (CD107a) were significantly higher (p < 0.05) in kidney and lungs with values of 3.63 ± 1.95 and 5.55 ± 0.74 % of total cells respectively when compared to those animals that received PBS only (0.07 ± 0.03 and 1.25 ± 0.34 and 1.71 ± 0.01 % of total cells for kidney and lungs respectively). CD107a positive cells were also more prevalent in all organ samples of the animals injected with DT + anti-PD-1 + anti-CTLA-4 when compared to PBS and DT only groups. As seen with CD3 and CD8 markers, administration of dexamethasone significantly decreased levels of CD107a positive cells.
Figure 5. Flow cytometry findings corroborates increased presence of immune cells and granzyme B in affected organs.
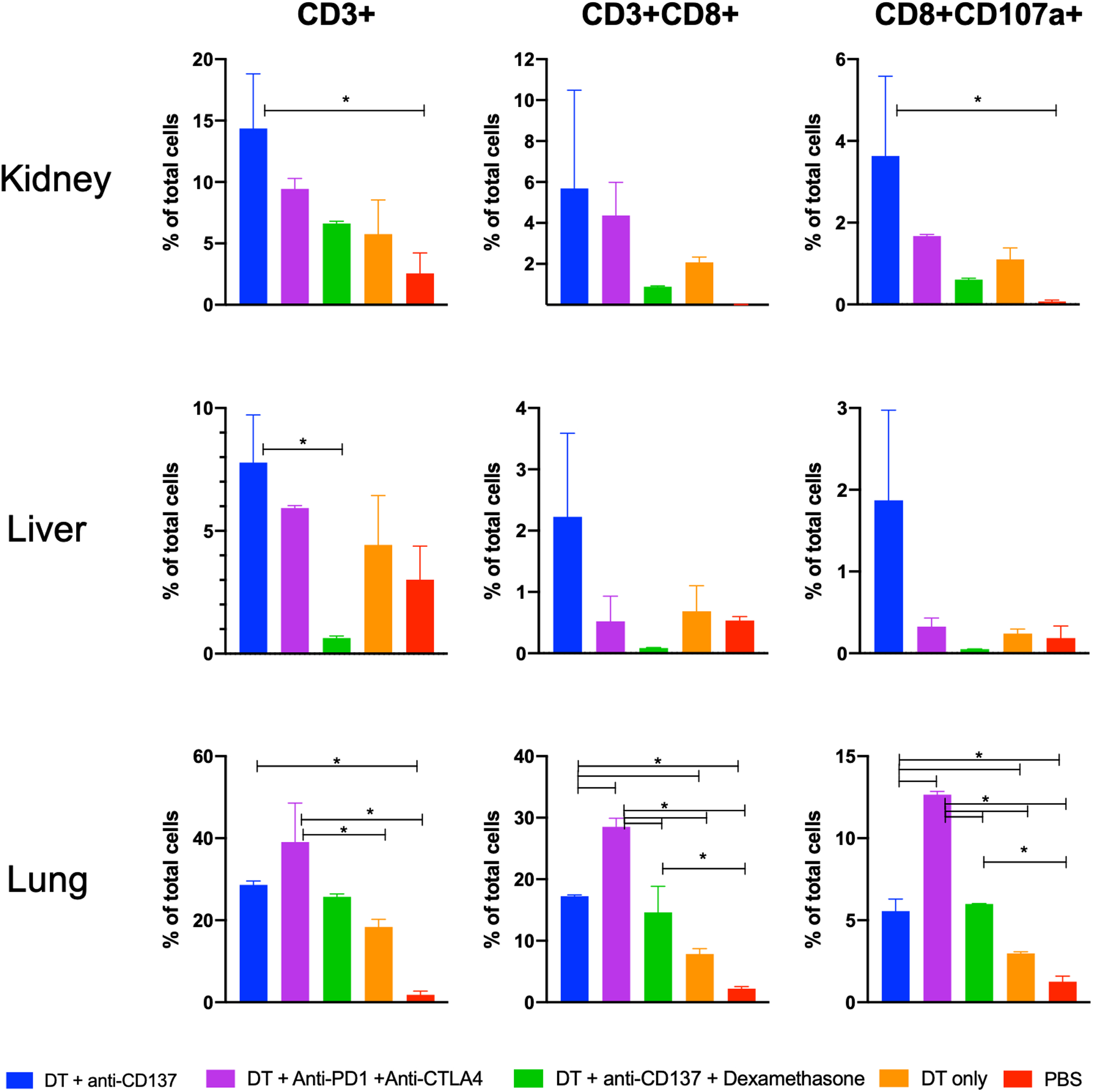
Data with the total number of CD3+, CD8+ and CD8+CD107a+ positive cells from single-cell suspensions generated from excised organs after last treatment.
Analysis of patient samples of established immune-related adverse events reveal granzyme B presence
Immunofluorescence staining to verify the presence of granzyme B was carried out in human colon and kidney samples of patients that presented with immune-checkpoint inhibitor associated colitis and nephritis, respectively (clinical information found on Tables S2 and S3). As observed in Figure 6A, granzyme B staining (in red) was increased in all patient kidney samples (n = 4) when compared to healthy kidney tissue sample (control). In addition, granzyme B signal was also identified in colon samples of all patients investigated (n =3) in higher levels when compared to control colon samples of patients that did not develop colitis as a result of receiving checkpoint inhibitor drugs.
Figure 6. Staining of kidney and colon samples of patients with immune-checkpoint inhibitor associated nephritis and colitis reveal presence of granzyme B.
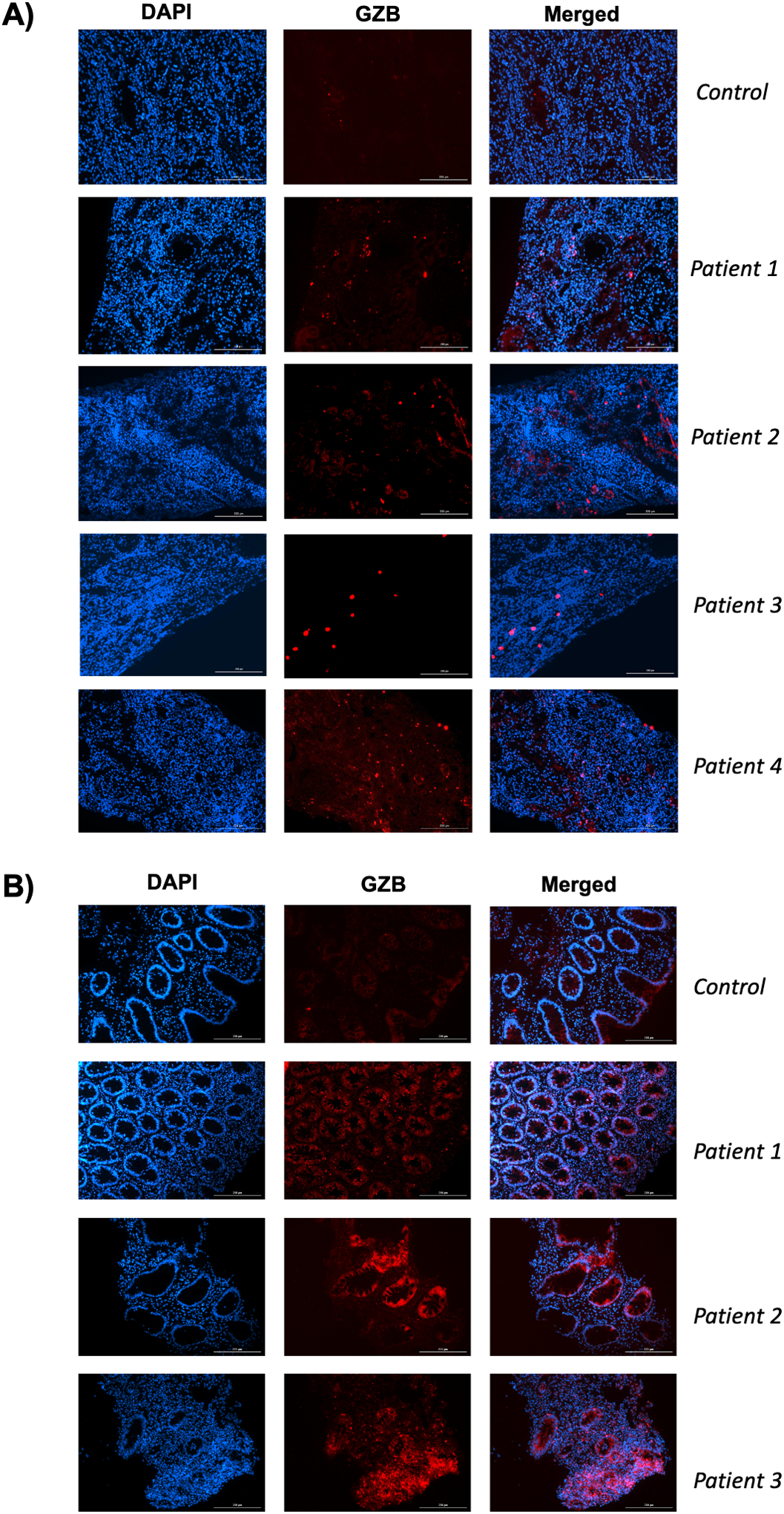
A) Immunofluorescence staining against Granzyme B (red) of a control and 4 kidney samples of clinical patients with immune-checkpoint inhibitor associated nephritis. B) Immunofluorescence staining against Granzyme B (red) of colon samples of clinical patients with (patients 1–3) and without (control) immune-checkpoint inhibitor associated colitis. Nuclei is stained with DAPI (blue).
DISCUSSION
The therapeutic benefits of checkpoint inhibitor drugs are indisputable. However, the overall safety of immune-checkpoint inhibitors is concerning, with an incidence of between 54–76% for the emergence of adverse events after monotherapy, with grade ≥3 adverse events (severe, life-threatening or death) (31) representing around 60% of the total irAEs (6,32). Substantial individual variations in the risk of irAEs development among patients and the idiosyncratic nature of irAEs contributes to difficulty in management (8). Diagnosis and management of irAEs should be done early to ensure minimal morbidity, to prevent life-threatening complications and to continue immunotherapy when the diagnosis of an irAE is excluded (1,23). However, diagnosis of irAEs relies mostly on biopsies and their inherent complications and usually require multidisciplinary efforts, with the clinical expertise of multiple specialties as well as multiple laboratory exams and imaging techniques (33). For that reason, high quality studies with a focus on irAEs are extremely important in order to improve early management and recognition of irAEs. Although the precise mechanisms of irAEs remain unclear and may differ depending on the drug used and the organ affected, it is known that checkpoint inhibitors increase cytotoxic T cell activation and proliferation while diminishing regulatory T cell functions, which can promote autoimmunity and, in turn, disturb immune homeostasis (34). In analysis of clinical specimens, CD8+ T cell abundance has been linked to the development of irAEs (35,36), and multiple reports have demonstrated intense CD8+ T cell infiltrates in biopsy specimens of patients with irAEs (37) .
Granzyme B, a serine-protease released by CD8+ T cells and natural killer cells during the cellular immune response, represents one of the two dominant mechanisms by which cytotoxic T cells mediate cancer cell death (38–40) and has been used as a T cell activation marker (24–26,41,42). Our group has previously demonstrated the ability of a peptide targeting granzyme B (GZP) imaging agent able to predict immunotherapy response by identifying those tumors with high levels of activated T cells (24,25). With the establishment of radiolabeled GZP as an indicator of T cell activation, and the fact that irAEs are linked to CD8+ T-cell-mediated organ infiltration (43), we explored the use of that imaging agent in identifying irAEs in vivo.
We first demonstrated the ability of radiolabeled GZP to be used as a non-invasive imaging agent for the identification of irAEs. PET imaging revealed the establishment of irAEs in multiple organs after administration of DT followed by administration of either anti-CD137 or anti-PD1 + anti-CTLA4. The development of irAEs such as dermatitis and enteritis were clear upon observation of overall health conditions of animals that received DT + anti-CD137 and DT + anti-PD-1 + anti-CTLA-4. In our study, the organ demonstrating the highest granzyme B positive signal was the colon. The tracer uptake in the colon reached more than 10% ID/g as calculated in biodistribution studies, even though clear quantification of signal in the colon was hampered by overlapping strong bladder signal in preclinical PET imaging. These findings are consistent with the fact that checkpoint inhibitor-induced colitis (CPI colitis) has been correlated with augmented activation and proliferation of cytotoxic effector CD8 positive T cells (37). In fact, these increased levels are so high that CD8+ T cell levels have been used to differentiated CPI colitis from other colon diseases such as ulcerative colitis and Crohńs disease. GZP uptake levels can therefore be potentially be used to discriminate CPI colitis from other inflammatory colon diseases and assist in therapeutic management decisions.
We also observed significantly higher tracer uptake in the kidneys of animals that received DT + anti-CD137, which could indicate the development of inflammatory disease related to the immunotherapy treatment. Although the most common tissues involved in irAEs are the skin, gastrointestinal tract and endocrine system, immune checkpoint-inhibitor associated acute kidney injury (CPI-AKI) has been reported (44). Non-invasive detection of CPI-AKI would be highly beneficial, as differentiating CPI-AKI from other causes of AKI is challenging and often requires a high-risk percutaneous renal biopsy (45).
We then sought to determine if the organs with higher GZP uptake had signs of immunotherapy-induced toxicity and corresponding granzyme B presence. Histologic and immunofluorescent assessment of the major organs confirmed the presence of immune-infiltrates, and granzyme B presence. Immune infiltrates were found in colon, kidney, skin and lung tissues of the animals injected with DT + anti-CD137 and colon, skin and lung tissues of animals injected with DT + anti-PD-1 + anti-CTLA-4. Immunofluorescent staining showed granzyme B expression in those same organs, confirmed radiotracer specificity. Colon, as correlated with PET uptake, had the highest degree of inflammatory infiltration and granzyme B expression. Interestingly, skin and lung samples of both group of animals also had very high levels of immune cell infiltrates. Even though tracer uptake in skin and lungs was not apparent in the PET scans, higher uptake levels were found for those groups when compared to all other groups when investigated through biodistribution studies, which further corroborates to the connection between granzyme B upregulation and adverse events establishment. Of note, while there is no specific irAE associated with increased T cell infiltrate in the spleen, we noted a marked increase in T cell infiltration and radiotracer accumulation in the spleen of mice treated with DT + anti-CD137, which speaks to overall up-regulation of the immune system. We also note a high-degree of granzyme B signal variability (both by PET tracer uptake and immunofluorescence) across the livers of animals injected with DT+ anti-CD137. We did see a correlation between PET uptake and immunofluorescence staining in the liver of animals from that group (i.e. the same animals that had higher immunofluorescence signal also had higher PET tracer uptake) The high degree of variability led to no statistically significant difference between this group and other treatment groups. While investigation of variability within each group is beyond the scope of this paper, we posit this could be due to a variable incidence of CPI-hepatitis within this treatment group.
Our flow cytometry analysis demonstrated a higher prevalence of CD3+, CD8+ and CD8+CD107a+ cells for the animals treated with DT + immunotherapy in all organs investigated when compared to PBS injected group. Levels of CD107a, which is a marker of CD8+ degranulation following stimulation (46), reached values 51 times higher for kidney and 10 times higher for liver of the animals that received DT + anti-CD137 when compared to those found for PBS injected group. In a similar fashion, the number of CD107a positive cells were 27 and 10 times higher for kidney and lungs of animals injected with DT + anti-PD-1 + anti-CTLA-4 when compared to PBS only group, respectively. Unfortunately, due to the difficulty in sample preparation for flow cytometry acquisition, colon and skin samples could not be analyzed. Altogether, findings from PET, biodistribution data and ex vivo studies demonstrate a direct association between granzyme B upregulation and irAEs and suggest its potential use as a marker of irAE development
Notably, we have found more robust and severe signs of irAEs in animals that received DT + anti-CD137 than DT + anti-PD-1 + anti-CTLA-4. These differences between treatment groups in this mouse model have been previously described (27), and can be explained by significant differences in proliferating CD8+ T cells and serum levels of IFNγ and TNF found among the groups. In fact, in the clinical settings, agonistic antibodies targeting CD137 induced dose-dependent fatal hepatotoxicity and other toxic effects that led to the termination of a number of clinical trials (47).
Seeking to understand the effects of immunosuppressant drugs on GZP tracer uptake, we selected the group demonstrating the most severe adverse events (DT + anti-CD137) and treated those animals with the steroid dexamethasone. Treatment with dexamethasone lowered radiotracer uptake in all organs investigated. This same pattern was observed for granzyme B expression, as assessed by immunofluorescence staining. This finding is especially important in the kidneys, since urinary excretion of tracer can partially obscure calculation of organ-specific radiotracer uptake. The fact that, upon dexamethasone administration, tracer uptake significantly diminished in the kidneys suggests that 68Ga-GZP can still potentially be used for the detection of irAEs in the kidneys. However, further studies focused on the pharmacokinetics of this radiotracer, including different imaging timepoints, are warranted to establish an optimal protocol for the detection of renal irAEs. Steroid dependent decrease in organ immune infiltrate was also observed through flow cytometry analysis and histological staining for all organs investigated. Interestingly, although granzyme B signal and the presence of immune infiltrates in colon diminished after administration of dexamethasone, granzyme B presence and immune infiltration can still be found in the colon samples of those animals. This may be consistent with the fact that dexamethasone is at times insufficient to resolve immune-related adverse events in clinical practice (48,49). Furthermore, we chose to evaluate the effect of steroids on resolving irAEs induced by a therapeutic regimen, anti-CD137, that has not been thoroughly evaluated in clinical practice, and the intensity of GZB uptake we observed may correlate to more severe irAE than typically observed with PD-1/CTLA4 inhibition. These can have important implications in the clinical practice since persistent high levels of granzyme B signal after steroids can indicate steroid resistance and suggest potential benefits in the utilization of other therapeutic targets such as anti-TNF drugs (50,51). Interestingly, through our tumor volume growth curve, it was possible to observe that after injection of dexamethasone, tumor size started to increase again, which may indicate that the anti-tumor efficacy of checkpoint inhibition was affected by immunosuppression. That hypothesis was enriched by reduced tumor tracer uptake as seen by PET and biodistribution data for the group injected with dexamethasone when compared to the group injected with DT and anti-CD137. The ability to monitor the immune response in both target tumor as well in affected organ during irAE treatment may potentially be helpful in guiding the decision to stop ICI drug administration and be invaluable in the clinical management of immunotherapy patients.
Finally, we sought to demonstrate the clinical correlate of our preclinical imaging findings by analyzing the presence of granzyme B expression in biopsy samples from immune-checkpoint inhibitor treated patients presenting with irAEs. The presence of granzyme B in those samples could indicate the clinical potential of the use of radiolabeled GZP for non-invasive detection and monitoring of irAEs. In fact, immunofluorescence staining of colon and kidney samples of patients presenting with immunotherapy-induced colitis and nephritis, demonstrates very high levels of granzyme B in all investigated specimens, with negligent staining of healthy kidney and colon samples. These findings further suggest that GZP could be used to non-invasively detect irAEs in CPI treated patients. Altogether, we believe that radiolabeled GZP has enormous potential in aiding clinicians through management of irAEs in two significant ways: (1) through diagnosis and monitoring of immune-related adverse events as herein demonstrated and (2) monitoring of tumor response throughout the course of irAE treatment to potentially identify patients that will experience reduced anti-tumor efficacy due to the use of immunosuppressants.
Contrasting our preclinical GZP-PET findings with the known clinical findings of FDG-PET may be helpful to highlight GZP-PETs future potential role for irAE assessment. GZP PET shows uniform low background uptake across organs susceptible to irAE, with the exception of kidneys due to urinary excretion. This stands in contrast to variable FDG uptake seen within organs including the colon and heart, that may make diagnosis of irAEs more challenging. From a mechanistic standpoint, numerous different have increased glucose uptake and utilization, potentially confounding FDG PET in the assessment of adverse events and lowering specificity of imaged FDG signal, whereas granzyme B PET signal should be specific to cytotoxic immune cells. Nonetheless, future clinical studies comparing these two imaging agents are warranted.
Even though the overarching goal of this paper was to non-invasively detect irAEs rather than to focus on the mechanistic details of irAE development, we understand that utilizing preclinical mouse models of irAEs imposes limitations. Unfortunately, the development of irAE events is uncommon in preclinical mouse models, even when multiple checkpoint blockade pathways are targeted. This might be explained by the fact that different mouse strains are more resistant to developing irAEs when compared to humans or simply by the fact that experiments using animals are usually carried out and monitored over shorter periods of time (43). To our knowledge, the only murine model specifically investigated for its ability to recapitulate symptomatology similar to clinical irAEs (27), is the Foxp3-DTR conditional knockout mouse model, in which transient Treg depletion is induced by intraperitoneal administration of Diphtheria toxin, lowering self-tolerance and thus allowing irAEs to be more easily induced following treatment with immunomodulatory antibodies. The authors found a significant increase in proliferating CD8+ T cells and serum levels of IFNγ and TNFalpha after administration of immunomodulatory antibodies in multiple organs, which correlated with irAE development and severity as well as anti-tumor effects. The correlation between CD8+ T cell abundance and irAEs development has been described in the clinical settings (35,36,52); and the known correlation between activated T cells and granzyme B presence (41,42,53) warranted our investigation of the use of a granzyme B targeted peptide to non-invasively detect irAEs. However, the mechanisms of irAE development in these animals and how they correlate with clinical irAEs still needs to be investigated, as this mouse model differs meaningfully from the clinical environment where no specific intervention on regulatory T cell activity is performed. In addition, clinical mechanistic details (54,55) and identification of potential biomarkers (36,56) of irAEs still need to be elucidated and efforts are being made to fully address this challenge.
In this proof-of-concept study we show in vitro, in vivo and ex vivo data which in aggregate suggests the possibility of detecting irAEs using a non-invasive imaging agent. This potential is further demonstrated by the fact that increased granzyme B expression was found in tissue samples of clinical patients that presented with CPI induced colitis and nephritis. More work remains to be done to determine optimal imaging time-points for irAE-specific imaging, and to understand whether granzyme B expression can serve as a biomarker in human tissues in other organ-specific irAEs. We do believe that clinical investigation of granzyme B PET imaging to evaluate clinical irAEs development is warranted to further answer these questions and determine the potential clinical value of this imaging agent.
CONCLUSION
In this study, we demonstrate an interconnection between the establishment of immune-related adverse events and granzyme B presence. Herein, to the best of our knowledge, we show for the first time the visualization of such events through PET imaging. We further demonstrate the presence of granzyme B in human tissue of clinical patients that presented with checkpoint inhibitor-associated adverse events, warranting the possibility of direct clinical translation. Taken together, our data suggest a direct role for granzyme B imaging in the clinical management of immunotherapy and immunotherapy-induced adverse events.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
TRANSLATIONAL RELEVANCE.
In this study we present a novel method for assessment of immune-related adverse events (irAEs) through PET imaging of granzyme B, which is a mediator of cytotoxic T-cell related cellular apoptosis. We demonstrate that granzyme B is abundantly present in tissues affected by irAEs, including in those of clinical patients presenting with checkpoint inhibitor-associated nephritis and colitis. Such imaging could be used as a tool for early detection and monitoring of irAEs in at-risk patients, reduce the chance of progression to life-threatening irAE by enabling early intervention, and guide treatment development and administration.
Financial Support:
This study was funded by a Melanoma Research Alliance award, NIH-R01CA214744 and NIH-R01DK123143 award to UM. PH supported by K08-CA249047.
Footnotes
Conflict of Interest: UM and EWK are cofounders, shareholders, and consultants (Scientific Advisory Board) of CytoSite BioPharma.
REFERENCES
- 1.Myers G Immune-related adverse events of immune checkpoint inhibitors: a brief review. Curr Oncol 2018;25(5):342–7 doi 10.3747/co.25.4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Esfahani K, Roudaia L, Buhlaiga N, Del Rincon SV, Papneja N, Miller WH Jr. A review of cancer immunotherapy: from the past, to the present, to the future. Curr Oncol 2020;27(Suppl 2):S87–S97 doi 10.3747/co.27.5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haslam A, Prasad V. Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw Open 2019;2(5):e192535 doi 10.1001/jamanetworkopen.2019.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70(1):7–30 doi 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 5.Pauken KE, Dougan M, Rose NR, Lichtman AH, Sharpe AH. Adverse Events Following Cancer Immunotherapy: Obstacles and Opportunities. Trends Immunol 2019;40(6):511–23 doi 10.1016/j.it.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu C, Chen YP, Du XJ, Liu JQ, Huang CL, Chen L, et al. Comparative safety of immune checkpoint inhibitors in cancer: systematic review and network meta-analysis. BMJ 2018;363:k4226 doi 10.1136/bmj.k4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Das S, Johnson DB. Immune-related adverse events and anti-tumor efficacy of immune checkpoint inhibitors. J Immunother Cancer 2019;7(1):306 doi 10.1186/s40425-019-0805-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramos-Casals M, Brahmer JR, Callahan MK, Flores-Chavez A, Keegan N, Khamashta MA, et al. Immune-related adverse events of checkpoint inhibitors. Nat Rev Dis Primers 2020;6(1):38 doi 10.1038/s41572-020-0160-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martins F, Sofiya L, Sykiotis GP, Lamine F, Maillard M, Fraga M, et al. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat Rev Clin Oncol 2019;16(9):563–80 doi 10.1038/s41571-019-0218-0. [DOI] [PubMed] [Google Scholar]
- 10.Wang DY, Salem JE, Cohen JV, Chandra S, Menzer C, Ye F, et al. Fatal Toxic Effects Associated With Immune Checkpoint Inhibitors: A Systematic Review and Meta-analysis. JAMA Oncol 2018;4(12):1721–8 doi 10.1001/jamaoncol.2018.3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fecher LA, Agarwala SS, Hodi FS, Weber JS. Ipilimumab and its toxicities: a multidisciplinary approach. Oncologist 2013;18(6):733–43 doi 10.1634/theoncologist.2012-0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weber JS, Kahler KC, Hauschild A. Management of immune-related adverse events and kinetics of response with ipilimumab. J Clin Oncol 2012;30(21):2691–7 doi 10.1200/JCO.2012.41.6750. [DOI] [PubMed] [Google Scholar]
- 13.Khoja L, Day D, Wei-Wu Chen T, Siu LL, Hansen AR. Tumour- and class-specific patterns of immune-related adverse events of immune checkpoint inhibitors: a systematic review. Ann Oncol 2017;28(10):2377–85 doi 10.1093/annonc/mdx286. [DOI] [PubMed] [Google Scholar]
- 14.Postow MA, Hellmann MD. Adverse Events Associated with Immune Checkpoint Blockade. N Engl J Med 2018;378(12):1165 doi 10.1056/NEJMc1801663. [DOI] [PubMed] [Google Scholar]
- 15.Yoest JM. Clinical features, predictive correlates, and pathophysiology of immune-related adverse events in immune checkpoint inhibitor treatments in cancer: a short review. Immunotargets Ther 2017;6:73–82 doi 10.2147/ITT.S126227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Passat T, Touchefeu Y, Gervois N, Jarry A, Bossard C, Bennouna J. [Physiopathological mechanisms of immune-related adverse events induced by anti-CTLA-4, anti-PD-1 and anti-PD-L1 antibodies in cancer treatment]. Bull Cancer 2018;105(11):1033–41 doi 10.1016/j.bulcan.2018.07.005. [DOI] [PubMed] [Google Scholar]
- 17.Afonina IS, Cullen SP, Martin SJ. Cytotoxic and non-cytotoxic roles of the CTL/NK protease granzyme B. Immunol Rev 2010;235(1):105–16 doi 10.1111/j.0105-2896.2010.00908.x. [DOI] [PubMed] [Google Scholar]
- 18.Cullen SP, Brunet M, Martin SJ. Granzymes in cancer and immunity. Cell Death Differ 2010;17(4):616–23 doi 10.1038/cdd.2009.206. [DOI] [PubMed] [Google Scholar]
- 19.Prizment AE, Vierkant RA, Smyrk TC, Tillmans LS, Nelson HH, Lynch CF, et al. Cytotoxic T Cells and Granzyme B Associated with Improved Colorectal Cancer Survival in a Prospective Cohort of Older Women. Cancer Epidemiol Biomarkers Prev 2017;26(4):622–31 doi 10.1158/1055-9965.EPI-16-0641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer 2016;16(5):275–87 doi 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bacac M, Colombetti S, Herter S, Sam J, Perro M, Chen S, et al. CD20-TCB with Obinutuzumab Pretreatment as Next-Generation Treatment of Hematologic Malignancies. Clin Cancer Res 2018;24(19):4785–97 doi 10.1158/1078-0432.CCR-18-0455. [DOI] [PubMed] [Google Scholar]
- 22.Bacac M, Klein C, Umana P. CEA TCB: A novel head-to-tail 2:1 T cell bispecific antibody for treatment of CEA-positive solid tumors. Oncoimmunology 2016;5(8):e1203498 doi 10.1080/2162402X.2016.1203498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Puzanov I, Diab A, Abdallah K, Bingham CO 3rd, Brogdon C, Dadu R, et al. Managing toxicities associated with immune checkpoint inhibitors: consensus recommendations from the Society for Immunotherapy of Cancer (SITC) Toxicity Management Working Group. J Immunother Cancer 2017;5(1):95 doi 10.1186/s40425-017-0300-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larimer BM, Bloch E, Nesti S, Austin EE, Wehrenberg-Klee E, Boland G, et al. The Effectiveness of Checkpoint Inhibitor Combinations and Administration Timing Can Be Measured by Granzyme B PET Imaging. Clin Cancer Res 2019;25(4):1196–205 doi 10.1158/1078-0432.CCR-18-2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Larimer BM, Wehrenberg-Klee E, Dubois F, Mehta A, Kalomeris T, Flaherty K, et al. Granzyme B PET Imaging as a Predictive Biomarker of Immunotherapy Response. Cancer Res 2017;77(9):2318–27 doi 10.1158/0008-5472.CAN-16-3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LaSalle T, Austin EE, Rigney G, Wehrenberg-Klee E, Nesti S, Larimer B, et al. Granzyme B PET imaging of immune-mediated tumor killing as a tool for understanding immunotherapy response. J Immunother Cancer 2020;8(1):e000291 doi 10.1136/jitc-2019-000291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu J, Blake SJ, Harjunpaa H, Fairfax KA, Yong MC, Allen S, et al. Assessing Immune-Related Adverse Events of Efficacious Combination Immunotherapies in Preclinical Models of Cancer. Cancer Res 2016;76(18):5288–301 doi 10.1158/0008-5472.CAN-16-0194. [DOI] [PubMed] [Google Scholar]
- 28.Aslani A, Snowdon GM, Bailey DL, Schembri GP, Bailey EA, Roach PJ. Gallium-68 DOTATATE Production with Automated PET Radiopharmaceutical Synthesis System: A Three Year Experience. Asia Ocean J Nucl Med Biol 2014;2(2):75–86 doi Fall;2(2):75–86. [PMC free article] [PubMed] [Google Scholar]
- 29.Mokaleng BB, Ebenhan T, Ramesh S, Govender T, Kruger HG, Parboosing R, et al. Synthesis, 68Ga-radiolabeling, and preliminary in vivo assessment of a depsipeptide-derived compound as a potential PET/CT infection imaging agent. Biomed Res Int 2015;2015:284354 doi 10.1155/2015/284354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi SR, Shi Y, Taylor CR. Antigen retrieval immunohistochemistry: review and future prospects in research and diagnosis over two decades. J Histochem Cytochem 2011;59(1):13–32 doi 10.1369/jhc.2010.957191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Basch E, Reeve BB, Mitchell SA, Clauser SB, Minasian LM, Dueck AC, et al. Development of the National Cancer Institute’s patient-reported outcomes version of the common terminology criteria for adverse events (PRO-CTCAE). J Natl Cancer Inst 2014;106(9) doi 10.1093/jnci/dju244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med 2017;377(14):1345–56 doi 10.1056/NEJMoa1709684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi J, Lee SY. Clinical Characteristics and Treatment of Immune-Related Adverse Events of Immune Checkpoint Inhibitors. Immune Netw 2020;20(1):e9 doi 10.4110/in.2020.20.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol 2020;20(11):651–68 doi 10.1038/s41577-020-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laubli H, Koelzer VH, Matter MS, Herzig P, Dolder Schlienger B, Wiese MN, et al. The T cell repertoire in tumors overlaps with pulmonary inflammatory lesions in patients treated with checkpoint inhibitors. Oncoimmunology 2018;7(2):e1386362 doi 10.1080/2162402X.2017.1386362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jing Y, Liu J, Ye Y, Pan L, Deng H, Wang Y, et al. Multi-omics prediction of immune-related adverse events during checkpoint immunotherapy. Nat Commun 2020;11(1):4946 doi 10.1038/s41467-020-18742-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luoma AM, Suo S, Williams HL, Sharova T, Sullivan K, Manos M, et al. Molecular Pathways of Colon Inflammation Induced by Cancer Immunotherapy. Cell 2020;182(3):655–71 e22 doi 10.1016/j.cell.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cullen SP, Martin SJ. Mechanisms of granule-dependent killing. Cell Death Differ 2008;15(2):251–62 doi 10.1038/sj.cdd.4402244. [DOI] [PubMed] [Google Scholar]
- 39.Russell JH, Ley TJ. Lymphocyte-mediated cytotoxicity. Annu Rev Immunol 2002;20(0732–0582 (Print)):323–70 doi 10.1146/annurev.immunol.20.100201.131730. [DOI] [PubMed] [Google Scholar]
- 40.Trapani JA, Sutton VR. Granzyme B: pro-apoptotic, antiviral and antitumor functions. Curr Opin Immunol 2003;15(5):533–43 doi 10.1016/s0952-7915(03)00107-9. [DOI] [PubMed] [Google Scholar]
- 41.Nowacki TM, Kuerten S, Zhang W, Shive CL, Kreher CR, Boehm BO, et al. Granzyme B production distinguishes recently activated CD8(+) memory cells from resting memory cells. Cell Immunol 2007;247(1):36–48 doi 10.1016/j.cellimm.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mellor-Heineke S, Villanueva J, Jordan MB, Marsh R, Zhang K, Bleesing JJ, et al. Elevated Granzyme B in Cytotoxic Lymphocytes is a Signature of Immune Activation in Hemophagocytic Lymphohistiocytosis. Front Immunol 2013;4:72 doi 10.3389/fimmu.2013.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu J, Blake SJ, Smyth MJ, Teng MW. Improved mouse models to assess tumour immunity and irAEs after combination cancer immunotherapies. Clin Transl Immunology 2014;3(8):e22 doi 10.1038/cti.2014.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murakami N, Motwani S, Riella LV. Renal complications of immune checkpoint blockade. Curr Probl Cancer 2017;41(2):100–10 doi 10.1016/j.currproblcancer.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gupta S, Cortazar FB, Riella LV, Leaf DE. Immune Checkpoint Inhibitor Nephrotoxicity: Update 2020. Kidney360 2020;1(2):130–40 doi 10.34067/kid.0000852019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alter G, Malenfant JM, Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods 2004;294(1–2):15–22 doi 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 47.Yonezawa A, Dutt S, Chester C, Kim J, Kohrt HE. Boosting Cancer Immunotherapy with Anti-CD137 Antibody Therapy. Clin Cancer Res 2015;21(14):3113–20 doi 10.1158/1078-0432.CCR-15-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Som A, Mandaliya R, Alsaadi D, Farshidpour M, Charabaty A, Malhotra N, et al. Immune checkpoint inhibitor-induced colitis: A comprehensive review. World J Clin Cases 2019;7(4):405–18 doi 10.12998/wjcc.v7.i4.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shivaji UN, Jeffery L, Gui X, Smith SCL, Ahmad OF, Akbar A, et al. Immune checkpoint inhibitor-associated gastrointestinal and hepatic adverse events and their management. Therap Adv Gastroenterol 2019;12:1756284819884196 doi 10.1177/1756284819884196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Swaminathan M, Olsson-Brown A, Subramaniam S, Pritchard M. PTU-064 immune checkpoint inhibitor colitis- a review of current management trends. Gut 2018;67(Suppl 1):A203 doi 10.1136/gutjnl-2018-BSGAbstracts.405. [DOI] [Google Scholar]
- 51.Weber JS. Challenging Cases: Management of Immune-Related Toxicity. Am Soc Clin Oncol Educ Book 2018;38(38):179–83 doi 10.1200/EDBK_209557. [DOI] [PubMed] [Google Scholar]
- 52.Kim KH, Hur JY, Cho J, Ku BM, Koh J, Koh JY, et al. Immune-related adverse events are clustered into distinct subtypes by T-cell profiling before and early after anti-PD-1 treatment. Oncoimmunology 2020;9(1):1722023 doi 10.1080/2162402X.2020.1722023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Boivin WA, Cooper DM, Hiebert PR, Granville DJ. Intracellular versus extracellular granzyme B in immunity and disease: challenging the dogma. Lab Invest 2009;89(11):1195–220 doi 10.1038/labinvest.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Urwyler P, Earnshaw I, Bermudez M, Perucha E, Wu W, Ryan S, et al. Mechanisms of checkpoint inhibition-induced adverse events. Clin Exp Immunol 2020;200(2):141–54 doi 10.1111/cei.13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weinmann SC, Pisetsky DS. Mechanisms of immune-related adverse events during the treatment of cancer with immune checkpoint inhibitors. Rheumatology (Oxford) 2019;58(Suppl 7):vii59–vii67 doi 10.1093/rheumatology/kez308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jia XH, Geng LY, Jiang PP, Xu H, Nan KJ, Yao Y, et al. The biomarkers related to immune related adverse events caused by immune checkpoint inhibitors. J Exp Clin Cancer Res 2020;39(1):284 doi 10.1186/s13046-020-01749-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.