

Key Points
Severe bone marrow failure was primarily observed in early childhood in children with biallelic SBDS mutations.
Absolute neutrophil counts were positively associated with age (P < .0001) in patients with biallelic SBDS mutations.
Visual Abstract

Abstract
Shwachman-Diamond syndrome (SDS) is an inherited bone marrow failure syndrome with leukemia predisposition. An understanding of the hematologic complications of SDS with age could guide clinical management, but data are limited for this rare disease. We conducted a cohort study of 153 subjects from 143 families with confirmed biallelic SBDS mutations enrolled on the North American Shwachman Diamond Registry or Bone Marrow Failure Registry. The SBDS c.258 + 2T>C variant was present in all but 1 patient. To evaluate the association between blood counts and age, 2146 blood counts were analyzed for 119 subjects. Absolute neutrophil counts were positively associated with age (P < .0001). Hemoglobin was also positively associated with age up to 18 years (P < .0001), but the association was negative thereafter (P = .0079). Platelet counts and marrow cellularity were negatively associated with age (P < .0001). Marrow cellularity did not correlate with blood counts. Severe marrow failure necessitating transplant developed in 8 subjects at a median age of 1.7 years (range, 0.4-39.5), with 7 of 8 requiring transplant prior to age 8 years. Twenty-six subjects (17%) developed a myeloid malignancy (16 myelodysplasia and 10 acute myeloid leukemia) at a median age of 12.3 years (range, 0.5-45.0) and 28.4 years (range, 14.4-47.3), respectively. A lymphoid malignancy developed in 1 patient at the age of 16.9 years. Hematologic complications were the major cause of mortality (17/20 deaths; 85%). These data inform surveillance of hematologic complications in SDS.
Introduction
Shwachman-Diamond syndrome (SDS) is a multisystem disorder that is characterized by bone marrow failure (BMF), exocrine pancreatic dysfunction, and predisposition to myeloid malignancies.1,2 Patients with SDS are at risk for severe cytopenias. Outcomes of myeloid malignancies are poor as a result of high treatment-related toxicities and resistant disease.3–5 An understanding of the natural history of hematologic complications with age would inform clinical management and surveillance, but data are sparse because of the rarity of SDS.
Biallelic mutations in SBDS account for >90% of cases of SDS.6 Rare patients with mutations in EFL1, DNAJC21, or SRP54 may also share clinical features of SDS.7–11 Genetic testing is now integrated into diagnostic evaluation to identify patients with cryptic presentations of inherited BMF disorders, as well as to avoid misdiagnosis due to clinical features overlapping with other disorders.12–15 Sequencing analysis may miss mutations in SBDS because of large deletions or difficulties with the highly homologous pseudogene, so further evaluation should be pursued when clinical suspicion is high.16,17
SDS registries in France and Italy recently reported characteristics of severe cytopenias, clonal cytogenetic abnormalities, and malignancies in patients with biallelic SBDS mutations. The French Neutropenia Registry, which included 102 patients with biallelic SBDS mutations, reported that the 20-year cumulative risk of nonmalignant and malignant severe cytopenias is 24.3%.18 Stratification for risk of malignant disease or severe cytopenias was proposed based on diagnosis at <3 months of age and abnormalities on the initial complete blood count (CBC). Hematologic complications were the leading cause of mortality.18 The Italian SDS Registry study of 121 patients with biallelic SBDS mutations found a cumulative incidence of 59.9% for severe neutropenia, 66.8% for thrombocytopenia, and 20.2% for anemia at 30 years of age. The 20-year cumulative incidence of myeloid malignancy and severe cytopenia was 9.8% and 9.9%, respectively.19
The objectives of this article are to evaluate the association of age with blood counts and bone marrow pathology and describe clinical outcomes of 153 patients with biallelic SBDS mutations.
Methods
Informed consent
The Shwachman-Diamond Registry and the Molecular and Genomic Studies of Bone Marrow Failure and Myelodysplastic Syndromes are prospective cohort studies enrolling patients with SDS. Informed consent was obtained in accordance with study protocols approved by the local Institutional Review Boards.
Data collection
Data were extracted from medical records and recorded in the Research Electronic Data Capture system.20 The cutoff date for data collection was 1 February 2020.
Longitudinal CBCs were collected. CBCs were included until the time of myelodysplasia (MDS) or leukemia diagnosis or until the time of hematopoietic stem cell transplant (HSCT), when applicable. Hemoglobin values were excluded during periods of red cell transfusions, platelet values were excluded during platelet transfusions, and absolute neutrophil counts (ANCs) were excluded while on granulocyte colony-stimulating factor (G-CSF).
Local bone marrow aspirate and biopsy reports were examined for reported cellularity (from biopsies), fluorescence in situ hybridization, karyotype, and flow cytometry data. Surveillance bone marrows were defined as bone marrow examinations performed in the absence of clinical symptoms. Bone marrows from the time of malignancy diagnosis or those performed post-HSCT were excluded from the surveillance marrow data.
Statistical analyses
Median and range or number and percentage are reported for continuous or categorical patient characteristic variables. Mean, standard deviation, median, and range are reported for blood counts. Box plots are also presented for blood counts. Scatter plots with locally estimated scatterplot smoothing are generated for blood counts and cellularity over age. We used 15 if the day of a month was missing.
The age at the time of blood counts varied, limiting our ability to evaluate changes within an individual over time. Therefore, the primary analysis evaluates the association between age and blood counts, adjusting for repeated measures.
To evaluate the association between age and blood counts and cellularity, we fit segmented mixed-effects models with random breakpoints. We assumed there is at most 1 breakpoint on each regression line, with the breakpoint defined as the point where the slope of the regression line changes abruptly. We first identified the breakpoint (when applicable) using the maximum likelihood method described by Muggeo et al.21 We then fit the mixed-effects model with a random intercept for a subject with the identified breakpoint. White blood cells (WBCs), platelets, and ANCs are log-transformed in this model. To avoid 0s, 0.1 is added to all laboratory values before the transformation (7 observations for WBCs and 37 observations for ANCs were 0).
The correlation between cellularity and blood counts was assessed using scatter plots and Spearman correlation coefficients. The χ2 test was used to compare the proportion of subjects with normal blood counts for those with low (<30%) vs high cellularity (≥30%). The survival distribution was estimated using the Kaplan-Meier method.
Results
Patient characteristics
One hundred and fifty-three individuals (143 families) with biallelic SBDS mutations are reported, including several short case descriptions illustrating the diversity of phenotypes in SDS. Ninety-two were male (60.1%). Median age at last follow-up was 10.4 years (range, 0.3-52.8). The cohort included 39 adults (25.5%) who were >18 years of age at the follow-up (Table 1).
Table 1.
Subject characteristics (N = 153)
| Characteristics | Data |
|---|---|
| Age at last follow-up | |
| Median (range), y | 10.4 (0.3-52.8) |
| >18 y | 39 (25.5) |
| ≤18 y | 114 (74.5) |
| Sex | |
| Male | 92 (60.1) |
| Female | 61 (39.9) |
| Disease manifestation | |
| Severe BMF | 8 (5.2) |
| Malignancy | |
| MDS | 16 (10.5) |
| AML | 10 (6.5) |
| Lymphoma | 1 (0.7) |
| Cause of death (n = 20) | |
| Malignancy | 15 (75) |
| BMF | 2 (10) |
| Asphyxiating thoracic dystrophy | 2 (10) |
| Liver failure | 1 (5) |
Unless otherwise noted, data are n (%).
SBDS genetics
Biallelic mutations in SBDS were identified in all 153 subjects in this cohort. SBDS mutations were also identified in 6 additional patients who lacked further clinical data for analysis. Of this combined group, 158 of 159 (99.4%) had ≥1 copy of the c.258 + 2T>C splice site mutation. The types and distribution of mutations are summarized in supplemental Table 1.
A single subject did not harbor the c.258 + 2T>C SBDS variant, a hypomorphic variant that expresses a low level of SBDS, but instead had 1 stop mutation and 1 missense mutation (Lys62*/Arg175Trp) in SBDS. This patient had a severe phenotype. She presented at birth with intrauterine growth retardation and neonatal severe aplastic anemia. She was transfusion dependent for platelets and red cells. Her severe neutropenia was unresponsive to G-CSF. She had clinodactyly, severe thoracic dystrophy, and exocrine pancreatic insufficiency requiring enzyme replacement. Irregular islands of cartilage surrounded by osteoid were observed in her bone marrow. Her bone-to-marrow space ratio was elevated, and the marrow contained a loose fibroblastic network surrounding the bone islands (Figure 1). She underwent a reduced intensity unrelated donor HSCT but succumbed to overwhelming bacterial infection prior to engraftment.
Figure 1.

Severe phenotype of the subject with SBDS c.183_184delTAinsCT/c.523C>T (p.Lys62*/p.Arg175Trp) and lacking the c.258 + 2T>C variant. (A-B) Bone marrow biopsy demonstrating irregular islands of cartilage surrounded by osteoid with increased bone-to-marrow space ratio and a loose fibroblastic network surrounding the bone islands. Original magnification ×40 (A), original magnification ×200 (B); hematoxylin and eosin stain.
Clinical testing platforms also sometimes miss SBDS mutations, which may be challenging to distinguish from the highly homologous SBDS pseudogene and require additional investigation. One patient presented with persistent neutropenia requiring G-CSF and mild transaminitis; initially, only a single heterozygous c.258 + 2T>C SBDS mutation was identified. Given the clinical suspicion for SDS, clinical microarray analysis was performed; a heterozygous 4-kb deletion was identified that included the entire exon 5 of the SBDS gene. Quantitative polymerase chain reaction analysis spanning the deletion breakpoint and using custom designed primers confirmed the exon 5 deletion. Molecular studies on the parents revealed the SBDS point mutation (c.258 + 2T>C) in the mother and the 4-kb deletion in the father.
Blood counts
A total of 2146 CBCs were available for 119 subjects, excluding blood counts from patients following development of malignancies or following an HSCT. Nine of 119 subjects (7.6%) developed severe trilineage cytopenias (hemoglobin < 8g/dL or transfusion dependent, platelet count < 50 000 per microliter or transfusion dependent, and ANC < 500 cells per microliter or on G-CSF) at a median age of 5.4 years (range, 0.2-29.2). Fifty-six subjects (47.1%) had normal blood counts (hemoglobin > 10 g/dL, platelet count > 150 000 per microliter, and ANC > 1500 cells per microliter) on ≥1 CBC.
To evaluate the association between blood counts and age, a piecewise regression model was fit for each lineage, allowing for 1 breakpoint (Figure 2). Significant breakpoints were detected for WBC, hemoglobin, and platelets (P ≤ .0021). LogANC was positively associated with age (slope, 0.0083; 95% confidence interval [CI], 0.0045-0.0122; P < .0001). Prior to age 18 years, hemoglobin was positively associated with age (slope, 0.0706; 95% CI, 0.053-0.0882; P < .0001); however; after age 18 years, the association was negative (slope, −0.0650; 95% CI, −0.1129 to −0.017; P = .0079). LogPlatelets was negatively associated with age (slope, −0.0226; 95% CI, −0.027 to −0.0181; P < .0001) before age 7 years, with a weaker negative association after 7 years of age (slope, −0.0133; 95% CI, −0.0157 to −0.011; P < .0001).
Figure 2.

Blood counts by age in subjects with SDS. A piecewise regression model (green shaded area) was used to determine the association between blood counts and age, allowing for a potential breakpoint for 2114 blood counts from 119 subjects. The locally estimated scatterplot smoothing (LOESS) curve is shown in purple. LogANC (A), hemoglobin (B), and log platelet count (C) as a function of age.
Eight subjects (5%) required an HSCT for severe BMF at a median age of 1.7 years (range, 0.4-39.5), with 7 of 8 patients transplanted prior to age 8 years. Median age at last follow-up for patients without severe BMF was 10.9 years (range, 0.3-52.8). iso7q was detected for 1 patient, whereas the others lacked cytogenetic abnormalities prior to HSCT. Three conditioning regimens were reported: alemtuzumab/fludarabine/melphalan (n = 4), fludarabine/alemtuzumab (n = 1), and treosulfan/fludarabine/ATG (n = 1). Data regarding the conditioning regimen were not available for 2 subjects. Acute or chronic graft-versus-host disease was seen in 0 and 1 patient, respectively (n = 5). There were 2 HSCT-related deaths. The patient described above who lacked the SBDS c.258 + 2T>C splice mutation died of infection on day 0. A second patient who underwent HSCT for BMF died of multiorgan failure on day +47.
Bone marrow pathology
Data were available for 547 marrows from 119 subjects, with a median of 3 marrows per subject (range, 1-21). Marrow cellularity data were available for 414 marrow examinations from 112 subjects. Median cellularity was 40% (range, 5-100). Reduced marrow cellularity was observed early in life, with only 35.3% (6/17) of subjects younger than 1 year having normocellular marrow (Figure 3). All marrows examined after age 16 years were hypocellular for age. Six subjects (5.3%) had a hypocellular marrow that transitioned to the normocellular range at least once during the follow-up period. Bone marrow cellularity < 30% was observed in 47 subjects (41.9%), and 4 (3.6%) had ≥1 marrow with <10% cellularity. Cellularity was negatively associated with age (P < .0001).
Figure 3.
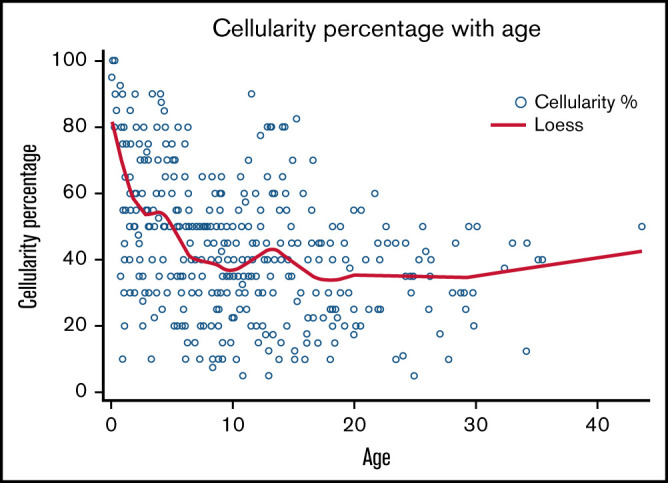
Marrow cellularity vs age in subjects with SDS. Scatter plot showing bone marrow cellularity vs age. Bone marrow was taken for 414 marrow examinations in 112 subjects.
To assess whether marrow cellularity correlated with blood counts, we examined 229 marrow reports for which an associated CBC had been drawn within 30 days for 81 subjects ≤30 years of age. The median marrow cellularity for this group was 40% (range, 5-100), which was consistent with the distribution of marrow cellularity across the entire cohort of 112 subjects. The Spearman correlation coefficient indicated a weak association between marrow cellularity and hemoglobin (−0.18; P = .0062) and platelets (0.22; P = .0009) but no significant association with ANC (−0.07; P = .4163). Of 47 subjects with marrow cellularity <30% and associated CBCs available, 12 had normal ANCs (33.3%), all had normal hemoglobins (100%), and 24 had normal platelet counts (52.2%). No statistical difference was observed between the proportion of patients with normal ANC, hemoglobin, and platelet counts in the group with marrow cellularity < 30% vs >30% (P = .9737, .3455, and .6479, respectively).
Cytogenetic clonal abnormalities
Marrow cytogenetic data were available for 111 subjects, of whom 41 (36.9%) had clonal abnormalities diagnosed by karyotype and/or fluorescence in situ hybridization. Nine subjects developed >1 cytogenetic abnormality. The most common cytogenetic abnormalities were del20q (19.8%; n = 22) and iso7q (9.9%; n = 11), including 2 patients who developed a del20q and a separate iso7q cytogenetic abnormality at different points in time and 1 patient who had a del20q clone existing at the same time as a separate iso7q clone. The cytogenetic clone became undetectable on subsequent marrow examinations for 15 clones, including 3 of 11 (27.3%) subjects with iso7q and 4 of 22 (18.2%) subjects with del20q. Clinical outcomes of patients with cytogenetic clonal abnormalities are described in Table 2 and supplemental Table 2.
Table 2.
Clonal abnormalities over time in patients with SDS
| Patient | Clonal abnormality | Outcome | MDS/AML karyotype |
|---|---|---|---|
| 1 | iso(7q) | unknown | |
| 2 | iso(7q), then separate add(mar), then separate i(X) | Transient clones | |
| 3 | iso(7q) | Transient clone | |
| 4 | iso(7q) | Persistent clone, then developed MDS | 46,XX,der(3;6)(q25;q13)[20]/45,dic(6;7)(q13;p11.2)[4]/46,XX[1] |
| 5 | iso(7q) | Unknown | |
| 6 | iso(7q) | Persistent clone | |
| 7 | iso(7q), separate del3q clone | Progressed to AML | Cytogenetics not available |
| 8 | iso(7q) | Persistent clone | |
| 9 | iso(X) and add X | Transient clones | |
| 10 | iso(X) | Persistent clone | |
| 11 | del(X) | Transient clone | |
| 12 | del(X) | Intermittent clone | |
| 13 | 46, XX [19]/27, XX, + mar[1] | unknown | |
| 14 | der(1) | Developed MDS | 46,XY+1,der(1;17)(q10;q10)[18] /46,XY[2] |
| 15 | inv(1), then add(7) | inv(1) was transient, add(7) unknown | |
| 16 | add(3q), del(8q) | Developed AML | 46,XX,add(3)(q27),del(8)(q13q22),der (10)t(10;15)(p11.2q15)[1], 43-44,der(2)t(2;12)(q33;q13), add(3)(q27),add(4)(q21),-5,-6,add(6)(q25),-7,del(8)(q13q22),-11,-12,der(18)t(11;19)(q13;p13.3)+2-3mar[6], 46,XX[13] |
| 17 | add(7q) | Transient clone | |
| 18 | add(7p) | unknown | |
| 19 | der(9) | Transient clone | |
| 20 | del20q | Persistent clone | |
| 21 | del20q | Transient clone | |
| 22 | add(Y), add(mar), del(20q) | add(Y) is constitutional, del20q persistent. Then developed MDS | 47,XYYc,del(20)(q11.2q13.2)[15]/47, XYYc |
| 23 | del20q | Persistent clone | |
| 24 | del20q | Persistent clone | |
| 25 | add (7p) then separate del(20q), and separate iso(7q), then aberrant complex karyotype in 1 cell (43,XY-1,+del(3)(p2?1),-5,-6,-7) | Both add(7p) and del(20q) disappeared, then progressed to MDS | 46, XY, del(20)(q11.2q13.1)[10]/46,XY,add(7)(q22)[7]/45, XY-7[3] |
| 26 | del20q | Persistent clone | |
| 27 | del20q | Intermittent clone | |
| 28 | del20q | Persistent clone | |
| 29 | del20q | Persistent clone | |
| 30 | del20q | Persistent clone | |
| 31 | del20q | Persistent clone | |
| 32 | del20q | Unknown | |
| 33 | del20q - developed 2 separate del20q clones (previously also developed iso7q) | Persistent clone, then developed MDS | 46XX,del(20)(q11.2q13.3)[9]/46,XX[11] |
| 34 | del20q | Transient clone | |
| 35 | del20q | Persistent clone | |
| 36 | del20q, separate del1p clone | Persistent clones | |
| 37 | del20q | Add 7p which disappeared, then del20q, then del12p developed and progressed to MDS | 46XY[5]/46, XY, del(12)(p11.1)[5]/35,idem,del(5)(q?22q?31),-7,del(9)(q21)[3]/44, idem, add(1)(p32),add(3)(p24),-5,-7,del(9)(q21)[7]/45, idem, add(1)(p32,add(3)(p24),-5,-7,del(9)(q21),=mar[4] |
| 38 | del20q, then separate iso7q | Unknown | |
| 39 | del20q | Unknown | |
| 40 | del20q | Persistent clone | |
| 41 | del20q | Persistent clone |
Malignancies
Twenty-six subjects (17%) developed a myeloid malignancy (MDS = 16, AML = 10) at a median age of 12.3 years (range, 0.5-45.0) and 28.4 years (range, 14.4-47.3), respectively (supplemental Table 2). Among those without a myeloid malignancy, the median age at last follow-up was 8.1 years (range, 0.3-39.5). Of the 16 subjects with MDS, data on G-CSF use was available for 15 of them. Three of those subjects were on G-CSF at the time of MDS diagnosis, and 1 had used G-CSF for 10 months 5 years prior to MDS diagnosis. Of the 10 subjects with AML, 9 had available data on prior G-CSF use, and 2 were taking G-CSF at the time of AML diagnosis.
Of the patients with AML, 90% (9/10) received upfront chemotherapy, and 1 proceeded straight to transplant. Of the patients with MDS, 62.5% (10/16) received upfront HSCT, 12.5% (2/16) received initial chemotherapy followed by HSCT, and 12.5% (2/16) received chemotherapy and died prior to HSCT. One subject did not receive treatment for MDS, and treatment data were not available for another subject. In those who underwent HSCT, acute and chronic GVHD was seen in 6 and 0 patients, respectively (n = 15).
One male subject with heterozygous mutations in SBDS (c.258 + 2T>C, c.653G>A (p.Arg218Gln) and exocrine pancreatic insufficiency requiring pancreatic enzyme supplementation developed primary mediastinal B-cell lymphoma at the age of 16.9 years. He had had regular blood counts that demonstrated neutropenia (400-700 cells per microliter) with normal hemoglobin and platelet count. No dysplasias had been noted on prior surveillance marrows. Following a transient episode of isolated severe neutropenia that resolved, the patient presented with diffuse pruritis, weakness, and shortness of breath. Imaging studies revealed a large anterior mediastinal mass measuring 15.0 × 8.5 × 13.5 cm compressing the left mainstem bronchus and invading the superior vena cava, with associated enlarged mediastinal lymph nodes and pleural effusion. A biopsy was diagnostic for a nongerminal center subtype of diffuse large B-cell lymphoma positive for CD20, CD23, MUM-1, Bcl-6, Bcl-2, and partial c-MYC (40-50%) but negative for CD10, TdT, CD30, and Epstein-Barr virus. Ki-67 proliferation index was high. A bone marrow demonstrated a del20q clone with karyotype 46,XY, del(20)(q11.2q13.1)[1]/46,XY[19] but was negative for lymphoma. He was treated with a rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP) regimen complicated by grade 3 sacral pressure ulcer, fever and neutropenia, reaccumulation of pleural effusions with loculations, deep vein thrombosis, and decreased ejection fraction by echocardiogram leading to omission of doxorubicin from subsequent cycles and initiation of carvedilol. He was in remission at last follow-up 15 months from diagnosis.
Survival
Median survival of the cohort was 38.2 years (95% CI, 29.5-48.5) (Figure 4). There were 20 deaths observed (12.6%) at a median age of 21.2 years (range, 0.3-52.8 years). Causes of death were MDS or AML (n = 15), followed by severe BMF requiring HSCT (n = 2), asphyxiating thoracic dystrophy (n = 2), and liver failure (n = 1).
Figure 4.

Overall survival of subjects with SDS. Kaplan-Meier curve showing overall survival of all subjects. Time is expressed in years since birth.
Discussion
The scarcity of experience with rare diseases, such as SDS, has driven the development of registries for the systematic longitudinal collection of clinical and biological data to advance our understanding of natural history, complications, and outcomes. Clinical care is typically siloed between pediatric and adult subspecialty providers for these complex conditions, further contributing to gaps in clinical knowledge and the expertise needed to provide lifelong care for these patients. The SDS Registry provided a critical platform for the analysis of hematologic complications with age for patients with biallelic SBDS mutations.
Analysis was restricted to subjects with biallelic SBDS mutations because of the limitations of clinical phenotype–based diagnoses. In our experience, a subset of patients with cardinal features of SDS, such as neutropenia with exocrine pancreatic dysfunction (eg, low trypsinogen/pancreatic isoamylase), lack mutations in any of the known SDS genes. Notably, these patients may not have SDS and are occasionally found to have a different genetic disorder altogether.
It is striking that all but 1 subject harbored ≥1 SBDS allele bearing the hypomorphic c.258 + 2T>C (IVS2 + 2) splice site mutation, which produces a scant amount of wild-type SBDS transcript. In our cohort, we observed the lack of this hypomorphic IVS2 + 2T>C splice mutation in only a single patient with compound heterozygous SBDS missense and stop mutations who had a particularly severe phenotype. Murine Sbds-mutant models of SDS carrying biallelic-null or missense mutations and lacking this hypomorphic splice mutation also exhibit early fulminant phenotypes of greater severity than those typical of patients with SDS.22–24 A patient with a p.Lys62X mutation and a p.Lys148Thr missense mutation has been reported.18 This patient, who lacked the IVS2 + 2 T>C mutation, presented with transient severe cytopenia at age 0.9 years and bone marrow that displayed dysmyelopoietic features and hemophagocytosis without clonal abnormalities. This patient remained clinically stable at 8.4 years of follow-up. One patient with 2 SBDS missense mutations was noted in the Italian SDS Registry, but no associated clinical data were reported.19 Further studies are needed to elucidate the potential clinical and biologic implications of the IVS2 + 2 T>C mutation.
Although data extraction from medical records provides information from real-world clinical practice, a limitation of a registry-based study is that the only available data are those that happen to be collected or recorded by the individual treating physician during clinical care. Variation in diagnostic evaluation and clinical management is particularly problematic for rare diseases for which provider experience is often limited and practice standards are unavailable or sporadically followed.
In our cohort, severe BMF requiring HSCT occurred predominantly in early childhood, with only 1 patient developing severe BMF in adulthood. This is consistent with data from the French Registry, which reported severe cytopenias at a median age of 0.13 years (range, 0.01-3.3).18 A European Society for Blood and Marrow Transplantation study reported that 7 of 61 patients with SDS and severe BMF were older than 18 years of age; however, SDS diagnosis was based on clinical features without genetic testing. A study from the Center for International Blood and Marrow Transplant Research reported 39 patients diagnosed with SDS who were transplanted for severe BMF at a median age of 7 years. Although the majority of patients (n = 23) with BMF were younger than 10 years, 11 patients were between the ages of 10 and 19 years, and 5 patients were ≥20 years old. Notably, only 49% of patients (n = 19) had been tested for SBDS mutations, and 14 of 39 (36%) had confirmed biallelic SBDS mutations. Standardized diagnostic criteria, including genetic testing, would strengthen future studies.
Correlation between the degree of marrow cellularity and cytopenias was poor. Low neutrophil count, the most common cytopenia in SDS, was positively associated with age of the subject. Assessment of marrow cellularity can be subjective and should be evaluated in comparison with prior marrows and in the context of the peripheral blood counts. Marrow cellularity in isolation does not constitute an indication for HSCT.
To our knowledge, this is the first report of a patient with biallelic SBDS mutations who developed a lymphoid malignancy. A patient with an acute lymphoblastic leukemia and SDS was reported previously; however, the diagnosis of SDS was based on postmortem pancreas histology, and genetic testing was not available at that time.25 Further study is needed to determine whether the risk of lymphoid malignancy is increased in SDS.
We previously reported our observation from the SDS Registry that surveillance strategies for SDS are heterogeneous in clinical practice.3 Though severe BMF requiring HSCT was observed predominantly in young patients, the median age for development of MDS and AML was higher, indicating the importance of ongoing surveillance. The rationale for surveillance is the abysmal prognosis once AML develops in patients with SDS.3 Improved strategies integrating somatic genomics to identify patients at high risk for malignant transformation are under investigation.26
Overall, only 3 subjects died of causes other than MDS, AML, or severe BMF requiring HSCT. No subject in our cohort died from bacterial, fungal, or severe viral infection outside of the context of malignancy or HSCT. Severe infections with bacterial sepsis, osteoarthritis, or meningitis and severe viral infections and infection-related mortality have been observed in other SDS registry reports, highlighting the importance of vigilant attention to neutropenia.18,19 Infections remain a significant cause of mortality in other neutropenia syndromes, such as severe congenital neutropenia.27
A recent study from our SDS Registry reported the development of clonal hematopoiesis with age in patients with SDS.26 Adaptive somatic loss-of-function mutations in EIF6 rescued the underlying ribosomal stress exerted by the SBDS mutations and were not associated with malignancy, whereas maladaptive somatic mutations in TP53 led to an increased risk for malignant transformation. Notably, patients who developed severe BMF, all at young ages, lacked clonal hematopoiesis. Larger patient numbers and mechanistic studies will be required to investigate the potential association between somatic mutations and risk of BMF in SDS. These studies raise the possibility that further investigation of adaptive somatic changes coupled with clinical studies might identify potential therapeutic targets to treat BMF in SDS.26,28
In conclusion, this SDS Registry study found that, although marrow cellularity decreased with age, as expected for a genetic BMF condition, blood counts unexpectedly improved with age. Severe BMF was observed in early childhood, whereas myeloid malignancy was observed in later childhood/early adulthood. Further study is needed to investigate the potential mechanistic link between clonal hematopoiesis and improvement in blood counts with age for patients with SDS. These data will guide clinicians in discussions with families and in clinical decision making, as well as inform further work addressing improved surveillance strategies.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank all patients and families participating in the SDS Registry, as well as physician colleagues who referred patients to the registry.
This work was supported in part by National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases grants R24 DK099808 (A.S. and M.F.) and 1RC2DK122533-01 (A.S., K.C.M., and M.F.) and National Institutes of Health National Heart, Lung, and Blood Institute grant T32 HL007574-36 (E.F.).
Authorship
Contributions: E.F., K.C.M., and A.S. designed the study and wrote the manuscript; E.F., K.C.M., M.M., J. Larson, A. Galvin, R.L., S. Loveless, L.M., K.Q., S.S., and A.S. designed the database and extracted data; E.F., K.C.M., S. Liu, A.Galvin, E.W., and A.S., analyzed data; and A.B., M.F., J.M.G., A.Geddis, R.H., S.K., B.L., J. Lipton, R.L., T.N., A.V., W.W., and S.D. contributed phenotype data and edited the manuscript.
Conflict-of-interest disclosure: J.M.G. is an employee of Alnylam Pharmaceuticals and holds stock and stock options in the company. The remaining authors declare no competing financial interests.
Correspondence: Akiko Shimamura, Boston Children’s Hospital, 1 Blackfan Circle, Karp 8210, Boston, MA 02115; e-mail: akiko.shimamura@childrens.harvard.edu.
References
- 1.Myers KC, Bolyard AA, Otto B, et al. Variable clinical presentation of Shwachman-Diamond syndrome: update from the North American Shwachman-Diamond Syndrome Registry. J Pediatr. 2014;164(4):866-870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nelson AS, Myers KC. Diagnosis, treatment, and molecular pathology of Shwachman-Diamond syndrome. Hematol Oncol Clin North Am. 2018;32(4):687-700. [DOI] [PubMed] [Google Scholar]
- 3.Myers KC, Furutani E, Weller E, et al. Clinical features and outcomes of patients with Shwachman-Diamond syndrome and myelodysplastic syndrome or acute myeloid leukaemia: a multicentre, retrospective, cohort study. Lancet Haematol. 2020;7(3):e238-e246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Myers K, Hebert K, Antin J, et al. Hematopoietic stem cell transplantation for Shwachman-Diamond syndrome. Biol Blood Marrow Transplant. 2020;26(8):1446-1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cesaro S, Pillon M, Sauer M, et al. Long-term outcome after allogeneic hematopoietic stem cell transplantation for Shwachman-Diamond syndrome: a retrospective analysis and a review of the literature by the Severe Aplastic Anemia Working Party of the European Society for Blood and Marrow Transplantation (SAAWP-EBMT) [published correction appears in Bone Marrow Transplant. 2020;55(9):1884]. Bone Marrow Transplant. 2020;55(9):1796-1809. [DOI] [PubMed] [Google Scholar]
- 6.Boocock GR, Morrison JA, Popovic M, et al. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet. 2003;33(1):97-101. [DOI] [PubMed] [Google Scholar]
- 7.Stepensky P, Chacón-Flores M, Kim KH, et al. Mutations in EFL1, an SBDS partner, are associated with infantile pancytopenia, exocrine pancreatic insufficiency and skeletal anomalies in a Shwachman-Diamond like syndrome. J Med Genet. 2017;54(8):558-566. [DOI] [PubMed] [Google Scholar]
- 8.Tan QK, Cope H, Spillmann RC, et al. ; Undiagnosed Diseases Network . Further evidence for the involvement of EFL1 in a Shwachman-Diamond-like syndrome and expansion of the phenotypic features. Cold Spring Harb Mol Case Stud. 2018;4(5):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tummala H, Walne AJ, Williams M, et al. DNAJC21 mutations link a cancer-prone bone marrow failure syndrome to corruption in 60S ribosome subunit maturation. Am J Hum Genet. 2016;99(1):115-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhanraj S, Matveev A, Li H, et al. Biallelic mutations in DNAJC21 cause Shwachman-Diamond syndrome. Blood. 2017;129(11):1557-1562. [DOI] [PubMed] [Google Scholar]
- 11.Carapito R, Konantz M, Paillard C, et al. Mutations in signal recognition particle SRP54 cause syndromic neutropenia with Shwachman-Diamond-like features. J Clin Invest. 2017;127(11):4090-4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang MY, Keel SB, Walsh T, et al. Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity. Haematologica. 2015;100(1):42-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghemlas I, Li H, Zlateska B, et al. Improving diagnostic precision, care and syndrome definitions using comprehensive next-generation sequencing for the inherited bone marrow failure syndromes. J Med Genet. 2015;52(9):575-584. [DOI] [PubMed] [Google Scholar]
- 14.Muramatsu H, Okuno Y, Yoshida K, et al. Clinical utility of next-generation sequencing for inherited bone marrow failure syndromes. Genet Med. 2017;19(7):796-802. [DOI] [PubMed] [Google Scholar]
- 15.Bluteau O, Sebert M, Leblanc T, et al. A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood. 2018; 131(7):717-732. [DOI] [PubMed] [Google Scholar]
- 16.Nicolis E, Bonizzato A, Assael BM, Cipolli M. Identification of novel mutations in patients with Shwachman-Diamond syndrome. Hum Mutat. 2005;25(4):410. [DOI] [PubMed] [Google Scholar]
- 17.Yamada M, Uehara T, Suzuki H, et al. Shortfall of exome analysis for diagnosis of Shwachman-Diamond syndrome: mismapping due to the pseudogene SBDSP1. Am J Med Genet A. 2020;182(7):1631-1636. [DOI] [PubMed] [Google Scholar]
- 18.Donadieu J, Fenneteau O, Beaupain B, et al. ; Associated investigators of the French Severe Chronic Neutropenia Registry* . Classification of and risk factors for hematologic complications in a French national cohort of 102 patients with Shwachman-Diamond syndrome. Haematologica. 2012;97(9):1312-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cesaro S, Pegoraro A, Sainati L, et al. A prospective study of hematologic complications and long-term survival of Italian patients affected by Shwachman-Diamond syndrome. J Pediatr. 2020;219:196-201.e1. [DOI] [PubMed] [Google Scholar]
- 20.Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Muggeo VRM, Atkins DC, Gallop RJ, Dimidjian S. Segmented mixed models with random changepoints: a maximum likelihood approach with application to treatment for depression study. Stat Model. 2014;14(4):293-313. [Google Scholar]
- 22.Zhang S, Shi M, Hui CC, Rommens JM. Loss of the mouse ortholog of the Shwachman-Diamond syndrome gene (Sbds) results in early embryonic lethality. Mol Cell Biol. 2006;26(17):6656-6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finch AJ, Hilcenko C, Basse N, et al. Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman-Diamond syndrome. Genes Dev. 2011;25(9):917-929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tourlakis ME, Zhong J, Gandhi R, et al. Deficiency of Sbds in the mouse pancreas leads to features of Shwachman-Diamond syndrome, with loss of zymogen granules. Gastroenterology. 2012;143(2):481-492. [DOI] [PubMed] [Google Scholar]
- 25.Strevens MJ, Lilleyman JS, Williams RB. Shwachman’s syndrome and acute lymphoblastic leukaemia. BMJ. 1978;2(6129):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kennedy AL, Myers KC, Bowman J, et al. Distinct genetic pathways define pre-malignant versus compensatory clonal hematopoiesis in Shwachman-Diamond syndrome. Nat Commun. 2021;12(1):1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenberg PS, Alter BP, Bolyard AA, et al. ; Severe Chronic Neutropenia International Registry . The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood. 2006;107(12):4628-4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan S, Kermasson L, Hilcenko C, et al. Somatic genetic rescue of a germline ribosome assembly defect. Nat Commun. 2021;12(1):5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.