

Abstract
Clinical manifestations and the need for treatment varies according to age in males with hypogonadism. Early foetal-onset hypogonadism results in disorders of sex development (DSD) presenting with undervirilised genitalia whereas hypogonadism established later in foetal life presents with micropenis, cryptorchidism and/or micro-orchidism. After the period of neonatal activation of the gonadal axis has waned, the diagnosis of hypogonadism is challenging because androgen deficiency is not apparent until the age of puberty. Then, the differential diagnosis between constitutional delay of puberty and central hypogonadism may be difficult. During infancy and childhood, treatment is usually sought because of micropenis and/or cryptorchidism, whereas lack of pubertal development and relative short stature are the main complaints in teenagers. Testosterone therapy has been the standard, although off-label, in the vast majority of cases. However, more recently alternative therapies have been tested: aromatase inhibitors to induce the hypothalamic-pituitary-testicular axis in boys with constitutional delay of puberty and replacement with GnRH or gonadotrophins in those with central hypogonadism. Furthermore, follicle-stimulating hormone (FSH) priming prior to hCG or luteinizing hormone (LH) treatment seems effective to induce an enhanced testicular enlargement. Although the rationale for gonadotrophin or GnRH treatment is based on mimicking normal physiology, long-term results are still needed to assess their impact on adult fertility.
Keywords: anorchia, cryptorchidism, delay of puberty, disorders of sex development, DSD, Kallmann syndrome, Klinefelter syndrome, micropenis
Introduction
The concept of male hypogonadism is often associated with low testosterone production by the testes. This notion derives from adult endocrinology; indeed, during adulthood, testosterone is the most conspicuous testicular hormone because of its circulating levels and target organ actions. Conversely, in paediatric endocrinology, basal serum testosterone determination is helpful only during the first months following birth and after mid-puberty. 1 During the rest of infancy and childhood, serum testosterone is physiologically below the detectable levels using classical assays, and anti-Müllerian hormone (AMH)2–4 and inhibin B 4 are more adequate biomarkers to initially screen testicular function (Figure 1).
Figure 1.

Ontogeny of the anatomical changes and serum levels of reproductive axis hormones in the male. In the early stages of the foetal period, testicular hormones begin to be produced before gonadotrophins and are responsible for the virilization of the internal and external genitalia. From the second trimester of foetal life onwards, luteinizing hormone (LH) regulates testosterone (T) secretion by Leydig cells, whereas follicle-stimulating hormone (FSH) controls Sertoli cell proliferation and AMH and inhibin B secretion. FSH induces testicular enlargement while T is involved in testicular descent to the scrotum and penile enlargement. During the 3–6 months following birth (also known as ‘mini-puberty’), the reproductive axis remains active, but then LH and T decline to undetectable levels during childhood. Conversely, Sertoli cells remain functional as revealed by high levels of anti- Müllerian hormone (AMH) and inhibin B. During puberty, gonadotrophins and T increase. The testes enlarge dramatically due to the proliferation of germ cells undergoing adult spermatogenesis. FSH and germ cells boost inhibin B secretion, whereas T is potent inhibitor of AMH. Reprinted, with permission, from Salonia et al. 5 © 2019 Springer Nature Limited.
The ontogeny of the hypothalamic-pituitary-testicular axis: its importance for the diagnosis of male hypogonadism
The hypothalamic-pituitary-testicular (HPT) axis is undoubtedly the pituitary axis that shows the most remarkable changes throughout life (Figure 1). The definition of male hypogonadism should take into account this ontogeny and reflect the inability of Sertoli, Leydig, and/or germ cells to fulfil their functions. 6 Primary hypogonadism, called hypergonadotrophic hypogonadism in adult endocrinology, reflects a primary defect of testicular components—Sertoli, germ and/or Leydig cells—that can affect all cell populations concomitantly or initiate with the impairment only one of them and affect the others secondarily. Secondary or central hypogonadism, called hypogonadotrophic hypogonadism in adult endocrinology, reflects a gonadal dysfunction that results from a hypothalamic-pituitary insufficiency.
The prenatal period: consequences of foetal-onset hypogonadism
During the embryonic and foetal periods, the testes differentiate before the gonadotroph is functional and secrete AMH and testosterone, which play essential roles in sexual differentiation of the genitalia. 7 Testosterone, secreted by Leydig cells in response to placental human chorionic gonadotrophin (hCG) in the first trimester of gestation and by foetal pituitary luteinising hormone (LH) thereafter, drives the differentiation of the Wolffian ducts into the epididymides, vasa deferentia and seminal vesicles. Upon transformation to dihydrotestosterone (DHT) by the action of the enzyme 5α-reductase in target tissues, it induces the formation of the prostate and the virilisation of the urethra and the external genitalia in the first trimester and the increase in the size of the genitalia, as well as testicular descent in the second and third trimesters of gestation. AMH, produced by Sertoli cells independently of follicle-stimulating hormone (FSH) action in the first trimester of foetal life, provokes the regression of Müllerian ducts, the primordia of the uterus and Fallopian tubes. Subsequently, FSH stimulates Sertoli cell proliferation resulting in testicular enlargement 8 and increased secretion of AMH9–11 and inhibin B.11,12 Note that the Sertoli cell population represents the major testicular component until the onset of puberty (Figure 1).
Foetal onset male hypogonadism has different clinical consequences according to the time of onset during the embryonic/foetal stage and to the testicular cell populations affected (Table 1). Very early foetal-onset hypogonadism can only be primary, since the gonadotroph is not yet functional. Androgen deficiency may result in a complete lack of virilisation and, thus, a female phenotype of the external genitalia at birth in the case of complete gonadal dysgenesis, a condition characterised by whole gonadal dysfunction and named dysgenetic disorder of sex development (DSD), 7 or of Leydig cell steroidogenic failure, characterised by specific cell dysfunction with normal Sertoli cell activity and known as non-dysgenetic DSD. 7 In these cases, newborns are assigned female and will not be discussed further in this review. Alternatively, the gonadal disorder may be partial, resulting in ambiguous genitalia with different degrees of virilisation, which may lead to male gender assignment at birth. 13
Table 1.
Clinical presentation of genitalia in male hypogonadism, according to type and age of initiation.
| Age of initiation | Type of hypogonadism | Whole testicular dysfunction | Leydig cell-specific dysfunction | Sertoli cell-specific dysfunction | |||
|---|---|---|---|---|---|---|---|
| Foetal 1st Trimester | Primary hypogonadism | Testicular dysgenesis | DSD, undervirilisation | Leydig cell hypoplasia Steroidogenic defects |
DSD, undervirilisation | FSH-R defects AMH mutation |
Micro-orchidism PMDS |
| Foetal 2nd – 3rd Trimesters | Primary hypogonadism | Testicular regression syndrome | Micropenis, Cryptorchidism | Inexistent | Inexistent | ||
| Central hypogonadism | Isolated central hypogonadism Multiple pituitary hormone deficiency |
Micropenis, Cryptorchidism, Micro-orchidism, Absent or
incomplete puberty Ibidem + hypothyroidism, cortisol deficiency, growth failure |
LHβ mutation | Micropenis, Cryptorchidism, Absent or incomplete puberty | FSHβ mutation | Micro-orchidism | |
| Postnatal | Primary hypogonadism | Bilateral gonadectomy Klinefelter syndrome Testicular radiotherapy |
Empty scrotum None, Cryptorchidism, Micro-orchidism Micro-orchidism |
Inexistent | Inexistent | ||
| Central hypogonadism | Surgery of the sellar/suprasellar region Pituitary tumours Cranial trauma High dose cranial radiotherapy Systemic diseases |
Absent or incomplete puberty | Inexistent | Inexistent | |||
| Constitutional delay of puberty | Absent puberty | Inexistent | Inexistent | ||||
AMH, anti-Müllerian hormone; DSD, Disorders of sex development; FSH, Follicle-stimulating hormone; FSHβ, FSH beta subunit; FSH-R, FSH receptor; LH, luteinising hormone; LHβ, LH beta subunit; PMDS: Persistent Müllerian Duct Syndrome.
Foetal-onset hypogonadism during the second or third trimesters, irrespective of its primary or central origin, is associated with a completely male phenotype of the external genitalia. Nonetheless, the androgen insufficiency may lead to micropenis and cryptorchidism. When the condition is of central origin, the FSH insufficiency results in reduced Sertoli cell numbers, leading to micro-orchidism and low serum AMH and inhibin B.11,14,15 If congenital hypogonadism goes unnoticed during the first 3 to 6 months of postnatal life, androgen insufficiency cannot be detected during childhood, and low Sertoli cell biomarkers3,4,16–18 and genetic tests19,20 are more helpful for the diagnosis (Figure 1). At the age of puberty, lack of pubertal signs prompts the assessment.
Childhood: male hypogonadism may remain unnoticed
As already mentioned, gonadotrophin and testosterone serum levels decline to very low or even undetectable values after the age of 3 to 6 months in the normal boy (Figure 1). 21 Therefore, the male hypogonadism established during childhood may be challenging not only in the case of central hypogonadism, because gonadotrophins and testosterone cannot be lower than in normal boys explaining why the term ‘hypogonadotrophic’ may be misleading at this age, 1 but also in face of cases of primary hypogonadism, because serum gonadotrophins show normal prepubertal values in a significant number of cases and the term ‘hypergonadotrophic’ may be confusing.1,22 A clear example is Klinefelter syndrome,23,24 the commonest aetiology of primary hypogonadism in males.
Adolescence: pubertal delay
The reactivation of GnRH secretion in the hypothalamus leads a to a progressive increase in LH and FSH pulsatile secretion. 25 FSH triggers again Sertoli cell proliferation, inducing a moderate but perceivable increase in testicular size, from 2 to 4 ml as measured by comparison to Prader’s orchidometer, or from 1.0 to 2.7 ml as measured by ultrasonography. 26 Concomitantly, LH acts on Leydig cells to induce a progressive increase in intratesticular androgen concentration during Tanner stages 2 and 3 of pubertal development, which provokes Sertoli cell maturation, 27 reflected in a decrease in serum AMH. 28 Then, Sertoli cells cease to proliferate and become capable of supporting adult spermatogenesis, the main responsible for the dramatic increase of testicular volume during puberty to reach an adult volume > 15 ml by orchidometer or > 17.5 ml by ultrasound. 26 Inhibin B levels represent a useful biomarker of Sertoli cell maturation and adequate spermatogenic progression. 29 Serum testosterone levels increase progressively from Tanner stage 3 to reach adult values in Tanner stage 5.28,30 The gradual increase in serum testosterone is associated with the development of secondary sex characteristics, especially penile growth which occurs from Tanner stage 3 onwards, 31 and a timely bone age maturation to attain adult height.
When there is a whole gonadal dysfunction, due to either primary or central hypogonadism, the lack of testosterone rise results in the lack of signs of pubertal development. For primary hypogonadism to be associated with complete lack of development of secondary sex characteristics, the testicular disorder must be severe, for example, complete testicular regression syndrome or gonadal removal. Otherwise, Leydig cell function often remains sufficient to provoke some signs of androgenisation, even if Sertoli cells are severely affected, for example, in many cases of partial gonadal dysgenesis 7 or long-standing cryptorchidism. 17
Apart from the already mentioned congenital form, central hypogonadism may be acquired during childhood, for example, due to tumours or other lesions of the central nervous system affecting the hypothalamic-pituitary region or to functional states such as chronic or acute illnesses that impair the HPT axis. 5 The severity of the condition may lead to a complete lack of pubertal onset or to a normal or delayed onset with incomplete progression.
In those cases where there is no personal or family history that could drive the diagnosis, constitutional delay of puberty is the commonest cause of lack of pubertal signs in a boy reaching the age of 14 years, that is, that corresponding to more than 2 standard deviations from normal pubertal age onset.32,33 The differential diagnosis may prove difficult: basal LH and testosterone levels are usually uninformative and several dynamic tests have been developed with controversial results. 34 Although neglected in males, the FSH-gonadal axis seems to be more informative.35–37
Pharmacotherapy for male hypogonadism
Medicines used
Androgenic drugs
The most frequently used formulations in adolescents are testosterone esters, such as enanthate, cypionate and propionate, available as oil-based compounds for slow-release IM injections. Testosterone enanthate is available in 200- or 250-mg ampoules or ready-to-use syringes. It has a half-life of 4.5 days and is cleared 90% through the kidneys and 10% through the bile (10%). Testosterone cypionate is available in 100-mg or 200-mg vials, and its half-life is 4 days. 38 A mixture of 100 mg decanoate, 60 mg isocaproate, 60 mg phenylpropionate and 30 mg propionate exists in 250-mg ampoules.39–41 IM testosterone undecanoate has not been used in paediatric patients owing to its long-acting period. 42
In adolescents > 12 years-old, the anabolic and androgenic effects induced by replacement therapy are expected to mimic the timing and tempo observed during normal pubertal development. In other words, androgen supplementation should start at low doses with progressive increases so that adult testosterone serum levels are attained 2 to 3 years later. Thus, with a progression of approximately one Tanner stage every year, a precocious early closure of the epiphyses is avoided. The most commonly used protocols with testosterone enanthate or cypionate consist of an initial dose of 50–100 mg every 4 weeks.5,43,44 Due to the pharmacokinetics of these formulations, circulating testosterone attains supraphysiological levels in the first week after IM injection and subphysiological levels in the fourth week (Figure 2),45,46 with interindividual variations.47,48 The dosage is progressively increased in 50- to 100-mg increments every 6 to 12 months to mimic the evolution of serum testosterone levels observed during pubertal stages, 28 to reach the adult dose of 250 mg every 4 weeks after approximately 2 to 3 years.
Figure 2.

Schematic representation of circulating levels of testosterone adult male patients with hypogonadism receiving testosterone enanthate or testosterone undecanoate IM. Time 0 represents before treatment; serum concentrations are indicated following IM injections of 250 mg of enanthate or 1000 mg of undecanoate. The grey area represents normal serum levels of testosterone for an adult male. Data obtained from references.46,49–51 Modified with permission from Rey and Grinspon. 52 © 2020 The Authors.
Transdermal testosterone formulations are the most frequently used in adults, 53 but like IM formulations, they have not yet been approved for use in paediatric patients. Only few studies are reported in the literature in prepubertal boys.
While the initial oral androgen formulations showed hepatotoxicity,53,54 testosterone undecanoate capsules avoid hepatic metabolism, and pilot studies have been reported in adolescents.55,56 Mucoadhesive tablets, designed to provide sustained testosterone release as it hydrates after inner cheek adhesion, 53 have not been used in children or adolescents. Similarly, no experience exists in paediatric patients with a testosterone gel for intranasal administration featuring easy delivery and low risk of transference.53,54
Side effects commonly described for androgenic drugs used for long periods include erythrocytosis, irritability, acne, persistent erections, and gynaecomastia due to aromatisation to oestrogens. 57 One particular concern in paediatric ages is the potential acceleration of bone age, which compromises adult height. Early pubarche can also be observed. Oral testosterone undecanoate shows lower risk for erythrocytosis than injectable formulations. 58 It should be kept in mind that the beneficial effects that systemic androgen therapy has on most organs contrast with their negative effects on the HPT axis. Indeed, exogenous androgens usually reach circulating levels that inhibit the gonadotroph. Because of the reduced secretion of LH, intratesticular androgen concentration remains low, spermatogenesis cannot go through meiosis and spermiogenesis, and testicular volume persists small.27,59
DHT, also known as stanolone or androstanolone, is the most potent natural androgen and cannot be aromatised to oestrogens. It is commercially available in some countries as 2.5% gels for transdermal delivery.60–62 DHT is typically applied directly to the external genitalia in patients with micropenis 63 and is indicated in patients with nondysgenetic DSD due to 5α-reductase deficiency.42,64
Nandrolone and oxandrolone are non-aromatisable drugs with a predominant anabolic action and are thus used when androgenic effects need to be avoided.57,65 Nandrolone exists in 1-ml vials at 200 mg/ml for IM injections. Oxandrolone is available as oral tablets of 2.5 and 10 mg. In patients with Klinefelter syndrome, positive effects have been reported on psychosocial and visual-motor functions 66 as well as on cardiometabolic health markers 67 after oxandrolone administration. The main side effect is hepatotoxicity, though rarely serious or irreversible. 57 Hepatic peliosis hepatocellular neoplasia have been described after long-term administration. In paediatric patients, even if its androgenic effects are reduced, oxandrolone may provoke precocious genital development. 68 Selective androgen receptor modulators (SARMs) with different androgen receptor binding capacity are under investigation. 69
Aromatase inhibitors, such as letrozole and anastrozole, repress the metabolisation of androgens to oestrogens and have been tested to boost androgen levels in patients with preserved androgen synthesis, that is, constitutional delay of puberty. 40
Gonadotrophin formulations
Several preparations containing hCG, LH or FSH, alone or in mixture, have existed in the market and have been used especially in adult reproductive endocrinology. 70 Here, I will focus on those formulations used in paediatric patients with hypogonadism.
Human chorionic gonadotrophin (hCG) has been available for several decades as solutions for IM or SC injection containing between 500 and 10,000 IU, obtained from the urine of pregnant women. LH and hCG bind the same receptor on Leydig cells, but hCG has a longer half-life 70 and can be given once or twice a week according to the therapeutic aim: in boys with cryptorchidism, hCG is usually administered at 500–1000 IU per week for 5 weeks, whereas in adolescents with delayed puberty the usual dosage is 1000 IU twice a week.
LH and FSH are also present in preparations obtained from urine: human menopausal gonadotrophin or menotropin (hMG) contains FSH, LH, and hCG, whereas immunological-based technologies have allowed to obtain highly purified urinary FSH preparations. However, almost no experience has been published in paediatric male patients with these preparations. Conversely, in the last two decades, recombinant gonadotrophin formulations have been developed, including r-LH, r-hCG, r-FSH, long-acting r-FSH (corifollitropin alfa), and a mixture of r-LH/r-FSH.70,71
Two r-FSH preparations, follitropins alfa and beta, are very similar and only differ in the technical approach using Chinese hamster ovary (CHO) cells resulting in slight differences in posttranslational changes. 71 Nonetheless, all recombinant follitropins obtained in CHO cells differ from the natural gonadotrophins in some of the attached glycans, which may affect their bioactivity. 70 In an attempt to solve this problem, recombinant follitropin delta was generated using a human cell line.71,72 Also more recently, corifollitropin alfa has been developed; it consists of r-FSH fused to the carboxyl-terminal peptide of the beta-subunit of hCG, which conveys a longer plasma half-life.71,73,74 Recombinant LH (lutropin alfa) and r-hCG are produced in CHO cells. 71 Although gonadotrophins may be given as IM or SC injections equivalently, SC administration is preferred by the overwhelming majority of patients and supports long-term adherence to treatment. 75
GnRH is available in a few countries for pulsatile administration using a SC pump delivering the drug at a rate of 25 ng/kg per pulse every 120 min. 76
Pharmacotherapy in neonates and infants
Irrespective of the aetiology of impaired gonadal hormone production, newborns or infants with male hypogonadism may require treatment for micropenis and/or cryptorchidism. However, those with central hypogonadism have a more favourable fertility potential, and treatment options should consider future spermatogenic development as an aim.
Treatment of newborns/infants with DSD
Newborns with dysgenetic DSD, including partial testicular dysgenesis, asymmetric gonadal differentiations (also known as mixed gonadal dysgenesis) and ovotesticular DSD, assigned male usually present with micropenis, hypospadias and cryptorchidism. While hypospadias can only be repaired surgically, micropenis and cryptorchidism may be subject to hormonal treatment. For penile enlargement and enhancement of scrotal trophism, the usual practice consists in the administration of 2 or 3 courses of IM testosterone enanthate 25–50 mg every 3–4 weeks.43,77–85 Longer courses may provoke bone age advancement and pubic hair development and should therefore be avoided. Although infrequent, side effects related to androgen excess are erections and acne; non-specific side effects are pain and infections in the injection site. In some countries, testosterone is available for percutaneous treatment: 0.2 g of 5% testosterone cream (i.e. 10 mg of testosterone) applied onto the phallus daily for 1 month showed efficacy (9-mm increase in penile length) and safe (no significant advancement in bone age).86,87 DHT gels, which are available in certain countries, may be even more efficacious in patients with DSD, and especially in those with 5α-reductase deficiency.64,88–91 Treatment consists of gel application onto the penis twice a day at a daily dose of 0.1–0.3 mg/kg/day, with caution not to exceed 5 mg/day, for a maximum period of 6 months.64,92
If LH levels are not too high, hCG treatment could be tried to induce testicular descent when the gonads are palpable in the lower part of the inguinal canal. However, this is infrequent and surgical orchiopexy is usually performed.
Treatment of newborns/infants with primary hypogonadism and completely virilised genitalia
Patients with testicular regression syndrome or testicular torsion occurring in the second half of gestation usually present at birth with micropenis and nonpalpable gonads. Once the lack of testicular tissue or its extreme scarcity is ascertained by the finding of undetectable or extremely low AMH or inhibin B in association with elevated FSH, together with low or undetectable testosterone and elevated LH, androgen replacement is indicated following the same protocol as that described above for patients with DSD.
Cryptorchidism may be associated with primary hypogonadism or with eugonadism. 17 Expectant behaviour during the first year is suggested by many authors, given that the testes may finalise their descent to the scrotum.93,94 However, others point to the importance of an adequate hormonal milieu for germ cell development during the neonatal activation period, also known as mini-puberty, and are prone to hCG or GnRH treatment without delay. 95 Three systematic reviews showed that the efficacy of hormone therapy is related to testis position –the lower, the better– rather than to age at treatment.96–98 Several protocols exist; 99 hCG given IM or SC 500–1000 IU once a week for 5 weeks is the most widely used, whereas native GnRH or GnRH analogues have also been used,99,100 but are not available in most countries.
Treatment of newborns/infants with central hypogonadism
Like patients with other forms of hypogonadism, those with central hypogonadism are brought to medical attention for micropenis and/or cryptorchidism. Micro-orchidism usually is underestimated or overlooked. These signs might represent a red flag for ruling out other pituitary hormone deficiencies, 15 which would need to be treated as a priority given their vital roles, for example, cortisol and thyroid hormone deficiencies.
Testosterone treatment has been classically used off-label to induce penile growth, as explained above, and the age at which treatment is installed does not seem to be critical. More recently, a few studies have adopted a physiology-based rationale to test gonadotrophin replacement during the first 6 months of life, in order to mimic the neonatal activation of the HPT axis. Combined treatment with recombinant FSH plus LH or hCG, or GnRH or a GnRH analogue, was expected to induce Sertoli cell proliferation–thus resolving micro-orchidism–and adequate germ cell development, together with Leydig cell androgen production promoting testicular descent and penile enlargement. Initial case reports showed that SC injections 101 or SC pump infusions16,102 of recombinant LH and recombinant FSH led to a significant increase (twofold to fourfold) in testicular volume and hormone (AMH, inhibin B and testosterone) levels, followed by an enlargement of penile size (from ~1 cm to 3–4 cm). LH was administered at 20–40 IU twice a week 101 or 56–75 IU/day,16,102 whereas FSH doses were 21.3 IU twice a week 101 or 67–75 IU/day.16,102 More recently, a case series study reported the results of recombinant LH 50 IU/day plus recombinant FSH 75–150 IU/day given through a SC pump, showing and increase in testicular size and penile length in the 8 patients included, with complete testicular descent in 6 of them. 11 While encouraging in the short term, the long-term effects of this strategy need to be validated.
Pharmacotherapy in childhood
Continued hormone replacement therapy does not seem necessary during childhood, based on the knowledge of the normal ontogeny of the HPT axis (Figure 1). Nonetheless, in boys with Klinefelter syndrome androgenic treatment has been assessed even though hypoandrogenism has not been unequivocally observed. In placebo-controlled trials, oxandrolone oral administration at a daily dose of 0.05–0.06 mg/kg for 24 months to boys aged 4–12 years showed improvements in patients’ cognition and behaviour, as well as in their motor and visual capacities. Clinically non-significant adverse events, such as minor advancement in bone age and decline in serum HDL, were more frequently observed than early pubarche and an increased risk of early testicular enlargement; HPT axis hormone levels were not affected.66–68 These results cannot be applied to patients with higher grade sex chromosome aneuploidies, such as 48, XXXY, 48, XXYY or 49, XXXXY, in whom testosterone treatment resulted in an earlier and persistent suppression of testicular hormone production. 103
One study assessed the effect of r-FSH treatment on 3 boys with central hypogonadism. 12 The underlying rationale was that FSH is important to provoke Sertoli cell proliferation before pubertal maturation induced by intratesticular testosterone. Subcutaneous r-FSH, administered for 12 months at 1.5 IU/kg 3 times a week, induced testicular enlargement and increased serum inhibin B levels, reflecting Sertoli cell stimulation, suggesting that FSH treatment is an option before pubertal maturation to induce Sertoli-cell proliferation with a potential of an enhanced sperm-producing capacity in adulthood.
Pharmacotherapy at pubertal age
Irrespective of the aetiology, androgen insufficiency needs to be treated at the age of puberty in order to tackle the lack of development of secondary sexual characteristics and the impaired growth spurt. Indeed, boys with hypoandrogenism maintain a childlike body aspect and become progressively shorter than their peers, which usually lead to psychosocial distress. 39 Androgen therapy may provoke androgenic and anabolic effects on target tissues. Androgenic effects include the development of male secondary sexual characteristics as well as of male sebaceous gland activity and hair growth pattern, whereas anabolic effects include greater skeletal muscle mass and bone metabolism.57,65
Treatment of patients with DSD or delayed puberty due to primary hypogonadism
As discussed for newborns, some forms of DSD reflect a primary gonadal insufficiency. 13 In many cases, especially those due to dysgenetic DSD with increased risk of gonadal tumour development, 104 orchiectomy is performed before pubertal age. 105 Therefore, androgen replacement is needed to induce the development of secondary sex characteristics, growth spurt and bone mass accrual typical of puberty. 106 The classic protocol of IM testosterone administration is used, starting at 50 mg every 4 weeks when bone age is at least 12 years-old, with progressive increases to reach 250 mg every 4 weeks approximately 3 years later. The same considerations are applicable to patients with primary hypogonadism not related to DSD but severely affecting testicular androgen secretion, such as testicular regression syndrome. Other forms of primary hypogonadism, for example, related to long-standing cryptorchidism, orchitis, chemotherapy, pelvic radiotherapy or Klinefelter syndrome, usually do not require androgen supplementation to induce pubertal changes, but may require it when testosterone levels are below normal. 52 In these cases, adult doses can be used directly, especially if near-adult height has already been achieved. A particular controversy exists regarding the initiation of androgen therapy in adolescents with Klinefelter syndrome. Testosterone production is most often within the normal range, which makes most authors recommend watchful waiting even if LH levels are elevated, limiting androgen replacement to those cases with low serum testosterone and clinical signs of hypogonadism.5,107 Others are prone to earlier exposure to androgen treatment based on the observation of improved physical and neurocognitive outcomes. 108 A clinical trial with a 1% testosterone gel showed that daily administration starting with 0.5 g and a progressive increase up to 5 g per day resulted in a two-fold increase in serum levels of testosterone and DHT, with no side effects. 109
Treatment of patients with constitutional delay of puberty or central hypogonadism
Patients with functional hypogonadism usually do not require hormonal treatment since normal activity of the HPT axis is re-established once the underlying condition is resolved. 110 Conversely, pharmacological treatment may be needed in boys with constitutional delay of puberty and is absolutely necessary in patients with central hypogonadism. Interestingly, approximately 20% of patients diagnosed with central hypogonadism may show a spontaneous increase in testicular volume and serum levels of gonadotrophins and testosterone when treatment is discontinued in adulthood.111–113
Androgen therapy
In boys with a well-established diagnosis of central hypogonadism, treatment should not be delayed beyond the age of 12 years to avoid the psychosocial burden 114 and the negative effect that the delay in exposure to sex steroids may have on the skeleton and pubertal growth spurt.115,116 Similarly, treatment is initiated even if a differential diagnosis between constitutional delay of puberty and central hypogonadism could not be solved. Although all current treatments are off-label, the protocol of IM testosterone administration with progressive increase already described for primary hypogonadism is the most widely used. 21 The doses (~50 mg every 4 weeks) used at the beginning are too low to inhibit the reactivation of the HPT axis in boys with constitutional delay of puberty, so that testosterone administration can be stopped if testicular volume reaches 4 ml. A frequently used alternative is to give testosterone for 6 months, then discontinue it for 3 months and watch whether testicular volume progresses. In a few cases, the patient feels satisfied with the androgenic effects and is not willing to discontinue testosterone administration. Treatment discontinuation can then be delayed until full development has been attained, approximately 3 years after initiation of treatment. In this case, a longer washout (~6 months) is needed to confirm that no spontaneous development occurs, given that the full dosage of 250 mg every 4 weeks provokes the inhibition of the HPT axis. While monitoring the potential side effects of androgen treatment on haematocrit and liver function is not critical when therapy is used for short periods like in boys with constitutional delay of puberty, the standard monitoring used in adults should be applied in boys receiving longer therapy. 39
In boys with constitutional delay of puberty, oral testosterone undecanoate has been tried for up to 15 months, at a daily dose of 10 to160 mg. Serum testosterone showed physiological levels for pubertal onset, in association with the beneficial effects of secondary sex characteristics and height velocity, while no excessive acceleration of bone age was noted. 56 Smaller case series or controlled trials had also been previously reported.55,117–119 Other androgen formulations have received little attention. A gel of 2% testosterone administered at 10 mg/day has shown good results in a small cohort of boys with constitutional delay of puberty. 120 Testosterone patches have shown variable efficacy and patient adherence.121,122
Aromatase inhibitors
Aromatase inhibitors, especially third-generation ones such as anastrozole and letrozole, have been used in the last two decades to increase adult height in boys, by delaying bone age progression. 123 However, a recent controlled clinical trial used letrozole to induce puberty in boys with constitutional delay. 40 Boys 14 years-old or older willing medical treatment and exhibiting the first signs of puberty received either letrozole 2.5 mg/day orally or testosterone 37.5–75 mg IM every 4 weeks for 6 months. Treatment with letrozole resulted in higher serum LH, FSH, testosterone and inhibin B, as well as greater testicular volume increase, suggesting that letrozole induces the activation of the HPT axis in boys with CDGP.
Gonadotrophins and GnRH
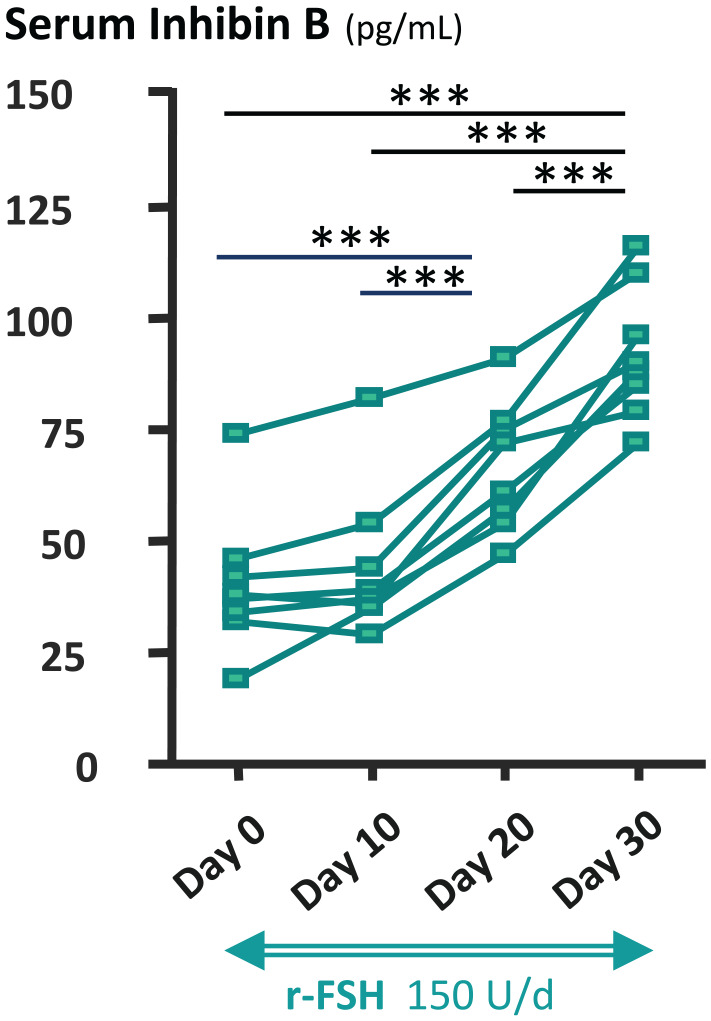
As proposed for newborns and infants with central hypogonadism, a more physiological option, aiming to mimic the normal development of testicular function during puberty, is the administration of gonadotrophins. Several treatment protocols have been reported, including hCG alone or in combination with r-FSH, hMG or GnRH. Initially, hCG treatment of adolescents with central hypogonadism showed a comparable effect to IM testosterone on secondary sex characteristics. 124 However, pulsatile administration of GnRH has showed better efficacy as compared to hCG in adolescents with central hypogonadism, underscoring the complementary role of FSH. 125 In fact, other studies showed the beneficial effect of FSH126–128 or hMG 129 addition to hCG. Nonetheless, to mimic physiological chronology, FSH proliferative effect on Sertoli cells should be induced before testosterone provokes Sertoli cell maturation inducing mitotic arrest. 27 Indeed, studies with r-FSH priming prior to hCG treatment showed enhanced testicular growth and increased circulating concentrations of AMH 130 (Figure 3) and inhibin B130,131 (Figure 4). Subsequent addition of hCG induced testosterone secretion by Leydig cells and Sertoli cell maturation, as reflected in AMH decline. 130 The increase in inhibin B levels is useful to monitor sperm production. 131 A randomised clinical trial in adolescents with central hypogonadism showed better results in patients pre-treated with r-FSH in terms of sperm production. 132 Typically, r-FSH is given 75–150 IU SC daily or every other day to attain target serum FSH levels of 7–9 IU/L during for 2–6 months, followed by combined r-FSH plus hCG starting at 250 IU weekly and progressively increasing by 250–500 IU weekly every 6 months to finally attain 1500 IU 3 times a week.76,132 Alternatively, hMG is given instead of r-FSH at 75–150 IU 2–3 times per week. 129 FSH priming may be especially needed in patients with small testes (e.g. volumes less than 5–6 ml), as compared to those with pubertal arrest or partial forms of central hypogonadism, who have larger testes volumes and are likely to only require hCG. Once full development has been obtained, treatment may be switched to testosterone administration for maintenance until the patient desires paternity.
Figure 4.
Effect of recombinant follicle stimulating hormone (r-FSH) priming followed by r-FSH plus human chorionic gonadotrophin (hCG) treatment on serum anti-Müllerian hormone (AMH), as a Sertoli cell biomarker, and testosterone (T), as a Leydig cell biomarker, in patients with central hypogonadism. Initial r-FSH treatment with 150 IU/day for 30 days increased Sertoli cell AMH secretion. The addition of hCG resulted in an increase of Leydig cell T secretion, which induced Sertoli cell maturation as reflected in the decline in AMH levels. The grey areas represent normal AMH levels for males with prepubertal T levels, corresponding to pubertal Tanner stage 1, on the left and to Tanner 4–5 levels on the right. *p < 0.05; **p < 0.01; ***p < 0.001. Modified with permission from Young et al. 130 © 2005 The Endocrine Society.
Figure 3.

Effect of recombinant follicle stimulating hormone (r-FSH) treatment on serum inhibin B, as a Sertoli cell biomarker in patients with central hypogonadism. ***p < 0.001. Modified with permission from Young et al. 130 © 2005 The Endocrine Society.
Pulsatile GnRH treatment is also a physiological approach in adolescents with central hypogonadism. GnRH is administered in a pulsatile manner using mini-infusion pump with an initial dose of 25 ng/kg per pulse every 2 h, with a subsequent titration to attain target serum testosterone.76,132 Although gonadotrophin and GnRH treatments have a physiology-based rationale in patients of pubertal age, whether fertility outcomes are improved as compared to using the more classical androgen therapy and waiting to administer these agents in adulthood still needs evidence.
Concluding remarks and research agenda
All treatments used in paediatric patients with hypogonadism are off-label. Testosterone administration IM is the most frequently used therapy in order to provoke genital enlargement in childhood or the full development of secondary sexual characteristics and growth spurt in adolescents. While this is the only possibility for patients with primary hypogonadism, the administration of gonadotrophins or GnRH may represent a more physiological therapy in boys with central hypogonadism. Clinical trials with long-term follow-up are needed to assess whether gonadotrophin treatment yields better results than initial androgen replacement followed by gonadotrophin treatment in adulthood when fertility is sought. Other possibilities based on recently developed technologies, such as Leydig cell 133 or spermatogenic 134 development in vitro, represent stimulating alternatives.
Footnotes
Author contributions: Rey, Rodolfo: Conceptualisation; Writing – original draft; Writing – review & editing.
Conflict of interest statement: The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: RAR has received honoraria from CONICET (Argentina) for technology services using the AMH ELISA and royalties derived from an agreement between INSERM (France) and Beckman-Coulter-Immunotech for the development of an AMH ELISA kit, until 2020, as well as lecture honoraria from Novo Nordisk and Sandoz, and travel grants from Biosidus, Merck, Novo Nordisk, Pfizer and Sandoz.
Funding: The author disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partially funded by Fondo para la Investigación Científica y Tecnológica (FONCYT, Argentina), grant numbers PIDC 32/2017 and PICT 2972/2018.
ORCID iD: Rodolfo A. Rey  https://orcid.org/0000-0002-1100-3843
https://orcid.org/0000-0002-1100-3843
References
- 1. Grinspon RP, Freire AV, Rey RA. Hypogonadism in pediatric health: adult medicine concepts fail. Trends Endocrinol Metab 2019; 30: 879–890. [DOI] [PubMed] [Google Scholar]
- 2. Josso N, Rey RA, Picard JY. Anti-müllerian hormone: a valuable addition to the toolbox of the pediatric endocrinologist. Int J Endocrinol 2013; 2013: 674105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weintraub A, Eldar-Geva T. Anti-mullerian hormone (AMH) determinations in the pediatric and adolescent endocrine practice. Pediatr Endocrinol Rev 2017; 14: 364–370. [DOI] [PubMed] [Google Scholar]
- 4. Condorelli RA, Cannarella R, Calogero AE, et al. Evaluation of testicular function in prepubertal children. Endocrine 2018; 62: 274–280. [DOI] [PubMed] [Google Scholar]
- 5. Salonia A, Rastrelli G, Hackett G, et al. Paediatric and adult-onset male hypogonadism. Nat Rev Dis Primers 2019; 5: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rey RA, Grinspon RP, Gottlieb S, et al. Male hypogonadism: an extended classification based on a developmental, endocrine physiology-based approach. Andrology 2013; 1: 3–16. [DOI] [PubMed] [Google Scholar]
- 7. Rey RA, Grinspon RP. Normal male sexual differentiation and aetiology of disorders of sex development. Best Pract Res Clin Endocrinol Metab 2011; 25: 221–238. [DOI] [PubMed] [Google Scholar]
- 8. Meroni SB, Galardo MN, Rindone G, et al. Molecular mechanisms and signaling pathways involved in sertoli cell proliferation. Front Endocrinol 2019; 10: 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lukas-Croisier C, Lasala C, Nicaud J, et al. Follicle-stimulating hormone increases testicular anti-Müllerian hormone (AMH) production through Sertoli cell proliferation and a nonclassical cyclic adenosine 5’-monophosphate-mediated activation of the AMH gene. Mol Endocrinol 2003; 17: 550–561. [DOI] [PubMed] [Google Scholar]
- 10. Lasala C, Schteingart HF, Arouche N, et al. SOX9 and SF1 are involved in cyclic AMP-mediated upregulation of anti-Mullerian gene expression in the testicular prepubertal Sertoli cell line SMAT1. Am J Physiol Endocrinol Metab 2011; 301: E539–E547. [DOI] [PubMed] [Google Scholar]
- 11. Lambert AS, Bougneres P. Growth and descent of the testes in infants with hypogonadotropic hypogonadism receiving subcutaneous gonadotropin infusion. Int J Pediatr Endocrinol 2016; 2016: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Raivio T, Toppari J, Perheentupa A, et al. Treatment of prepubertal gonadotrophin-deficient boys with recombinant human follicle-stimulating hormone. Lancet 1997; 350: 263–264. [DOI] [PubMed] [Google Scholar]
- 13. Grinspon RP, Bergadá I, Rey RA. Male hypogonadism and disorders of sex development. Front Endocrinol 2020; 11: 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grinspon RP, Loreti N, Braslavsky D, et al. Spreading the clinical window for diagnosing fetal-onset hypogonadism in boys. Front Endocrinol 2014; 5: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Braslavsky D, Grinspon RP, Ballerini MG, et al. Hypogonadotropic hypogonadism in infants with congenital hypopituitarism: a challenge to diagnose at an early stage. Horm Res Paediatr 2015; 84: 289–297. [DOI] [PubMed] [Google Scholar]
- 16. Bougnères P, François M, Pantalone L, et al. Effects of an early postnatal treatment of hypogonadotropic hypogonadism with a continuous subcutaneous infusion of recombinant follicle-stimulating hormone and luteinizing hormone. J Clin Endocrinol Metab 2008; 93: 2202–2205. [DOI] [PubMed] [Google Scholar]
- 17. Grinspon RP, Gottlieb S, Bedecarras P, et al. Anti-Müllerian hormone and testicular function in prepubertal boys with cryptorchidism. Front Endocrinol 2018; 9: 182–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hildorf S, Dong L, Thorup J, et al. Sertoli cell number correlates with serum inhibin B in infant cryptorchid boys. Sex Dev 2019; 13: 74–82. [DOI] [PubMed] [Google Scholar]
- 19. Xu C, Lang-Muritano M, Phan-Hug F, et al. Genetic testing facilitates prepubertal diagnosis of congenital hypogonadotropic hypogonadism. Clin Genet 2017; 92: 213–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grinspon RP, Castro S, Brunello FG, et al. Diagnosis of male central hypogonadism during childhood. J Endocr Soc 2021; 5: bvab145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mason KA, Schoelwer MJ, Rogol AD. Androgens during infancy, childhood, and adolescence: physiology and use in clinical practice. Endocr Rev 2020; 41: 1–36. [DOI] [PubMed] [Google Scholar]
- 22. Grinspon RP, Ropelato MG, Bedecarrás P, et al. Gonadotrophin secretion pattern in anorchid boys from birth to pubertal age: pathophysiological aspects and diagnostic usefulness. Clin Endocrinol 2012; 76: 698–705. [DOI] [PubMed] [Google Scholar]
- 23. Bastida MG, Rey RA, Bergadá I, et al. Establishment of testicular endocrine function impairment during childhood and puberty in boys with Klinefelter syndrome. Clin Endocrinol 2007; 67: 863–870. [DOI] [PubMed] [Google Scholar]
- 24. Pacenza N, Pasqualini T, Gottlieb S, et al. Clinical presentation of Klinefelter’s syndrome: differences according to age. Int J Endocrinol 2012; 2012: 324835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Spaziani M, Tarantino C, Tahani N, et al. Hypothalamo-Pituitary axis and puberty. Mol Cell Endocrinol 2021; 520: 111094. [DOI] [PubMed] [Google Scholar]
- 26. Madsen A, Oehme NB, Roelants M, et al. Testicular ultrasound to stratify hormone references in a cross-sectional norwegian study of male puberty. J Clin Endocrinol Metab 2020; 105: 1888–1898. [DOI] [PubMed] [Google Scholar]
- 27. Rey RA. Mini-puberty and true puberty: differences in testicular function. Ann Endocrinol 2014; 75: 58–63. [DOI] [PubMed] [Google Scholar]
- 28. Grinspon RP, Bedecarrás P, Ballerini MG, et al. Early onset of primary hypogonadism revealed by serum anti-Müllerian hormone determination during infancy and childhood in trisomy 21. Int J Androl 2011; 34: e487–e498. [DOI] [PubMed] [Google Scholar]
- 29. Kelsey TW, Miles A, Mitchell RT, et al. A normative model of serum inhibin B in young males. PLoS One 2016; 11: e0153843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kelsey TW, Li LQ, Mitchell RT, et al. A validated age-related normative model for male total testosterone shows increasing variance but no decline after age 40 years. PLoS One 2014; 9: e109346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marshall WA, Tanner JM. Variations in the pattern of pubertal changes in boys. Arch Dis Child 1970; 45: 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Howard SR, Dunkel L. Delayed puberty-phenotypic diversity, molecular genetic mechanisms, and recent discoveries. Endocr Rev 2019; 40: 1285–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jonsdottir-Lewis E, Feld A, Ciarlo R, et al. Timing of pubertal onset in girls and boys with constitutional delay. J Clin Endocrinol Metab 2021; 106: e3693–e3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Harrington J, Palmert MR. Clinical review: distinguishing constitutional delay of growth and puberty from isolated hypogonadotropic hypogonadism: critical appraisal of available diagnostic tests. J Clin Endocrinol Metab 2012; 97: 3056–3067. [DOI] [PubMed] [Google Scholar]
- 35. Grinspon RP, Ropelato MG, Gottlieb S, et al. Basal follicle-stimulating hormone and peak gonadotropin levels after gonadotropin-releasing hormone infusion show high diagnostic accuracy in boys with suspicion of hypogonadotropic hypogonadism. J Clin Endocrinol Metab 2010; 95: 2811–2818. [DOI] [PubMed] [Google Scholar]
- 36. Chaudhary S, Walia R, Bhansali A, et al. FSH-stimulated inhibin B (FSH-iB): a novel marker for the accurate prediction of pubertal outcome in delayed puberty. J Clin Endocrinol Metab 2021; 106: e3495–e3505. [DOI] [PubMed] [Google Scholar]
- 37. Grinspon RP, Urrutia M. The importance of follicle-stimulating hormone in the prepubertal and pubertal testis. Curr Opin Endocr Metab Res 2020; 14: 137–144. [Google Scholar]
- 38. Bi Y, Perry PJ, Ellerby M, et al. Population pharmacokinetic/pharmacodynamic modeling of depot testosterone cypionate in healthy male subjects. CPT Pharmacometrics Syst Pharmacol 2018; 7: 259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stancampiano MR, Lucas-Herald AK, Russo G, et al. Testosterone therapy in adolescent boys: the need for a structured approach. Horm Res Paediatr 2019; 92: 215–228. [DOI] [PubMed] [Google Scholar]
- 40. Varimo T, Huopio H, Kariola L, et al. Letrozole versus testosterone for promotion of endogenous puberty in boys with constitutional delay of growth and puberty: a randomised controlled phase 3 trial. Lancet Child Adolesc Health 2019; 3: 109–120. [DOI] [PubMed] [Google Scholar]
- 41. Vandewalle S, Van Caenegem E, Craen M, et al. Growth, sexual and bone development in a boy with bilateral anorchia under testosterone treatment guided by the development of his monozygotic twin. J Pediatr Endocrinol Metab 2018; 31: 361–367. [DOI] [PubMed] [Google Scholar]
- 42. Aydogdu A, Swerdloff RS. Emerging medication for the treatment of male hypogonadism. Expert Opin Emerg Drugs 2016; 21: 255–266. [DOI] [PubMed] [Google Scholar]
- 43. Ladjouze A, Donaldson M. Primary gonadal failure. Best Pract Res Clin Endocrinol Metab 2019; 33: 101295. [DOI] [PubMed] [Google Scholar]
- 44. Rogol AD. Pubertal androgen therapy in boys. Pediatr Endocrinol Rev 2005; 2: 383–390. [PubMed] [Google Scholar]
- 45. Schurmeyer T, Nieschlag E. Comparative pharmacokinetics of testosterone enanthate and testosterone cyclohexanecarboxylate as assessed by serum and salivary testosterone levels in normal men. Int J Androl 1984; 7: 181–187. [DOI] [PubMed] [Google Scholar]
- 46. Di Luigi L, Sgrò P, Romanelli F, et al. Urinary and serum hormones profiles after testosterone enanthate administration in male hypogonadism: concerns on the detection of doping with testosterone in treated hypogonadal athletes. J Endocrinol Invest 2009; 32: 445–453. [DOI] [PubMed] [Google Scholar]
- 47. Ekström L, Schulze JJ, Guillemette C, et al. Bioavailability of testosterone enanthate dependent on genetic variation in the phosphodiesterase 7B but not on the uridine 5’-diphospho-glucuronosyltransferase (UGT2B17) gene. Pharmacogenet Genomics 2011; 21: 325–332. [DOI] [PubMed] [Google Scholar]
- 48. Zitzmann M. Mechanisms of disease: pharmacogenetics of testosterone therapy in hypogonadal men. Nat Clin Pract Urol 2007; 4: 161–166. [DOI] [PubMed] [Google Scholar]
- 49. Morgentaler A, Dobs AS, Kaufman JM, et al. Long acting testosterone undecanoate therapy in men with hypogonadism: results of a pharmacokinetic clinical study. J Urol 2008; 180: 2307–2313. [DOI] [PubMed] [Google Scholar]
- 50. Nieschlag E, Behre HM. Testosterone therapy. In: Nieschlag E, Behre HM, Nieschlag S. (eds) Andrology. Berlin: Springer, 2010, pp. 437–455. [Google Scholar]
- 51. Zhang GY, Gu YQ, Wang XH, et al. A pharmacokinetic study of injectable testosterone undecanoate in hypogonadal men. J Androl 1998; 19: 761–768. [PubMed] [Google Scholar]
- 52. Rey RA, Grinspon RP. Androgen treatment in adolescent males with hypogonadism. Am J Mens Health 2020; 14: 1557988320922443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Shoskes JJ, Wilson MK, Spinner ML. Pharmacology of testosterone replacement therapy preparations. Transl Androl Urol 2016; 5: 834–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McBride JA, Carson CC, Coward RM. Diagnosis and management of testosterone deficiency. Asian J Androl 2015; 17: 177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Albanese A, Kewley GD, Long A, et al. Oral treatment for constitutional delay of growth and puberty in boys: a randomised trial of an anabolic steroid or testosterone undecanoate. Arch Dis Child 1994; 71: 315–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lawaetz JG, Hagen CP, Mieritz MG, et al. Evaluation of 451 Danish boys with delayed puberty: diagnostic use of a new puberty nomogram and effects of oral testosterone therapy. J Clin Endocrinol Metab 2015; 100: 1376–1385. [DOI] [PubMed] [Google Scholar]
- 57. Orr R, Fiatarone Singh M. The anabolic androgenic steroid oxandrolone in the treatment of wasting and catabolic disorders: review of efficacy and safety. Drugs 2004; 64: 725–750. [DOI] [PubMed] [Google Scholar]
- 58. Jick SS, Hagberg KW. The risk of adverse outcomes in association with use of testosterone products: a cohort study using the UK-based general practice research database. Br J Clin Pharmacol 2013; 75: 260–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Coviello AD, Bremner WJ, Matsumoto AM, et al. Intratesticular testosterone concentrations comparable with serum levels are not sufficient to maintain normal sperm production in men receiving a hormonal contraceptive regimen. J Androl 2004; 25: 931–938. [DOI] [PubMed] [Google Scholar]
- 60. De Lignieres B. Transdermal dihydrotestosterone treatment of ‘andropause’. Ann Med 1993; 25: 235–241. [DOI] [PubMed] [Google Scholar]
- 61. Swerdloff RS, Dudley RE, Page ST, et al. Dihydrotestosterone: biochemistry, physiology, and clinical implications of elevated blood levels. Endocr Rev 2017; 38: 220–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cailleux-Bounacer A, Rohmer V, Lahlou N, et al. Impact level of dihydrotestosterone on the hypothalamic-pituitary-leydig cell axis in men. Int J Androl 2009; 32: 57–65. [DOI] [PubMed] [Google Scholar]
- 63. Becker D, Wain LM, Chong YH, et al. Topical dihydrotestosterone to treat micropenis secondary to partial androgen insensitivity syndrome (PAIS) before, during, and after puberty – a case series. J Pediatr Endocrinol Metab 2016; 29: 173–177. [DOI] [PubMed] [Google Scholar]
- 64. Xu D, Lu L, Xi L, et al. Efficacy and safety of percutaneous administration of dihydrotestosterone in children of different genetic backgrounds with micropenis. J Pediatr Endocrinol Metab 2017; 30: 1285–1291. [DOI] [PubMed] [Google Scholar]
- 65. Wu C, Kovac JR. Novel uses for the anabolic androgenic steroids nandrolone and oxandrolone in the management of male health. Curr Urol Rep 2016; 17: 72. [DOI] [PubMed] [Google Scholar]
- 66. Ross JL, Kushner H, Kowal K, et al. Androgen treatment effects on motor function, cognition, and behavior in boys with Klinefelter syndrome. J Pediatr 2017; 185: 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Davis SM, Cox-Martin MG, Bardsley MZ, et al. Effects of oxandrolone on cardiometabolic health in boys with Klinefelter syndrome: a randomized controlled trial. J Clin Endocrinol Metab 2017; 102: 176–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Davis SM, Lahlou N, Cox-Martin M, et al. Oxandrolone treatment results in an increased risk of gonadarche in prepubertal boys with Klinefelter syndrome. J Clin Endocrinol Metab 2018; 103: 3449–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Solomon ZJ, Mirabal JR, Mazur DJ, et al. Selective androgen receptor modulators: current knowledge and clinical applications. Sex Med Rev 2019; 7: 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ulloa-Aguirre A, Lira-Albarran S. Clinical applications of gonadotropins in the male. Prog Mol Biol Transl Sci 2016; 143: 121–174. [DOI] [PubMed] [Google Scholar]
- 71. Lunenfeld B, Bilger W, Longobardi S, et al. The development of gonadotropins for clinical use in the treatment of infertility. Front Endocrinol 2019; 10: 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Bosch E, Nyboe Andersen A, Barri P, et al. Follicular and endocrine dose responses according to anti-Mullerian hormone levels in IVF patients treated with a novel human recombinant FSH (FE 999049). Clin Endocrinol 2015; 83: 902–912. [DOI] [PubMed] [Google Scholar]
- 73. Fauser BC, Mannaerts BM, Devroey P, et al. Advances in recombinant DNA technology: corifollitropin alfa, a hybrid molecule with sustained follicle-stimulating activity and reduced injection frequency. Hum Reprod Update 2009; 15: 309–321. [DOI] [PubMed] [Google Scholar]
- 74. Croxtall JD, McKeage K. Corifollitropin alfa: a review of its use in controlled ovarian stimulation for assisted reproduction. Biodrugs 2011; 25: 243–254. [DOI] [PubMed] [Google Scholar]
- 75. Boehm U, Bouloux PM, Dattani MT, et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism – pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 2015; 11: 547–564. [DOI] [PubMed] [Google Scholar]
- 76. Young J, Xu C, Papadakis GE, et al. Clinical management of congenital hypogonadotropic hypogonadism. Endocr Rev 2019; 40: 669–710. [DOI] [PubMed] [Google Scholar]
- 77. Guthrie RD, Smith DW, Graham CB. Testosterone treatment for micropenis during early childhood. J Pediatr 1973; 83: 247–252. [DOI] [PubMed] [Google Scholar]
- 78. Burstein S, Grumbach MM, Kaplan SL. Early determination of androgen-responsiveness is important in the management of microphallus. Lancet 1979; 2: 983–986. [DOI] [PubMed] [Google Scholar]
- 79. Bin-Abbas B, Conte FA, Grumbach MM, et al. Congenital hypogonadotropic hypogonadism and micropenis: effect of testosterone treatment on adult penile size why sex reversal is not indicated. J Pediatr 1999; 134: 579–583. [DOI] [PubMed] [Google Scholar]
- 80. Ishii T, Sasaki G, Hasegawa T, et al. Testosterone enanthate therapy is effective and independent of SRD5A2 and AR gene polymorphisms in boys with micropenis. J Urol 2004; 172: 319–324. [DOI] [PubMed] [Google Scholar]
- 81. Rogol AD. New facets of androgen replacement therapy during childhood and adolescence. Expert Opin Pharmacother 2005; 6: 1319–1336. [DOI] [PubMed] [Google Scholar]
- 82. Zenaty D, Dijoud F, Morel Y, et al. Bilateral anorchia in infancy: occurence of micropenis and the effect of testosterone treatment. J Pediatr 2006; 149: 687–691. [DOI] [PubMed] [Google Scholar]
- 83. Hatipogˇlu N, Kurtogˇlu S. Micropenis: etiology, diagnosis and treatment approaches. J Clin Res Pediatr Endocrinol 2013; 5: 217–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Nerli RB, Guntaka AK, Patne PB, et al. Penile growth in response to hormone treatment in children with micropenis. Indian J Urol 2013; 29: 288–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wisniewski AB, Batista RL, Costa EMF, et al. Management of 46, XY differences/disorders of sex development (DSD) throughout life. Endocr Rev 2019; 40: 1547–1572. [DOI] [PubMed] [Google Scholar]
- 86. Ben-Galim E, Hillman RE, Weldon VV. Topically applied testosterone and phallic growth. Am J Dis Child 1980; 134: 296–298. [DOI] [PubMed] [Google Scholar]
- 87. Arisaka O, Hoshi M, Kanazawa S, et al. Systemic effects of transdermal testosterone for the treatment of microphallus in children. Pediatr Int 2001; 43: 134–136. [DOI] [PubMed] [Google Scholar]
- 88. Odame I, Donaldson MD, Wallace AM, et al. Early diagnosis and management of 5 alpha-reductase deficiency. Arch Dis Child 1992; 67: 720–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Choi SK, Han SW, Kim DH, et al. Transdermal dihydrotestosterone therapy and its effects on patients with microphallus. J Urol 1993; 150: 657–660. [DOI] [PubMed] [Google Scholar]
- 90. Charmandari E, Dattani MT, Perry LA, et al. Kinetics and effect of percutaneous administration of dihydrotestosterone in children. Horm Res 2001; 56: 177–181. [DOI] [PubMed] [Google Scholar]
- 91. Bertelloni S, Scaramuzzo RT, Parrini D, et al. Early diagnosis of 5alpha-reductase deficiency in newborns. Sex Dev 2007; 1: 147–151. [DOI] [PubMed] [Google Scholar]
- 92. Kaya C, Bektic J, Radmayr C, et al. The efficacy of dihydrotestosterone transdermal gel before primary hypospadias surgery: a prospective, controlled, randomized study. J Urol 2008; 179: 684–688. [DOI] [PubMed] [Google Scholar]
- 93. Ghirri P, Ciulli C, Vuerich M, et al. Incidence at birth and natural history of cryptorchidism: a study of 10,730 consecutive male infants. J Endocrinol Invest 2002; 25: 709–715. [DOI] [PubMed] [Google Scholar]
- 94. Boisen KA, Kaleva M, Main KM, et al. Difference in prevalence of congenital cryptorchidism in infants between two Nordic countries. Lancet 2004; 363: 1264–1269. [DOI] [PubMed] [Google Scholar]
- 95. Hadziselimovic F, Zivkovic D, Bica DTG, et al. The importance of mini-puberty for fertility in cryptorchidism. J Urol 2005; 174: 1536–1539; discussion 1538–1539. [DOI] [PubMed] [Google Scholar]
- 96. Pyörälä S, Huttunen NP, Uhari M. A review and meta-analysis of hormonal treatment of cryptorchidism. J Clin Endocrinol Metab 1995; 80: 2795–2799. [DOI] [PubMed] [Google Scholar]
- 97. Henna MR, Del Nero RG, Sampaio CZ, et al. Hormonal cryptorchidism therapy: systematic review with metanalysis of randomized clinical trials. Pediatr Surg Int 2004; 20: 357–359. [DOI] [PubMed] [Google Scholar]
- 98. Penson D, Krishnaswami S, Jules A, et al. Effectiveness of hormonal and surgical therapies for cryptorchidism: a systematic review. Pediatrics 2013; 131: e1897–e1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Esposito C, De Lucia A, Palmieri A, et al. Comparison of five different hormonal treatment protocols for children with cryptorchidism. Scand J Urol Nephrol 2003; 37: 246–249. [DOI] [PubMed] [Google Scholar]
- 100. Hadziselimovic F. Successful treatment of unilateral cryptorchid boys risking infertility with LH-RH analogue. Int Braz J Urol 2008; 34: 319–326; discussion 327–328. [DOI] [PubMed] [Google Scholar]
- 101. Main KM, Schmidt IM, Toppari J, et al. Early postnatal treatment of hypogonadotropic hypogonadism with recombinant human FSH and LH. Eur J Endocrinol 2002; 146: 75–79. [DOI] [PubMed] [Google Scholar]
- 102. Sarfati J, Bouvattier C, Bry-Gauillard H, et al. Kallmann syndrome with FGFR1 and KAL1 mutations detected during fetal life. Orphanet J Rare Dis 2015; 10: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Spaziani M, Tarantino C, Pozza C, et al. Adverse pathophysiological influence of early testosterone therapy on the testes of boys with higher grade sex chromosome aneuploidies (HGAs): a retrospective, cross-sectional study. J Endocrinol Invest 2021; 44: 1483–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hersmus R, van Bever Y, Wolffenbuttel KP, et al. The biology of germ cell tumors in disorders of sex development. Clin Genet 2017; 91: 292–301. [DOI] [PubMed] [Google Scholar]
- 105. Lucas-Herald AK, Bryce J, Kyriakou A, et al. Gonadectomy in conditions affecting sex development: a registry-based cohort study. Eur J Endocrinol 2021; 184: 791–801. [DOI] [PubMed] [Google Scholar]
- 106. Stancampiano MR, Lucas-Herald AK, Bryce J, et al. Testosterone therapy and its monitoring in adolescent boys with hypogonadism: results of an international survey from the I-DSD registry. Sex Dev 2021; 15: 236–243. [DOI] [PubMed] [Google Scholar]
- 107. Gravholt CH, Chang S, Wallentin M, et al. Klinefelter syndrome: integrating genetics, neuropsychology, and endocrinology. Endocr Rev 2018; 39: 389–423. [DOI] [PubMed] [Google Scholar]
- 108. Chan YM, Feld A, Jonsdottir-Lewis E. Effects of the timing of sex-steroid exposure in adolescence on adult health outcomes. J Clin Endocrinol Metab 2019; 104: 4578–4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Rogol AD, Swerdloff RS, Reiter EO, et al. A multicenter, open-label, observational study of testosterone gel (1%) in the treatment of adolescent boys with Klinefelter syndrome or anorchia. J Adolesc Health 2014; 54: 20–25. [DOI] [PubMed] [Google Scholar]
- 110. Grob F, Zacharin M. Puberty in chronic inflammatory conditions. Curr Opin Endocr Metab Res 2020; 14: 29–36. [Google Scholar]
- 111. Raivio T, Falardeau J, Dwyer A, et al. Reversal of idiopathic hypogonadotropic hypogonadism. N Engl J Med 2007; 357: 863–873. [DOI] [PubMed] [Google Scholar]
- 112. Sidhoum VF, Chan YM, Lippincott MF, et al. Reversal and relapse of hypogonadotropic hypogonadism: resilience and fragility of the reproductive neuroendocrine system. J Clin Endocrinol Metab 2014; 99: 861–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Dwyer AA, Raivio T, Pitteloud N. MANAGEMENT OF ENDOCRINE DISEASE: reversible hypogonadotropic hypogonadism. Eur J Endocrinol 2016; 174: R267–R274. [DOI] [PubMed] [Google Scholar]
- 114. Dwyer AA. Psychosexual effects resulting from delayed, incomplete, or absent puberty. Curr Opin Endocr Metab Res 2020; 14: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Raivio T, Miettinen PJ. Constitutional delay of puberty versus congenital hypogonadotropic hypogonadism: genetics, management and updates. Best Pract Res Clin Endocrinol Metab 2019; 33: 101316. [DOI] [PubMed] [Google Scholar]
- 116. Harrington J, Palmert MR. Distinguishing self-limited delayed puberty from permanent hypogonadotropic hypogonadism: how and why? J Clin Endocrinol Metab 2021; 106: e5264–e5266. [DOI] [PubMed] [Google Scholar]
- 117. Gregory JW, Greene SA, Thompson J, et al. Effects of oral testosterone undecanoate on growth, body composition, strength and energy expenditure of adolescent boys. Clin Endocrinol 1992; 37: 207–213. [DOI] [PubMed] [Google Scholar]
- 118. Butler GE, Sellar RE, Walker RF, et al. Oral testosterone undecanoate in the management of delayed puberty in boys: pharmacokinetics and effects on sexual maturation and growth. J Clin Endocrinol Metab 1992; 75: 37–44. [DOI] [PubMed] [Google Scholar]
- 119. Brown DC, Butler GE, Kelnar CJ, et al. A double blind, placebo controlled study of the effects of low dose testosterone undecanoate on the growth of small for age, prepubertal boys. Arch Dis Child 1995; 73: 131–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Chioma L, Papucci G, Fintini D, et al. Use of testosterone gel compared to intramuscular formulation for puberty induction in males with constitutional delay of growth and puberty: a preliminary study. J Endocrinol Invest 2018; 41: 259–263. [DOI] [PubMed] [Google Scholar]
- 121. Mayo MW, Baldwin AS. The transcription factor NF-kappaB: control of oncogenesis and cancer therapy resistance. Biochim Biophys Acta 2000; 1470: M55–M62. [DOI] [PubMed] [Google Scholar]
- 122. Mason A, Wong SC, McGrogan P, et al. Effect of testosterone therapy for delayed growth and puberty in boys with inflammatory bowel disease. Horm Res Paediatr 2011; 75: 8–13. [DOI] [PubMed] [Google Scholar]
- 123. Hero M, Varimo T, Raivio T. Aromatase inhibitors in puberty. Curr Opin Endocr Metab Res 2020; 14: 37–41. [Google Scholar]
- 124. Bistritzer T, Lunenfeld B, Passwell JH, et al. Hormonal therapy and pubertal development in boys with selective hypogonadotropic hypogonadism. Fertil Steril 1989; 52: 302–306. [DOI] [PubMed] [Google Scholar]
- 125. Gong C, Liu Y, Qin M, et al. Pulsatile GnRH is superior to hCG in therapeutic efficacy in adolescent boys with hypogonadotropic hypogonadodism. J Clin Endocrinol Metab 2015; 100: 2793–2799. [DOI] [PubMed] [Google Scholar]
- 126. Barrio R, De Luis D, Alonso M, et al. Induction of puberty with human chorionic gonadotropin and follicle-stimulating hormone in adolescent males with hypogonadotropic hypogonadism. Fertil Steril 1999; 71: 244–248. [DOI] [PubMed] [Google Scholar]
- 127. Rohayem J, Hauffa BP, Zacharin M, et al. Testicular growth and spermatogenesis: new goals for pubertal hormone replacement in boys with hypogonadotropic hypogonadism? a multicentre prospective study of hCG/rFSH treatment outcomes during adolescence. Clin Endocrinol 2017; 86: 75–87. [DOI] [PubMed] [Google Scholar]
- 128. Zacharin M, Sabin MA, Nair VV, et al. Addition of recombinant follicle-stimulating hormone to human chorionic gonadotropin treatment in adolescents and young adults with hypogonadotropic hypogonadism promotes normal testicular growth and may promote early spermatogenesis. Fertil Steril 2012; 98: 836–842. [DOI] [PubMed] [Google Scholar]
- 129. Liu Y, Ren XY, Peng YG, et al. Efficacy and safety of human chorionic gonadotropin combined with human menopausal gonadotropin and a gonadotropin-releasing hormone pump for male adolescents with congenital hypogonadotropic hypogonadism. Chin Med J 2021; 134: 1152–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Young J, Chanson P, Salenave S, et al. Testicular anti-mullerian hormone secretion is stimulated by recombinant human FSH in patients with congenital hypogonadotropic hypogonadism. J Clin Endocrinol Metab 2005; 90: 724–728. [DOI] [PubMed] [Google Scholar]
- 131. Raivio T, Wikström AM, Dunkel L. Treatment of gonadotropin-deficient boys with recombinant human FSH: long-term observation and outcome. Eur J Endocrinol 2007; 156: 105–111. [DOI] [PubMed] [Google Scholar]
- 132. Dwyer AA, Sykiotis GP, Hayes FJ, et al. Trial of recombinant follicle-stimulating hormone pretreatment for GnRH-induced fertility in patients with congenital hypogonadotropic hypogonadism. J Clin Endocrinol Metab 2013; 98: E1790–E1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Zhou J, Hou Y, Zhang Z, et al. Conversion of human fibroblasts into functional Leydig-like cells by small molecules and a single factor. Biochem Biophys Res Commun 2019; 516: 1–7. [DOI] [PubMed] [Google Scholar]
- 134. Sharma S, Wistuba J, Pock T, et al. Spermatogonial stem cells: updates from specification to clinical relevance. Hum Reprod Update 2019; 25: 275–297. [DOI] [PubMed] [Google Scholar]