

Abstract
Background
The kallikrein‐kinin system is involved in many (patho)physiological processes and kinin peptides are considered potential clinical biomarkers. Variance in blood specimen collection and processing, artificial ex vivo bradykinin formation, and rapid degradation of kinins have contributed to divergence in published plasma levels, therefore limiting their significance. Thus, reliable preanalytical settings are highly required.
Objectives
This study aimed to develop and evaluate a standardized preanalytical procedure for reliable kinin quantification. The procedure was based on identification of the most impactful variables on ex vivo plasma level alterations.
Methods
Suitable protease inhibitors and blood specimen collection and handling conditions were systematically investigated. Their influence on plasma levels of seven kinins was monitored using an established in‐house liquid chromatography–tandem mass spectrometry platform.
Results
In nonstandardized settings, ex vivo rise of bradykinin was found to already occur 30 seconds after blood sampling with high interindividual variation. The screening of 17 protease inhibitors resulted in a customized seven‐component protease inhibitor, which efficiently stabilized ex vivo kinin levels. The reliability of kinin levels was substantially jeopardized by prolonged rest time until centrifugation, phlebotomy methodology (eg, straight needles, catheters), vacuum sampling technique, or any time delays during venipuncture. The subsequently developed standardized procedure was applied to healthy volunteers and proved it significantly limited interday and interindividual kinin level variability.
Conclusion
The developed procedure for blood specimen collection and handling is feasible in clinical settings and allows for determination of reliable kinin levels. It may contribute to further elucidating the role of the kallikrein‐kinin system in diseases like angioedema, sepsis, or coronavirus disease 2019.
Keywords: blood specimen collection, bradykinin, factor XII, kallikrein‐kinin system, phlebotomy
Essentials.
Current divergency of published plasma kinin levels impede the significance of studies.
Here, impactful preanalytical variables regarding reliable kinin quantitation were identified.
A standardized procedure was developed that was proven to limit interday and interindividual variability.
Applicability in clinical settings facilitates study of kinins in, for example, angioedema or sepsis.
1. BACKGROUND
The kallikrein‐kinin system (KKS) is an endogenous cascade of several bioactive kinin peptides that promote vasodilation, diuresis, inflammation, vascular permeability, and angiogenesis. 1 Thus, kinins are considered potential biomarkers in disorders like coronavirus disease 2019 (COVID‐19), cancer, sepsis, or angioedema. 2 , 3 , 4 However, bioanalysis of endogenous kinin levels is highly sensitive to artificial alterations hindering their reliable quantification and interpretation of study results. These alterations in the KKS intertwine closely with the intrinsic coagulation cascade, whereby contact between blood and either pathophysiologic (eg, neutrophil extracellular traps, long‐chain polyphosphates) or artificial (eg, glass) surfaces initiates autoactivation of factor XII (FXII). 5 Activated FXII (FXIIa) can liberate bradykinin (BK) from high‐molecular‐weight kininogen after activating prekallikrein to plasma kallikrein (PKa). For bioanalysis, this process of so‐called contact activation increases the risk of inaccurate results.
It is well established that preanalytical variables considerably impact the sample quality and reliability of results, with reported rates between 32% and 75%. 6 , 7 Bioanalytical assays related to the coagulation system, particularly peptide analysis, are most prone to variations caused by inadequate specimen handling. 8 , 9 Insufficient control of contact activation during blood sampling and processing introduces a potential source of error in kinin quantitation. 5 Additionally, BK and kallidin (KBK) have short half‐lives (<1 minute). 10 Thus, inadequate phlebotomy conditions and downstream processes may alter kinin levels artificially, impeding reliable bioanalysis.
Published approaches to determining kinins vary regarding the protease inhibitor used, and reporting on preanalytical variables is limited. 11 , 12 , 13 , 14 Consequently, results have yielded diverging levels of BK, ranging from low picograms‐per‐milliliter to high nanograms‐per‐milliliter levels in healthy volunteers. 12 , 13 , 15 , 16 , 17 Even in healthy individuals, this large variability hinders the comparison and interpretation of study results. 18 Some research groups have therefore focused on measuring the more stable (half‐life ≈ 90 minutes) but inactive metabolite BK‐(1‐5) only, which is less prone to artificial changes. 11 , 19 However, monitoring only individual kinin levels renders the profiles of kinin level alterations within the KKS in pathophysiological processes incomplete. The versatility of metabolic processes and the numerous active kinins within the KKS highlight this lack of comprehensive evidence.
Recently, optimization of mass spectrometric sensitivity of kinins enabled the detection of several kinin peptides simultaneously. 20 , 21 However, despite a capable protease inhibitor stabilizing kinins, 22 kinin level variability persisted in preliminary experiments, signifying the influence of additional preanalytical variables.
Therefore, this study aimed to enable robust kinin quantification in plasma by first identifying critical preanalytical variables contributing to artificial changes in plasma kinin levels. Therefore, plasma levels of the kinins BK, KBK, BK‐(1‐8), KBK‐(1‐8), BK‐(1‐7), BK‐(2‐9) and BK‐(1‐5) were monitored using mass spectrometry. Subsequently, this study sought to develop a standardized procedure combining the findings from preanalytical blood specimen collection and handling and appropriate inhibition of kinin formation and degradation.
2. MATERIALS AND METHODS
2.1. Human samples
Fresh blood samples were collected from healthy adult volunteers. The study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of the Medical Faculty at Heinrich‐Heine University (study no. 6112). All participants gave their written informed consent before enrollment.
2.2. Effect of the protease inhibitor on kinin quantification
Figure 1 provides an overview of the investigated preanalytical variables and the general workflow in the following sections.
FIGURE 1.
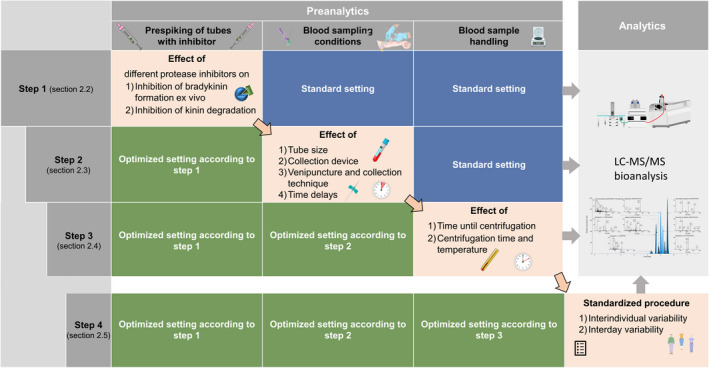
Overview of the conducted investigation regarding the preanalytical variables. The preanalytical conditions were optimized step by step based on the results of the previous step (top‐down). The general workflow was consistent for all experiments, including fresh blood samples and direct analysis without long‐term storage. LC‐MS/MS, liquid chromatography–tandem mass spectrometry
2.2.1. Extent and time course of artificial kinin alterations after phlebotomy
Initially, the extent of artificial kinin formation via contact activation in a routine blood collection without protease inhibition was assessed (n = 4). Blood was collected freshly into EDTA (1.6 mg/mL S‐Monovettes (Sarstedt, Nümbrecht, Germany), centrifuged, and immediately analyzed within 30 minutes.
Additionally, ex vivo formation and degradation of BK and its metabolites in freshly collected blood samples (EDTA and citrate) were monitored over time to investigate the latency of this process. At predefined time points after blood sampling, a customized protease inhibitor 22 was added to whole‐blood aliquots to stop reactions (15, 30, 45, 60, 90, 120, 180, 240, and 300 seconds; and 10, 15, and 30 minutes). Next, whole blood was centrifuged and plasma was analyzed directly.
2.2.2. Evaluation of plasma kinin stability using protease inhibitors
Although the final composition of the protease inhibitor has already been published, 22 the development and rationale driving the recently introduced protease inhibitor have been included here. The protease inhibitor’s development entailed addressing ex vivo BK formation and kinin degradation separately, owing to the numerous enzymes involved in kinin metabolism (Figure 2).
FIGURE 2.
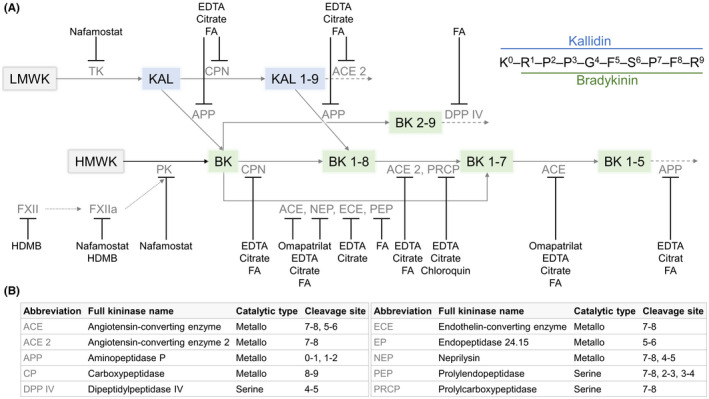
Addressed targets by the customized protease inhibitor within the kallikrein‐kinin system (A). Targets were assigned based on known characteristics of inhibitors. The kininases involved in kinin metabolism are shown with their catalytic type and cleavage site in (B). BK, bradykinin; FA, formic acid; FXIIa, activated factor XII; HDMB, hexadimethrine bromide; HK, high‐molecular‐weight kininogen; KBK, kallidin; LK, low‐molecular‐weight kininogen; PKa, plasma kallikrein, TK, tissue kallikrein
First, the effective prevention of artificial ex vivo BK formation only was examined in greater detail. BK, the first kinin released during contact activation mediated by the serine proteases FXIIa and PKa, was monitored in the presence of several serine protease inhibitors. In identifying suitable protease inhibitors, it was assumed that samples with the best protease inhibition would yield the lowest BK levels. Blood was freshly sampled into S‐Monovettes prespiked with one of the following inhibitors: 0.3 µM aprotinin (Sigma‐Aldrich [catalog no. A1153]), 1 mg/mL hexadimethrine bromide (HDMB [≥94%]; Sigma‐Aldrich [catalog no. H9268]), 19.8 µM nafamostat mesylate ([≥98%] Sigma‐Aldrich [catalog no. N2089]), 100 µM leupeptin hemisulfate ([>90%]; Sigma‐Aldrich [catalog no. L2884]), 200 µg/mL chicken‐egg trypsin inhibitor (Sigma‐Aldrich [catalog no. T2011]), 100 µM phenylmethanesulfonyl fluoride (PMSF; Sigma‐Aldrich [catalog no. 78330]), and 2 mM 4‐(2‐aminoethyl) benzenesulfonyl fluoride (AEBSF [≥97.0%]; Sigma‐Aldrich [catalog no. A8456]). BK levels were measured immediately (reference value), 1.5 and 4.5 hours after sampling to rate the inhibition of artificial formation. Combinations of the most promising inhibitors were further studied. Uninhibited plasma was examined for reference.
After achieving effective inhibition of BK formation in plasma, inhibition of the degradation of the seven kinins was examined. The following protease inhibitors (catalytic type in parentheses) were prespiked based on identified kininases (Figure 2B): 43 to 83 mM EDTA disodium dihydrate (metallo [≥99%]; Carl Roth, Karlsruhe, Germany [catalog no. 8043.2]), 10 to 20 mM trisodium citrate dihydrate (metallo [>99.5%]; Fisher Scientific, Loughborough, UK [catalog no. BP327‐1]), 0.1% to 1% formic acid ([FA] unspecific; Merck, Darmstadt, Germany [catalog no. 1.00264]), 1% ammonia (unspecific; VWR, Langenfeld, Germany [catalog no. 1133.1000]), 200 nM enalaprilat dihydrate (angiotensin‐converting enzyme (ACE)‐specific [100%]; EDQM, Strasburg, France [catalog no. Y0000615]), 10 mM sodium hydroxide (unspecific; VWR [catalog no. 28244.295]), 5 to 50 mM 1,10‐phenanthroline (metallo [≥99%]; Sigma‐Aldrich [catalog no. 131377]), 10 to 1000 nM omapatrilat (ACE and neprilysin‐specific [≥98%]; Sigma‐Aldrich [catalog no. BM0002]), 0.1 to 1 mM benzamidine (serine [≥99.0%]; SERVA Electrophoresis GmbH, Heidelberg, Germany [catalog no. 14525.01]), 1 mM chloroquine diphosphate (prolylcarboxypeptidase‐specific [≥98%]; Sigma‐Aldrich [catalog no. C6628]), and 0.1 to 1 mM PMSF (serine). Except for spiked kinins (1 ng/mL), all tubes were prespiked with 3 mg/mL HDMB and 18.3 µM nafamostat to prevent BK formation. Blood was sampled freshly and plasma aliquots of each inhibitor combination were analyzed immediately and after 3 hours (n = 3).
The developed inhibitor’s performance was evaluated through comparison against commercially available inhibitors and samples without any protease inhibitors. BD P100 and P800 blood collection tubes (Becton Dickinson [BD], Heidelberg, Germany) were applied. These tubes are precoated with BD’s proprietary protease inhibitors (unknown exact composition) and designed to minimize preanalytical variability for protein analysis. Blood was freshly drawn into respective tubes prespiked with kinins (1 ng/mL) to monitor their degradation or formation. Plasma was then analyzed immediately and 3 hours after blood sampling (n = 3).
2.3. Effect of blood specimen collection on kinin quantification
The impact of the blood sampling technique on measured kinin levels was investigated to account for the contact activation‐mediated artificial rises of kinin levels. Blood was sampled into EDTA tubes prespiked with the customized protease inhibitor in the upright position. 22 The tubing was prefilled to avoid insufficient filling of the tubes by air volume. Three tubes were drawn sequentially per setting and volunteer to assess kinin level consistency better. In comparing blood collection systems, a distinct peripheral arm vein (left or right median cubital, cephalic, or basilic) was used to avoid impacts of venipuncture or time delays. An interexperimental control was drawn in triplicate using 21G Safety Multifly with 200 mm tubing (0.8 × 19 mm) (Sarstedt) and 1.2 mL S‐Monovettes (Sarstedt). Blood samples were centrifuged immediately after collection, and plasma was then analyzed.
2.3.1. Blood collection system
Seven different collection devices were examined: conventional straight 21G needles, 0.8 × 25.4 mm (S‐Monovette Safety needle; Sarstedt) and 0.8 × 32 mm (BD Eclipse BD); butterfly winged 21G Safety Multifly needles with 200 mm tubing (0.8 × 19 mm; Sarstedt) and 80 mm tubing (0.8 × 19 mm; Sarstedt); 25G Safety Multifly® needles with 200 mm tubing (0.5 × 19 mm; Sarstedt); 18G Vasofix Safety peripheral intravenous catheters (13 × 45 mm; B. Braun, Melsungen, Germany) and Micro‐Needles 21G (0.8 × 19 mm; Sarstedt). Using the Micro‐Needle, blood was sampled without the adapter or tubing by directly dripping the blood in the opened prespiked S‐Monovette.
2.3.2. Blood collection tube
Four distinct blood collection tubes were assessed: EDTA S‐Monovettes (three diameters: 1.2 [with mixing ball], 2.7, and 9 mL; Sarstedt) and EDTA Vacutainers (2 mL; BD). All were compared using 21G Safety Multifly needles with 200 mm tubing. Glass tubes were excluded due to increased FXII autoactivation on negatively charged surfaces 23 and nonspecific adsorption of BK. 20
2.3.3. Blood collection technique
First, time delay effects were assessed, by resting the blood flow for 2 minutes in the butterfly tubing and 5 minutes in the peripheral catheter. Second, blood collection via aspiration was compared against the vacuum technique. Third, venipunctures were conducted in antegrade (punctured with blood flow, drawn against blood flow) and retrograde (vice versa) directions to analyze potential differences in shear forces. Fourth, the influence of a tourniquet application was examined. All approaches were investigated applying 21G Safety Multifly needles with 200 mm tubing into 1.2‐mL S‐Monovettes under aspiration, unless otherwise stated.
2.4. Effect of specimen handling on kinin quantification
2.4.1. Time until centrifugation
In clinical blood collection settings, blood processing and centrifugation delays often occur (e.g. unavailability of centrifuges on the ward). During this dwell‐time increased metabolic activities via additional proteolytic enzymes located on cells 24 , 25 , 26 may alter peptide metabolism, affecting the reliability of results. In assessing this impact on kinin levels, whole blood was sampled freshly into tubes containing the customized protease inhibitor and 1 ng/mL of kinins. Subsequently, the blood was centrifuged immediately (reference [≈ 30 minutes]), 75 and 110 minutes after blood sampling, and stability was analyzed (n = 3).
2.4.2. Centrifugation method
For each centrifugation protocol, spiked kinin peptides in a concentration of 150 pg/mL (n = 3) were evaluated. Three centrifugation methods were applied at 21°C. First, the laboratory’s standard protocol of single centrifugation at 2,000 g for 10 minutes was used; second, single centrifugation at 2,000 g for 20 minutes; and third, two‐step centrifugation at 2,000 g for 10 minutes followed by 16,100 g for 10 minutes. Additionally, whole blood was sampled on ice and centrifuged at 2,000 g for 10 minutes at 4°C to assess temperature influence.
2.5. Validation and applicability of the standardized procedure
A standardized procedure for kinin collection, processing, and quantification was developed on the basis of identified impactful preanalytical variables. In determining the reliability of optimized preanalytical conditions, the repeatability of sampling was assessed. Based on the findings presented, the following standardized procedure was applied: Venous blood was collected in an antegrade fashion into three consecutive 1.2 mL EDTA S‐Monovettes prespiked with the customized protease inhibitor using 21G Safety Multifly needles with 200 mm tubing (Sarstedt) under aspiration. Phlebotomy was conducted in the upright position between 11 am and 1 pm using a tourniquet. Samples were centrifuged at 2,000 g for 10 minutes at 21°C. Kinin levels were determined in one female volunteer on five different days (interday variability). In addition, venous blood was collected from seven healthy volunteers (interindividual variability).
2.6. Liquid chromatography–tandem mass spectrometry monitoring of kinins
Kinins were measured using a previously established in‐house liquid chromatography–tandem mass spectrometry (LC‐MS/MS) platform, 21 which had been validated according to the bioanalytical guideline of the US Food and Drug Administration (FDA) 27 using the customized protease inhibitor. 22 Lower limits of quantification (LLOQ) of this method are presented in Table 1.
TABLE 1.
Lower limits of quantification of the mass spectrometric kinin platform
| Analyte | Lower limit of quantification (pg/mL) |
|---|---|
| Bradykinin | 2.0 |
| Kallidin | 3.9 |
| Des‐Arg(9)‐bradykinin | 2.0 |
| Des‐Arg(10)‐kallidin | 2.0 |
| Bradykinin 1–7 | 2.0 |
| Bradykinin 2–9 | 7.8 |
| Bradykinin 1–5 | 15.6 |
2.7. Quality control system
In proving the validity and collection of high‐quality data, a quality control system directed the handling of all measurements. This audited quality control system 28 required processing validation and applicability runs with calibration standards and quality control samples. Before each analysis, a system suitability test ensured accurate and reproducible LC‐MS/MS performance. Additionally, an interexperimental control was drawn in triplicate (Section 2.3) to correct for biological variability.
2.8. Statistical analysis
Descriptive statistics (mean ± standard deviation [SD] or median [interquartile range]) and box‐whisker‐plots depicted kinin level data. Outcomes were analyzed using the Mann‐Whitney U‐test or Levene test for equality of variance. Statistical analyses were performed with the two‐sided alternative hypothesis at the 5% significance level. Calculations and graphics were made using OriginPro 2021 (9.8.0.200).
3. RESULTS
3.1. Effect of the protease inhibitor on kinin quantification
3.1.1. Extent and time course of artificial kinin formation after phlebotomy
Levels of BK and its metabolites (mean ± SD) sampled without the protease inhibitor were in the high picograms‐per‐milliliter to high nanograms‐per‐milliliter range with high interindividual variation in EDTA plasma (n = 4 volunteers): 3130.8 ± 2921.0 pg/mL for BK, 2318.5 ± 1951.6 pg/mL for BK‐(1‐8), 1356.8 ± 834.3 pg/ml for BK‐(1‐7), 753.8 ± 943.9 pg/mL for BK‐(2‐9), and 779.0 ± 53.3 pg/mL for BK‐(1‐5). KBK and KBK‐(1‐8), unaffected by contact activation, fell below the LLOQ.
Monitoring artificial kinin formation revealed increased BK levels 30 seconds after blood sampling, rising continuously over the observed 30‐minute period in EDTA plasma (Figure 3). BK metabolites emerged between 30 seconds and 3 minutes. After 30 minutes, BK‐(1‐8) was the primary metabolite detected. An equilibrium between formation and degradation occurred for BK‐(1‐7) and BK‐(2‐9) (59% and 24% of BK formation), but not for BK‐(1‐5) or BK‐(1‐8). These rapid artificial ex vivo changes in plasma highlighted the need for direct sampling into suitable protease inhibitors and the urgency for investigating phlebotomy conditions. A change in kinin levels over time was also detectable in citrate plasma, although the extent was less than in EDTA plasma.
FIGURE 3.
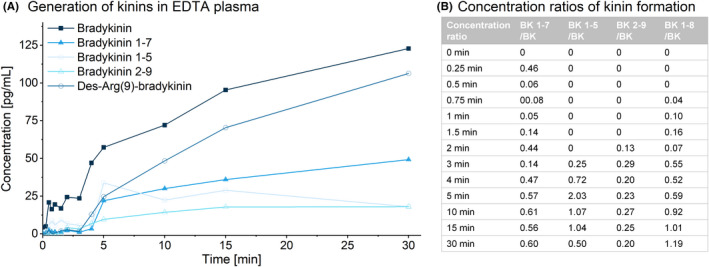
Self‐formation of bradykinin and formation of its metabolites after blood sampling. Spontaneous contact activation without in vitro activation in the absence of inhibitors in healthy volunteer in EDTA S‐Monovettes (A) was monitored. The ratios of metabolite to bradykinin formation from 5 to 30 minutes are further displayed (B)
3.1.2. Evaluation of plasma kinin stability using protease inhibitors
To prevent artificial ex vivo BK formation, screening of serine protease inhibitors revealed similar or higher BK levels using aprotinin, PMSF, or AEBSF compared with uninhibited plasma (Figure 4), indicating improper control of artificial BK formation. In contrast, nafamostat, leupeptin (0.5/1.5 h) and chicken‐egg trypsin‐inhibitor (0.5 hours) reduced BK formation; however, BK levels were increased after 4.5 hours. BK levels were lowest using HDMB, which prevented BK formation most effectively and accounted for only 0.4% of uninhibited plasma (Figure 4). The combination of 19.8 µM nafamostat and 3 mg/mL HDMB rendered BK undetectable, indicating control of artificial BK formation.
FIGURE 4.
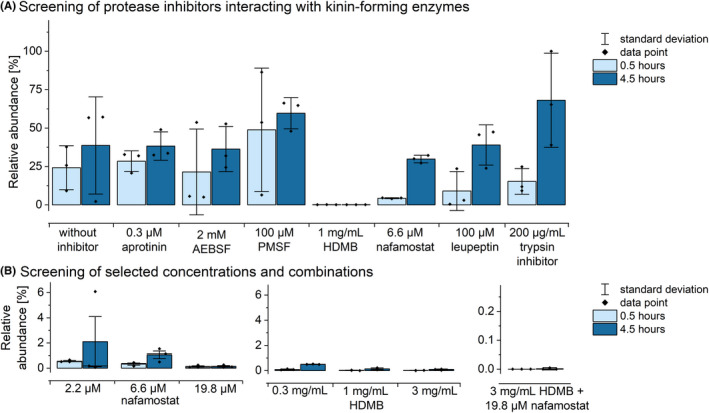
Effects of distinct inhibitors on the formation of bradykinin in citrate plasma. The relative abundance of bradykinin 0.5 and 4.5 hours after blood sampling is shown. (A) Results of the screening of multiples protease inhibitors is shown. (B) The evaluation of distinct concentrations of the best‐performing inhibitors HDMB and nafamostat and their combination are depicted. Mean values with standard deviation and individual data points (n = 3)
The BK formation inhibiting combination of HDMB and nafamostat was insufficient to prevent kinin degradation, as demonstrated by the degradation of all spiked kinins (Figure 5). While adding enalaprilat (200 nM) did not improve kinin stability, kinin degradation decreased to <27%, except for BK‐(2‐9), when including the metalloprotease inhibitors EDTA (83 mM) and citrate (20 mM). Unspecific protease inhibition via basification using ammonia or sodium hydroxide did not improve the stability of BK‐(2‐9) (−88%/−96%). In contrast, acidification reduced BK‐(2‐9) degradation, whereby higher amounts of FA were beneficial (degradation of 61% for 0.1% FA vs 32% for 1% FA). The addition of omapatrilat reduced between‐sample variability (coefficient of variation [CV]) and stabilized kinin levels to ≤15% relative error (RE), except for BK‐(1‐8) and BK‐(1‐5). To achieve stabilization of the latter two, additional serine protease inhibitors were investigated, of which chloroquine best prevented their degradation, resulting in RE and CV ≤15% for all kinins. These limits are consistent with the FDA’s bioanalytical guideline. 27 Thus, the customized protease inhibitor consisted of a final concentration of 3 mg/mL HDMB, 19.8 µM nafamostat, 83 mM EDTA, 20 mM citrate, 1% FA, 1 µM omapatrilat and 1 mM chloroquine in the filled tube. Its respective enzymatic targets are presented in Figure 2.
FIGURE 5.
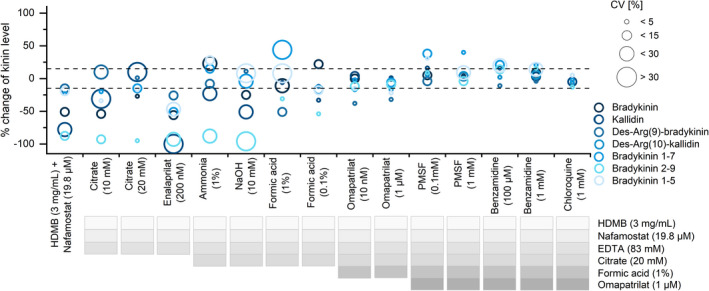
Development of the customized protease inhibitor. The percentage change of spiked kinin levels (1 ng/mL) in plasma after 3 h of benchtop storage using different protease inhibitor approaches is shown. Mean values (n = 3) with their coefficient of variations (CV) are presented. The dashed line represents the maximum deviation of ±15% according to the US Food and Drug Administration guideline. 27 HDMB, hexadimethrine bromide
While the customized protease inhibitor stabilized kinin levels within the FDA limits, the absence of the inhibitor resulted in marked changes in spiked plasma kinin levels. Besides a significant degradation in the observed period (−65.7% for BK‐[1‐7] up to −100% for KBK), increases in BK and derived metabolites occurred within 1 hour after blood sampling. The commercially available P100 and P800 tubes did not contribute to any substantial improvement (degradation of 41% for BK‐[1‐7] up to 100% for KBK), but P800 tubes limited BK formation as far as could be assessed given the spiked concentrations.
3.2. Effect of blood specimen collection on kinin quantification
3.2.1. Blood collection devices
When comparing blood collection systems, no significant difference was found between the 21G butterfly‐winged systems with 80 or 200 mm tubing (Figure 6C). However, collection with 21G straight needles varied significantly (Eclipse, P = .009; S‐Monovette Safety,P = .03). When sampling with the 21G Micro Needle, BK levels (median, 32.0 [13.0‐67.7] pg/mL) were detected besides an artificial formation of BK‐(1‐8) (median, 3.7 [1.4‐6.4] pg/mL).
FIGURE 6.
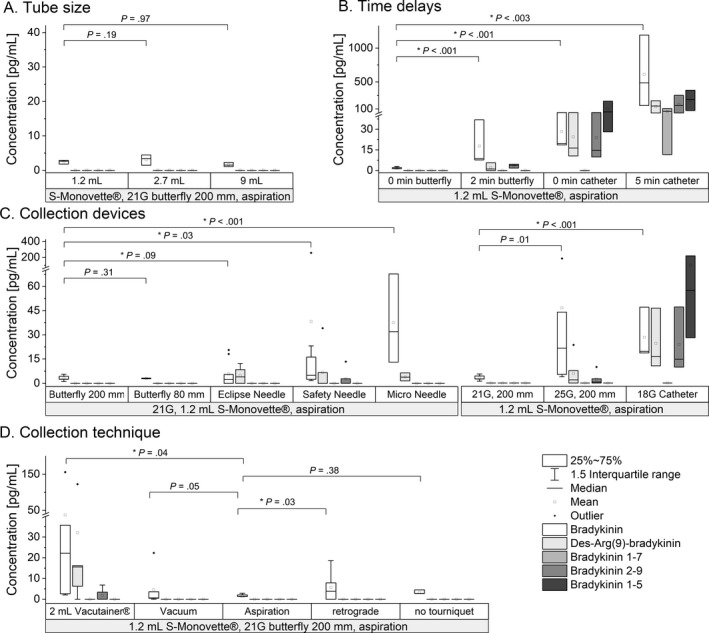
Impact of blood sampling conditions on kinin levels. Box‐whisker‐plots show the impact of the tube size (A), time delays (B), collection devices (C) and the collection technique (D). Experiments were carried out at least in triplicate (A: n = 3; B: n = 3 or n = 6, respectively; C: n = 3, n = 6 or n = 9; D: n = 6). An interexperimental control using Safety Multifly needles 21G with 200 mm tubing into 1.2‐mL S‐Monovettes under aspiration was run for each experiment
In comparing needle sizes, the smaller needle size (25G) significantly negatively impacted BK levels (P = .01). In addition, BK‐(1‐8) and BK‐(1‐7) were detectable using the smaller needle size. Use of the peripheral catheter resulted in highest BK levels (19.2 [18.6‐47.0] pg/mL; P < .001) and increased levels of BK‐(1‐8) (16.4 [10.6–46.4] pg/mL), BK‐(1‐7) (14.7 [9.8–47.9] pg/mL), and BK‐(1‐5) (57.4 [28.1‐216.0] pg/mL).
3.2.2. Blood collection tube
The altered blood‐to‐surface ratio due to distinct diameters of S‐Monovettes did not compromise kinin levels, when sampled under aspiration (Figure 6A). Interestingly, comparing S‐Monovettes and Vacutainer tubes revealed differences in kinin levels; the latter showed higher variability in kinin levels (P = .04; Figure 6D).
3.2.3. Blood collection technique
The resting of blood in the collection device during blood sampling must be avoided, as kinin levels significantly increased for both butterfly (8‐fold during 2‐minute rest) and peripheral catheter (25‐fold during 5‐minute rest) sampling (P < .001 and P = .003; Figure 6B).
Comparison between Vacutainers and S‐Monovettes indicated that blood should be collected using the aspiration technique. BK levels differed significantly with vacuum collection using Vacutainers (P = .04). This tendency was also detectable for vacuum collection using S‐Monovettes (P = .05) (Figure 6D). The evaluation of potential shear forces affected by antegrade or retrograde cannulation showed significantly elevated BK levels when inserted in a retrograde fashion (P = .03).
3.3. Effect of blood sample handling on kinin quantification
3.3.1. Time until centrifugation
Exogenously added kinins demonstrated increased degradation in whole blood despite using the customized protease inhibitor. In comparing time to centrifugation, centrifugation within 30 minutes resulted in REs ≤15% compared to a fresh standard for all kinins. Delayed centrifugation after 75 and 120 minutes caused a decline of kinin levels (up to −62% for BK‐[1‐8]). BK‐(1‐5) was the exception; levels increased up to 26% (120 minutes), indicating accumulation of the most stable kinin due to degradation of more short‐lived upstream kinins. Additionally, variability between the tubes increased over time.
3.3.2. Centrifugation
Comparison of centrifugation methods did not yield significant differences (P > .38) in kinin levels.
3.4. Validation and applicability of the standardized procedure
Table 2 presents a standardized blood sampling and handling procedure developed from the findings. Using this procedure successfully validated and applied the evaluated preanalytical conditions. The consecutive sampling of three tubes in seven healthy subjects (29 [27–54] years) revealed kinin levels in a similar range (mean levels for subjects, 0–4.3 pg/mL; Figure 7). Only one sample from one female volunteer produced levels of 2.4 ± 0.4 pg/mL for KBK‐(1‐8), whereas all other kinin levels fell below the LLOQ. Samples from one healthy volunteer (27 years) yielded mean ± SD values of 2.5 ± 1.5 pg/mL BK on five distinct days. Intertube variations (n = 3) were <1.2 pg/mL from the mean for the seven volunteers and <1.7 pg/mL from the mean for one volunteer on 5 days.
TABLE 2.
Standardized procedure for preanalytical conditions of kinin quantification
| Preanalytical variable | Finding | Advised procedure |
|---|---|---|
| Protease inhibitor |
The use of suitable protease inhibitors largely influences plasma kinin levels. |
A mixture of 3 mg/mL hexadimethrine bromide, 19.8 µM nafamostat, 83 mM EDTA, 20 mM citrate, 1% formic acid, 1 µM omapatrilat, and 1 mM chloroquine was found to be effective. 22 |
| Blood collection system | Sampling using catheter, Micro‐Needle or straight needles jeopardized kinin levels. | Butterfly‐winged needles with a needle size of 21G are advisable. |
| Blood collection tube | No major influence if aspiration technique is applicable. | Polypropylene tubes with size from 1.2 up to 9 mL possible. |
| Blood collection technique | Vacuum sampling and time‐delays increased variability of kinin levels. | Use the aspiration technique applying a constant move, rapidly perform sampling of blood and avoid any time delays after venipuncture. |
| Whole‐blood storage | Increased degradation of kinins in whole blood. | Centrifuge within 30 min after blood collection. |
| Centrifugation method | No major influence on kinin levels. | Apply 2,000 g for 10 min at 21°C. |
| Plasma storage |
confirmed using the developed protease inhibitor. 21 |
FIGURE 7.
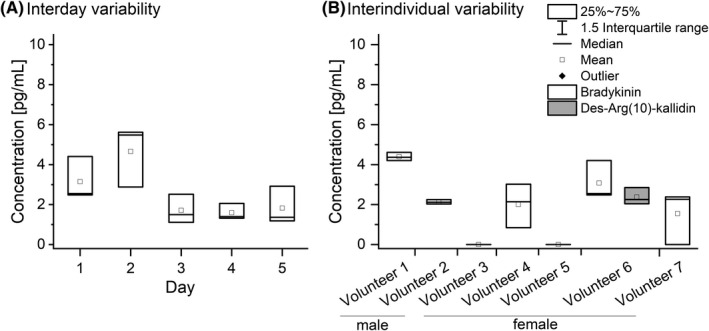
Box‐whisker‐plots of the kinin levels for interday (A) and interindividual (B) variability using the developed standardized procedure. Only kinin levels above the lower limit of quantification were shown. N = 3
4. DISCUSSION
This study identified impactful preanalytical variables (eg, sampling technique, blood collection devices, protease inhibitor) on plasma kinin levels. Controlling kinin formation and preventing degradation during blood sampling and specimen handling were addressed by developing a standardized procedure. Applying this procedure, coupled with a validated LC‐MS/MS platform, proved its applicability for robust and reliable kinin determination in human plasma.
Strong dependence on sample collection methods has hindered reliable and accurate kinin quantification. 5 , 29 This obstacle has contributed to significant differences in previous research, with kinin levels ranging from 0.4 pg/mL to 162 ng/mL of BK in healthy volunteers, 12 , 15 , 16 , 17 rendering identification of a dysregulated KKS in disease difficult. However, the combination of HDMB—an inhibitor of FXII activation and FXIIa 30 —and nafamostat—an inhibitor of FXIIa and PKa, 31 successfully prevented contact activation‐induced artificial BK formation.
Further, the comprehensive monitoring of kinins other than BK, especially active kinins that interact with BK receptors, is critical when considering possible therapeutic targets. Thus far, protease inhibitors have shown their potential but did not sufficiently stabilize all kinins investigated within this study. 22 Moreover, previously published approaches have failed to show comprehensive proof of stability or disclosed deviations exceeding FDA guidance limits. 11 , 14 , 32 In addition to HDMB and nafamostat preventing BK formation, adding five additional protease inhibitors inhibited kinin degradation effectively The chelators citrate and EDTA inhibit metalloproteases, such as BK‐degrading ACE or carboxypeptidases. 18 FA reduces enzyme activity by shifting the blood pH toward suboptimal enzyme conditions. 33 Specifically, omapatrilat inhibits ACE and Neprilysin, being among the most relevant kinin‐degrading enzymes. 18 Finally, chloroquine inhibits degradation of BK‐(1‐8) by prolylcarboxypeptidase 34 and contributed toward kinin stabilization in plasma according to FDA bioanalytical guideline stability criteria. 27
The phlebotomy setting severely impacted the reliability and accuracy of kinin levels. For coagulation assays, research has suggested that conventional straight needles were superior to butterfly needles or peripheral catheters due to reduced hemostatic alteration and decreased surface area for contact activation. 35 However, others have found no differences among these three collection systems. 36 , 37 In this study, consistent kinin levels were obtained only using butterfly tubing. This finding may be attributable to additional handling space due to the tubing, thus avoiding needle pressure on the vessel wall during blood sampling tube exchanges. Kinin levels were highest when measured using catheters, indicating that their use is not practical. The time for catheter positioning likely played a significant role in kinin formation, but kinin levels also rose sharply during time delays while the catheter was in the vein. Therefore, it is always advisable to puncture anew when taking repeated blood samples to determine kinin values and, in addition, to use veins that have not been punctured previously. For hemostatic investigations, it is further recommended to minimize trauma during blood draws, to avoid producing bubbles or foam, and to reduce shear stress. 38 In line, blood sampling in Vacutainers or S‐Monovettes under vacuum produced more variable results than blood sampling under aspiration. Consequently, reduction of shear forces and stress on the vessel in addition to rapid sampling facilitated the most consistent kinin level measurements.
While the sample collection settings were most crucial in controlling kinin formation, specimen handling was predominantly decisive regarding loss of kinins. Higher availability of degrading enzymes on blood cell surfaces, for example, aminopeptidases or ACE2, presumably limited kinin stability in whole blood (30 minutes) in comparison to plasma (1.5 hours). 21 , 24 , 25 , 26 Furthermore, reducing nonspecific peptide adsorption during sample handling minimized variability of recovery, particularly in extracted samples due to reduced saturation of protein‐binding surfaces. 39 In addition to using protein low‐binding tubes, high acidic amounts and organic solvents using polypropylene materials reduced kinin adsorption and contributed to increased method robustness. 20
Finally, using the implemented standardized procedure demonstrated repeatability of measured kinin levels. This study exemplified the limitation of interday and interindividual variability (<1.7 pg/mL). The sample collection and on‐ward preparation using the standardized procedure in this study allow for implementation into clinical routine to further investigate kinins in diseases like angioedema or sepsis. The developed procedure combined with the LC‐MS/MS platform 21 enables improved study of diseases related to the KKS, such as COVID‐19, angioedema, sepsis, Alzheimer disease, epilepsy, cancer, and strokes. Reliable kinin quantification facilitates investigation of kinin profile alterations in health and disease and therefore may help identify promising new therapeutic targets.
RELATIONSHIP DISCLOSURE
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
TG and BBB designed the concept and performed the experiments. TG analyzed the data and wrote the first draft of the paper. BBB advised on the data analysis and revised the manuscript draft. Both authors read and approved the final version of the manuscript.
ACKNOWLEDGMENTS
The authors thank Prof. Holger Schwender (Mathematics, Heinrich‐Heine University Düsseldorf) for his statistical advice for evaluation of the study results.
Gangnus T, Burckhardt BB. Reliable measurement of plasma kinin peptides: Importance of preanalytical variables. Res Pract Thromb Haemost. 2022;6:e12646. doi: 10.1002/rth2.12646
Handling Editor: Prof. Yotis Senis
REFERENCES
- 1. Campbell DJ. Bradykinin Peptides. In: Kastin AJ, editor. Handbook of biologically active peptides, 2nd edn. Elsevier; 2013. p. 1386‐1393. [Google Scholar]
- 2. Garvin MR, Alvarez C, Miller JI, et al. A mechanistic model and therapeutic interventions for COVID‐19 involving a RAS‐mediated bradykinin storm. Elife. 2020;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kashuba E, Bailey J, Allsup D, Cawkwell L. The kinin‐kallikrein system: physiological roles, pathophysiology and its relationship to cancer biomarkers. Biomarkers. 2013;18:279‐296. [DOI] [PubMed] [Google Scholar]
- 4. Nicola H. The role of contact system in septic shock: the next target? An overview of the current evidence. J Intensive Care. 2017;5:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hofman Z, de Maat S, Hack CE, Maas C. Bradykinin: inflammatory product of the coagulation system. Clin Rev Allergy Immunol. 2016;51:152‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carraro P, Plebani M. Errors in a stat laboratory: types and frequencies 10 years later. Clin Chem. 2007;53:1338‐1342. [DOI] [PubMed] [Google Scholar]
- 7. Bonini P, Plebani M, Ceriotti F, Rubboli F. Errors in laboratory medicine. Clin Chem. 2002;48:691‐698. [PubMed] [Google Scholar]
- 8. Lawrence JB. Preanalytical variables in the coagulation laboratory. Laboratory Medicine. 2003;34:49‐57. [Google Scholar]
- 9. Debunne N, de Spiegeleer A, Depuydt D, et al. Influence of blood collection methods and long‐term plasma storage on quorum‐sensing peptide stability. ACS Omega. 2020;5:16120‐16127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Decarie A. Serum interspecies differences in metabolic pathways of bradykinin and [des‐Arg9]BK: influence of enalaprilat. American Physiological Society. 1996;271(4):H1340–H1347. [DOI] [PubMed] [Google Scholar]
- 11. Seip KF, Bjerknes KC, Johansen HT, Nielsen EW, Landrø L, Reubsaet L. Bradykinin analysis revived–a validated method for determination of its stable metabolite in whole blood by LC‐MS/MS. J Chromatogr B. 2014;947–948:139‐144. [DOI] [PubMed] [Google Scholar]
- 12. Duncan AM, Kladis A, Jennings GL, Dart AM, Esler M, Campbell DJ. Kinins in humans. Am J Physiol Regul Integr Comp Physiol. 2000;278:R897‐R904. [DOI] [PubMed] [Google Scholar]
- 13. Nussberger J, Cugno M, Amstutz C, Cicardi M, Pellacani A, Agostoni A. Plasma bradykinin in angio‐oedema. The Lancet. 1998;351:1693‐1697. [DOI] [PubMed] [Google Scholar]
- 14. Lindström M, Valkonen M, Tohmola N, et al. Plasma bradykinin concentrations during septic shock determined by a novel LC‐MS/MS assay. Clin Chim Acta. 2019;493:20‐24. [DOI] [PubMed] [Google Scholar]
- 15. van den Broek I, Sparidans RW, Schellens JHM, Beijnen JH. Quantitative assay for six potential breast cancer biomarker peptides in human serum by liquid chromatography coupled to tandem mass spectrometry. J Chromatogr B. 2010;878:590‐602. [DOI] [PubMed] [Google Scholar]
- 16. Wheelock KM, Cai J, Looker HC, et al. Plasma bradykinin and early diabetic nephropathy lesions in type 1 diabetes mellitus. PLoS One. 2017;12:e0180964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pellacani A, Brunner HR, Nussberger J. Antagonizing and measurement: approaches to understanding of hemodynamic effects of kinins. J Cardiovasc Pharmacol. 1992;20(Suppl 9):S28‐34. [DOI] [PubMed] [Google Scholar]
- 18. Blais C, Drapeau G, Raymond P, Lamontagne D, Venneman I, Adam A. Contribution of angiotensin‐converting enzyme to the cardiac metabolism of bradykinin: an interspecies study. American Physiology Society. 1997;273(5):H2263‐H2271. [DOI] [PubMed] [Google Scholar]
- 19. Murphey LJ, Hachey DL, Vaughan DE, Brown NJ, Morrow JD. Quantification of BK1‐5, the stable bradykinin plasma metabolite in humans, by a highly accurate liquid‐chromatographic tandem mass spectrometric assay. Anal Biochem. 2001;292:87‐93. [DOI] [PubMed] [Google Scholar]
- 20. Gangnus T, Burckhardt BB. Improving sensitivity for the targeted LC‐MS/MS analysis of the peptide bradykinin using a design of experiments approach. Talanta. 2020;218: 121134. [DOI] [PubMed] [Google Scholar]
- 21. Gangnus T, Burckhardt BB. Targeted LC‐MS/MS platform for the comprehensive determination of peptides in the kallikrein‐kinin system. Anal Bioanal Chem. 2021;413:2971‐2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gangnus T, Burckhardt BB. Stabilization of short‐lived peptides of the kallikrein‐kinin system in human plasma to facilitate use as promising biomarkers. Clin Chem. 2021;67:1287‐1289. [DOI] [PubMed] [Google Scholar]
- 23. Schmaier AH. The contact activation and kallikrein/kinin systems: Pathophysiologic and physiologic activities. J Thromb Haemost. 2016;14:28‐39. [DOI] [PubMed] [Google Scholar]
- 24. O'Leary H, Ou X, Broxmeyer HE. The role of dipeptidyl peptidase 4 in hematopoiesis and transplantation. Curr Opin Hematol. 2013;20:314‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rutkowska‐Zapała M, Suski M, Szatanek R, et al. Human monocyte subsets exhibit divergent angiotensin I‐converting activity. Clin Exp Immunol. 2015;181:126‐132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hendriks D, de Meester I, Umiel T, et al. Aminopeptidase P and dipeptidyl peptidase IV activity in human leukocytes and in stimulated lymphocytes. Clin Chim Acta. 1991;196:87‐96. [DOI] [PubMed] [Google Scholar]
- 27. U.S. Food and Drug Administration . Bioanalytical Method Validation: Guidance for Industry. https://www.fda.gov/files/drugs/published/Bioanalytical‐Method‐Validation‐Guidance‐for‐Industry.pdf, 2018.
- 28. Suessenbach FK, Makowski N, Feickert M, et al. A quality control system for ligand‐binding assay of plasma renin activity: Proof‐of‐concept within a pharmacodynamic study. J Pharm Biomed Anal. 2020;181:113090. [DOI] [PubMed] [Google Scholar]
- 29. Kaplan AP, Maas C. The search for biomarkers in hereditary angioedema. Front Med (Lausanne). 2017;4:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eisen V. Effect of hexadimethrine bromide on plasma kinin formation, hydrolysis of p‐Tosyl‐L‐argnine methyl ester and fibrinolysis. Br J Pharmacol. 1964;22(1):87‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hitomi Y, Ikari N, Fujii S. Inhibitory effect of a new synthetic protease inhibitor (FUT‐175) on the coagulation system. Haemostasis. 1985;15:164‐168. [DOI] [PubMed] [Google Scholar]
- 32. Campbell DJ, Kladis A, Duncan AM. Bradykinin peptides in kidney, blood, and other tissues of the rat. Hypertension. 1993;21:155‐165. [DOI] [PubMed] [Google Scholar]
- 33. Maguire GA, Price CP. A kinetic fluorimetric assay for the measurement of angiotensin‐converting enzyme in human serum. Ann Clin Biochem. 1984;21(Pt 5):372‐377. [DOI] [PubMed] [Google Scholar]
- 34. Kumamoto K, Stewart TA, Johnson AR, Erdös EG. Prolylcarboxypeptidase (angiotensinase C) in human lung and cultured cells. J Clin Invest. 1981;67:210‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Spronk HMH, Dielis AWJH, Panova‐Noeva M, et al. Monitoring thrombin generation: is addition of corn trypsin inhibitor needed? Thromb Haemost. 2009;101:1156‐1162. [PubMed] [Google Scholar]
- 36. Loeffen R, Kleinegris M‐CF, Loubele STBG, et al. Preanalytic variables of thrombin generation: towards a standard procedure and validation of the method. J Thromb Haemost. 2012;10:2544‐2554. [DOI] [PubMed] [Google Scholar]
- 37. Lippi G, Salvagno GL, Guidi GC. No influence of a butterfly device on routine coagulation assays and D‐dimer measurement. J Thromb Haemost. 2005;3:389‐391. [DOI] [PubMed] [Google Scholar]
- 38. Magnette A, Chatelain M, Chatelain B, ten Cate H, Mullier F. Pre‐analytical issues in the haemostasis laboratory: guidance for the clinical laboratories. Thrombosis Journal. 2016;14:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hoofnagle AN, Whiteaker JR, Carr SA, et al. Recommendations for the generation, quantification, storage, and handling of peptides used for mass spectrometry–based assays. Clin Chem. 2016;62:48‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]