

Abstract
A novel class of peptide bioreporters was developed to perform multiplexed measurements of the activation of epidermal growth factor receptor kinase (EGFR), Akt kinase (Akt/protein kinase B), and proteases/peptidases in single cells. The performance characteristics of three reporters were assessed by measuring the reporter’s proteolytic stability, kinetic constants for EGFR and Akt and dephosphorylation rate. The reporter displaying optimal performance was comprised of 6-carboxyfluorescein (6-FAM) on the peptide N-terminus, Akt substrate sequence employing a threonine phosphorylation site for Akt, followed by tri-D arginine linker, and finally an EGFR substrate sequence bearing a phosphatase-resistant 7-(S)-hydroxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid (L-Htc) residue as the EGFR phosphorylation site. Importantly use of a single electrophoretic condition separated the mono- and di-phosphorylated products as well as proteolytic forms permitting the quantitation of multiple enzyme activities simultaneously using a single reporter. Since the Akt and EGFR substrates were linked, a known ratio (EGFR:Akt) of reporter was loaded into cells. A photoactivatable version of the reporter was synthesized by adding two 4,5-dimethoxy 2-nitrobenzyl (DMNB) moieties to mask the EGFR and Akt phosphorylation sites. The DMNB moieties were readily photocleaved following exposure to 360 nm light, unmasking the phosphorylation sites on the reporter. The new photoactivatable reporter permitted multiplexed measurements of kinase signaling, and proteolytic degradation in single cells in a temporally controlled manner. This work will facilitate the development of a new generation of multiplexed activity-based reporters capable of light-initiated, measurement of enzymatic activity in single cells.
Graphical Abstract
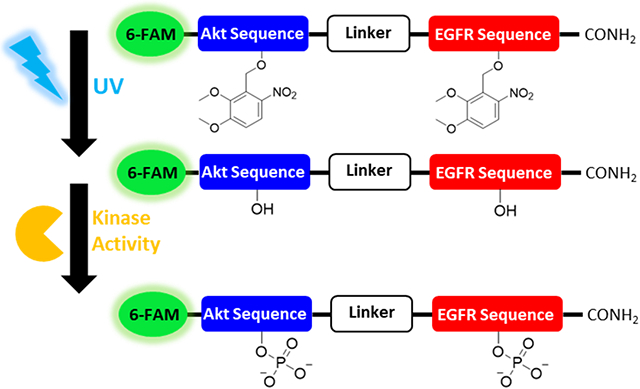
Aberrant epidermal growth factor receptor kinase (EGFR) and Akt activity are important drivers of cancer, particularly epithelial cell tumors such as lung, breast, head and neck, and bladder.1-5 Activation of EGFR by EGF initiates an array of downstream intracellular signal transduction events including activation of phosphatidylinositol-3-kinase (PI3-K), and Akt.6 Akt exerts its activity by phosphorylating downstream targets, such as transcription factors which facilitate cell survival.7 A significant challenge to performing measurements of EGFR and Akt kinase activity is the high degree of phenotypic heterogeneity present within populations of cancer cells.8 Phenotypic heterogeneity is an inherent characteristic in human cancer cells and is believed to be critical for enabling cells to overcome the multitude of selective pressures faced in the tumor microenvironment.9-11 The resulting phenotypic heterogeneity can result in widely different enzyme activity amongst individual cells within a cell population and which differs significantly from the bulk averaged activity obtained from thousands to millions of cells. The high degree of signaling variability, necessitates the development of technologies to interrogate enzyme activity at the level of individual cells, to better resolve signaling trends that are typically obfuscated by traditional bulk cell assays.9,12
Several methods exist to quantify the spatial and temporal differences in enzyme activity in biological systems. Often times these analytical methods employ artificial substrates to react with target classes of enzymes in tissues, intact or lysed bulk cells, and increasingly at the level of single cells.13,14 Matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) methods have seen a dramatic growth in popularity for quantifying enzyme activity using both natural and artificial substrates at the level of tissues and even single cells but remains limited to high abundance species.15-18 Optical techniques often employ genetically encoded fluorescent substrates, and/or small molecule dye reporters to infer enzyme behavior in cell-based assays. Genetically encoded reporters take advantage of binding events between phosphorylated substrate sequences and phosphopeptide binding domains to bring two fluorescent proteins sufficiently close for energy transfer (typically <10 nm).19,20 While genetic probes have led to many advances in quantitation of intracellular enzyme activity, the technology has several limitations including: 1) the need to transfect cells; 2) a large background fluorescence due to the presence of non-functional reporter (mis-folded and/or degraded); 3) altered enzyme-substrate interactions due to the large sizes of the fluorescent moiety (>28 kDa for GFP-based reporters).28 Conversely, small molecule fluorogenic substrates often display favorable cellular uptake due to the inherent hydrophobicity of the aromatic fluorophores, coupled with the small size of the dye molecules themselves (often <1 kDa). Drawbacks of small molecule reporters are that they tend to provide information on general classes of enzyme activity and can undergo non-enzymatic hydrolysis resulting in high background fluorescence. Additionally, the lack of a separation method precludes interrogation of the diversity of enzymatic modifications to the reporters.21-25
Chemical cytometry employs high-sensitivity analytical tools to characterize analytes within single cells.26,32 To date, a diverse array of analytical platforms have been adapted to perform chemical cytometry measurements including, capillary electrophoresis employing fluorescence detection (CE-F).26 CE-F analytical platforms have been used to characterize enzyme activity in biological samples from the tissue level down to single cells, and typically focus on the analysis of small molecule metabolites and peptides to infer enzymatic behavior.26 Advantages of CE-F are its high limits of detection (often exceeding 10−20 mol for fluorescent peptides and lipids), high peak capacity enabling many reporter modifications to be resolved, and low complexity readout since only fluorescently-labeled analytes are detected.27-31 Traditionally, investigations employing CE-F have utilized a reporter which contains a single enzyme substrate sequence.12,26 Such reporters have been used to profile Akt, EGFR, and peptidase activity within single cells. However, it has been challenging to perform tyrosine kinase and serine/threonine kinase activity measurements simultaneously at the level of single cells due to the difficulty of co-loading two reporters, lack of control over the ratio of the two reporters in each cell, and the often-distinct electrophoretic conditions needed for separation of the chemically distinct reporters. Additionally, the inclusion of photoactivatable protecting groups is desirable since photocleavage enables a common start time for initiation of enzymatic modifications to the reporter.
The goal of this work was to develop a novel class of photoactivable dual kinase reporter to facilitate the simultaneous quantitation of EGFR, Akt, and protease/peptidase activity within single human cells. We designed a test set of three dual-substrate reporters and assessed the ability to separate the parent, phosphorylated, and proteolytic reporter fragments by CE-F. We further characterized the reporters by assessing their affinities for the target kinases, resistance to protein tyrosine phosphatase (PTP) activity, and peptidase resistance. The lead reporter was assessed in detail by measurement of phosphorylation in cell lysates as well as single cells. Additionally, a photoactivatable analogue of the lead reporter was synthesized and assayed for photocleavage by UV light. This light-activatable reporter was used to measure EGFR and Akt activity as well as proteases in the presence of recombinant kinases, and cell lysates. UV-induced DNA damage within cells during reporter photoactivation was also assayed. Finally, the ability to assay EGFR, Akt and peptidase activity using the photoactivable reporter was assessed in single cells.
EXPERIMENTAL SECTION
Reporter Synthesis.
Detailed information on synthesis of peptides can be found in the Supporting Information. The sequences of the dual-substrate bioreporters are as follows: 24A (6FAM-GRP-MeR-AFTF-MeA-rrr-EDDEYEEV-NH2), 24B (6FAM-GRP-MeR-AFTF-MeA-Ahx-EDDEYEEV-NH2), 24C (6FAM-GRP-MeR-AFTF-MeA-rrr-EDDE-Htc-EEV-NH2), where r = (D)Arg, Ahx = aminohexanoic acid, MeR = N(Me)Arg, MeA = N(Me)Ala, Htc = 7-hydroxy-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid, and 6FAM = 6-carboxyfluorescein. The sequence of the photoactivatable dual-substrate reporter is as follows: C24C [6FAM-GRP-MeR-AF-T(ODMNB)-FMeA-rrr-EDDE-Htc(ODMNB)-EEV-CONH2].
Immortalized Cell Culture.
Detailed information regarding the culture of A431 and PANC-1 cells, and WST-8 proliferation assays can be found in the Supporting Information.
In Vitro Assays.
Akt and EGFR in vitro phosphorylation assays were performed with commercially available recombinant enzymes as described in the Supporting Information. Proteolytic degradation assays were performed with commercially available protease/peptidases including: pronase E, proteinase K, and endoproteinase-Glu-C, as described in the Supporting Information.
Assays Using Cell Lysates.
Confluent monolayers of A431 cells in polystyrene T-75 culture flasks (70-90% confluency, < 10 passages) were rinsed with phosphate buffered saline (PBS), and trypsinized (0.05% trypsin + EDTA, Gibco). Trypsinized cells were then washed with 5 mL extracellular buffer supplemented with 10 mM D-glucose (ECB) and pelleted. Next, M-PER Mammalian Protein Extraction Reagent (Thermo Scientific) was added to the cell pellets (100 μL per 1x106 cells) and shaken gently at 20 °C for 10 min with occasional inversion. Cell lysates were clarified by centrifugation at 4 °C for 15 min at 14000 g. The protein content of all cell lysates was quantified using a BCA assay (Fisher, Pierce™ BCA Protein Assay Kit), and normalized between all treatment groups. All cell lysate assays were performed within 1 h following lysate preparation. Details of lysis buffer additives, and specific optimizations for phosphorylation, and proteolysis assays are available in the Supporting Information.
Intracellular Loading of Reporters by Pinocytosis.
A solution of the desired reporter, at a final concentration of 5 - 200 μM, was prepared in hypertonic medium (ECB, RPMI-1640, or DMEM) at a maximum dilution of 10% (Influx, Pinocytosis Kit), and 1-2x106 cells were incubated in 30 μL hypertonic medium at either 37 °C or 20 °C for 20 min. Careful attention was given to ensure that sufficient hypertonic medium was added to completely cover the apical surface of the cells, and cells were agitated every 10 min by gently tapping the side of the tube for 3 s. The hypertonic loading solution was aspirated and replaced with 1 mL of hypotonic loading medium (60:40 (v/v) serum-free culture medium: deionized H2O). The cells were incubated at 20°C for 2 min, then the cells were pelleted, and the hypotonic medium was gently aspirated via pipette. Next, the cells were washed twice with 1 mL ECB, pelleted, aspirated, and re-equilibrated in 1 mL ECB at 37 °C prior to analysis. Cells for bulk analysis were pelleted and resuspended in 30 μL PBS just prior to thermally lysing the cells by incubation at 90 °C for 5 min. The cell lysate was clarified at 14,000 g for 15 min at 4°C and the supernatant was used immediately for assays. Fluorescent images of pinocytically loaded live cells were obtained by confocal microscopy on an Olympus FV3000 microscope. Cells to be imaged were counterstained with 2 μg/mL Hoechst (Invitrogen) during reporter pinocytosis to stain cellular DNA. Additional information on image processing is available in the Supporting Information.
Reporter Photoactivation and Measurement of DNA-Damage.
C24C in a solution of ECB, or within pinocytically loaded cells, was photoactivated by illuminating the sample with one of the following light sources: a) 360 nm solid-state laser with a source irradiance of 10 mW/cm2, b) Lumencor Sola III Light Engine with a maximum source irradiance of 14.5 mW/cm2 at 360 nm. Samples were exposed through a Nikon UV-A bandpass filter (360 ± 20 nm). To determine the maximum safe biological exposure for C24C-loaded A431 cells, the formation of DNA lesions in the form of cyclopyrimidine dimers (CPDs) was measured following UV exposure. A CPD ELISA kit (Cell Biolabs Inc.) was used to assess the formation of UV-induced DNA lesions following exposure to 360 ± 20 nm light. A431 cells were seeded in 96-well plates at a density of 20,000 cells/well and incubated for 18 h (37 °C, 5% CO2). Next, cells were exposed to UV radiation (14 mW/cm2) for a cumulative exposure of 0 - 3.0 J/cm2. Cells were then fixed, and then assayed for CPDs following the manufacturer’s protocol. CPD formation was assessed measured by measuring absorbance at 420 nm using a SpectraMax 5M plate-reader (Molecular Devices). Background absorbance was subtracted using a control sample that was not incubated with the primary antibody to account for non-specific binding of the secondary antibody. Exposure of cells to a germicidal lamp (significant output below 300 nm) for 10 min was used as a positive DNA damage control. Assay data were analyzed using a one-way ANOVA with Tukey’s multiple comparison’s analysis in GraphPad Prism 9.0. Additional information on reporter photoactivation assays can be found in the Supporting Information.
CE-F.
Capillary electrophoretic analyses of reporter samples incubated with recombinant enzymes, and cell lysates were performed using commercial Sciex P/ACE MDQ Plus instruments outfitted with 488 nm solid-state lasers (Coherent). Electrophoretic separations were performed in a 30 μm i.d. (360 μm o.d.) bare silica capillary with an electrophoretic buffer (0.1 M Tris/Tricine, pH 8.2, 3.4 mS, containing either 50, 100 or 150 mM Tween 20® (Millipore Sigma)) under a 500 V cm−1 applied electric field. Prior to analysis, samples were diluted to 200 nM reporter in DI H2O or 50 mM HEPES (pH 7.3, 1.2 mS) to promote field-amplified sample stacking. Data was analyzed using custom CE-analysis software (GigaCE) and Origin 9.0.
Single-Cell CE-F.
A431 cells were pinocytically loaded with reporter as described above. Next, loaded cells were seeded onto single cell O-ring chambers (2.5 cm2) at an approximate density of 10,000 cells/mL. Cells were incubated for 18 - 24 h in low-serum media containing 0.2% FBS (v/v). Next, cells were trypsinized, split into an aliquot containing approximately 1 - 2x106 cells, and incubated with 500 μL of 37 °C low-serum DMEM containing 200 ng/mL recombinant human epidermal growth factor (hEGF) (Calbiochem). Inhibitor (10 μM gefitinib (Millipore Sigma), was incubated with the cells for 30 min or 60 minin a humidified atmosphere of 37 ° in 5% CO2. The cells were then placed into 500 μL of 37 °C ECB. The cell chamber was placed on the microscope stage of a customized single-cell CE-F analysis system.34-36 Individual cells were identified and lysed with a 532 nm ND:YAG pulsed microbeam and the cell contents then electrokinetically injected (3 s, 200 V cm−1) into the capillary. The capillary inlet was moved into a reservoir of electrophoretic buffer (100 mM Tris/Tricine, 100 mM Tween 20®, 1% DMSO (v/v), pH 8.2, 3.4 mS). Cell contents were electrokinetically separated under an applied field of (600 V cm−1). Data was analyzed using custom Python software (GigaCE), and Origin 9.0. Data from the basal, stimulated, and inhibitor-treated cells were compared using a Mann U Whitney Test, and Cohen’s d effect sizes were calculated for each comparison.
RESULTS AND DISCUSSION
Dual-Substrate Reporter Design.
The goal of this work was to develop a photoactivatable, peptidase-resistant, dual substrate reporter that facilitated the simultaneous quantitation of EGFR and Akt activity within single cells upon the application of a photostimulus, Figure 1A. We based the dual-substrate reporters on previously optimized individual reporters that were developed to measure Akt and EGFR activity, Figure 1B; Table S1.30,33 The Akt reporter sequence incorporated nonnative N-methyl residues which were previously demonstrated to improve resistance to intracellular peptidases.12,37,38 Similarly, the highly protease resistant EGFR reporter sequence incorporated either a tyrosine (reporters 24A and 24B) or the conformationally constrained tyrosine analogue, L-Htc (reporter 24C).30 The L-Htc residue is readily phosphorylated by EGFR but, unlike a native phospho-tyrosyl residue, is highly resistant to dephosphorylation by phosphatases.30 A linker was used to connect, but spatially separate, the Akt and EGFR substrate sequences, while incorporating additional nonnative modifications to further improve peptidase resistance. The two linkers employed were tri-D-arginine (rrr, for reporters 24A and 24C) and aminohexanoic acid (Ahx, reporter 24B). The rrr linker is highly cationic at physiological pH and was expected to improve substrate affinity for basophilic kinases such as Akt while, by virtue of the D stereochemistry, improve resistance against proteolytic degradation.39 The Ahx linker is neutral, and consists of a highly flexible alkane backbone which was hypothesized to enhance kinase access to the target residues. For reporters 24A-C, the N-terminus was capped by 6-FAM further protecting the reporter from amino-terminal peptidases and enabling fluorescence detection. The reporter components were ordered as follows from N-terminus to C-terminus: fluorescein, Akt substrate, linker, and EGFR substrate. It was postulated that this design would ensure that most reporter fragments would display a higher net-cationic charge and lower molecular weight relative to the parent reporter. This is due to the presence of several arginine residues on the Akt substrate and linker regions, while the C-terminal EGFR substrate was rich in aspartic and glutamic acid residues. This reporter design increases the likelihood that reporter fragments would migrate faster relative to the parent reporter and its intact phosphorylated forms, thus facilitating the separation and quantitation of the intact and phosphorylated reporter variants.
FIGURE 1.

Schematic of dual-substrate reporters. (A) Illustration of the photoactivation and subsequent phosphorylation of the photo-caged EGFR/Akt reporter with UV light to yield the phosphorylated reporter. (B) Chemical structures of dual substrate EGFR/Akt reporters. 6-FAM is indicated in green, followed by Akt substrate sequence in blue, linker sequence in black, and EGFR substrate sequence in red.
Measuring Reporter Affinity for Kinases.
The reporters 24A,B,C were incubated with purified recombinant active EGFR and Akt to verify that the phosphorylation sites (T for Akt and Y or L-Htc for EGFR) could be phosphorylated by their respective enzymes, Figure 2A; Figure S1-S2. The kinetic properties of EGFR and Akt were then measured for each of the substrates (Table 1). No significant difference in KM for Akt was observed between 24A, 24B, and 24C. Surprisingly, the 24B reporter, with its neutral Ahx linker, displayed a significantly poorer KM for EGFR relative to that of reporter 24A. It is possible that the increased conformational flexibility of the Ahx linker enabled the highly positively charged Akt sequence to interact with the negatively charged EGFR sequence, causing a significant loss in affinity for EGFR.
FIGURE 2.

Phosphorylation of 24C by EGFR and Akt. (A) Purified EGFR and Akt were incubated with 24C and then the mixture separated. PEGFR marks the reporter peak with only the L-Htc site phosphorylated. PAkt marks the reporter peak with only the T or Akt site phosphorylated. PDi marks the reporter peak with both the EGFR and Akt sites phosphorylated. (B) PTP resistance of EGFR phosphorylated standards of reporters 24A and 24C in A431 lysates in the presence and absence of 100 μM pervanadate (PV) after a 5 min incubation at 37°C ( n = 3). ****p ≤ 0.0001. (C) 24C phosphorylation at the EGFR site by A431 cell lysates after 1 h in the presence and absence of 50 μM gefitinib (n = 3, ***p ≤ 0.0002). (D) Phosphorylation at the Akt site by PANC-1 lysates in the presence and absence of supplemented active Akt (n =3, ***p ≤0.0002).
Table 1.
Kinetics parameters of 24A, B, C phosphorylation by recombinant EGFR and Akt, and reporter lifetime in A431 lysates (All values are n = 3 for 24A, B, C, *p ≤ 0.0486).
| Reporter Structure | Akt Kinetics |
EGFR Kinetics |
A431 Lysate | ||||
|---|---|---|---|---|---|---|---|
| Name | Linker | EGFR Site | KM (μM) | kcat (s−1) | KM (μM) | kcat (s−1) | Half-Life (min) |
| 24A | rrr | Y | 14 ± 3 | 0.7 ± 0.1 | 82 ± 22* | 7.7 ± 1.2 | 267 ± 26 |
| 24B | Ahx | Y | 28 ± 16 | 1.0 ± 0.4 | 812 ± 459* | 19.8 ± 10.2 | 222 ± 21 |
| 24C | rrr | L-Htc | 17 ± 5 | 0.8 ± 0.1 | 365 ± 201 | 15.0 ± 6.8 | 248 ± 23 |
Reporter Performance in the Presence of Cell Lysates.
Once loaded into cells, enzyme reporters face a number of challenges such as degradation by intracellular peptidases and rapid dephosphorylation by phosphatases.12 Therefore, the resistance to degradation and dephosphorylation was assessed in cell lysates before performing experiments in intact live cells. Reporters 24A,B,C and a control peptide comprised of naturally occurring amino acids (6FAM-GRPRAATFAEG-NH2) were incubated with lysates prepared from A431 cells, Table S1. All dual-substrate reporters all displayed a significantly greater resistance to proteolysis relative to that of the control reporter, Table 1. All dual reporters displayed half-lives of greater than 200 min compared to 3±1 min for the control peptide, and there were no significant differences observed in the A431 lysates for the dual reporters.
Since intracellular protein tyrosine phosphatases (PTP) are known to rapidly dephosphorylate phosphoryl-tyrosine residues, the ability of the nonnative L-Htc residue on 24C to- resist dephosphorylation relative to the native Y on 24A and 24B was assessed.40 EGFR-phosphorylated standards of 24A,B,C were incubated in A431 lysates. After incubation of EGFR-phosphorylated 24A,B,C in A431 lysates, over 95% of the initially phosphorylated 24A,B was dephosphorylated in 5 min whereas only 10% of the phosphorylated 24C was dephosphorylated at 5 min, Figure 2B. To demonstrate that the loss of 24A phosphorylation was the result of endogenous PTP activity, A431 lysates were treated with pervanadate, a potent PTP inhibitor.41 A significantly greater quantity of 24A,B retained the phosphoryl-tyrosine moiety compared to reactions without pervanadate. In contrast pervanadate did not alter the level of phosphorylation at the L-Htc site of 24C. These results support previous findings that that L-Htc residues are resistant to PTP activity relative to that of the native tyrosine.30 Consequently 24C was used in all subsequent experiments.
Next, the ability of 24C to be phosphorylated by A431 cell lysates was assessed by incubating intact cells with EGF, and then incubation of 24C with a lysate from these activated cells. EGF-activated cell lysates phosphorylated 24C and this phosphorylation was shown to be blocked by gefitinib, an EGFR inhibitor (p ≤ 0.0002), Figure 2C. 24C was also phosphorylated when co-incubated with PANC-1 lysates enriched with active Akt (p ≤ 0.0002) but not in lysates without added active Akt consistent with prior reports, Figure 2D (since endogenous phosphatases deactivate Akt in cell lysates).31 These data suggest that active EGFR and Akt could phosphorylate 24C within a cellular environment.
Phosphorylation of Reporter 24C within Live Cells.
Reporter 24C was loaded into serum-starved cells using pinocytosis. Loaded cells displayed a diffuse fluorescence when imaged by confocal microscopy suggesting that the reporter was distributed throughout the cytosol, Figure S3. Additionally, a cell proliferation assay indicated that A431 cells loaded with 24C did not display a significant reduction in proliferation over 24 h when compared to cells that underwent mock pinocytosis in the absence of peptide (n =3, p ≥ 0.5083). 24C-loaded A431 cells were incubated with EGF in the presence and absence of gefitinib. In the absence of gefitinib, 10.5 ± 4.6% of the 24C reporter was phosphorylated on the L-Htc site while exposure to this drug reduced phosphorylation to 2.5 ± 1.6% (n =3, **p ≤ 0.006), Figure 3. Phosphorylation of 24C at the threonine residue (Akt site) was also observed following EGF stimulation and the level of phosphorylation was independent of gefitinib treatment (n =3, p ≥ 0.99). The amount of degraded reporter was not significantly different between the basal and EGF stimulation (79.2 ±2.0%, 71.2 ± 1.1%, n = 3, p ≥ 0.19) and/or between the EGF and gefitinib treatments (71.2±1.1%, 59.9 ± 8.1%, n = 3, p ≥ 0.06). However, a significant reduction in proteolysis was observed for the gefitinib and basal treatment conditions (59.9 ± 8.1%, 79.1± 2.0%, n = 3, *p ≤ 0.0197).
FIGURE 3.
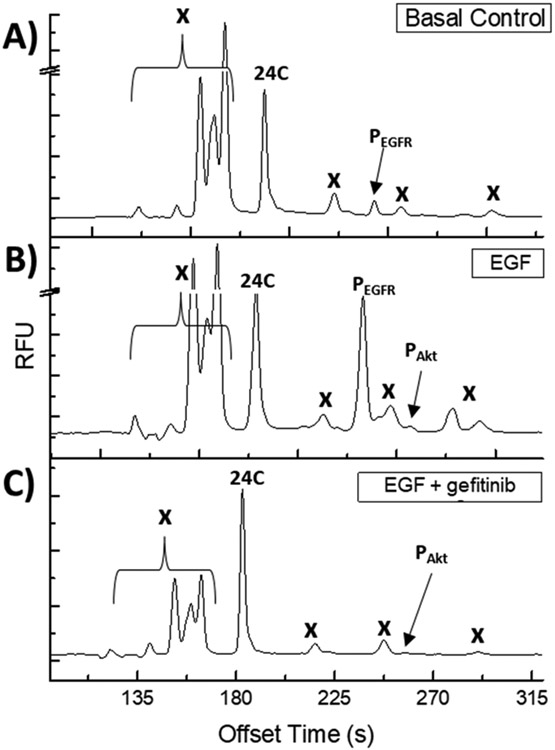
Electropherograms of A431 cells loaded with 24C, lysed, and the lysate separated by CE-F. (A) Unstimulated cells. (B) Cells stimulated with 200 ng/mL EGF. (C) Cells stimulated with 200 ng/mL EGF in the presence of 10 μM gefitinib; “x” denotes marks peaks due to proteolytic fragments of 24C.
To determine where the possible proteolytic fragments of 24C might migrate on the electropherogram, a complex mixture of partially degraded reporter was generated using commercially available proteases and peptidases, Figures S4-5. Under these conditions, 24C and phosphorylated 24C peaks were resolved from their peptide fragments insuring accurate quantification of 24C and its various phosphorylated forms.
Characterizing C24C Photoactivation in Solution & Live A431 Cells.
To gain temporal control over the initiation of cellular reactions with 24C, a photoactivatable variant of 24C, termed C24C (for photocaged-24C), was synthesized. C24C possessed 2-4,5-dimethoxy 2-nitrobenzyl (DMNB) protecting groups on both the Akt (T) and EGFR (L-Htc) phosphorylation sites, respectively, Figure 1A. C24C, like native 24C, was efficiently loaded into cells and displayed a diffuse distribution throughout the cytosol, with significantly greater green fluorescence relative to an unloaded control, Figure S6. Cell proliferation was not significantly impacted by C24C loading relative to that of unloaded cells, Figure S7. Next photoactivation of C24C in solution was demonstrated, Figure S8. When C24C was incubated with recombinant Akt and EGFR and without application of a 360 nm photostimulus, minimal levels of phosphorylation of reporter C24C was observed over 60 min for Akt (3.3 ± 0.9 %) and/or for EGFR (0 %). However, following photoactivation, a significant increase in reporter phosphorylation was observed on both sites after 60 min of 87.1 ± 1.8% (n =3, ****p ≤ 0.0001) for Akt and 56.8 ± 0.4% (n =3, ****p ≤ 0.0001) for EGFR. Live A431 cells were loaded with C24C and in the absence of photoactivation, no phosphorylation or proteolytic degradation of the reporter was observed over the course of 30 min (0±0% phosphorylation 0±0%, degradation 0±0%), Figure 4A-B. However, when C24C-loaded A431 cells were exposed to a 1.5 J/cm2 photostimulus at 360 nm, 84.6 ± 5.2% of the reporter was photoactivated, resulting in a more complex electropherogram that resembled the results of the 24C studies in A431 cell lysates, Figure 4C. UV-induced A431 cell damage was assessed by measurement of CPDs in A431 cells following irradiation. Under the conditions used, the quantity of CPDs formed was not significantly different in the UV-exposed cells relative to that in the unexposed control cells up to 3.0 J/cm2 (n =3, p ≥ 0.9952), Figure S9. These data suggest that the DMNB protecting groups on C24C prevented enzymatic modifications by kinases and proteases/peptidases prior to photoactivation, and that photoactivation of reporter loaded cells was performed without resulting in significant cell damage.
FIGURE 4.
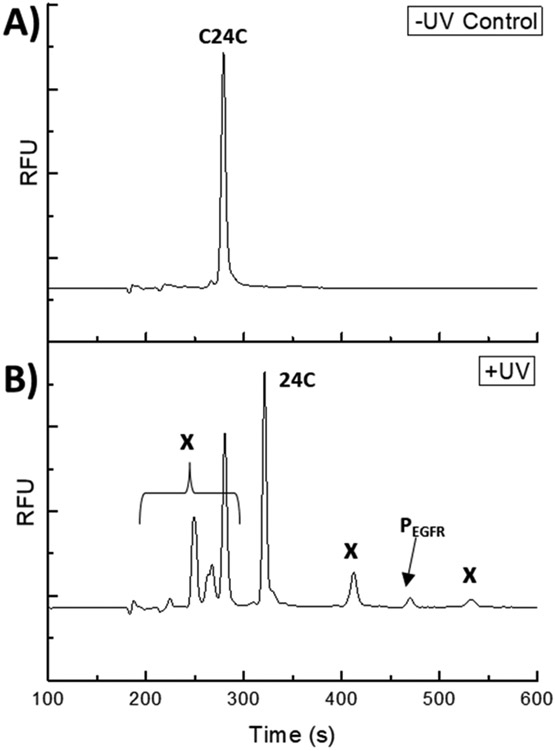
Photoactivation of C24C-loaded into A431 cells. (A) C24C loaded cells after 30 min at 37°C and without UV exposure; (B) C24C-loaded cells exposed to UV, and then incubated for 20 min at 37°C. “x” marks peaks due to proteolytic fragments of 24C.
Measurements of Enzyme Activation in Single A431 Cells Loaded with Either 24C or C24C.
Single cells were loaded with 24C or C24C, stimulated with EGF and then assayed for peptidase, kinase activity. However, due to the reduced peak resolution for the single cell relative to that of lysate assays, reporter phosphorylation on either the Akt and EGFR sites was pooled as cumulative phosphorylation. When 24C loaded cells were treated with EGF in the presence of gefitinib, there was a significant reduction in total reporter phosphorylation supporting the results from the bulk cell studies, which indicate the majority of observed 24C phosphorylation in A431 cells is due to RTK activity, Figure 5A, B. Conversely, for reporter C24C at times up to 4 h post-loading without photoactivation, all cells (100%, n = 9) displayed 2 peaks on their electropherogram corresponding to a proteolytic product, and the intact parent reporter, Figure 5C. Following photoactivation, all cells (100%, n = 11) loaded with C24C displayed electropherograms with peaks that were similar to electropherograms of A431 cells loaded with 24C, Figure 5D. After photoactivation C24C-loaded single cells, displayed a significant increase in total reporter phosphorylation, Figure 5E. This was supported when comparing single cell data for the non-photoactivatable reporter 24C, to photoactivated C24C as no significant difference in total reporter phosphorylation was observed under the same treatment conditions and experimental timescale, Figure 5E. Additionally, there was a significant increase in reporter proteolysis for reporter 24C relative to that of photoactivated C24C in loaded cells over similar timescales, Figure 5E. In live single cells, the DMNB groups enabled the reporter to resist enzymatic modifications such as proteolysis and phosphorylation prior to the application of a photostimulus. Further, the photoactivated form of C24C behaved similarly in cells to that of the native 24C reporter, suggesting that the photoactivation process did not interfere with the targeted enzymes.
FIGURE 5.

Measurement of enzyme activation in single A431 cells loaded with 24C or C24C. (A) Representative electropherogram of a 24C-loaded A431 cell following EGF stimulation. (B) Representative electropherogram of a 24C-loaded A431 cell following EGF stimulation in the presence of gefitinib. (C) A representative electropherogram of a cell loaded with C24C in the absence of photoactivation. “x” marks peaks due to proteolytic fragments of the reporter. (D) Representative electropherogram of a cell loaded with C24C followed by C24C photoactivation in the presence of EGF. (E) Cumulative phosphorylation and proteolysis data for single A431 cells loaded with either 24C or C24C. Cumulative phosphorylation is defined as total EGFR and Akt phosphorylation on the reporter C24C-loaded cells were assayed with (n = 11), or without (n = 9) a photostimulus. 24C loaded cells were analyzed in the presence (n = 22), and absence of gefitinib (n = 22). Statistics: change in C24C phosphorylation following photoactivation: Mann Whitney U = 15, n1 = 11, n2 = 9, z = 2.58311 ***p ≤ 0.00988, d = 1.5); change in C24C proteolysis following photoactivation (Mann Whitney U = 7, n1 = 11, n2 = 9, **p = 0.0142, d = 1.9); Difference in total phosphorylation between photoactivated C24C and native 24C: Mann Whitney U = 105, n1 = 9, n2 = 22, z = −0.59144, p ≥ 0.5552, d = 0.43); Difference in reporter proteolysis between photoactivated C24C and native 24C: Mann Whitney U = 66.5, n1 = 9, n2 = 22, z = 2.06223, *p ≤ 0.0394, d = 0.95; Difference in 24C phosphorylation following gefitinib treatment: Mann-Whitney U= 77, n1 = 22, n2 = 24, ****p < 0.0001, d =1.57).
CONCLUSIONS
In conclusion, the feasibility of using a novel dual-substrate reporter for performing simultaneous measurements of kinase, and protease/peptidase activity in human epidermoid carcinoma cells was demonstrated. The 24C reporter was readily phosphorylated by EGFR and Akt, displayed resistance to proteolysis and PTP activity in cell lysates, and was phosphorylated at the L-Htc site in A431 cell lysates. Importantly, the structure of 24C enabled robust electrophoretic separations by ensuring that the majority of generated peptide fragments migrated earlier than the intact phosphorylated forms of 24C. Additionally, the L-Htc moiety on the EGFR substrate of reporter 24C displayed a significantly greater resistance to dephosphorylation than that of a native tyrosine residue on 24A. This enabled the poorer KM of 24C for EGFR to be offset by this reporter’s ability to retain higher levels of phosphorylation on the EGFR and act as a substrate within cells. The photoactivatable reporter, C24C, was resistant to both kinase and peptidase activity prior to photoactivation. Importantly, the photolysis reaction was demonstrated to proceed efficiently in living cells without significantly damaging cellular DNA. In single A431 cells, enzymatic modifications were also observed to be mitigated prior to photoactivation, and small populations of photoactivated A431 cells displayed similar activity profiles to those observed in single cells loaded with native 24C. Continued improvement and integration of non-proteinogenic reporter modifications are expected to improve the temporal control, resistance to degradation, and resolution of reporter species for quantitation of enzyme activity at the single cell level.
Supplementary Material
ACKNOWLEDGMENTS
We would like to acknowledge funding support from the NIH in the form of grants F31HL147500 (M.M.A.), R01CA203032 (D.S.L.), R01CA177993 (N.L.A.). Special thanks to Dr. Brae Petersen for his deep knowledge of chemical cytometry instrumentation and programming to aid in the progression of single cell experiments. Special thanks to Professor Ilona Jaspers for her expertise in cell biology and kinase signaling and for always displaying a high degree of enthusiasm for this project during our conversations.
Footnotes
Supporting Information
The supporting information contains additional experimental details and methods regarding reporter synthesis, characterization, and cell-based assays including electropherograms, images, and mass spectra. (File type, PDF)
The Supporting Information is available free of charge on the ACS Publications website.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest for the work involved in preparing this manuscript.
REFERENCES
- (1).Normanno N; De Luca A; Bianco C; Strizzi L; Mancino M; Maiello MR; Carotenuto A; De Feo G; Caponigro F; Salomon DS Epidermal Growth Factor Receptor (EGFR) Signaling in Cancer. Gene 2006, 366 (1), 2–16. 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- (2).Kumar A; Rajendran V; Sethumadhavan R; Purohit R AKT Kinase Pathway: A Leading Target in Cancer Research. Sci. World J 2013, 2013, e756134. 10.1155/2013/756134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Manning BD; Toker A AKT/PKB Signaling: Navigating the Network. Cell 2017, 169 (3), 381–405. 10.1016/j.cell.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Sigismund S; Avanzato D; Lanzetti L Emerging Functions of the EGFR in Cancer. Mol. Oncol 2018, 12 (1), 3–20. 10.1002/1878-0261.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Lemmon MA; Schlessinger J; Ferguson KM The EGFR Family: Not So Prototypical Receptor Tyrosine Kinases. Cold Spring Harb. Perspect. Biol 2014, 6 (4), a020768. 10.1101/cshperspect.a020768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Garay C; Judge G; Lucarelli S; Bautista S; Pandey R; Singh T; Antonescu CN Epidermal Growth Factor–Stimulated Akt Phosphorylation Requires Clathrin or ErbB2 but Not Receptor Endocytosis. Mol. Biol. Cell 2015, 26 (19), 3504–3519. 10.1091/mbc.E14-09-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Hers I; Vincent EE; Tavaré JM Akt Signalling in Health and Disease. Cell. Signal 2011, 23 (10), 1515–1527. 10.1016/j.cellsig.2011.05.004. [DOI] [PubMed] [Google Scholar]
- (8).Mainz ER; Serafin DS; Nguyen TT; Tarrant TK; Sims CE; Allbritton NL Single Cell Chemical Cytometry of Akt Activity in Rheumatoid Arthritis and Normal Fibroblast-like Synoviocytes in Response to Tumor Necrosis Factor α. Anal. Chem 2016, 88 (15), 7786–7792. 10.1021/acs.analchem.6b01801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lawson DA; Kessenbrock K; Davis RT; Pervolarakis N; Werb Z Tumour Heterogeneity and Metastasis at Single-Cell Resolution. Nat. Cell Biol 2018, 20 (12), 1349–1360. 10.1038/s41556-018-0236-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Schmidt F; Efferth T Tumor Heterogeneity, Single-Cell Sequencing, and Drug Resistance. Pharmaceuticals 2016, 9 (2), 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Arriaga EA Determining Biological Noise via Single Cell Analysis. Anal. Bioanal. Chem 2009, 393 (1), 73–80. 10.1007/s00216-008-2431-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Proctor A; Wang Q; Lawrence DS; Allbritton NL Selection and Optimization of Enzyme Reporters for Chemical Cytometry. In Methods in Enzymology; Academic Press, 2019. 10.1016/bs.mie.2019.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kovarik ML; Allbritton NL Measuring Enzyme Activity in Single Cells. Trends Biotechnol. 2011, 29 (5), 222–230. 10.1016/j.tibtech.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ou Y; Wilson RE; Weber SG Methods of Measuring Enzyme Activity Ex Vivo and In Vivo. Annu. Rev. Anal. Chem 2018, 11 (1), 509–533. 10.1146/annurev-anchem-061417-125619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Neumann EK; Comi TJ; Rubakhin SS; Sweedler JV Lipid Heterogeneity between Astrocytes and Neurons Revealed by Single-Cell MALDI-MS Combined with Immunocytochemical Classification. Angew. Chem. Int. Ed 2019, 58 (18), 5910–5914. 10.1002/anie.201812892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Comi TJ; Makurath MA; Philip MC; Rubakhin SS; Sweedler JV MALDI MS Guided Liquid Microjunction Extraction for Capillary Electrophoresis–Electrospray Ionization MS Analysis of Single Pancreatic Islet Cells. Anal. Chem 2017, 89 (14), 7765–7772. 10.1021/acs.analchem.7b01782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Yang M; Nelson R; Ros A Toward Analysis of Proteins in Single Cells: A Quantitative Approach Employing Isobaric Tags with MALDI Mass Spectrometry Realized with a Microfluidic Platform. Anal. Chem 2016, 88 (13), 6672–6679. 10.1021/acs.analchem.5b03419. [DOI] [PubMed] [Google Scholar]
- (18).Jansson ET; Comi TJ; Rubakhin SS; Sweedler JV Single Cell Peptide Heterogeneity of Rat Islets of Langerhans. ACS Chem. Biol 2016, 11 (9), 2588–2595. 10.1021/acschembio.6b00602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hu H-Y; Gehrig S; Reither G; Subramanian D; Mall MA; Plettenburg O; Schultz C FRET-Based and Other Fluorescent Proteinase Probes. Biotechnol. J 2014, 9 (2), 266–281. 10.1002/biot.201300201. [DOI] [PubMed] [Google Scholar]
- (20).Zhou X; Herbst-Robinson KJ; Zhang J Visualizing Dynamic Activities of Signaling Enzymes Using Genetically Encodable FRET-Based Biosensors: From Designs to Applications. Methods Enzymol. 2012, 504, 317–340. 10.1016/B978-0-12-391857-4.00016-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Fu Y; Finney NS Small-Molecule Fluorescent Probes and Their Design. RSC Adv. 2018, 8 (51), 29051–29061. 10.1039/c8ra02297f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Komatsu T; Urano Y Evaluation of Enzymatic Activities in Living Systems with Small-Molecular Fluorescent Substrate Probes. Anal. Sci 2015, 31 (4), 257–265. 10.2116/analsci.31.257. [DOI] [PubMed] [Google Scholar]
- (23).Chan J; Dodani SC; Chang CJ Reaction-Based Small-Molecule Fluorescent Probes for Chemoselective Bioimaging. Nat. Chem 2012, 4 (12), 973–984. 10.1038/nchem.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Yang NJ; Hinner MJ Getting Across the Cell Membrane: An Overview for Small Molecules, Peptides, and Proteins. In Site-Specific Protein Labeling; Springer New York, 2015; pp 29–53. 10.1007/978-1-4939-2272-7_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Liu H-W; Chen L; Xu C; Li Z; Zhang H; Zhang X-B; Tan W Recent Progresses in Small-Molecule Enzymatic Fluorescent Probes for Cancer Imaging. Chem. Soc. Rev 2018, 47 (18), 7140–7180. 10.1039/c7cs00862g. [DOI] [PubMed] [Google Scholar]
- (26).Vickerman BM; Anttila MM; Petersen BV; Allbritton NL; Lawrence DS Design and Application of Sensors for Chemical Cytometry. ACS Chem. Biol 2018, 13 (7), 1741–1751. 10.1021/acschembio.7b01009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Essaka DC; Prendergast J; Keithley RB; Palcic MM; Hindsgaul O; Schnaar RL; Dovichi NJ Metabolic Cytometry: Capillary Electrophoresis with Two-Color Fluorescence Detection for the Simultaneous Study of Two Glycosphingolipid Metabolic Pathways in Single Primary Neurons. Anal. Chem 2012, 84 (6), 2799–2804. 10.1021/ac2031892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Phillips RM; Bair E; Lawrence DS; Sims CE; Allbritton NL Measurement of Protein Tyrosine Phosphatase Activity in Single Cells by Capillary Electrophoresis. Anal. Chem 2013, 85 (12), 6136–6142. 10.1021/ac401106e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Keithley RB; Rosenthal AS; Essaka DC; Tanaka H; Yoshimura Y; Palcic MM; Hindsgaul O; Dovichi NJ Capillary Electrophoresis with Three-Color Fluorescence Detection for the Analysis of Glycosphingolipid Metabolism. The Analyst 2013, 138 (1), 164–170. 10.1039/c2an36286d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Turner AH; Lebhar MS; Proctor A; Wang Q; Lawrence DS; Allbritton NL Rational Design of a Dephosphorylation-Resistant Reporter Enables Single-Cell Measurement of Tyrosine Kinase Activity. ACS Chem. Biol 2016, 11 (2), 355–362. 10.1021/acschembio.5b00667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Mainz ER; Wang Q; Lawrence DS; Allbritton NL An Integrated Chemical Cytometry Method: Shining a Light on Akt Activity in Single Cells. Angew. Chem. Int. Ed 2016, 55 (42), 13095–13098. 10.1002/anie.201606914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Dovichi NJ; Hu S Chemical Cytometry. Curr. Opin. Chem. Biol 2003, 7 (5), 603–608. 10.1016/j.cbpa.2003.08.012. [DOI] [PubMed] [Google Scholar]
- (33).Proctor A; Herrera-Loeza SG; Wang Q; Lawrence DS; Yeh JJ; Allbritton NL Measurement of Protein Kinase B Activity in Single Primary Human Pancreatic Cancer Cells. Anal. Chem 2014, 86 (9), 4573–4580. 10.1021/ac500616q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sims CE; Meredith GD; Krasieva TB; Berns MW; Tromberg BJ; Allbritton NL Laser–Micropipet Combination for Single-Cell Analysis. Anal. Chem 1998, 70 (21), 4570–4577. 10.1021/ac9802269. [DOI] [PubMed] [Google Scholar]
- (35).Rau KR; Quinto-Su PA; Hellman AN; Venugopalan V Pulsed Laser Microbeam-Induced Cell Lysis: Time-Resolved Imaging and Analysis of Hydrodynamic Effects. Biophys. J 2006, 91 (1), 317–329. 10.1529/biophysj.105.079921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lai H-H; Quinto-Su PA; Sims CE; Bachman M; Li GP; Venugopalan V; Allbritton NL Characterization and Use of Laser-Based Lysis for Cell Analysis on-Chip. J. R. Soc. Interface 2008, 5 (Suppl 2), S113–S121. 10.1098/rsif.2008.0177.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Proctor A; Zigoneanu IG; Wang Q; Sims CE; Lawrence DS; Allbritton NL Development of a Protease-Resistant Reporter to Quantify BCR–ABL Activity in Intact Cells. Analyst 2016, 141 (21), 6008–6017. 10.1039/C6AN01378C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kasperkiewicz P; Gajda AD; Drąg M Current and Prospective Applications of Non-Proteinogenic Amino Acids in Profiling of Proteases Substrate Specificity. Biol. Chem 2012, 393 (9), 843–851. 10.1515/hsz-2012-0167. [DOI] [PubMed] [Google Scholar]
- (39).Meloni BP; Mastaglia FL; Knuckey NW Cationic Arginine-Rich Peptides (CARPs): A Novel Class of Neuroprotective Agents With a Multimodal Mechanism of Action. Front. Neurol 2020, 11. 10.3389/fneur.2020.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Smoly I; Shemesh N; Ziv-Ukelson M; Ben-Zvi A; Yeger-Lotem E An Asymmetrically Balanced Organization of Kinases versus Phosphatases across Eukaryotes Determines Their Distinct Impacts. PLOS Comput. Biol 2017, 13 (1), e1005221. 10.1371/journal.pcbi.1005221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Huyer G; Liu S; Kelly J; Moffat J; Payette P; Kennedy B; Tsaprailis G; Gresser MJ; Ramachandran C Mechanism of Inhibition of Protein-Tyrosine Phosphatases by Vanadate and Pervanadate. J. Biol. Chem 1997, 272 (2), 843–851. 10.1074/jbc.272.2.843. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.