

Abstract
Tropism of neural stem cells (NSCs) to hypoxic tumor areas provides an opportunity for the drug delivery. Here, we demonstrate that NSCs effectively transport antisense oligonucleotides (ASOs) targeting oncogenic and tolerogenic signal transducer and activator of transcription 3 (STAT3) protein into glioma microenvironment. To enable spontaneous, scavenger receptor-mediated endocytosis by NSCs, we used previously described CpG-STAT3ASO conjugates. Following uptake and endosomal escape, CpG-STAT3ASO colocalized with CD63+ vesicles and later with CD63+CD81+ exosomes. Over 3 days, NSCs secreted exosomes loaded up to 80% with CpG-STAT3ASO. Compared to native NSC exosomes, the CpG-STAT3ASO-loaded exosomes potently stimulated immune activity of human dendritic cells or mouse macrophages, inducing nuclear factor κB (NF-κB) signaling and interleukin-12 (IL-12) production. Using orthotopic GL261 tumors, we confirmed that NSC-mediated delivery improved oligonucleotide transfer from a distant injection site into the glioma microenvironment versus naked oligonucleotides. Correspondingly, the NSC-delivered CpG-STAT3ASO enhanced activation of glioma-associated microglia. Finally, we demonstrated that NSC-mediated CpG-STAT3ASO delivery resulted in enhanced antitumor effects against GL261 glioma in mice. Peritumoral injections of 5 × 105 NSCs loaded ex vivo with CpG-STAT3ASO inhibited subcutaneous tumor growth more effectively than the equivalent amount of oligonucleotide alone. Based on these results, we anticipate that NSCs and NSC-derived exosomes will provide a clinically relevant strategy to improve delivery and safety of oligonucleotide therapeutics for glioma treatment.
Keywords: extracellular vesicles, exosomes, antisense oligonucleotides, neural stem cells, glioma, cancer immunotherapy, STAT3, TLR9, CpG
Graphical abstract
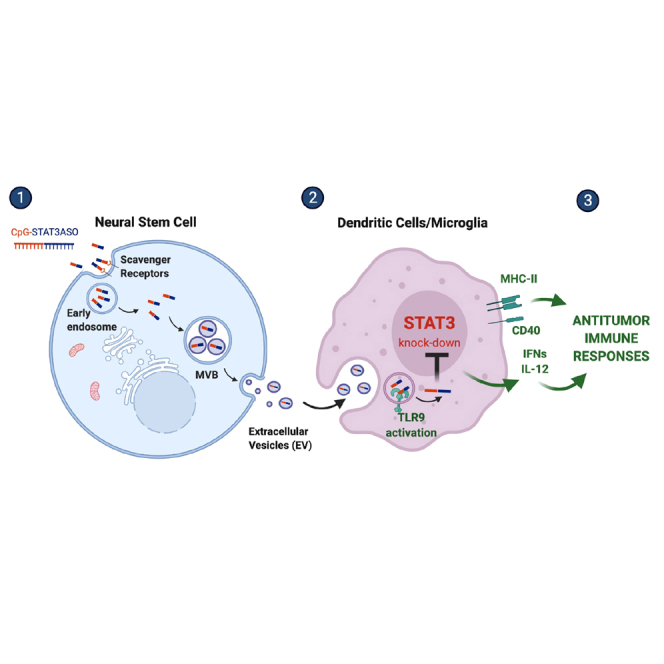
Here, we demonstrate that tropism of neural stem cells to hypoxic tumor areas allows for oligonucleotide delivery. NSCs secreting exosome-encapsulated CpG-STAT3ASOs induced antitumor effects against glioma in mice. We believe that NSC-mediated transfer of oligonucleotide therapeutics is an opportunity for safer and more effective treatment of brain tumors.
Introduction
Malignant gliomas (MGs) are rapidly fatal despite multimodal treatments.1 The oncogenic signal transducer and activator of transcription 3 (STAT3) is a transcription factor and an essential promoter of tumorigenesis and resistance to therapies in a variety of human cancers, including malignant glioma.2, 3, 4 STAT3 is also a central immune checkpoint regulator in human cancers and in tumor-associated myeloid cells, such as dendritic cells (DCs), macrophages and microglia, and myeloid-derived suppressor cells (MDSCs).2,5,6 Direct downstream gene targets of STAT3 include immunosuppressive proteins, such as PD-L1 and arginase-1, while indirectly, STAT3 interferes with expression and processing of major histocompatibility complex (MHC) class II molecules in antigen-presenting cells (APCs).3 The lack of US Food and Drug Administration (FDA)-approved, small-molecule STAT3 inhibitors prompted the development of oligonucleotide-based STAT3 inhibitors, such as STAT3 small interfering RNA (siRNA),7 decoy,8 or antisense9 oligonucleotides (ASOs).8,9 To improve the in vivo targeting of STAT3ASO into the microenvironment of solid tumors, we recently generated CpG-STAT3ASO conjugates that undergo spontaneous uptake via scavenger receptors on target cells.10 Target cell populations include Toll-like receptor-9 (TLR9)-expressing myeloid cells in the glioma microenvironment, such as macrophages and microglia cells and granulocytic MDSCs,11,12 as well as glioma stem-like cells.13 TLR9 stimulation using intracerebral injections of CpG oligodeoxynucleotides (ODNs) were tested in earlier clinical studies in MG patients. However, CpG ODNs as a monotherapy were not effective and are limited by a potential risk of inducing dangerous inflammatory responses with potential for brain swelling.14
Neural stem cells (NSCs) emerged as an attractive cellular strategy for treatment of central nervous system diseases. NSCs show ability to traffic into hypoxic areas of tumors, as initially described in glioma and in secondary brain metastases and later in several other solid tumors.15,16 The follow-up studies explored the potential of using NSCs for the delivery of antitumor agents, such as chemotherapeutic drugs, small-molecule inhibitors, antibodies, cytokines, and even oncolytic viruses, but not oligonucleotide therapeutics (ONTs). The NSC-mediated delivery improved tumor selectivity and half-lives of antitumor agents, especially in preclinical models of brain tumors.17 The clinical grade NSCs are human leukocyte antigen (HLA) class II-negative and chromosomally and functionally stable over multiple passages. The safety and tumor selectivity of this approach was validated in the recent first-in-human clinical trial in high-grade glioma patients, which demonstrated their clinical safety without any dose-limiting toxicities, immunotoxicities, or humoral responses.18 The study confirmed that these NSCs migrated to distant tumor areas, resulting in tumor-localized chemotherapy production. Importantly, the autopsy results from patients that died from progressive disease after over 1 or 2 months after NSC injection did not find any evidence of NSC tumorigenicity.18 While relatively little is known about mechanisms of transport of therapeutic cargo from these NSCs to target cells, it is likely to involve secretory, extracellular vesicles. Recent studies indicated that NSCs are sensitive to various TLR ligands, including CpG ODNs, and thus contribute to immune activity in the brain.19,20
Exosomes are secreted by cells through endosomal pathway, and they play a key role in intercellular communication by shuttling biomolecules, such as proteins, lipids, mRNAs, and microRNAs (miRNAs), between cells.21, 22, 23 In particular, increasing evidence suggests that tumor-derived exosomes play a pivotal role in promoting tumorigenesis by mediating metastasis and immune responses.24 Recent studies have revealed that tumor exosomes promote cancer metastasis and systemically suppress the immune system using immune checkpoint inhibition via PD-L1/PD1 or induction of STAT3 signaling.25, 26, 27 Moreover, with a size range of 40–200 nm, exosomes emerged as promising carriers for anticancer drugs and immunotherapeutics.28, 29, 30 One of the early proof-of-concept studies demonstrated that dendritic cell-derived exosomes can be engineered to deliver therapeutic siRNA specifically to cellular targets in mouse brain.31 However, the challenges of engineering exosomes for specific cellular targets and in large-scale manufacturing so far hindered their therapeutical application.32 Here, we assessed the feasibility of using clinically tested NSCs18 and NSC-derived exosomes as vehicles for the delivery of ONTs into the glioma microenvironment.
Results
NSCs encapsulate CpG-STAT3ASO into extracellular vesicles
We previously generated a strategy for the targeted delivery of ONTs, such as siRNA or ASO, to various types of immune and malignant cells as conjugates with CpG ODNs.6,10 As demonstrated earlier, phosphorothioated CpG conjugates with siRNA or ASO are rapidly internalized via scavenger-receptor-mediated endocytosis into target cells, followed by endosomal escape and target knockdown.10,33 To assess whether human NSCs could serve as a vehicle for CpG-ASO conjugate transport, we investigated the uptake and intracellular fate of these oligonucleotides chemically modified for protection from rapid nuclease degradation.10 In fact, NSCs efficiently internalized fluorescently labeled CpG-STAT3ASOCy3 already at 3 h, and the intracellular levels of the oligonucleotide increased further for up to 24 h as measured using flow cytometry (Figure 1A). Confocal microscopy imaging indicated that the internalized CpG-STAT3ASOCy3 colocalized partly with CD63+ cellular vesicles in NSC cytoplasm, which to a large extent represent multivesicular bodies (MVBs) (Figure 1B).34 Since MVBs are a well-known step in the biogenesis of exosomes, this colocalization may suggest that CpG-STAT3ASO undergoes intracellular sorting into the exosome-producing cell compartments. We next isolated and characterized extracellular vesicles (EVs) present in the media of CpG-STAT3ASO-treated or naive NSCs following the criteria as previously described.35,36 Compared to naive EVs, the size of EVs derived from NSCs incubated with CpG-STAT3ASO was minimally increased as determined by NanoSight (Figure 1C), but both types of EVs had similar morphology as assessed using electron microscopy (Figure 1D). The flow cytometric analysis of EVs indicated expression of key exosomal markers CD63 and CD81, but not cell membrane-derived CD9, which is associated predominantly with ectosomes (Figure 1E).37 Altogether, these results underline the ability of NSCs to encapsulate CpG-STAT3ASO into extracellular vesicles.
Figure 1.

Neural stem cells rapidly internalize and process CpG-STAT3ASO, which results in secretion of oligonucleotide-containing exosomes
(A) NSCs were incubated with CpG-STAT3ASOCy3 (500 nM), and the oligonucleotide uptake was assessed using flow cytometry at various times. (B) Intracellular colocalization of CpG-STAT3ASOCy3 with CD63+ vesicles in NSC cytoplasm. Confocal microscopic imaging of NSCs treated with CpG-STAT3ASOCy3 (250 nM) for 3 h; scale bar represents 10 μm. (C–E) Characterization of NSC-derived exosomes with or without prior incubation with 2 μM of CpG-STAT3ASOCy3 for 24 h. After washing and replacing media, cells were incubated for 48 h before harvesting media and isolating EVs. Size and concentration of EVs isolated using ultracentrifugation were measured using NanoSight NS300 (C), and EV morphology was assessed using transmission electron microscopy (D); scale bars represent 100 μm. The isolated EVs were captured using anti-CD63 beads, and the expression of exosome-specific surface antigens was assessed using flow cytometry (E). Shown are representative results from one of three independent experiments.
Secretion of exosomes carrying CpG-STAT3ASO by NSCs is time and dose dependent
Mammalian cells, such as mesenchymal stem cells, can be genetically engineered for long-term production of exosomes containing non-endogenous nucleic acids, such as miRNA and siRNA.38 Given that synthetic CpG-ASO conjugates only transiently accumulate in target NSCs, we assessed the kinetics and efficacy of oligonucleotide loading into exosomes using flow cytometry. After 1 day of NSCs incubation with CpG-STAT3ASOCy3 (5 μM), the percentage of fluorescently labeled EVs isolated from the culture media reached ∼80% at 24 h and decreased over time from ∼55% at 48 h to ∼25% at 96 h (Figure 2A). The EV loading efficacy as measured at 48 h clearly increased in correlation with the oligonucleotide concentration from 0.5 to 5 μM (Figure 2B). NSCs treated with 2–5 μM CpG-STAT3ASOCy3 produced 33%–65% of Cy3+ EVs. At all tested oligonucleotide concentrations, CpG-STAT3ASOCy3 was found almost exclusively in CD63+CD81+ exosome population (Figure 2C). As expected, the amount of encapsulated EV(CpG-STAT3ASO) derived from 1 × 106 NSCs was correlated with the NSC loading dose as measured using spectrophotometer (Figure 2D). Even at the maximum amount of the exosome-encapsulated oligonucleotide (300 ng/1 × 106 NSCs), we did not observe significant changes in the total exosomal protein content (Figure 2E). Importantly, the NSC-dependent method of EV loading was not selective to CpG-STAT3ASO, as demonstrated by effective EV encapsulation of CpG conjugated to an unrelated AON as measured using two independent methods (Figure S1). In parallel studies, we also confirmed that other human and mouse cells also have ability to spontaneously load CpG conjugates into exosomes, indicating broader applicability of this approach (Figure S2). Overall, our findings underscored feasibility and simplicity of using NSCs as a source of encapsulated therapeutic EV(CpG-STAT3ASO).
Figure 2.

Time- and dose-dependent secretion of exosomes carrying CpG-STAT3ASO by NSCs
(A–C) NSCs were treated with various concentrations of CpG-STAT3ASOCy3 for 24 h and then culture media were replaced and EVs were isolated using ultracentrifugation and anti-CD63 beads at indicated times. Time- (A) and dose-dependent (B) effects on the percentage of the oligonucleotide-loaded exosomes secreted by NSCs; means ± SD (n = 3). (C) NSC-derived CD63+CD81+ exosomes are a major vesicle population carrying CpG-STAT3ASOCy3 as assessed by flow cytometry. (D and E) The amounts of encapsulated CpG-STAT3ASOCy3 (D) or the secreted total EV proteins (E) were measured using absorbance (564 nm) or colorimetric BCA assay, respectively, and normalized for 1 × 106 NSCs; means ± SD (n = 3).
Exosome-encapsulated CpG-STAT3ASO retains immunostimulatory potential
While immune properties of NSC-derived exosomes are less characterized, recent studies suggested that they have similar anti-inflammatory properties to MSC-derived exosomes.39 Thus, we decided to investigate whether loading NSC exosomes with a synthetic and potently immunostimulatory CpG-STAT3ASO10 will change their immune properties. First, we tested the effect of native and CpG-STAT3ASO-loaded exosomes on human monocyte-derived dendritic cells (mDCs). The mDCs were differentiated from human monocytes and then incubated for 24 h with exosomes carrying CpG-STAT3ASO (∼100 nM oligonucleotide concentration), native exosomes, or naked CpG-STAT3ASO (500 nM). We measured mRNA levels of IL12, as a critical indicator of DC activation, and STAT3 gene target using real-time qPCR. As shown in Figure 3A, EV(CpG-STAT3ASO) showed enhanced stimulation of IL12 expression compared with native EVs as well as naked CpG-STAT3ASO in the equimolar amount. Both encapsulated and naked CpG-STAT3ASO significantly reduced target STAT3 expression, although the knockdown effect of EV(CpG-STAT3ASO) was slightly reduced (Figure 3A). We further evaluated the efficacy of NSC-derived, EV-encapsulated CpG-STAT3ASO on mouse immune cells by measuring maturation of bone-marrow-derived-macrophages using flow cytometry. Macrophages were treated for 24 h with EV(CpG-STAT3ASO) (∼50–110 nM equivalent oligonucleotide concentration), native exosomes, or naked oligonucleotide (250 nM) and lipopolysaccharide (LPS) as a positive control. EV(CpG-STAT3ASO) induced significant and dose-dependent immune activation as measured by an increased percentage of CD11b+MHCHICD40+ macrophages similar to the effect of the naked CpG-STAT3ASO and LPS (Figure 3B). Treatment with naive EVs appeared to inhibit macrophage activation compared with untreated cells, suggesting immunosuppressive properties of NSC-derived exosomes. The immunostimulatory effects of EV(CpG-STAT3ASO) correlated also with the enhanced stimulation of immunostimulatory nuclear factor κB (NF-κB) signaling compared with naive NSC EVs. As measured using RAW-Blue macrophages, stably expressing NF-κB-driven reporter gene, we observed dose-dependent increase of NF-κB activation by EV(CpG-STAT3ASO) for up to 10-fold upregulation comparable to the effect of naked oligonucleotide (Figure 3C). Altogether, these experiments provide evidence that the CpG-STAT3ASO encapsulation did not reduce immunostimulatory properties of this molecule but rather enhanced the potency.
Figure 3.

EV-encapsulated CpG-STAT3ASO retains biological activity
(A) The effect of EVs (isolated using precipitation-based method) with or without CpG-STAT3ASO on human immune cells. Human mDCs were incubated for 24 h with CpG-STAT3ASO-loaded EVs (200 μg/mL, with oligonucleotide concentration ∼100 nM), 200 μg/mL of empty exosomes, or naked CpG-STAT3ASO (500 nM). The expression of IL12 (left) and STAT3 mRNAs (right) were measured using real-time qPCR. Shown are representative results using mDCs derived from two donors in triplicates; means + SD; ∗p < 0.05, ∗∗∗p < 0.001. (B) The encapsulated CpG-STAT3ASO dose-dependently activates mouse bone-marrow-derived macrophages. Differentiated macrophages were treated for 24 h as indicated before flow cytometric assessment of immune activation markers, such as MHC class II complexes and CD40 costimulatory molecule. Naked CpG-STAT3ASO (250 nM) and LPS (100 ng/mL) were used as positive controls; means + SD.; ∗∗∗p < 0.001 (C) The encapsulated CpG-STAT3ASO dose-dependently activates NF-κB signaling in macrophages. RAW-Blue macrophages expressing NF-κB-SEAP reporter gene were treated for 24 h using EV(CpG-STAT3ASO) at 50 or 100 μg/mL protein (containing ∼25 or 50 nM oligonucleotide concentrations) or naked CpG-STAT3ASO (250 nM). The NF-κB activity was assessed colorimetrically; shown are means + SD; ∗∗∗p < 0.001.
NSC/EV-delivered CpG-STAT3ASO activates glioma-associated myeloid cells and inhibits tumor progression in mice
The NSC ability to migrate into hypoxic tumor areas is an attractive opportunity for drug delivery, especially to brain tumors.16 To determine the feasibility of using NSCs for the delivery of encapsulated EV(CpG-STAT3ASO), we first assessed the effect of the oligonucleotide alone on NSC viability and migration in vitro. As shown in Figure 4A, CpG-STAT3ASO was not cytotoxic to NSCs at any tested concentration. This is expected since the immortalized NSCs used in our studies (HB1.F3.CD) stably express v-myc.17 Therefore, they can maintain myc-dependent survival signaling, even in the absence of STAT3, which is a key upstream regulator of this pathway in stem cells.2,40 In addition, the NSC migration triggered by U251 glioma cell-conditioned media or by SDF-141 was not significantly affected for up to 5 μM dose of CpG-STAT3ASO, as measured using transwell assays (Figures 4B and S3). Thus, we selected to load NSCs using 2 μM CpG-STAT3ASO concentration in further studies on the EV(CpG-STAT3ASO) activity in vivo.
Figure 4.

NSCs deliver CpG-STAT3ASO to glioma-associated myeloid cells and inhibit growth of GL261 tumors in vivo
(A and B) CpG-STAT3ASO loading does not affect NSC viability but can influence cell migration. The NSC viability (A) was measured using flow cytometry after 24 h incubation with CpG-STAT3ASO at various dosing. (B) NSCs migration was assessed using transwell assay; means ± SEM (n = 4); ∗∗p < 0.01, ∗∗∗p < 0.001. (C) NSCs are an effective vehicle for CpG-STAT3ASO delivery into the glioma-associated myeloid cells. GL261 glioma-bearing mice were injected peritumorally with 1 × 106 NSCs loaded for 24 h with CpG-STAT3ASOCy3 or with the corresponding amount of oligonucleotide alone (0.05 mg/kg). After 1 day, the percentage of CpG-STAT3ASOCy3 uptake was assessed in glioma-associated myeloid cells using flow cytometry; means ± SEM (n = 3); ∗p < 0.05, ∗∗p < 0.01. (D) CpG-STAT3ASOCy3 is detectable in Iba1+ glioma-associated microglia after NSC-mediated intracranial delivery. Mice were treated as in (A), and brains were harvested after 24 h for immunofluorescent staining and confocal imaging. Shown are representative images (n = 4); scale bars represent 100 μm (20 μm in insets). (E) NSC delivery of CpG-STAT3ASO results in immune activation of glioma-associated microglia (CD11b+CD45hi); shown are means ± SEM (n = 5); ∗∗p < 0.01. (F) Mice with established GL261 tumors were treated using every other day i.t. injections of 1 × 106 NSCs loaded with CpG-STAT3ASO, native NSCs, CpG-STAT3ASO alone at the equivalent amount (0.05 mg/kg, 20× lower than the effective dose for a naked oligonucleotide), or PBS; shown are means ± SEM (n = 5); ∗p < 0.05.
Our recent studies demonstrated superior effects of CpG-STAT3ASO compared with STAT3ASO alone or TLR9 stimulation alone against solid tumor models.10 CpG-STAT3ASO was also effective against GL261 glioma when used at high 5 mg/kg intratumoral dosing (Figure S4). To test the feasibility of NSC-mediated delivery of CpG-STAT3ASO into the intracranial glioma microenvironment, we first compared biodistribution of fluorescently labeled oligonucleotides loaded into NSCs or injected naked at a similar estimated amount (∼150 ng based on the results in Figure 2D). NSCs or oligonucleotides were injected peritumorally into the brain parenchyma in the vicinity of orthotopic GL261 tumors. As shown in Figure 4C, the percentage of CpG-STAT3ASOCy3 in the tumor-associated myeloid cells, such as microglia, macrophages, and MDSCs, was significantly elevated by the NSC-mediated oligonucleotide delivery compared with the injection of oligonucleotide alone. In contrast, no detectable fluorescent signal from either treatment was detected in the opposite tumor-free brain hemisphere. The successful NSC-mediated delivery of CpG-STAT3ASOCy3 to glioma-associated microglia was also verified using confocal microscopy (Figure 4D). Peritumoral injection of NSCs loaded with CpG-STAT3ASOCy3 resulted in detectable localization of fluorescently labeled oligonucleotide with Iba-1+ microglia within 24 h after injection. The treatment with NSCs loaded with CpG-STAT3ASO resulted in significant increase of CD40 and CD80 activation markers on glioma-associated microglia by about 60% and 80%, respectively (Figures 4E and S5).
Since repeated injections of NSCs into an intact brain parenchyma in mice were not possible, we used subcutaneous model of GL261 glioma for the initial proof-of-concept efficacy study. Mice with established subcutaneous tumors were treated every other day with peritumoral injections of 1 × 106 NSCs loaded with CpG-STAT3ASO, 1 × 106 naive NSCs, CpG-STAT3ASO alone (in equivalent amount, 0.05 mg/kg, 100× lower than the standard effective dosing in Figure S4), or PBS. As shown in Figure 4F, CpG-STAT3ASO-loaded NSCs significantly inhibited GL261 tumor growth compared with the lack of antitumor effects of native NSCs or the suboptimal dose of CpG-STAT3ASO alone. Altogether, these results strongly suggest the feasibility and potential of using NSC-mediated delivery of encapsulated CpG-STAT3ASO for glioma immunotherapy.
Discussion
The clinical translation of ONTs faces multiple challenges, including the limited circulatory half-life for small oligomers and potential for exacerbated inflammatory effects.42 The latter is a primary concern for the use of ONTs in brain tumor therapy.43 Hence, there is significant interest in utilizing nanoparticle, vesicle, or cellular vehicles for ONT delivery. Hereby presented results are, to our knowledge, the first demonstration of spontaneous encapsulation of synthetic ONTs into extracellular vesicles, including exosomes. The phosphorothioated CpG conjugates provide a simple and scalable EV loading strategy that is not limited to NSCs but could be adopted to a variety of myeloid cells, B cells, and cancer cells, which were shown to efficiently internalize CpG conjugates with siRNA, antisense, or DNA decoy molecules.6 It does not require stable expression of RNA cargo or ex vivo manipulation, such as electroporation or lipofection, which often fail to efficiently load ONTs into exosomes.44,45 Also, the dose-dependent loading of synthetic CpG conjugates allows for efficient and reliable EV loading, while endogenous RNA and DNA molecules are known to be differentially distributed into various types of EVs and non-vesicular extracellular matter.46
Native and engineered exosomes are increasingly being explored for cancer immunotherapy.24,44 However, their success depends largely on the effective trafficking to tumor tissues and/or lymphoid organs as opposed to liver, which is the major site of exosome accumulation after systemic delivery.47,48 The well-defined NSC tropism to hypoxic tumor areas provides an alternative cellular vehicle for the targeted transport and release of EV-encapsulated ONTs. The recent study demonstrated a successful delivery of NSC-carried nanoparticles into target tumors within hours after intraperitoneal injection.49 Even when injected into a brain hemisphere or ventricle distant from tumor, the NSCs were found accumulated in glioma on the opposite site within 2 days.50 As demonstrated in this study, NSCs can deliver sufficient amounts of CpG-STAT3ASO into orthotopic glioma microenvironment to activate the major tumor-associated immunosuppressive cell subsets, such as microglia, tumor-associated macrophages (TAMs), and MDSCs. Furthermore, peritumoral injections of NSC(CpG-STAT3ASO) were effective in reducing growth of both subcutaneous and orthotopic glioma. In addition to more effective intratumoral delivery of EV-encapsulated versus naked CpG-STAT3ASO, our results suggest that EV(CpG-STAT3ASO) has increased immunostimulatory potential compared with the equivalent amount of free oligonucleotide. This is consistent with previous reports suggesting that CpG ODNs formulated into nanoparticles can elicit immunostimulatory and antitumor effects at much lower dosing than nonencapsulated ODNs.51 It is yet unclear whether these effects result from the enhanced uptake of encapsulated oligonucleotides, the increased intracellular immune signaling triggered by the complexed TLR9 agonists, or both. Importantly, the immunostimulatory properties of CpG-STAT3ASO10,52 seem to completely alleviate the inherent immunomodulatory functions ascribed to NSCs as well as to NSC-derived exosomes.39 The immunosuppressive effects of protein and miRNA content of NSC-derived exosomes and soluble protein mediators secreted by NSCs fueled interest in their application to treat chronic inflammatory brain disorders through M2 polarization of microglia.39 The low effective dose of encapsulated CpG-STAT3ASO together with any residual immunoregulatory effect of NSCs and their exosomes are likely to have additional neuroprotective effects and reduce chances of unwanted immunostimulatory effects beyond the glioma microenvironment. While further studies will focus on the optimization of NSC(CpG-STAT3ASO) dosing and delivery routes, our results so far underscore safety of this approach in intracranial administration. As demonstrated for another cellular glioma therapy, it is clinically feasible and safe to administer to glioma patients repeated injections of glioma-cell-specific CAR T cells into a post-surgical intracranial cavity using an implanted Rickham device.53 A similar method of intracranial and intracavitary infusions was also used in clinical trials to deliver unformulated, immunostimulatory CpG oligonucleotides to patients with brain tunors.14,54 NSCs have already proven clinically safe and effective in the delivery of chemotherapeutic drugs to brain tumors.18 Loaded with the bifunctional, immunostimulatory, and tolerance-breaking CpG-STAT3ASO, the NSCs provide a promising strategy for therapy of immune “cold” gliomas. We believe that NSC-mediated delivery of CpG-STAT3ASO creates an opportunity for safer and more effective oligonucleotide-based immunotherapy of glioma and potentially other brain tumors.
Materials and methods
Cell culture
PBMCs from anonymous healthy donors were collected in accordance with the Declaration of Helsinki under the institutional review board (IRB) protocol 13378 (City of Hope). Human U251, mouse GL261 glioma cells, and RAW264.7 macrophages were from American Type Culture Collection (ATCC) (Manassas, VA, USA), whereas RAW-Blue cells were purchased from Invivogen (San Diego, CA). HB1.F3 NSCs (passage 18–24) were kindly provided by Dr. Karen Aboody and previously characterized for clinical use.15,17 Cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS). All tested cells were cultured for less than 6 months before experiments and tested bimonthly from mycoplasma infections.
For the generation of human mDCs, peripheral blood mononuclear cells (PBMCs) were cultured for 6 days in RPMI1640 medium with 10% FBS in a presence of 100 ng/mL granulocyte-macrophage colony-stimulating factor (GM-CSF) and 50 ng/mL interleukin-4 (IL-4) (Peprotech, Cranbury, NJ). Next, cells were seeded on a 12-well plate and treated with EV(PBS) and EV(CpG-STAT3ASO) or EV(CpG-STAT3ASOCy3), respectively, for 24 h before further studies. For generation of the bone-marrow-derived macrophages (BMMs), bone marrow cells from C57BL/6 mice were plated at 2 × 106 cells/mL in triplicates in RPMI1640, 20% FBS supplemented with 50 ng/mL M-CSF. The medium was replaced on day 4, and the culture was continued until day 7.
Oligonucleotide synthesis, NSC loading, and isolation of extracellular vesicles
The human- and mouse-specific CpG-STAT3ASO conjugates were synthesized in the DNA/RNA Synthesis Core (COH) by linking CpG-ODNs to STAT3 ASO as previously described.10 For the internalization studies, oligonucleotides were labeled on 3′ ends using Cy3 fluorochrome.
NSCs were cultured overnight in DMEM with exosome-free 10% FBS (exosomes removed by ultracentrifugation at 110,000×g for 16 h). The next day, cells were incubated with or without 0.5–5 μM CpG-STAT3ASO for 24 h and then medium was replaced and cells were cultured for 24–96 h, depending on the experiment. Next, culture media were collected, briefly centrifuged at 2000×g, filtered through 0.22 μm filter (Millipore), followed by EV isolation using either ultracentrifugation using standard method55 (100,00×g/20 min at 4°C followed by two-step 1,100,00×g/1 h centrifugation with an intermittent PBS wash) or using Total Exosome Isolation Reagent (Invitrogen, Waltham, MA) following the manufacturer’s protocol. EV pellets were resuspended in filtered PBS and then used for downstream applications or stored at 4°C for up to 1 week. The concentration and size distribution profile of the isolated EVs were evaluated using a NanoSight NS300 instrument and NTA 3.2 software. Videos were recorded at camera level 15. Samples were diluted 1:100 in PBS to achieve a concentration of 1–5 × 108 particles/mL. For each sample, three 60-s recordings were analyzed in the batch-processing mode. The molar concentrations of the encapsulated oligonucleotide were estimated using spectrophotometric measurements at 545/575 nm using Cytation5 multimode absorbance reader (BioTek, Winooski, VT) in relation to the standard curve based on serial dilutions of CpG-STAT3ASOCy3 with non-loaded EVs as a blank control for background subtraction.
Glioblastoma mouse models
C57BL/6 mice, aged between 6 and 8 weeks, were purchased from the Jackson Laboratory (Bar Harbor, ME). Mouse care and experimental procedures were performed under pathogen-free conditions in accordance with established institutional guidance and approved protocols from Institutional Animal Care and Use Committees. For assessment of NSCs and EVs delivery and activity in brain tumor microenvironment (TME), 1 × 105 GL261 were injected intracranially (IC) into frontal lobe. After 7 days, mice were injected IC (caudal-lateral to tumor) with PBS (4 μL), CpG-STAT3ASO alone (75 ng/4 μL), NSCs alone, or NSCs loaded with CpG-STAT3ASO (5 × 105 cells/4 μL). Single-cell suspension was prepared using Brain Dissociation Kit (Miltenyi Biotec), according to manufacturer’s instructions, and then analyzed using flow cytometry. For tumor growth experiments, 5 × 105 GL261 cells were injected subcutaneously (s.c.). When tumors reached ∼50 mm3 size, mice were treated intratumorally (i.t.) with PBS, 1 × 106 cells NSCs alone, 1 × 106 cells NSCs treated with 2 μM of CpG-STAT3ASO, or equivalent amount of CpG-STAT3ASO alone. Tumor size was measured every other day.
Confocal microscopy
NSCs were seeded on 18-well Nunc Lab-Tec chamber slides (Sigma-Aldrich, St. Louis, MO) and allowed to attach overnight. The next day, cells were incubated with various CpG-STAT3ASOCy3 concentrations for indicated times. After washing, cells were fixed in 4% paraformaldehyde (EMS), permeabilized with Triton-X, blocked with 5% normal serum, and incubated with primary antibodies (anti-CD63; Abcam, Waltham, MA) for 18 h. For tissue sections, formalin-fixed tumors were impregnated with 30% sucrose and then cut into 10-μm sections using a cryostat followed by the incubation with Iba-1-specific antibody (Abcam). Next, samples were labeled with Alexa-488-conjugated secondary antibody, stained with Hoechst33342, and mounted in Vectashield Hard-Set medium (Vector Laboratories, Burlingame, CA). Slides were visualized on an LSM880 confocal microscope (Zeiss, White Plains, NY) and analyzed using LSM ImageBrowser (Zeiss).
Electron microscopy
The EVs were placed onto 300-mesh carbon-formvar-coated grids and allowed to absorb to the formvar for a minimum of 1 min. Grids were rinsed with double-distilled water and stained for contrast using 1% uranyl acetate. The samples were viewed with an FEI Tecnai T12 transmission electron microscope at 120 keV, and images were taken with a Gatan Ultrascan 2K charge-coupled device (CCD) camera.
Flow cytometry
Mouse cells staining was performed using fluorochrome-labeled antibodies specific to MHC class II, CD11b, F4/80, Gr-1, CD40, CD45, CD80, and CD86 (Thermo Fisher Scientific, Irwindale, CA). Unspecific binding was blocked with anti-FcγIII/IIR (Thermo Fisher Scientific). For viability assessment, cells were stained using APC Annexin V apoptosis detection kit I (BD Biosciences, Franklin Lakes, NJ). For EVs phenotyping, isolated vesicles were incubated with anti-CD63 beads (Thermo Fisher Scientific) according to manufacturer’s instruction and then stained with fluorochrome-labeled CD9, CD63, and CD81 antibodies (Thermo Fisher Scientific). Fluorescence data were acquired on Attune NxT Flow Cytometer (Thermo-Fisher) and analyzed using FlowJo software (TreeStar).
Migration assay
NSCs were treated with indicated doses of CpG-STAT3ASO for 24–72 h. Next, cells were seeded on gelatin-coated transwell inserts (Corning, Corning, NY) in DMEM-supplemented 0.5% BSA. The lower chamber was filled with DMEM 10% FBS, U251 conditioned media, or SDF-1 (100 ng/mL) and then cells were permitted to migrate for 4 h. Next, cells were fixed in 4% paraformaldehyde and stained with 0.2% crystal violet. Images were acquired on Observer Z1 Live Cell (Zeiss), and the number of migrated cells was quantified using ImageJ (NIH) from at least 4 view fields.
Quantitative real-time PCR
Total RNA was extracted from cultured or in vivo grown tumor cells using Maxwell system (Promega, Madison, WI) and then transcribed into cDNAs using iScript cDNA Synthesis kit (Bio-Rad, Hercules, CA). The qPCR was carried out using specific primers for IL12A, STAT3, ACTB, and UBQ using CFX96 Real-Time PCR Detection System (Bio-Rad), as previously described.10
Statistics
Unpaired t test was used to calculate two-tailed p value to estimate statistical significance of differences between two treatment groups. One- or two-way ANOVA followed by Bonferroni post-test were applied to assess the differences between multiple groups or in tumor growth kinetics experiments. Statistically significant p values were indicated in figures as follows: ∗∗∗p < 0.001; ∗∗p < 0.01; and ∗p < 0.05. Data were analyzed using Prism software v.6.01 (GraphPad).
Acknowledgments
We are grateful to the staff at Analytical Cytometry, Light Microscopy, DNA/RNA Synthesis, Pathology, and Animal Resource Cores (COH). This work was supported in part by the National Cancer Institute/National Institutes of Health award numbers R01CA215183 (M.K.) and P30CA033572 (COH) and the Department of Defense award number W81XWH1920852 (M.K.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions
Conceptualization, M.K.; methodology, M.K., S.N., K.S.A., M.H., L.F., K.M., and P.S.; investigation, T.A., C.Y., S.N., and Q.Z.; writing – original draft, T.A., C.Y., and M.K.; writing – review & editing, M.K.; resources, M.K. and K.S.A.; supervision, M.K.
Declaration of interests
M.K. and K.S.A. are inventors on the patent application submitted by COH that covers the design of the NSC-mediated delivery strategy presented in this report. M.K. serves on the Scientific Advisory Board of Scopus Biopharma and its subsidiary Duet Therapeutics with stock options. All other authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2021.12.029.
Supplemental information
5
References
- 1.Weller M., Wick W., Aldape K., Brada M., Berger M., Pfister S.M., Nishikawa R., Rosenthal M., Wen P.Y., Stupp R., et al. Glioma. Nat. Rev. Dis. Primers. 2015;1:1–18. doi: 10.1038/nrdp.2015.17. [DOI] [PubMed] [Google Scholar]
- 2.Huynh J., Chand A., Gough D., Ernst M. Therapeutically exploiting STAT3 activity in cancer - using tissue repair as a road map. Nat. Rev. Cancer. 2019;19:82–96. doi: 10.1038/s41568-018-0090-8. [DOI] [PubMed] [Google Scholar]
- 3.Su Y.-L., Banerjee S., White S.V., Kortylewski M. STAT3 in tumor-associated myeloid cells: multitasking to disrupt immunity. Int. J. Mol. Sci. 2018;19:1–14. doi: 10.3390/ijms19061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferguson S.D., Srinivasan V.M., Heimberger A.B. The role of STAT3 in tumor-mediated immune suppression. J. Neurooncol. 2015;123:385–394. doi: 10.1007/s11060-015-1731-3. [DOI] [PubMed] [Google Scholar]
- 5.Yu H., Kortylewski M., Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 6.Kortylewski M., Moreira D. Myeloid cells as a target for oligonucleotide therapeutics: turning obstacles into opportunities. Cancer Immunol. Immunother. 2017;66:979–988. doi: 10.1007/s00262-017-1966-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kortylewski M., Swiderski P., Herrmann A., Wang L., Kowolik C., Kujawski M., Lee H., Scuto A., Liu Y., Yang C., et al. In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat. Biotechnol. 2009;27:925–932. doi: 10.1038/nbt.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sen M., Paul K., Freilino M.L., Li H., Li C., Johnson D.E., Wang L., Eiseman J., Grandis J.R. Systemic administration of a cyclic signal transducer and activator of transcription 3 (STAT3) decoy oligonucleotide inhibits tumor growth without inducing toxicological effects. Mol. Med. 2014;20:46–56. doi: 10.2119/molmed.2013.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hong D.S., Kurzrock R., Woessner R., Younes A.F., Nemunaitis J., Fowler N., Zhou T., Schmidt J., Jo M., Lee S.J., et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015;31:314–326. doi: 10.1126/scitranslmed.aac5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moreira D., Adamus T., Zhao X., Su Y.-L., Zhang Z., White S.V., Swiderski P., Lu X., DePinho R.A., Pal S.K., et al. STAT3 inhibition combined with CpG immunostimulation activates antitumor immunity to eradicate genetically distinct castration-resistant prostate cancers. Clin. Cancer Res. 2018;24:5948–5962. doi: 10.1158/1078-0432.CCR-18-1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.da Fonseca A.C.C., Badie B. Microglia and macrophages in malignant gliomas: recent discoveries and implications for promising therapies. Clin. Dev. Immunol. 2013;2013:264124. doi: 10.1155/2013/264124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meng Y., Kujas M., Marie Y., Paris S., Thillet J., Delattre J.-Y., Carpentier A.F. Expression of TLR9 within human glioblastoma. J. Neurooncol. 2008;88:19–25. doi: 10.1007/s11060-008-9536-2. [DOI] [PubMed] [Google Scholar]
- 13.Herrmann A., Cherryholmes G., Schroeder A., Phallen J., Alizadeh D., Xin H., Wang T., Lee H., Lahtz C., Swiderski P., et al. TLR9 is critical for glioma stem cell maintenance and targeting. Cancer Res. 2014;74:5218–5228. doi: 10.1158/0008-5472.CAN-14-1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carpentier A., Metellus P., Ursu R., Zohar S., Lafitte F., Barrié M., Meng Y., Richard M., Parizot C., Laigle-Donadey F., et al. Intracerebral administration of CpG oligonucleotide for patients with recurrent glioblastoma: a phase II study. Neuro Oncol. 2010;12:401–408. doi: 10.1093/neuonc/nop047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aboody K.S., Brown A., Rainov N.G., Bower K.A., Liu S., Yang W., Small J.E., Herrlinger U., Ourednik V., Black P.M., et al. Neural stem cells display extensive tropism for pathology in adult brain: evidence from intracranial gliomas. Proc. Natl. Acad. Sci. U S A. 2000;97:12846–12851. doi: 10.1073/pnas.97.23.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mooney R., Hammad M., Batalla-Covello J., Abdul Majid A., Aboody K.S. Concise review: neural stem cell-mediated targeted cancer therapies. Stem Cells Transl. Med. 2018;7:740–747. doi: 10.1002/sctm.18-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aboody K.S., Najbauer J., Metz M.Z., D’Apuzzo M., Gutova M., Annala A.J., Synold T.W., Couture L.A., Blanchard S., Moats R.A., et al. Neural stem cell-mediated enzyme/prodrug therapy for glioma: preclinical studies. Sci. Transl. Med. 2013;5:184–195. doi: 10.1126/scitranslmed.3005365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Portnow J., Synold T.W., Badie B., Tirughana R., Lacey S.F., D’Apuzzo M., Metz M.Z., Najbauer J., Bedell V., Vo T., et al. Neural stem cell-based anticancer gene therapy: a first-in-human study in recurrent high-grade glioma patients. Clin. Cancer Res. 2017;23:2951–2960. doi: 10.1158/1078-0432.CCR-16-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu H.-M., Zhang L.-F., Ding P.-S., Liu Y.-J., Wu X., Zhou J.-N. Microglial activation mediates host neuronal survival induced by neural stem cells. J. Cell Mol. Med. 2014;18:1300–1312. doi: 10.1111/jcmm.12281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alvarado A.G., Lathia J.D. Taking a toll on self-renewal: tlr-mediated innate immune signaling in stem cells. Trends Neurosci. 2016;39:463–471. doi: 10.1016/j.tins.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 21.Tkach M., Théry C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164:1226–1232. doi: 10.1016/j.cell.2016.01.043. [DOI] [PubMed] [Google Scholar]
- 22.Janas T., Janas M.M., Sapoń K., Janas T. Mechanisms of RNA loading into exosomes. FEBS Lett. 2015;589:1391–1398. doi: 10.1016/j.febslet.2015.04.036. [DOI] [PubMed] [Google Scholar]
- 23.Lötvall J., Hill A.F., Hochberg F., Buzás E.I., Di Vizio D., Gardiner C., Gho Y.S., Kurochkin I.V., Mathivanan S., Quesenberry P., et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles. 2014;3:26913. doi: 10.3402/jev.v3.26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu R., Rai A., Chen M., Suwakulsiri W., Greening D.W., Simpson R.J. Extracellular vesicles in cancer - implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 2018;15:617–638. doi: 10.1038/s41571-018-0036-9. [DOI] [PubMed] [Google Scholar]
- 25.Chen G., Huang A.C., Zhang W., Zhang G., Wu M., Xu W., Yu Z., Yang J., Wang B., Sun H., et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560:382–386. doi: 10.1038/s41586-018-0392-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoshino A., Costa-Silva B., Shen T.-L., Rodrigues G., Hashimoto A., Tesic Mark M., Molina H., Kohsaka S., Di Giannatale A., Ceder C., et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329–335. doi: 10.1038/nature15756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chalmin F., Ladoire S., Mignot G., Vincent J., Bruchard M., Remy-Martin J.-P., Boireau W., Rouleau A., Simon B., Lanneau D., et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Invest. 2010;120:457–471. doi: 10.1172/JCI40483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferguson S.W., Nguyen J. Exosomes as therapeutics: the implications of molecular composition and exosomal heterogeneity. J. Control Release. 2016;228:179–190. doi: 10.1016/j.jconrel.2016.02.037. [DOI] [PubMed] [Google Scholar]
- 29.Bell B.M., Kirk I.D., Hiltbrunner S., Gabrielsson S., Bultema J.J. Designer exosomes as next-generation cancer immunotherapy. Nanomedicine. 2016;12:163–169. doi: 10.1016/j.nano.2015.09.011. [DOI] [PubMed] [Google Scholar]
- 30.Tominaga N., Yoshioka Y., Ochiya T. A novel platform for cancer therapy using extracellular vesicles. Adv. Drug Deliv. Rev. 2015;95:50–55. doi: 10.1016/j.addr.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 31.Alvarez-Erviti L., Seow Y., Yin H., Betts C., Lakhal S., Wood M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011;29:341–345. doi: 10.1038/nbt.1807. [DOI] [PubMed] [Google Scholar]
- 32.Elahi F.M., Farwell D.G., Nolta J.A., Anderson J.D. Preclinical translation of exosomes derived from mesenchymal stem/stromal cells. Stem Cells. 2020;38:15–21. doi: 10.1002/stem.3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nechaev S., Gao C., Moreira D., Swiderski P., Jozwiak A., Kowolik C.M., Zhou J., Armstrong B., Raubitschek A., Rossi J.J., et al. Intracellular processing of immunostimulatory CpG-siRNA: Toll-like receptor 9 facilitates siRNA dicing and endosomal escape. J. Control Release. 2013;170:307–315. doi: 10.1016/j.jconrel.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalluri R., LeBleu V.S. The biology, function, and biomedical applications of exosomes. Science. 2020;367:eaau6977. doi: 10.1126/science.aau6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taylor D.D., Shah S. Methods of isolating extracellular vesicles impact down-stream analyses of their cargoes. Methods. 2015;87:3–10. doi: 10.1016/j.ymeth.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 36.Mateescu B., Kowal E.J.K., van Balkom B.W.M., Bartel S., Bhattacharyya S.N., Buzás E.I., Buck A.H., de Candia P., Chow F.W., Das S., et al. Obstacles and opportunities in the functional analysis of extracellular vesicle RNA - an ISEV position paper. J. Extracell. Vesicles. 2017;6:1–32. doi: 10.1080/20013078.2017.1286095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mathieu M., Névo N., Jouve M., Valenzuela J.I., Maurin M., Verweij F.J., Palmulli R., Lankar D., Dingli F., Loew D., et al. Specificities of exosome versus small ectosome secretion revealed by live intracellular tracking of CD63 and CD9. Nat. Commun. 2021;12:4389. doi: 10.1038/s41467-021-24384-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vakhshiteh F., Atyabi F., Ostad S.N. Mesenchymal stem cell exosomes: a two-edged sword in cancer therapy. Int. J. Nanomed. 2019;14:2847–2859. doi: 10.2147/IJN.S200036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vogel A., Upadhya R., Shetty A.K. Neural stem cell derived extracellular vesicles: attributes and prospects for treating neurodegenerative disorders. EBioMedicine. 2018;38:273–282. doi: 10.1016/j.ebiom.2018.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galoczova M., Coates P., Vojtesek B. STAT3, stem cells, cancer stem cells and p63. Cell. Mol. Biol. Lett. 2018;23:12. doi: 10.1186/s11658-018-0078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao D., Najbauer J., Garcia E., Metz M.Z., Gutova M., Glackin C.A., Kim S.U., Aboody K.S. Neural stem cell tropism to glioma: critical role of tumor hypoxia. Mol. Cancer Res. 2008;6:1819–1829. doi: 10.1158/1541-7786.MCR-08-0146. [DOI] [PubMed] [Google Scholar]
- 42.Levin A.A. Treating disease at the RNA level with oligonucleotides. N. Engl. J. Med. 2019;380:57–70. doi: 10.1056/NEJMra1705346. [DOI] [PubMed] [Google Scholar]
- 43.Khvorova A., Watts J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017;35:238–248. doi: 10.1038/nbt.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wiklander O.P.B., Brennan M.Á., Lötvall J., Breakefield X.O., El Andaloussi S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019;11:eaav8521. doi: 10.1126/scitranslmed.aav8521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kooijmans S.A.A., Stremersch S., Braeckmans K., de Smedt S.C., Hendrix A., Wood M.J.A., Schiffelers R.M., Raemdonck K., Vader P. Electroporation-induced siRNA precipitation obscures the efficiency of siRNA loading into extracellular vesicles. J. Control Release. 2013;172:229–238. doi: 10.1016/j.jconrel.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 46.Jeppesen D.K., Fenix A.M., Franklin J.L., Higginbotham J.N., Zhang Q., Zimmerman L.J., Liebler D.C., Ping J., Liu Q., Evans R., et al. Reassessment of exosome composition. Cell. 2019;177:428–445.e18. doi: 10.1016/j.cell.2019.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wiklander O.P.B., Nordin J.Z., O’Loughlin A., Gustafsson Y., Corso G., Mäger I., Vader P., Lee Y., Sork H., Seow Y., et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. J. Extracell. Vesicles. 2015;4:26316. doi: 10.3402/jev.v4.26316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smyth T., Kullberg M., Malik N., Smith-Jones P., Graner M.W., Anchordoquy T.J. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. J. Control Release. 2015;199:145–155. doi: 10.1016/j.jconrel.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mooney R., Abidi W., Batalla-Covello J., Ngai H.W., Hyde C., Machado D., Abdul-Majid A., Kang Y., Hammad M., Flores L., et al. Allogeneic human neural stem cells for improved therapeutic delivery to peritoneal ovarian cancer. Stem Cell Res. Ther. 2021;12:205. doi: 10.1186/s13287-021-02226-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gutova M., Flores L., Adhikarla V., Tsaturyan L., Tirughana R., Aramburo S., Metz M., Gonzaga J., Annala A., Synold T.W., et al. Quantitative evaluation of intraventricular delivery of therapeutic neural stem cells to orthotopic glioma. Front. Oncol. 2019;9:68. doi: 10.3389/fonc.2019.00068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buss C.G., Bhatia S.N. Nanoparticle delivery of immunostimulatory oligonucleotides enhances response to checkpoint inhibitor therapeutics. Proc. Natl. Acad. Sci. U S A. 2020;117:13428–13436. doi: 10.1073/pnas.2001569117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moreira D., Sampath S., Won H., White S.V., Su Y.-L., Alcantara M., Wang C., Lee P., Maghami E., Massarelli E., et al. Myeloid cell-targeted STAT3 inhibition sensitizes head and neck cancers to radiotherapy and T cell-mediated immunity. J. Clin. Invest. 2020;131:e137001. doi: 10.1172/JCI137001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brown C.E., Alizadeh D., Starr R., Weng L., Wagner J.R., Naranjo A., Ostberg J.R., Blanchard M.S., Kilpatrick J., Simpson J., et al. Regression of glioblastoma after Chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 2016;375:2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ursu R., Carpentier A., Metellus P., Lubrano V., Laigle-Donadey F., Capelle L., Guyotat J., Langlois O., Bauchet L., Desseaux K., et al. Intracerebral injection of CpG oligonucleotide for patients with de novo glioblastoma-A phase II multicentric, randomised study. Eur. J. Cancer. 2017;73:30–37. doi: 10.1016/j.ejca.2016.12.003. [DOI] [PubMed] [Google Scholar]
- 55.Théry C., Amigorena S., Raposo G., Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006;Chapter 3:Unit 3.22. doi: 10.1002/0471143030.cb0322s30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
5