

Abstract
Myosin VI(MYO6) is an unconventional myosin that is vital for auditory and vestibular function. Pathogenic variants in the human MYO6 gene cause autosomal-dominant or -recessive forms of hearing loss. Effective treatments for Myo6 mutation causing hearing loss are limited. We studied whether adeno-associated virus (AAV)-PHP.eB vector-mediated in vivo delivery of Staphylococcus aureus Cas9 (SaCas9-KKH)-single-guide RNA (sgRNA) complexes could ameliorate hearing loss in a Myo6WT/C442Y mouse model that recapitulated the phenotypes of human patients. The in vivo editing efficiency of the AAV-SaCas9-KKH-Myo6-g2 system on Myo6C442Y is 4.05% on average in Myo6WT/C442Y mice, which was ∼17-fold greater than editing efficiency of Myo6WT alleles. Rescue of auditory function was observed up to 5 months post AAV-SaCas9-KKH-Myo6-g2 injection in Myo6WT/C442Y mice. Meanwhile, shorter latencies of auditory brainstem response (ABR) wave I, lower distortion product otoacoustic emission (DPOAE) thresholds, increased cell survival rates, more regular hair bundle morphology, and recovery of inward calcium levels were also observed in the AAV-SaCas9-KKH-Myo6-g2-treated ears compared to untreated ears. These findings provide further reference for in vivo genome editing as a therapeutic treatment for various semi-dominant forms of hearing loss and other semi-dominant diseases.
Keywords: hearing loss, gene therapy, myosin VI, CRISPR-Cas9, semi-dominant hearing loss
Graphical abstract
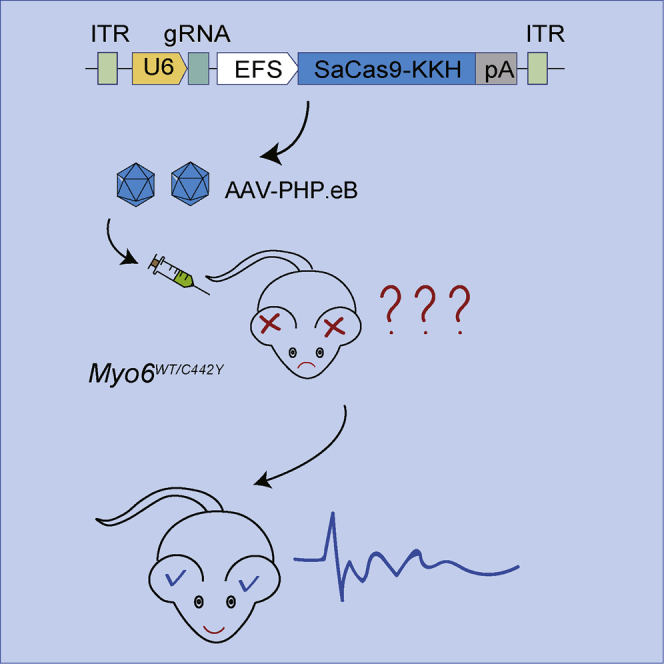
A newly identified and highly efficient AAV-PHP.eB vector carrying Staphylococcus aureus Cas9 (SaCas9-KKH)-single-guide RNA (sgRNA) complexes was used to knock out the Myo6C442Y mutant allele, which could rescue the auditory function in Myosin VI p.C442Y semi-dominant mouse model of genetic hearing loss.
Introduction
MYO6 is an unconventional myosin of 1,265 amino acids.1 It consists of an N-terminal motor domain that is involved in actin binding and movement, a calmodulin-interacting neck domain, and a C-terminal tail domain that often connects to various cargo-associated proteins.2 MYO6 is expressed in the hair cells of the inner ear and is localized to the base of the stereocilia in the cuticular plate and thus is assumed to play important roles in anchoring the stereocilia to the cuticular plate.3 Pathogenic variants in the MYO6 gene can cause either autosomal dominant inherited non-syndromic hearing loss (DFNA22) or autosomal recessive inherited non-syndromic hearing loss (DFNB37).3, 4, 5, 6, 7 Recently, a large-scale genetic analysis of Japanese patients with hearing loss found that 2.4% of autosomal dominant sensorineural hearing loss cases were caused by MYO6 mutations.8
The MYO6 p.C442Y mutation causes DFNA22. Carriers of the Myo6C442Y mutation begin to develop progressive hearing loss during childhood and show profound sensorineural hearing loss by middle age.5 We previously established a Myo6WT/C442Y mouse model that recapitulates postlingual progressive sensorineural deafness in humans. This mouse model exhibits hearing loss starting from 3 weeks after birth and progresses to profound deafness accompanied by degeneration of hair cells and disorganization of the stereocilia in the organ of Corti with a semi-dominant inheritance pattern.9 Although MYO6 mutations are known to play an important role in hereditary deafness, currently there is still a lack of effective treatment and prevention methods.
Gene therapy holds a promise for treating hearing loss or promoting the survival of spiral ganglion neurons after injury,10 and multiple preclinical trials of adeno-associated virus (AAV)-mediated gene overexpression therapy in recessive mouse models of human forms of deafness have reported successful prevention of hearing impairment.11, 12, 13, 14, 15, 16, 17, 18, 19 However, such strategies have the disadvantages of a short time window for their effectiveness and a lack of controlled gene expression,20 and such therapies might not be appropriate for semi-dominant inheritance mutations like that seen for MYO6 p.C442Y. Fortunately, the CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats, CRISPR-associated protein 9) system provides a powerful genome-editing tool that can precisely correct mutations in the inner ear.21, 22, 23, 24
MYO6 is expressed in both outer hair cells (OHCs) and inner hair cells (IHCs), particularly at the base of the stereocilia in the cuticular plate.3,4 Further studies also revealed that MYO6 is involved in the morphological and functional maturation of IHC ribbon synapses and is required for efficient Ca2+-dependent exocytosis.4,25 Several Myo6 mutations are known to exhibit an autosomal recessive inheritance pattern.1,6,26,27 For instance, an intronic point mutation (IVS5 G>A) results in skipping of exon 5 (130 bp) from the Myo6 transcript and results in a frameshift that introduces a stop codon at amino acid 95. Auditory brainstem response (ABR) testing showed that this heterozygous mutation had similar hearing threshold levels as wild-type (WT) mice when tested at 4, 8, and 24 weeks of age.27 One Myo6WT allele appears to be sufficient to maintain normal hearing, and we hypothesized that specifically knocking out the mutated Myo6C442Y allele in heterozygous Myo6WT/C442Y mice can restore hearing.
In order to verify this hypothesis, the Myo6C442Y allele was specifically knocked out using SaCas9-KKH and a newly identified and highly efficient AAV-PHP.eB delivery system for hair cells.28 The results demonstrated that knocking out the Myo6C442Y mutant allele could rescue hearing loss, and these observations support the use of this therapeutic strategy for most other semi-dominant diseases.
Results
Specific disruption of the Myo6C442Y allele in vitro using SaCas9-KKH
The Myo6WT/C442Y mice have a G to A change (NM_001039546.1: c.1325G>A), and Cas9 must distinguish the Myo6C442Y allele from the Myo6 WT allele; the shorter SaCas9-KKH was chosen to enable successful packaging within one AAV vector. Two sgRNAs were designed to specifically target the mutant Myo6C442Y allele (Figure 1A). In order to evaluate the allele specificity of the genomic DNA modification, we performed lipid-mediated delivery of the Cas9-sgRNA-GFP plasmid into cultured mouse embryonic stem cells (mESCs) derived from Myo6WT/WT, Myo6WT/C442Y, and Myo6C442Y/C442Y mice (Figure 1B). The sgRNA target site was amplified by PCR, and the editing efficiency was analyzed by next-generation sequencing (NGS).
Figure 1.

The genome-editing strategy to disrupt the Myo6C442Y mutant allele
(A) SaCas9-KKH sgRNA design. The Myo6 p.C442Y mutation site is highlighted in red (a G to A transition at position 1325 of the Myo6 cDNA). Nucleotides of the PAM sites are marked by green. (B) Schematic for the in vitro studies in Myo6C442Y/C442Y ESCs. Lipofectamine 3000-mediated delivery of the SaCas9-KKH sgRNA therapy vector into ESCs. Cells were sorted by GFP expression. (C and D) Indel percentages of g1 and g2 in Myo6WT/WT, Myo6C442 /C442Y, and Myo6WT/C442Y mESCs were determined by targeted NGS and analysis using Seqkit. Individual values (n = 3) are shown, and horizontal lines and error bars represent the mean values ± SD of three independent biological replicates. Each dot represents the DNA from one sample. (E) The indels causing in-frame versus frameshift mutations were calculated after SaCas9-KKH-Myo6-g2 transfection in Myo6C442Y/ C442Y ESCs. The data were obtained from the average of three independent repeated experiments. (F) Indel profiles from SaCas9-KKH-Myo6-g2-transfected Myo6C442Y/C442Y ESCs. Negative numbers represent deletions, and positive numbers represent insertions. Sequences without indels (value = 0) are not included in the chart. (G) Indel frequency at the Myo6C442Y mutation locus (On) and at each of the off-target (OT) loci in SaCas9-KKH-Myo6-g2-treated Myo6C442Y/C442Y ESCs. Blue, samples treated with DNA plasmids encoding SaCas9-KKH-Myo6-g2; orange, control samples with SaCas9-KKH-Myo6-NT-sg.
A higher rate of targeted insertions or deletions (indels) in mutant Myo6C442Y/C442Y mESCs (23.22% ± 1.76%) was observed with Myo6-g2 compared to Myo6-g1 (0.27% ± 0.06%) (Figure 1C). The Myo6-g2 editing efficiency of the Myo6C442Y allele in Myo6C442Y/C442Y was ∼21-fold greater than editing efficiency of the Myo6WT allele (1.13% ± 0.25%) in Myo6WT/WT mESCs. The editing efficiency of Myo6-g2 was also higher for the Myo6C442Y allele in Myo6WT/C442Y mESCs (23.7% ± 2.04%) than that of Myo6-g1 (0.06% ± 0.04%) (Figure 1D). The Myo6-g2 editing efficiency of the Myo6C442Y allele in Myo6WT/C442Y was ∼19-fold greater than editing efficiency of the Myo6WT allele (1.23% ± 0.25%) in Myo6WT/C442Y mESCs. Myo6-g2 specifically edited Myo6C442Y alleles both in Myo6C442Y/C442Y and Myo6WT/C442Y mice, which is consistent with the report that the 1-bp mismatch in the protospacer adjacent motif (PAM) was sufficient to distinguish between the mutant and WT alleles.22 The indel profile indicated that majority of the Myo6-g2-induced variants were deletions, and the frameshift mutation ratios were 83.3% (Figures 1E and 1F; Data S1). In the in silico off-target analysis of Myo6-g2 using the Cas-OFFinder software,29 we examined up to 15 off-target sites in mESCs transfected with Myo6-g2 or control sgRNA (non-targeted sgRNA, NT-sg), and no obvious indel mutations were observed (Figure 1G; Data S2). These results suggested that SaCas9-KKH-Myo6-g2 was a better strategy when compared to SaCas9-KKH-Myo6-g1, and SaCas9-KKH-Myo6-g2 was used in the subsequent in vivo experiments.
In vivo genome editing with SaCas9-KKH-Myo6-g2
SaCas9-KKH-Myo6-g2 was packaged into AAV-PHP.eB, which was reported in our previous work to be highly effective at infecting hair cells (Figure S1).28 The AAV-SaCas9-KKH-Myo6-g2 was injected into the inner ears of Myo6C442Y/C442Y, Myo6WT/C442Y, and Myo6WT/WT post-natal day (P) 0–2 mice (Figure 2A).
Figure 2.

In vivo genome editing with the AAV.PHP.eB-SaCas9-KKH-Myo6-g2 system
(A) Schematic of the AAV vectors (control and g2) and the experimental overview of the in vivo studies. (B and C) The indel percentages at P14 in Myo6C442Y/C442Y, Myo6WT/C442Y, and Myo6WT/WT in the sensory epithelia were determined by targeted NGS. The samples were from ears treated with or without AAV.PHP.eB-SaCas9-KKH-Myo6-g2 at P0–2 via the scala media (n = 3–7). Data are presented as the mean ± SD. (D) The indels causing in-frame versus frameshift mutations were calculated after AAV.PHP.eB-SaCas9-KKH-Myo6-g2 infection in Myo6C442Y/C442Y and Myo6WT/C442Y mice. (E) Indel profiles from SaCas9-KKH-Myo6-g2-transfected Myo6C442Y/C442Y and Myo6WT/C442Y mice. Negative numbers represent deletions, and positive numbers represent insertions. Myo6C442Y and Myo6WT reads are plotted separately. Sequences without indels (value = 0) are omitted from the chart. (F) The most abundant reads in the AAV.PHP.eB-SaCas9-KKH-Myo6-g2-treated Myo6C442Y/C442Y and Myo6WT/C442Y mice are shown.
PCR products of target sites were analyzed by NGS at P14, and we observed an indel frequency of 3.51% ± 3.12% in AAV-SaCas9-KKH-Myo6-g2-treated Myo6C442Y/C442Y ears but effectively no indels (0.01%) in the untreated ears (Figure 2B). For Myo6WT/WT mice, there was no significant difference in indel frequency between the AAV-SaCas9-KKH-Myo6-g2-treated and untreated groups (0.015% for treated versus 0.012% for untreated), which was consistent with the in vitro results in Figure 1C. In Myo6WT/C442Y mice, Myo6-g2 edited the Myo6C442Y alleles specifically (4.05% ± 2.38% from six independent cochlea), which was ∼17-fold greater than the editing efficiency of Myo6WT alleles (0.24% ± 0.16% from four independent cochlea) (Figure 2C and Data S1).
The indel profile showed that the majority of AAV-SaCas9-KKH-Myo6-g2-induced variants in the Myo6C442Y allele were deletions, and the frameshift mutation rates were 89.0% and 92.3% for Myo6C442Y/C442Y and Myo6WT/C442Y, respectively (Figure 2D). The most common mutation variants were −1 bp (0.81%/3,690 reads in Myo6WT/C442Y, 0.64%/7,221 reads in Myo6C442Y/C442Y), −2 bp (0.89%/4,077 reads in Myo6WT/C442Y, 0.56%/6,328 reads in Myo6C442Y/C442Y), and +1 bp (0.51%/2,350 reads in Myo6WT/C442Y, 0.28%/3,115 reads in Myo6C442Y/C442Y) frameshifts (Figures 2E and 2F; Data S1).
In AAV-SaCas9-KKH-Myo6-g2-treated Myo6WT/C442Y mice, we also observed indel formation for Myo6C442Y alleles at the mRNA level at P15 (0.8% ± 0.01%), which was ∼10-fold greater than the editing efficiency of the Myo6WT alleles (0.08%; Figure S2A). We observed the proportion of Myo6C442Y mRNA allele reads decreased in the treated group relative to the untreated group (51.3% versus 52.4%, n = 4) (Figure S2B).
In order to determine whether the gene editing was stable for a long time, we chose 15 weeks, which is one of the best time points for hearing rescue performance according to our study, to analyze the gene-editing efficacy. Because the sensory epithelium is difficult to separate at 15 weeks, the whole soft cochlear tissue was used instead. In the whole cochlea, in which supporting cells vastly outnumber the AAV-SaCas9-KKH-Myo6-g2-targeted hair cells, we could still observe an indel frequency of 3.63%, which was similar to 4.05% at P14. However, editing in WT alleles was also observed (0.17%; Figure S3), which was consistent with a previous report.22 This might be caused by the inability of the single G or A to accurately distinguish the Myo6WT from Myo6C442Y allele for indel analysis. However, the ratio of indel frequencies in AAV-SaCas9-KKH-Myo6-g2-treated Myo6WT alleles and untreated Myo6WT (background) was not increased even after 15 weeks (6.36-fold at P14 versus 2.62-fold at 15 weeks) (Figure 2C; Figure S3; Data S1). These results indicated that AAV-SaCas9-KKH-Myo6-g2 could specifically edit the Myo6C442Y alleles in vivo for a long time.
Allele-specific knockout of Myo6C442Y improves hearing function
To study the effect of the AAV-SaCas9-KKH-Myo6-g2 system on hearing function in Myo6WT/C442Y mice, we recorded ABRs, which represent the sound-evoked neural output of the cochlea from treated and untreated ears to evaluate the extent of hearing recovery across frequencies ranging from 4 kHz to 32 kHz. We first evaluated whether the injection of the vectors affected hearing function. We found that there was no ABR threshold elevation in the AAV-SaCas9-KKH-Myo6-g2-treated ears compared with the untreated ears (16 kHz: treated, 31.1 ± 1.8 dB, n = 9; untreated, 31.1 ± 1.8 dB, n = 9, p > 0.05) in Myo6WT/WT mice (Figure S4A). No hair cell loss was detected along the tonotopic axis of the cochlea in the AAV-SaCas9-KKH-Myo6-g2-treated group (Figures S4B and S4C; p > 0.05), which confirmed that the AAV-SaCas9-KKH-Myo6-g2 therapeutic system does not affect the hearing function of WT mice.
At 10 weeks, the ABR thresholds were significantly decreased at all the tested frequencies in AAV-SaCas9-KKH-Myo6-g2-treated ears compared to the untreated ears (16 kHz: treated, 43.3 ± 2.7 dB, n = 26; untreated, 65.2 ± 3.8 dB, n = 26, p < 0.0001) (Figure 3A). The wave 1 latencies stimulated by 90 dB SPL (sound pressure level) sound at 8 kHz and 16 kHz were shorter in the AAV-SaCas9-KKH-Myo6-g2-treated ears compared to the untreated ears (8 kHz: treated, 1.9 ± 0.1 ms, n = 15; untreated, 2.4 ± 0.1 ms, n = 13, p < 0.01; 16 kHz: treated, 1.8 ± 0 ms, n = 15; untreated, 2.5 ± 0.1 ms, n = 15, p < 0.0001) (Figure 3B), but we did not detect significant changes in ABR wave I amplitudes in the AAV-SaCas9-KKH-Myo6-g2-treated ears compared with the untreated ears at 10 weeks (Figure 3C).
Figure 3.

Effects of the AAV-PHP.eB-SaCas9-KKH-g2 virus on the inner ear ABR thresholds in Myo6WT/C442Y mice
(A) ABR thresholds in Myo6WT/C442Y ears treated with AAV.PHP.eB-SaCas9-KKH-Myo6-g2 (blue), untreated Myo6WT/C442Y ears (orange), and WT ears (black) after 10 weeks, 15 weeks, and 5 months. At 10 months, the dashed line of the same orange color denotes the mean data of untreated Myo6WT/C442Y ears, and the solid lines (purple, black, and blue) denote the three individual AAV-SaCas9-KKH-Myo6-g2-treated ears showing mild hearing improvement. Statistical analysis between untreated and AAV.PHP.eB-SaCas9-KKH-Myo6-g2-treated Myo6WT/C442Y ears was by one-way ANOVA: ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05. The statistical analysis between WT and AAV.PHP.eB-SaCas9-KKH-Myo6-g2-treated Myo6WT/C442Y ears was by one-way ANOVA: ###p < 0.001; ##p < 0.01; #p < 0.05. Values and error bars reflect the mean ± SD. (B) Latency of ABR wave 1 at 8 kHz and 16 kHz at 10 weeks. (C) Amplitude of ABR wave 1 at 8 kHz and 16 kHz at 10 weeks. AAV.PHP.eB-SaCas9-KKH-Myo6-g2-treated Myo6WT/C442Y ears (blue) were compared with untreated Myo6WT/C442Y ears (orange). Values and error bars reflect the mean ± SEM. Statistical analysis was by unpaired two-tailed Student’s t tests. ∗∗∗p < 0.001.
At 15 weeks, the ABR thresholds were also significantly decreased in AAV-SaCas9-KKH-Myo6-g2-treated ears compared to the untreated ears (16 kHz: treated, 53.3 ± 17.3 dB, n = 20; untreated, 70 ± 22.2 dB, n = 29, p < 0.0001) (Figure 3A). However, the magnitude of the improvement at 5 months was reduced at 4 out of 5 frequencies and persisted only at 8 kHz (treated, 70.8 ± 17.7 dB, n = 12; untreated, 89.6 ± 12.5 dB, n = 12, p < 0.01) (Figure 3A). With continuous observation up to 10 months, untreated Myo6WT/C442Y mice showed no detectable ABR thresholds, indicating profound deafness. In contrast, 3 out of 5 mice showed mild hearing improvements in the AAV-SaCas9-KKH-Myo6-g2-treated group (Figure 3A).
From the ABR threshold comparison between WT controls and AAV-SaCas9-KKH-Myo6-g2-treated Myo6WT/C442Y ears, there also appeared to be significant differences at 24 kHz and 32 kHz at 10 weeks; at 4 kHz, 24 kHz, and 32 kHz at 15 weeks; and at all frequencies at 5 months. There was significant hearing loss at 5 months. The ABR thresholds of individual mice at 10 weeks, 15 weeks, 5 months, and 10 months are shown in Figure S5, which indicates the variations in individual hearing function in semi-dominant mouse models. The results demonstrated that knocking out the Myo6C442Y mutant allele could initially improve auditory function, but the auditory function began to decline at 5 months, and by 10 months the auditory performance was severely degraded. It is likely that the waning of ABR thresholds at 10 months was a continuation of the decline in responsivity seen at 5 months.
In contrast with the injection of AAV-SaCas9-KKH-Myo6-g2, none of the mice treated with the control AAV-SaCas9-KKH virus recovered auditory function at 10–12 weeks (16 kHz: treated, 62.5 ± 20.7 dB, n = 10; untreated, 65 ± 20.1 dB, n = 10, p > 0.05) (Figure S6A), and the hair cell survival rate was also not improved in the AAV-SaCas9-KKH-treated group (p > 0.05 at all tested frequencies) (Figures S6B and S6C). This suggested that the specific gene editing is necessary for auditory rescue.
We also found that the DPOAE thresholds in AAV-SaCas9-KKH-Myo6-g2-treated Myo6WT/C442Y mice showed recovery at 5, 10, and 12 weeks (Figure S7), which reflected the recovery of OHCs’ amplification function. At 10 weeks, the DPOAE threshold of the AAV-SaCas9-KKH-Myo6-g2-treated ears was significantly decreased at 8 kHz and 16 kHz compared with the untreated ears. The untreated ears showed almost no detectable DPOAE thresholds following 80 dB SPL stimulation at high-frequency ranges (24–32 kHz), while the treated ears showed detectable DPOAE thresholds. From the DPOAE threshold comparison between WT controls and AAV-SaCas9-KKH-Myo6-g2-treated Myo6WT/C442Y, it seemed that OHC function in the treated group was still significantly different from that of WT mice. Although the ABR thresholds of the AAV-SaCas9-KKH-Myo6-g2-treated ears at 24–32 kHz were decreased significantly compared to the untreated ears (p < 0.05), the treated ears show mild DPOAE recovery at the corresponding frequencies. We suspect that the large improvement in ABR thresholds at 24–32 kHz in the treated ears might be mainly due to the protection of IHC function. Together, these results indicate that the auditory function of Myo6 WT/C442Y was partially recovered by injection of AAV-SaCas9-KKH-Myo6-g2 into the inner ears, but the effect was gradually lost over time.
Allele-specific knockout of Myo6C442Y improves hair cell survival rates
Myo6WT/C442Y mice exhibited substantial degeneration of OHCs as previously described.9 To assess the effect of gene editing by AAV-SaCas9-KKH-Myo6-g2 on hair cell survival, we counted the hair cells at 5 months and 10 months. At 5 months, the number of IHCs in Myo6WT/C442Y mice was not significantly different from WT, and the number of IHCs in the AAV-SaCas9-KKH-Myo6-g2-treated ears did not change (Figure 4A; Figures S4B and S6B). There were more OHCs in the AAV-SaCas9-KKH-Myo6-g2-treated ears (40.09 ± 0.34 and 35.18 ± 1.41 cells/100 μm in the middle and basal turns of the cochlea, respectively) than in the untreated ears (34.36 ± 0.98 and 17.73 ± 2.43 cells/100 μm in the middle and basal turns of the cochlea, respectively) (Figure 4B). The survival of OHCs was significantly enhanced in the middle and basal turns of the cochlea in the AAV-SaCas9-KKH-Myo6-g2-treated ears. Representative images of hair cells at 5 months are shown in Figure S8.
Figure 4.

Effects of the AAV-PHP.eB-SaCas9-KKH-g2 virus on cell survival rates in Myo6WT/C442Y mice
(A and B) The number of IHCs (A) and OHCs (B) per 100-μm section for four untreated and four AAV-PHP.eB-SaCas9-KKH-g2-treated Myo6WT/C442Y cochleae from four mice at 5 months. (C and D) The numbers of IHCs (C) and OHCs (D) per 100-μm section for three untreated and three AAV-PHP.eB-SaCas9-KKH-g2-treated Myo6WT/C442Y cochleae from three mice at 10 months. Values and error bars reflect the mean ± SEM. Statistical analysis was by two-tailed Student’s t tests. ∗∗∗p < 0.001. NS, not significant. (E) Representative confocal images of cochleae harvested at 10 months from the AAV-PHP.eB-SaCas9-KKH-g2-treated and untreated ears of Myo6WT/C442Y mice. The apical, middle, and basal turns were dissected and stained with MYO7A (red) for hair cells and with DAPI (blue) for nuclei. Scale bar, 50 μm.
At 10 months, more IHCs in the middle and basal turns were observed in the AAV-SaCas9-KKH-Myo6-g2-treated ears (11.33 ± 0.55 and 6 ± 0.41 cells/100 μm in the middle and basal turns of the cochlea, respectively) than in the untreated ears (6.11 ± 0.98 and 1.11 ± 0.31 cells/100 μm in the middle and basal turns of the cochlea, respectively) (Figures 4C and 4E). More OHCs were observed in the middle turn of the AAV-SaCas9-KKH-Myo6-g2-treated ears (33.78 ± 1.65 cells/100 μm) compared with untreated ears (13.44 ± 3.27 cells/100 μm) (Figures 4D and 4E). Almost no OHCs were observed in the basal turns of the two groups (Figure 4D). These results indicated that the survival rate of hair cells was significantly improved by knockout of the Myo6C442Y allele, and the hair cell loss in Myo6WT/C442Y mice progressed from OHCs to IHCs and from the basal turn to the apical turn.
Allele-specific knockout of Myo6C442Y improves hair bundle morphology
Stereocilia bundles on hair cells are responsible for sound detection, and morphological defects in the stereocilia can cause deafness. In our previous study,9 morphological abnormalities in cochlear hair bundles in Myo6WT/C442Y mice were observed at 6 weeks. Most hair bundles anchored on the remaining OHCs and IHCs retained normal morphology, but some were disorganized, with fused stereocilia and rotated polarity. At 12 weeks, the number of hair bundles in WT mice was approximately twice that of the Myo6WT/C442Y heterozygous mice along the middle and basal tonotopic axes. Thus, in present study the hair bundle morphology was evaluated by scanning electron microscopy in the AAV-SaCas9-KKH-Myo6-g2-treated and untreated ears of Myo6WT/C442Y mice at 10 weeks of age. In WT control ears, the hair bundles were arranged into two to three rows in a staircase pattern in OHCs and IHCs (Figure 5). In Myo6WT/C442Y mice, the surviving IHCs and OHCs showed significant disorganization, with fused stereocilia and rotated polarity in some regions, which was consistent with a previous report.6 When AAV-SaCas9-KKH-Myo6-g2 was delivered into the Myo6WT/C442Y cochlea, we observed more organized hair bundles in OHCs and IHCs. The scanning electron microscopy results indicated that hair bundle defects caused by Myo6WT/C442Y mutation were amenable to restoration by injection of AAV-SaCas9-KKH-Myo6-g2.
Figure 5.

Effects of the AAV-PHP.eB-SaCas9-KKH-g2 virus on hair bundle morphology of Myo6WT/C442Y mice
Scanning electron microscope images of the cochlea sensory epithelium showing the hair bundle morphology for untreated Myo6WT/WT (left), untreated Myo6WT/C442Y (middle), and AAV.PHP.eB-SaCas9-KKH-Myo6-g2-treated Myo6WT/C442Y mice (right) at 10 weeks. Representative bundles (arrows) of OHCs and IHCs were enlarged in the corresponding lower panel. Scale bar in low-magnification figures, 10 μm. Scale bar in high-magnification figures, 4 μm. High, high magnification; Low, low magnification.
Electrophysiology showed recovery of IHC Ca2+ current amplitudes after treatment with AAV-SaCas9-KKH-Myo6-g2
Previous studies have shown that knockout of Myo6 affects Ca2+ current amplitude (ICa) and leads to capacitance changes in IHCs.25,30 A net increase in whole-cell membrane capacitance (ΔCm) indicates sustained Ca2+-dependent exocytosis of synaptic vesicles in IHCs, which will activate the auditory neuron. Therefore, we explored whether the ICa and ΔCm in IHCs of Myo6WT/C442Y mice were affected by SaCas9-KKH-Myo6-g2-mediated disruption of the Myo6C442Y allele. We isolated the apical turn of the sensory epithelium directly from the Myo6WT/WT and SaCas9-KKH-Myo6-g2-treated and untreated Myo6WT/C442Y cochleae at P15–16. We found that the Myo6C442Y mutation resulted in a decrease in ICa current (WT ears, −235.1 ± 22.9 pA; untreated Myo6WT/C442Y ears, −145.8 ± 70.5 pA), and the ICa of SaCas9-KKH-Myo6-g2-treated Myo6WT/C442Y ears was recovered to −194.3 ± 65.1 pA (Figures 6A and 6B). However, under our experimental conditions we found that the Ca2+-induced exocytosis of IHCs from untreated mice was not affected by the Myo6C442Y mutation (WT ears, 115.4 ± 105.9 fF; untreated ears, 75.7 ± 57.6 fF, p > 0.05). For the treated Myo6WT/C442Y IHCs, the ΔCm was 82.2 ± 71.9 fF (n = 26), which was not significantly different from WT (p > 0.05) (Figures 6C and 6D). The ΔCm results indicated that the Myo6C442Y mutation did not affect the exocytosis of IHCs.
Figure 6.

Effects of the AAV-PHP.eB-SaCas9-KKH-g2 virus on the electrophysiology of Myo6WT/C442Y mice
(A) Representative ICa recordings in apical IHCs at P15–16 from untreated Myo6WT/C442Y mice (orange), WT mice (black), and Myo6WT/C442Y mice injected with AAV.PHP.eB-SaCas9-KKH-Myo6-g2 at P0–2 (blue). Untreated Myo6WT/C442Y = −150.84 pA. Treated Myo6WT/C442Y = −191.71 pA. WT = −291.77 pA. (B) ICa values were recorded from a holding potential of −90 mV to +70 mV in 300 ms at P15–16 in IHCs from untreated ears (orange) and AAV.PHP.eB-SaCas9-KKH-Myo6-g2-treated ears (blue). Values and error bars reflect the mean ± SEM. Statistical analysis was by one-way ANOVA. ∗∗p < 0.01; ∗p < 0.05. (C) Representative Cm measurements in IHCs, one from each group. Untreated Myo6WT/C442Y = 73.91 fF. AAV.PHP.eB-SaCas9-KKH-Myo6-g2-treated Myo6WT/C442Y = 96.46 fF. WT = 162.16 fF. (D) The net increase in Cm before and after stimulation (ΔCm) was used to quantify exocytosis from the same IHCs as ICa. Values and error bars reflect the mean ± SEM. Statistical analysis was by one-way ANOVA. ns, not significant.
Discussion
In this study, we explored a possible treatment approach for MYO6 gene mutations, which are a frequent cause of autosomal-dominant hearing loss. We used a gene therapy system that included highly efficient hair-cell-targeting AAV.PHP.eB virus vector and CRISPR-SaCas9-KKH-Myo6-g2. The therapeutic system was delivered into Myo6WT/ C442Y mouse ears at P0–2, where it specifically disrupted the Myo6C442Y mutant allele. Consequently, the genome editing specifically of Myo6C442Y alleles significantly improved the auditory function in Myo6WT/C442Y mice even though there was a marked decline from 5 months. This outcome together with previous reports of successful auditory restoration demonstrated the feasibility of gene therapy for hereditary deafness.11, 12, 13,15, 16, 17, 18,22, 23, 24,31, 32, 33, 34, 35, 36, 37 All of these preclinical gene therapy studies that have focused on different gene mutation sites will contribute to the development of precise treatments for deafness.38
Several factors were associated with the improvement in hearing seen in this work. First, it is widely accepted that OHCs are much more difficult to transfect with AAV compared to IHCs. Here, we used the highly infectious PHP.eB virus that even at a relatively low dose ensured that sufficient numbers of IHCs and OHCs would be transduced to enable assessment of whether or not there was rescue by the proposed strategy (Figure S1).28 The high-titer AAVs AAV2.7m8, Anc80L65, and PHP.B have been fully studied in OHCs,32,39, 40, 41, 42 but this newly identified and highly efficient AAV-PHP.eB vector has not been previously used to deliver CRISPR-SaCas9 into the inner ear. Furthermore, the treatment approach via the scala media was found to be safe. Audiometry measurements and hair cell counting were performed 4 weeks after AAV-SaCas9-KKH-Myo6-g2 was injected into WT P0–2 mice, and no ABR threshold shifts or hair cell loss was observed, indicating that the injection of AAV-PHP.eB-SaCas9-KKH-g2 did not disrupt hearing function (Figures S4A–S4C). Finally, the progressive hearing loss in the Myo6WT/C442Y mouse model provided a sufficient treatment time window for P0–2 injection. According to a previous electrophysiology study of Myo6sv/sv mutant mice lacking Myo6 gene expression, ionic current and ribbon synapse maturation of IHCs could still proceed normally until P6, which indicated that the therapeutic time window of P0–2 we used to correct Myo6C442Y mutation would be effective in mice.
In this study, the in vitro work for testing genome editing efficiency on mESCs was done using lipid-mediated delivery. It is well accepted that for genome editing efficiency studies, the specific values of guide RNA (gRNA) efficiency using lipid-mediated delivery or AAV-mediated delivery would be different. However, when we used the same delivery vector to compare different gRNA efficiencies in the same cell type, the sequence of gRNA was the main factor affecting the editing efficiency. Correspondingly, the highly efficient gRNAs screened using lipid-mediated delivery in vitro should also have relatively high editing efficiency in vivo when using an AAV-mediated delivery system.43 Meanwhile, the use of lipid-mediated delivery systems for gRNA screening is well accepted in the field.24
We observed gene editing in all AAV-SaCas9-KKH-Myo6-g2-treated cochleae, and to a lesser extent in the contralateral ears of AAV-SaCas9-KKH-Myo6-g2-treated mice, and this was likely because AAV vectors can diffuse to the contralateral ear of the same mouse through the cochlear aqueduct.44
It has been reported that MYO6 is located at the synaptic active zone of IHCs and is required for efficient Ca2+-dependent exocytosis in mature IHC ribbon synapses.4,25 Therefore, we also focused on the ICa and ΔCm in IHCs to study the effect of the C442Y mutation in Myo6. Compared with WT mice, we found that the untreated Myo6WT/C442Y inner ear showed decreased ICa, and AAV-SaCas9-KKH-Myo6-g2 treatment could rescue the decrease in ICa, which reflected the partially improved IHC function. We found that neither the exocytosis of IHCs (Figure 6D) nor the ABR wave I amplitude (Figure 3C) showed significant differences between treated and untreated ears. However, the two experiments were performed with extremely strong stimulation (500 ms voltage step to 0 mV, or 90 dB tone), which is likely to have obscured the subtle changes expected from the minimal and short stimulations used for determining the hearing thresholds. Our understanding of how the Myo6C442Y mutation causes reduced electrophysiological performance is still very limited, and future work will help to fully understand the mechanisms behind these effects.
DPOAEs are an indicator of OHC function, and here we found that the DPOAE thresholds in AAV-SaCas9-KKH-Myo6-g2-treated Myo6WT/C442Y mice showed partial recovery at 5, 10, and 12 weeks (Figure S7), which was not completely consistent with the ABR results. We also observed differences in the number of hair cells at 5 months and 10 months that also were not completely consistent with the ABR results. These observations suggest that the improvement of one index alone might not fully reflect the improvement of hearing function, and the improvement should instead be the result of the synergistic effect of several indexes. The morphological and physiological changes included increased hair cell numbers combined with partially rescued hair bundle arrangements in both IHCs and OHCs, partially recovered voltage-gated Ca2+ current in IHCs, and recovered DPOAE in OHCs, and these are together likely to account for the overall hearing improvement we observed in AAV-SaCas9-KKH-Myo6-g2-treated ears.
Despite the positive results in our study, there were some limitations that should be noted. First, for the AAV-SaCas9-KKH-Myo6-g2 safety evaluation, 15 off-target sites were predicated by CasOFFinder and tested by NGS. Although CasOFFinder is widely accepted to predict off-target sites,29 it is still necessary to further evaluate the specificity and safety of gRNA by genome-wide detection of off-target sites such as GUIDE-Seq, CIRCLE-seq, or GOTI before possible clinical application.45,46 The second limitation of this study is that we analyzed only AAV-Cas9 infection of WT mice at 4 weeks but not at longer times. In this study, we found that knocking out the Myo6C442Y mutant allele improved auditory function initially, while the auditory function began to decline from 5 months onward, and the ABR performance was severely degraded to the untreated control level at 10 months. The phenomenon that auditory function decreases with time is common in previous other gene therapy studies of hearing loss.23,37 It would be interesting and important to study the mechanism behind why the treatment effect declined with the prolongation of time. On the one hand, the AAV-PHP.eB virus we used could efficiently infect the IHCs and the OHCs throughout the cochlea (Figure S1), it is possible that the gene editing efficiency for Myo6C442Y allele cannot rescue all mutations to maintain proper cellular function, and the ongoing, irreversible hair cell damage caused by mutation might be one of the possible reasons for this unsatisfactory situation. Our results indicated that improving the editing efficiency by optimizing or discovering new AAV-Cas9 therapy systems will potentially improve hearing more significantly and more consistently in the future. On the other hand, further studies of AAV-Cas9 infection of WT mice lasting 10 months or more are also required to confirm that AAV-SaCas9-KKH does not disrupt hearing function and that the hair cells can tolerate the expression of AAV-Cas9 for a long time, which would provide valuable information for illustrating the mechanism behind the phenomenon. Finally, it is important to note that neonatal mouse inner ears at P0–2, as used here, are only partially developed and continue to undergo structural maturation until the onset of hearing at P14–15.47 The cochlea in P1–2 mice is temporally equivalent to the human cochlea prior to 26 weeks’ gestational age,48 so studies in older mice are needed before these findings can be translated to humans.
In conclusion, this study provides a proof of concept for in vivo genome editing as a therapeutic treatment for various semi-dominant forms of deafness and other semi-dominant human diseases.
Materials and methods
Animals
Myo6WT/C442Y and Myo6C442Y/C442Y (Myo6WT/WT as control) mice were established by Beijing Biocytogen.9 Briefly, Cas9 mRNA and sgRNAs along with the targeting vector containing the C442Y allele were microinjected into fertilized C57BL/6J oocytes to generate F0 founders. Since C57BL/6J mice have age-related hearing loss due to the homozygous Cdh23ahl allele, the Myo6-C442Y mice were crossed with CBA/CaJ mice, and offspring without Cdh23ahl were selected. Male and female Myo6-C442Y heterozygous mice were intercrossed to breed WT, heterozygous, and homozygous progenies. This animal study was approved by the Institutional Animal Care and Use Committee of Fudan University and the Shanghai Medical Experimental Animal Administrative Committee. All animals were bred and housed in the Department of Laboratory Animal Science of Fudan University, and all experimental procedures were conducted in accordance with the policies and ethics for animal research.
Plasmids and cloning
Two sgRNAs (Myo6-g1: 5′-aagatgtttcaaaaggaaagTactgat-3′; Myo6-g2: 5′-aataagaagatgtttcaaaaggaaagT-3′) that targeted the Myo6C442Y allele and differed in their length and distance between the mutation and PAM site were tested. A gene fragment of SaCas9-KKH together with the specific sgRNA was cloned into the pX601 backbone plasmid (Addgene).
ESC culture and transfection
The ESCs’ derivation and maintenance were in ESC medium with feeder cells and cultured at 37°C in a 5% CO2 atmosphere. The ESC medium was DMEM-high glucose (Millipore, SLM-220-M) supplemented with 15% (v/v) fetal bovine serum (FBS) (Gibco, 10099-141), 1× penicillin-streptomycin (Gibco, 15140-122), 1× GlutaMAX (Gibco, 35050-061), 1× MEM NEAA (non-essential amino acids) (Gibco, 11140-050), 1× 2-mercaptoethanol (Millipore, ES-007-E), 1× nucleosides (Millipore, ES-008-D), 1000 U ml-1 LIF (Leukemia inhibitory factor), and 2i (MEK inhibitor PD0325901, 1 mM; GSK3 inhibitor CHIR99021, 3 mM).49 For transfection, ESCs were plated onto 12-well plates and plasmids were mixed with Lipofectamine 3000 (Life Technologies) in Opti-MEM (Life Technologies) according to the manufacturer’s instructions. A total of 2 μg SaCas9-KKH-Myo6-sg1/sg2/NT-sg expression plasmid was transfected into the ESCs. GFP-positive cells were sorted by fluorescence-activated cell sorting (FACS) at 48 h post-transfection.
Mouse genomic DNA/RNA isolation and PCR
For the in vitro experiments, after GFP-positive ESCs were sorted by FACS, genomic DNA was isolated from the ESCs using the Quick Extract DNA Extraction Solution (Epicenter) following the manufacturer’s instructions. A total of 500–1,400 ng of genomic DNA was used for NGS. Two strategies were used for the in vivo experiments, either the sensory epithelium was dissected at P7 and cultured for another 7 days in vitro or it was directly extracted from the whole cochlea soft tissue at 15 weeks after injection of the AAV-PHP.eB-SaCas9-KKH-Myo6-g2 system into the inner ear at P0–2. For RNA analysis, the RNA was extracted from the whole cochlea soft tissue at P15. Nested PCR was used for the amplification of target sites, and the PCR amplification products were purified with a Gel Extraction Kit (QIAGEN) for NGS.
Off-target analysis
CasOFFinder software was used to predict the potential off-target sites for SaCas9-KKH-Myo6-g2, and specific primers were designed for the potential off-target sites. Off-target genomic sites were amplified from the edited genomic DNA, and the PCR products were sequenced on a MiSeq high-throughput DNA sequencer (Illumina). The primers are specified in Data S2.
AAV virus production
The SaCas9-KKH plasmid with or without target sgRNA was sequenced before packaging into AAV-PHP.eB, and the AAV vectors were packaged by OBIO (Shanghai, China) or from Yang’s lab.28 The vector titer was 3.73 × 1013 and 1.12 × 1013 genome copies/mL as determined by qPCR specific for the inverted terminal repeat of the AAV-PHP.eB-SaCas9-KKH-Myo6-g2 and control virus.
Inner ear injections
Myo6C442Y/WT, Myo6C442Y/C442Y, and Myo6WT//WT mouse pups were anesthetized by immersion in ice water at P0–2. Upon anesthesia, a post-auricular incision was made to expose the otic bulla and the stapedial artery. A total of 500 nL of AAV-PHP.eB-SaCas9-KKH-Myo6-g2 virus per cochlea was injected into the scala media through the soft cochlear lateral wall at a rate of 169 nL/min using glass micropipettes on a Nanoliter Microinjection System (WPI, Sarasota, FL, USA). The location of the basal cochlear turn is distinguished by its anatomy in relation to the stapedius artery.23,41,50, 51, 52 After injection, a suture was used to close the incision, and the mice were placed on a 42°C heating pad for recovery. The pups were returned to their mother after they fully recovered within ∼10 min. Standard post-operative care was applied after surgery.
Hearing tests
ABR and DPOAE measurements were performed using an RZ6 Acoustic System (Tucker-Davis Technologies, Alachua, FL, USA). ABRs and DPOAEs were recorded during the same session. Mice of either sex were anesthetized with intraperitoneal injection of xylazine (10 mg/kg) and ketamine (100 mg/kg). One subcutaneous needle electrode was inserted into the subcutaneous tissues of the vertex as the reference electrode, one was inserted into the mastoid portion as the recording electrode, and one was inserted into the rump of the animal as the ground electrode. ABR potentials were evoked and subsequently amplified 10,000 times with 1,024 responses and bandpass filtered at 300 Hz–3 kHz at each sound pressure level. Tone burst sound stimuli were presented at 4, 8, 16, 24, and 32 kHz to test the frequency-specific hearing thresholds. The threshold of a certain frequency was determined as the lowest SPL at which any ABR wave could be detected upon visual inspection and could be repeated. The amplitude of wave 1 of the ABR was measured from the peak of wave 1 to the following trough.53 The mice were placed on a heating pad covered by a sterile drape to maintain their body temperature during the testing. Data were analyzed and plotted using GraphPad Prism 8, and threshold averages ± SEM or SD are presented unless otherwise stated.
Confocal microscopy and cell counting
We used Myosin7a as the marker for mature hair cells. The temporal bones of adult mice were harvested, cleaned, and placed in 4% formaldehyde for 1 h at room temperature, followed by decalcification for 36–72 h with 10% EDTA (pH 7.4). The sensory epithelium was then dissected and stored in PBS until staining. Tissues were permeabilized with 0.1% Triton X-100 for 1 h, blocked with 10% normal donkey serum, and then incubated with anti-MYO7A primary antibody (1:500 dilution, Proteus BioSciences) overnight, and appropriate Alexa-conjugated secondary antibodies were used for detection. DAPI was used to label the nuclei (1:1,000 dilution, Sigma). Images were acquired on a Leica TCS SP8 laser scanning confocal microscope and a 40/63× objective. Images were edited by Adobe Photoshop and the ImageJ plugin. The tools in the ImageJ software were used for counting the number of Myosin7a-positive hair cells per 100 μm length along the sensory epithelia in the apical, middle, and basal turns of the cochlea. At least two 100 μm regions from at least three independent cochleae were counted, and the results are presented as the mean ± SEM.
Scanning electron microscopy
The temporal bones of 10-week-old adult mice were harvested, and cleaned temporal bones were placed in 2.5% glutaraldehyde in 0.1 M PBS at 4°C overnight. After the samples were sufficiently decalcified in 10% EDTA, the sensory epithelia were carefully dissected. The samples were then washed with PBS and immersed in a 1% OsO4 solution in the dark for 4 h. After dehydration through an ethanol gradient, the samples were dehydrated with a critical point dryer (Samdri-795, BioCryo Tousimis) according to the manufacturer’s instructions. The mounted tissues were coated with a 10 nm layer of gold (JFC-1100, Jeol, Japan), and an electron microscope (Hitachi S-4700 FESEM) was used for imaging.9
Electrophysiology
To assess IHC function, we performed patch-clamp recordings in IHCs from the apical turn of the sensory epithelium from both Myo6WT/WT and Myo6WT/C442Y mice at P15–16 directly according to a protocol described previously.54 Briefly, the apical turns of the cochlea were excised from AAV-SaCas9-KKH-Myo6-g2-treated and untreated Myo6WT/C442Y mice or from Myo6WT/WT of either sex and then bathed in an oxygenated extracellular solution containing 125 mM NaCl, 5.8 mM KCl, 5 mM CaCl2, 0.9 mM MgCl2, 10 mM HEPES, 5.6 mM D-glucose, 0.7 mM NaH2PO4·H2O, and 2 mM Na-pyruvate (∼290 mOsm [pH 7.2]). The tissue was visualized through a 60× water-immersion objective on an upright microscope (Olympus), and patch-clamp recordings were made through an EPC10 amplifier (HEKA Electronics, Lambrecht Pfalz, Germany) driven by the Patchmaster software (HEKA Electronics). Recording pipettes were pulled from borosilicate glass capillaries (Sutter) to a resistance of 5–6 MΩ, coated with dental wax and then filled with an intracellular solution containing 135 mM Cs-methane sulfonate, 10 mM CsCl, 10 mM TEA-Cl, 2 mM EGTA, 10 mM HEPES, 3 mM Mg-ATP, and 0.5 mM Na-GTP (∼290 mOsm [pH 7.2]). All patch-clamp experiments were carried out at room temperature, and the liquid junction potential was corrected offline. To measure ICa, we applied a voltage ramp from a holding potential of −90 mV to +70 mV in 0.3 s and recorded the resulting current. To measure exocytosis, we performed whole-cell Cm measurements with the lock-in feature and the “Sine + DC” method in Patchmaster (HEKA). Sine waves of 1 kHz and 50 mV (peak-to-peak) were superposed onto the holding potential before and after a step depolarization (0 mV, 500 ms), and the resulting current responses were used to determine Cm. The net increase of capacitance before and after stimulation (ΔCm) was used to assess exocytosis of synaptic vesicles from IHCs.
Statistical analysis
We used GraphPad Prism v.8.0 for statistical analysis. Two groups were compared using unpaired two-tailed Student’s t tests. To compare more than two groups of ABRs, one-way analysis of variance (ANOVA) was performed followed by Tukey’s post hoc test or Dunn’s multiple comparisons test (to compare every mean to every other mean), and Holm-Sidak (or Bonferroni) post hoc analyses were performed to compare selected pairs of means. The adjusted p < 0.05 was considered statistically significant. At least 234 mice (P0–2) of either sex were used for injections. The mice were randomly assigned to different test groups. Data are presented as the mean ± SD or SEM as noted in the text and figure legends.
Data and materials availability
All data and materials associated with this study are presented in the paper or the Supplemental information. Raw sequencing files have been uploaded to NCBI’s Sequence Read Archive (SRA) (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA684466). Information on the AAV-PHP.eB and SaCas9-KKH constructs is available from Addgene.
Acknowledgments
We thank W. Li for assistance with sensory epithelia dissection, C. Cui for assistance with animal breeding, C.J. Lai for help with the ABR test, and H.H. Tang and Y.X. Chen for helping with the manuscript. This work was supported by grants from the National Natural Science Foundation of China (nos. 81822011, 81771013, 81620108005, and 31922048), the National Key R&D Program of China (nos. 2017YFA0103900, 2020YFA0908201, and 2016YFC0905200), the Science and Technology Commission of Shanghai Municipality (no. 21S11905100), the Central Public-interest Scientific Institution Basal Research Fund to E.Z., the China Postdoctoral Science Foundation (no. 2020M670545), and the National Institutes of Health (nos. R01DC016875 and UG3TR002636).
Author contributions
Y.X., X.H., D.W., and D.L. designed and performed the experiments, analyzed the data, generated the figures, and wrote the manuscript. Y.X. performed in vivo injections. X.H., D.W., and D.L. contributed to genome editing. Y.X., J.Z., Z.X., and X.G. contributed to hearing function experiments. F.W., Y.L., and G.L. performed the electrophysiology experiment and analyzed the data. Y.X. and J.W. contributed to confocal microscopy and SEM experiments. M.H., R.C., J.S., Z.C., and G.L. helped design the experiments, analyze the data, and write the manuscript. E.Z. and H.Y. assisted with genome editing and helped with the sequencing data analysis. Y.S., Z.C., E.Z., and H.L. conceived the project, designed and supervised the research, and wrote the manuscript. All authors reviewed, edited, and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.06.015.
Contributor Information
Huawei Li, Email: hwli@shmu.edu.cn.
Erwei Zuo, Email: erweizuo@163.com.
Yilai Shu, Email: yilai_shu@fudan.edu.cn.
Supplemental information
References
- 1.Avraham K.B., Hasson T., Steel K.P., Kingsley D.M., Russell L.B., Mooseker M.S., Copeland N.G., Jenkins N.A. The mouse Snell’s waltzer deafness gene encodes an unconventional myosin required for structural integrity of inner ear hair cells. Nat. Genet. 1995;11:369–375. doi: 10.1038/ng1295-369. [DOI] [PubMed] [Google Scholar]
- 2.Spink B.J., Sivaramakrishnan S., Lipfert J., Doniach S., Spudich J.A. Long single α-helical tail domains bridge the gap between structure and function of myosin VI. Nat. Struct. Mol. Biol. 2008;15:591–597. doi: 10.1038/nsmb.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmed Z.M., Morell R.J., Riazuddin S., Gropman A., Shaukat S., Ahmad M.M., Mohiddin S.A., Fananapazir L., Caruso R.C., Husnain T., et al. Mutations of MYO6 are associated with recessive deafness, DFNB37. Am. J. Hum. Genet. 2003;72:1315–1322. doi: 10.1086/375122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hasson T., Gillespie P.G., Garcia J.A., MacDonald R.B., Zhao Y., Yee A.G., Mooseker M.S., Corey D.P. Unconventional myosins in inner-ear sensory epithelia. J. Cell Biol. 1997;137:1287–1307. doi: 10.1083/jcb.137.6.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Melchionda S., Ahituv N., Bisceglia L., Sobe T., Glaser F., Rabionet R., Arbones M.L., Notarangelo A., Di Iorio E., Carella M., et al. MYO6, the human homologue of the gene responsible for deafness in Snell’s waltzer mice, is mutated in autosomal dominant nonsyndromic hearing loss. Am. J. Hum. Genet. 2001;69:635–640. doi: 10.1086/323156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mochizuki E., Okumura K., Ishikawa M., Yoshimoto S., Yamaguchi J., Seki Y., Wada K., Yokohama M., Ushiki T., Tokano H., et al. Phenotypic and expression analysis of a novel spontaneous myosin VI null mutant mouse. Exp. Anim. 2010;59:57–71. doi: 10.1538/expanim.59.57. [DOI] [PubMed] [Google Scholar]
- 7.Sanggaard K.M., Kjaer K.W., Eiberg H., Nürnberg G., Nürnberg P., Hoffman K., Jensen H., Sørum C., Rendtorff N.D., Tranebjaerg L. A novel nonsense mutation in MYO6 is associated with progressive nonsyndromic hearing loss in a Danish DFNA22 family. Am. J. Med. Genet. A. 2008;146A:1017–1025. doi: 10.1002/ajmg.a.32174. [DOI] [PubMed] [Google Scholar]
- 8.Oka S.I., Day T.F., Nishio S.Y., Moteki H., Miyagawa M., Morita S., Izumi S., Ikezono T., Abe S., Nakayama J., et al. Clinical Characteristics and In Vitro Analysis of MYO6 Variants Causing Late-Onset Progressive Hearing Loss. Genes (Basel) 2020;11:273. doi: 10.3390/genes11030273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J., Shen J., Guo L., Cheng C., Chai R., Shu Y., Li H. A humanized mouse model, demonstrating progressive hearing loss caused by MYO6 p.C442Y, is inherited in a semi-dominant pattern. Hear. Res. 2019;379:79–88. doi: 10.1016/j.heares.2019.04.014. [DOI] [PubMed] [Google Scholar]
- 10.Leake P.A., Akil O., Lang H. Neurotrophin gene therapy to promote survival of spiral ganglion neurons after deafness. Hear. Res. 2020;394:107955. doi: 10.1016/j.heares.2020.107955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akil O., Seal R.P., Burke K., Wang C., Alemi A., During M., Edwards R.H., Lustig L.R. Restoration of hearing in the VGLUT3 knockout mouse using virally mediated gene therapy. Neuron. 2012;75:283–293. doi: 10.1016/j.neuron.2012.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang Q., Wang J., Li Q., Kim Y., Zhou B., Wang Y., Li H., Lin X. Virally mediated Kcnq1 gene replacement therapy in the immature scala media restores hearing in a mouse model of human Jervell and Lange-Nielsen deafness syndrome. EMBO Mol. Med. 2015;7:1077–1086. doi: 10.15252/emmm.201404929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chien W.W., Isgrig K., Roy S., Belyantseva I.A., Drummond M.C., May L.A., Fitzgerald T.S., Friedman T.B., Cunningham L.L. Gene Therapy Restores Hair Cell Stereocilia Morphology in Inner Ears of Deaf Whirler Mice. Mol. Ther. 2016;24:17–25. doi: 10.1038/mt.2015.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delmaghani S., Defourny J., Aghaie A., Beurg M., Dulon D., Thelen N., Perfettini I., Zelles T., Aller M., Meyer A., et al. Hypervulnerability to Sound Exposure through Impaired Adaptive Proliferation of Peroxisomes. Cell. 2015;163:894–906. doi: 10.1016/j.cell.2015.10.023. [DOI] [PubMed] [Google Scholar]
- 15.Dulon D., Papal S., Patni P., Cortese M., Vincent P.F., Tertrais M., Emptoz A., Tlili A., Bouleau Y., Michel V., et al. Clarin-1 gene transfer rescues auditory synaptopathy in model of Usher syndrome. J. Clin. Invest. 2018;128:3382–3401. doi: 10.1172/JCI94351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Emptoz A., Michel V., Lelli A., Akil O., Boutet de Monvel J., Lahlou G., Meyer A., Dupont T., Nouaille S., Ey E., et al. Local gene therapy durably restores vestibular function in a mouse model of Usher syndrome type 1G. Proc. Natl. Acad. Sci. USA. 2017;114:9695–9700. doi: 10.1073/pnas.1708894114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Isgrig K., Shteamer J.W., Belyantseva I.A., Drummond M.C., Fitzgerald T.S., Vijayakumar S., Jones S.M., Griffith A.J., Friedman T.B., Cunningham L.L., Chien W.W. Gene Therapy Restores Balance and Auditory Functions in a Mouse Model of Usher Syndrome. Mol. Ther. 2017;25:780–791. doi: 10.1016/j.ymthe.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan B., Askew C., Galvin A., Heman-Ackah S., Asai Y., Indzhykulian A.A., Jodelka F.M., Hastings M.L., Lentz J.J., Vandenberghe L.H., et al. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat. Biotechnol. 2017;35:264–272. doi: 10.1038/nbt.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taiber S., Cohen R., Yizhar-Barnea O., Sprinzak D., Holt J.R., Avraham K.B. Neonatal AAV gene therapy rescues hearing in a mouse model of SYNE4 deafness. EMBO Mol. Med. 2021;13:e13259. doi: 10.15252/emmm.202013259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minoda R., Miwa T., Ise M., Takeda H. Potential treatments for genetic hearing loss in humans: current conundrums. Gene Ther. 2015;22:603–609. doi: 10.1038/gt.2015.27. [DOI] [PubMed] [Google Scholar]
- 21.Zuris J.A., Thompson D.B., Shu Y., Guilinger J.P., Bessen J.L., Hu J.H., Maeder M.L., Joung J.K., Chen Z.Y., Liu D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015;33:73–80. doi: 10.1038/nbt.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.György B., Nist-Lund C., Pan B., Asai Y., Karavitaki K.D., Kleinstiver B.P., Garcia S.P., Zaborowski M.P., Solanes P., Spataro S., et al. Allele-specific gene editing prevents deafness in a model of dominant progressive hearing loss. Nat. Med. 2019;25:1123–1130. doi: 10.1038/s41591-019-0500-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao X., Tao Y., Lamas V., Huang M., Yeh W.H., Pan B., Hu Y.J., Hu J.H., Thompson D.B., Shu Y., et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature. 2018;553:217–221. doi: 10.1038/nature25164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeh W.H., Shubina-Oleinik O., Levy J.M., Pan B., Newby G.A., Wornow M., Burt R., Chen J.C., Holt J.R., Liu D.R. In vivo base editing restores sensory transduction and transiently improves auditory function in a mouse model of recessive deafness. Sci. Transl. Med. 2020;12:eaay9101. doi: 10.1126/scitranslmed.aay9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roux I., Hosie S., Johnson S.L., Bahloul A., Cayet N., Nouaille S., Kros C.J., Petit C., Safieddine S. Myosin VI is required for the proper maturation and function of inner hair cell ribbon synapses. Hum. Mol. Genet. 2009;18:4615–4628. doi: 10.1093/hmg/ddp429. [DOI] [PubMed] [Google Scholar]
- 26.Deol M.S., Green M.C. Snell’s waltzer, a new mutation affecting behaviour and the inner ear in the mouse. Genet. Res. 1966;8:339–345. doi: 10.1017/s0016672300010193. [DOI] [PubMed] [Google Scholar]
- 27.Williams L.H., Miller K.A., Dahl H.H., Manji S.S. Characterization of a novel ENU-generated myosin VI mutant mouse strain with congenital deafness and vestibular dysfunction. Hear. Res. 2013;299:53–62. doi: 10.1016/j.heares.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 28.Hu X., Wang J., Yao X., Xiao Q., Xue Y., Wang S., Shi L., Shu Y., Li H., Yang H. Screened AAV variants permit efficient transduction access to supporting cells and hair cells. Cell Discov. 2019;5:49. doi: 10.1038/s41421-019-0115-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heidrych P., Zimmermann U., Kuhn S., Franz C., Engel J., Duncker S.V., Hirt B., Pusch C.M., Ruth P., Pfister M., et al. Otoferlin interacts with myosin VI: implications for maintenance of the basolateral synaptic structure of the inner hair cell. Hum. Mol. Genet. 2009;18:2779–2790. doi: 10.1093/hmg/ddp213. [DOI] [PubMed] [Google Scholar]
- 31.Akil O., Dyka F., Calvet C., Emptoz A., Lahlou G., Nouaille S., Boutet de Monvel J., Hardelin J.P., Hauswirth W.W., Avan P., et al. Dual AAV-mediated gene therapy restores hearing in a DFNB9 mouse model. Proc. Natl. Acad. Sci. USA. 2019;116:4496–4501. doi: 10.1073/pnas.1817537116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Askew C., Rochat C., Pan B., Asai Y., Ahmed H., Child E., Schneider B.L., Aebischer P., Holt J.R. Tmc gene therapy restores auditory function in deaf mice. Sci. Transl. Med. 2015;7:295ra108. doi: 10.1126/scitranslmed.aab1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lentz J.J., Jodelka F.M., Hinrich A.J., McCaffrey K.E., Farris H.E., Spalitta M.J., Bazan N.G., Duelli D.M., Rigo F., Hastings M.L. Rescue of hearing and vestibular function by antisense oligonucleotides in a mouse model of human deafness. Nat. Med. 2013;19:345–350. doi: 10.1038/nm.3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lentz J.J., Pan B., Ponnath A., Tran C.M., Nist-Lund C., Galvin A., Goldberg H., Robillard K.N., Jodelka F.M., Farris H.E., et al. Direct Delivery of Antisense Oligonucleotides to the Middle and Inner Ear Improves Hearing and Balance in Usher Mice. Mol. Ther. 2020;28:2662–2676. doi: 10.1016/j.ymthe.2020.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nist-Lund C.A., Pan B., Patterson A., Asai Y., Chen T., Zhou W., Zhu H., Romero S., Resnik J., Polley D.B., et al. Improved TMC1 gene therapy restores hearing and balance in mice with genetic inner ear disorders. Nat. Commun. 2019;10:236. doi: 10.1038/s41467-018-08264-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shibata S.B., Ranum P.T., Moteki H., Pan B., Goodwin A.T., Goodman S.S., Abbas P.J., Holt J.R., Smith R.J.H. RNA Interference Prevents Autosomal-Dominant Hearing Loss. Am. J. Hum. Genet. 2016;98:1101–1113. doi: 10.1016/j.ajhg.2016.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoshimura H., Shibata S.B., Ranum P.T., Moteki H., Smith R.J.H. Targeted Allele Suppression Prevents Progressive Hearing Loss in the Mature Murine Model of Human TMC1 Deafness. Mol. Ther. 2019;27:681–690. doi: 10.1016/j.ymthe.2018.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rudman J.R., Mei C., Bressler S.E., Blanton S.H., Liu X.Z. Precision medicine in hearing loss. J. Genet. Genomics. 2018;45:99–109. doi: 10.1016/j.jgg.2018.02.004. [DOI] [PubMed] [Google Scholar]
- 39.Landegger L.D., Pan B., Askew C., Wassmer S.J., Gluck S.D., Galvin A., Taylor R., Forge A., Stankovic K.M., Holt J.R., Vandenberghe L.H. A synthetic AAV vector enables safe and efficient gene transfer to the mammalian inner ear. Nat. Biotechnol. 2017;35:280–284. doi: 10.1038/nbt.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.György B., Meijer E.J., Ivanchenko M.V., Tenneson K., Emond F., Hanlon K.S., Indzhykulian A.A., Volak A., Karavitaki K.D., Tamvakologos P.I., et al. Gene Transfer with AAV9-PHP.B Rescues Hearing in a Mouse Model of Usher Syndrome 3A and Transduces Hair Cells in a Non-human Primate. Mol. Ther. Methods Clin. Dev. 2018;13:1–13. doi: 10.1016/j.omtm.2018.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shu Y., Tao Y., Wang Z., Tang Y., Li H., Dai P., Gao G., Chen Z.Y. Identification of Adeno-Associated Viral Vectors That Target Neonatal and Adult Mammalian Inner Ear Cell Subtypes. Hum. Gene Ther. 2016;27:687–699. doi: 10.1089/hum.2016.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Isgrig K., McDougald D.S., Zhu J., Wang H.J., Bennett J., Chien W.W. AAV2.7m8 is a powerful viral vector for inner ear gene therapy. Nat. Commun. 2019;10:427. doi: 10.1038/s41467-018-08243-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang D., Zhang C., Wang B., Li B., Wang Q., Liu D., Wang H., Zhou Y., Shi L., Lan F., Wang Y. Optimized CRISPR guide RNA design for two high-fidelity Cas9 variants by deep learning. Nat. Commun. 2019;10:4284. doi: 10.1038/s41467-019-12281-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kho S.T., Pettis R.M., Mhatre A.N., Lalwani A.K. Safety of adeno-associated virus as cochlear gene transfer vector: analysis of distant spread beyond injected cochleae. Mol. Ther. 2000;2:368–373. doi: 10.1006/mthe.2000.0129. [DOI] [PubMed] [Google Scholar]
- 45.Zuo E., Sun Y., Wei W., Yuan T., Ying W., Sun H., Yuan L., Steinmetz L.M., Li Y., Yang H. GOTI, a method to identify genome-wide off-target effects of genome editing in mouse embryos. Nat. Protoc. 2020;15:3009–3029. doi: 10.1038/s41596-020-0361-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zuo E., Sun Y., Wei W., Yuan T., Ying W., Sun H., Yuan L., Steinmetz L.M., Li Y., Yang H. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science. 2019;364:289–292. doi: 10.1126/science.aav9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ehret G. Development of absolute auditory thresholds in the house mouse (Mus musculus) J. Am. Audiol. Soc. 1976;1:179–184. [PubMed] [Google Scholar]
- 48.Hall J.W., 3rd Development of the ear and hearing. J. Perinatol. 2000;20:S12–S20. doi: 10.1038/sj.jp.7200439. [DOI] [PubMed] [Google Scholar]
- 49.Czechanski A., Byers C., Greenstein I., Schrode N., Donahue L.R., Hadjantonakis A.K., Reinholdt L.G. Derivation and characterization of mouse embryonic stem cells from permissive and nonpermissive strains. Nat. Protoc. 2014;9:559–574. doi: 10.1038/nprot.2014.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y., Sun Y., Chang Q., Ahmad S., Zhou B., Kim Y., Li H., Lin X. Early postnatal virus inoculation into the scala media achieved extensive expression of exogenous green fluorescent protein in the inner ear and preserved auditory brainstem response thresholds. J. Gene Med. 2013;15:123–133. doi: 10.1002/jgm.2701. [DOI] [PubMed] [Google Scholar]
- 51.Yu Q., Wang Y., Chang Q., Wang J., Gong S., Li H., Lin X. Virally expressed connexin26 restores gap junction function in the cochlea of conditional Gjb2 knockout mice. Gene Ther. 2014;21:71–80. doi: 10.1038/gt.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gu X., Wang D., Xu Z., Wang J., Guo L., Chai R., Li G., Shu Y., Li H. Prevention of acquired sensorineural hearing loss in mice by in vivo Htra2 gene editing. Genome Biol. 2021;22:86. doi: 10.1186/s13059-021-02311-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bramhall N.F., Niemczak C.E., Kampel S.D., Billings C.J., McMillan G.P. Evoked Potentials Reveal Noise Exposure-Related Central Auditory Changes Despite Normal Audiograms. Am. J. Audiol. 2020;29:152–164. doi: 10.1044/2019_AJA-19-00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu H., Lu J., Wang Z., Song L., Wang X., Li G.L., Wu H. Functional alteration of ribbon synapses in inner hair cells by noise exposure causing hidden hearing loss. Neurosci. Lett. 2019;707:134268. doi: 10.1016/j.neulet.2019.05.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.