

Abstract
Adrenoleukodystrophy (ALD) is caused by various pathogenic mutations in the X-linked ABCD1 gene, which lead to metabolically abnormal accumulations of very long-chain fatty acids in many organs. However, curative treatment of ALD has not yet been achieved. To treat ALD, we applied two different gene-editing strategies, base editing and homology-independent targeted integration (HITI), in ALD patient-derived fibroblasts. Next, we performed in vivo HITI-mediated gene editing using AAV9 vectors delivered via intravenous administration in the ALD model mice. We found that the ABCD1 mRNA level was significantly increased in HITI-treated mice, and the plasma levels of C24:0-LysoPC (lysophosphatidylcholine) and C26:0-LysoPC, sensitive diagnostic markers for ALD, were significantly reduced. These results suggest that HITI-mediated mutant gene rescue could be a promising therapeutic strategy for human ALD treatment.
Keywords: adrenoleukodystrophy, ABCD1, very long-chain fatty acid, homology-independent targeted integration, base editing, CRISPR, genome editing, gene therapy
Graphical abstract
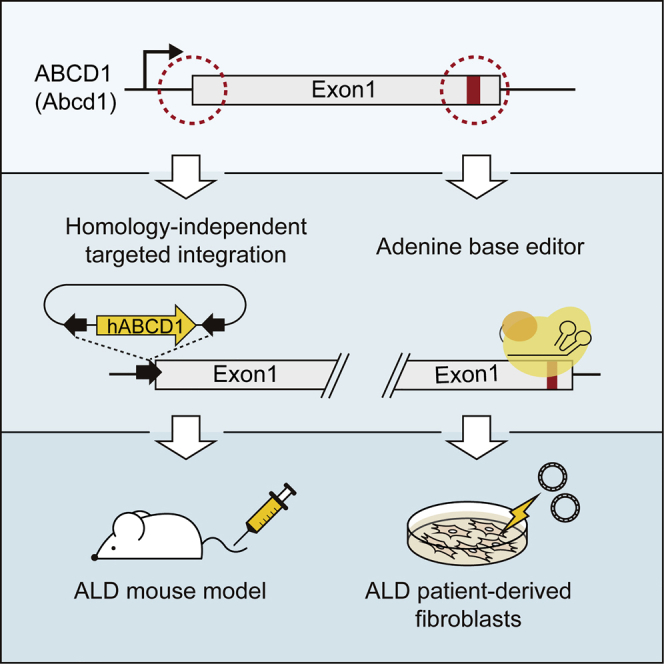
Hong et al. demonstrate adenine base editing and a homology-independent targeted integration (HITI) method for adrenoleukodystrophy (ALD) treatment in ALD patient-derived fibroblasts. They ultimately show in vivo HITI-mediated gene editing using a dual AAV9 system in ALD model mice, resulting in reducing the level of very long-chain fatty acids.
Introduction
X-linked adrenoleukodystrophy (ALD) is a genetic disease that results from a deficiency in the ALD-associated protein (ALDP), which is encoded by the ATP-binding cassette sub-family D member 1 (ABCD1) gene that is localized to chromosome Xq28 in humans.1 ALDP is a peroxisomal transmembrane protein that is thought to play a role in transporting very long-chain fatty acids (VLCFAs) into peroxisomes. ALDP defects cause impaired peroxisomal β-oxidation of saturated straight-chain VLCFAs (≥C22:0); these VLCFAs accumulate not only in plasma, but also in most tissues of the human body, including the brain, spinal cord, and adrenal gland. Thus, excessive VLCFA accumulation is a diagnostic hallmark of ALD.2, 3, 4 This metabolically abnormal accumulation damages myelin nerve sheaths and the adrenal cortex, ultimately resulting in seizures, hyperactivity, loss of fine motor control, and adrenal insufficiency.5, 6, 7
The clinical manifestations of ALD are highly variable. ALD is considered to be a disease with two distinct phenotypes: either rapidly progressive cerebral demyelination, known as cerebral ALD, or slowly progressive myeloneuropathy, known as adrenomyeloneuropathy (AMN).8 The clinical characteristics also vary by sex. Male patients with ALD usually exhibit adrenal insufficiency in childhood and develop symptoms of myeloneuropathy in adulthood,9 although progressive cerebral ALD usually develops in childhood.10 Most women with ALD develop mild symptoms of myeloneuropathy by the age of 60 years.11 Because the probability of experiencing these symptoms increases with age, ALD is a progressive metabolic disease.11
However, no curative treatment for ALD is currently established, although many approaches have been attempted. Although dietary therapy with Lorenzo’s oil, which restricts the intake of saturated VLCFA, has been attempted to normalize VLCFA concentrations in the body,12,13 its effectiveness for treating disease manifestations is still controversial. Allogeneic bone marrow transplantation or hematopoietic stem cell transplantation (HSCT) for childhood-onset cerebral ALD patients has been shown to stop demyelination at an early stage,14,15 but the usefulness of HSCT is limited to an early stage in childhood, and the prognosis is poor for advanced cerebral ALD. Furthermore, human leukocyte antigen-matched donors are not always available. As an effective alternative to allogeneic HSCT, autologous transplantation of genetically engineered CD34+ hematopoietic stem cells, which have been transduced with ABCD1 DNA using a lentiviral vector (Lenti-D), has been performed in cerebral ALD patients.16, 17, 18, 19 However, lentiviral gene delivery is associated with potential problems, such as insertional mutagenesis resulting from viral vector integration into the host genome.20,21
Alternatively, in vivo gene transfer strategies, leading to the expression of normal ABCD1, have been applied.22 A previous study reported that intravenous or intracerebroventricular injection of adeno-associated virus 9 (AAV9)-borne human ABCD1 into an ALD mouse model reduced VLCFA levels in the mouse brain and spinal cord.23 Despite the effectiveness of this method, the exogenous transferred gene would be constitutively expressed at levels that differ from that of the endogenous gene regardless of the cell-cycle stage, which might mean that the approach is not ideal for treating genetic disorders, including ALD. Rather, targeted gene correction of the endogenous ABCD1 gene is expected to be a more reliable and fundamental approach for ALD treatment.
Currently, precise gene editing or correction is realized by using the CRISPR-Cas system, derived from an adaptive immune response of bacteria and archaea.24 Initially, CRISPR-Cas9 nucleases were harnessed to induce DNA double-strand breaks (DSBs) in a single-guide RNA (sgRNA)-specific manner. The resulting DSB is repaired by the cells’ repair pathways: nonhomologous end joining (NHEJ), typically resulting in gene knockout (KO), or homology-directed repair (HDR) in the presence of donor DNAs, provoking gene knockin. Although HDR-mediated gene correction is a practical strategy, its editing efficacy is generally low, and it is of limited use in non-dividing cells because HDR is active only in the late S and G2 phases of the cell cycle. An alternative, NHEJ-mediated knockin method, also called homology-independent targeted integration (HITI),25,26 can be used for gene correction in vivo. Basically, HITI-mediated gene correction has no limitation with respect to cell type; previous studies showed that HITI led to robust and effective DNA knockin in both dividing and non-dividing cells, including neurons, in vivo.27, 28, 29, 30, 31 In addition to CRISPR nuclease-based gene correction tools, cytosine base editors (CBEs) and adenine base editors (ABEs) were subsequently developed to convert cytosine to thymine (C-to-T) or adenine to guanine (A-to-G), respectively.32, 33, 34 Both CBEs and ABEs enable highly efficient, direct base substitutions without generating DNA cleavage or a requirement for template donor DNA.
Here, we demonstrated both HITI and base editing methods for ABCD1 rescue in ALD patient-derived fibroblasts. Furthermore, we applied the HITI method to an ALD mouse model and achieved targeted integration of human ABCD1 near the 5′ untranslated region (UTR) of the Abcd1 gene in vivo by the delivery of two AAV vectors, one encoding CRISPR components and the other containing a HITI donor. Our results suggest that this method would be a suitable and reliable approach for treating human ALD.
Results
In silico investigation of applicable targets for base editing approaches in ALD-associated databases
We first inspected all targetable ALD-associated variations registered in three different databases, which include the genome aggregation database (gnomAD),35 the ALD Mutation Database, and the Clinvar database, to investigate in silico how many genetic variations in ALD could potentially be treated by base editing technologies. Of the 1,659 mutations in the databases, missense mutations account for 42.74%, nonsense for 5.85%, small insertions and deletions (indels) for 18.32%, and others (synonymous and noncoding) for 33.09% (Figure 1A). Because base editing tools cannot correct indel patterns, we then calculated the frequency of missense and nonsense mutations that are targetable by CBE and ABE. We found that, among the missense mutations, 9.9% are targetable by conventional CBEs that recognize a canonical protospacer adjacent motif (PAM), 5′-NGG-3′, and 17.3% are targetable by engineered CBEs recognizing an expanded PAM, 5′-NG-3′ (Figure 1B), whereas 22.0% and 36.8% are targetable by ABEs that recognize NGG and NG PAMs, respectively. Among the nonsense mutations, 24.7% and 42.3% are respectively targetable by ABEs that recognize NGG and NG PAMs, whereas approaches based on ABE-mediated read-through of premature termination codons (previously named CRISPR-Pass36) could respectively target 80.4% and 100% when ABEs that recognize NGG and NG PAMs are used.
Figure 1.

Database of ABCD1 mutations in ALD patients
(A) The distribution of mutation types. (B) The percentages of missense and nonsense mutations that are targetable using CBE, ABE, or CRISPR-Pass.
Thus, although base editing tools are a promising therapeutic approach, they can be used to correct less than 50% of ALD-associated variants. In contrast, the HITI method enables targeted integration of full ABCD1 coding sequences (CDSs) within the 5′ UTR, regardless of the mutation pattern or length, suggesting that HITI would be an effective treatment approach for most ALD-associated variants.
Application of base editing and HITI methods to patient-derived fibroblasts
We sought to demonstrate the use of a base editing strategy in fibroblasts derived from a male patient with ALD who harbored a point mutation in the ABCD1 gene (c.796G>A, p.Gly266Arg) (Figure 2A). For correcting the disease-causing mutation, adenine to guanine, we used ABEmax, an optimized ABE variant. We designed a reliable sgRNA (named ABE-sg1) for which the target A was positioned at the fifth nucleotide in the editing activity window (which spans the fourth to eighth nucleotides, counting from the 5′ end of sgRNA) (Figure S1A). Then, we transfected the patient-derived fibroblasts with two plasmids, one expressing ABE and the other expressing the sgRNA. Genomic DNA from a bulk population of fibroblasts was subjected to high-throughput sequencing to assess the base editing efficiency. The results showed that in about 14% of the sequences, the target A was precisely converted to G (Figure 2B), indicating that ABE can be an effective tool for ABCD1 gene correction.
Figure 2.

Mutant ABCD1 gene rescue in patient-derived fibroblasts using ABE and HITI
(A) Schematic of WT and patient sequences. The disease-causing mutation is marked in red. (B) A sequence table showing the percentages of nucleotides at each position in the target sequence in patient-derived fibroblasts transfected with ABE. The PAM sequence is shown in green. (C) Scheme for integration of the normal hABCD1 cDNA near the endogenous 5′ UTR of ABCD1 by HITI. The blue arrows indicate the primers used to amplify the junctions between endogenous ABCD1 and the HITI donor after integration, and the green arrows indicate the primers used for RT-PCR. (D) Results from PCR analysis in which the integration junctions between endogenous ABCD1 and the HITI donor were amplified. (E) RT-PCR to confirm transcription from the HITI donor after integration. Expected band size of RT-PCR product for GAPDH is 536 bp. (+) RT, reverse transcriptase-positive sample; (−) RT, reverse transcriptase-negative sample.
Next, to demonstrate a HITI strategy in the patient-derived fibroblasts, we integrated the full ABCD1 complementary DNA (cDNA) containing 5′ UTR, full ABCD1 CDS, and 3′ UTR without a promoter into the 5′ UTR of ABCD1 to express normal and functional ABCD1 protein (Figure 2C). We designed four sgRNAs targeting sites near the 5′ UTR and selected the one (named HITI-sg3) that was associated with the highest editing activity (76.2%) (Figure S1B). We generated a HITI plasmid that contained the ABCD1 cDNA flanked by HITI-sg3 target sequences, so that ABCD1 could be integrated into the endogenous target site after it was cleaved by SpCas9 (Cas9 from Streptococcus pyogenes). The HITI-sg3 target sequences in the HITI plasmid were oriented in the opposite direction compared with that in the endogenous target sequence to prevent a recurrence of Cas9-mediated DNA cleavage after the donor fragment was inserted in the intended direction. In addition, we included sequences encoding the red fluorescent protein (mCherry) so that it would be fused to the ABCD1 protein via T2A to mark cells that contained the desired integration event. Then, we co-transfected SpCas9- and sgRNA-encoding plasmids along with the HITI plasmid into the fibroblasts.
To examine whether the full ABCD1 sequence was correctly integrated into the target site, we performed PCR with two pairs of primers that spanned the junctions between the endogenous sequences and the integrated donor DNA. The primers can amplify the target DNA only when the donor DNA is inserted, whereas they cannot amplify normal endogenous sequences. The PCR data showed the successful insertion of the ABCD1 sequence at the desired site (Figure 2D), and it was supported by Sanger sequencing (Figure S2). We further confirmed the expression of the inserted HITI donor by detecting the mRNAs using reverse-transcription PCR (RT-PCR) (Figure 2E). Because the HITI donor did not have a promoter, it was supposed that the integrated ABCD1 cDNA would be expressed by the endogenous ABCD1 promoter. However, we could not detect the red fluorescent mCherry signal, perhaps because of the low ABCD1 expression level in fibroblasts; ABCD1 is known to be abundantly expressed in brain and liver.37,38
In vivo mutant gene rescue using the HITI method in an ALD mouse model
We ultimately demonstrated in vivo gene editing in a mouse model of ALD. To this end, we purchased Abcd1 KO mice from The Jackson Laboratory (stock number: 003716),39 in which a large part of exon 1, including a translational initiation codon, was eliminated, and about 150 bp of 5′ UTR was left. This strain shows few symptoms; no early neurological deficits or pathological changes in behavior have been detected in mice up to 6 months in age. However, VLCFA levels have been observed to increase in various tissues and plasma within 3 months; thus, the onset of ALD can be determined by measurement of C26:0-LysoPC (lysophosphatidylcholine) or C26:0.39, 40, 41 Because the Abcd1 KO mice have a large exon 1 deletion, we used the HITI method as our approach to treating ALD (Figure 3A). For in vivo delivery of the HITI components, we designed two AAV9 vectors: one vector encoded CRISPR components (i.e., Cas9 and sgRNA), and the other contained a HITI donor (Figure 3B). We chose AAV9 because its intravenous delivery could lead to widespread transduction, to brain and spinal cord as well as liver.23 In addition, we used SaCas9 (Cas9 from Staphylococcus aureus), which recognizes a 5′-NNGRRT-3′ PAM, because of its relatively small size, an advantage when generating the AAV9 construct. We tested two sgRNAs targeted to sites near the 5′ UTR of Abcd1 by transfecting the sgRNA-encoding plasmids into mouse NIH 3T3 cells and selected one (named HITI-Sa-sg2) that was associated with higher editing activity (Figures S3A and S3B). Next, we tested integration of the HITI donor in NIH 3T3 cells and confirmed that this event occurred using PCR primers spanning the junctions between endogenous and donor sequences (Figure S3C).
Figure 3.

Mutant Abcd1 gene rescue in the ALD mouse model using HITI
(A) Scheme for HITI of the normal hABCD1 cDNA near the endogenous 5′ UTR of Abcd1 in the ALD model mouse. The start codon and most of exon 1 is replaced with a neomycin resistance gene in the mouse model. The blue arrows indicate the primers used to amplify the junction between endogenous Abcd1 and the HITI donor after integration. (B) Schematic representation of the AAV constructs used in this study. AAV9 #1 and #2 carry SaCas9-sgRNA and the HITI donor, respectively. (C) Timeline of the experiment. AAV9s were injected into 6-week-old mice, which were then euthanized at 9 weeks of age. (D) T7E1 assay to detect SaCas9 activity. Indel frequencies are indicated at the bottom of each lane. (E) Results from PCR analysis in which the integration junctions between endogenous Abcd1 and the HITI donor were amplified. Genomic DNA was extracted from tissues from two groups of three ALD model mice, transduced with either AAV9 #1+#2 or AAV9-EGFP; labels for the individual mice (1, 2, 3, 7, 8, and 9) are displayed at the top of each lane.
Following these positive results in NIH 3T3 cells, we next used intravenous injection to deliver the two AAV9 vectors (i.e., AAV9-SaCas9-sgRNA and AAV9-HITI donor, hereafter AAV9 #1+#2) into 6-week-old Abcd1 KO mice at a titer of ∼1–2 × 1012 genome copies (gc)/mouse (100 μL) (Figure 3C). As a negative control, we injected an AAV9 vector expressing enhanced green fluorescent protein (EGFP; named AAV9-EGFP) or phosphate-buffered saline (PBS). Three weeks after injection, we collected plasma from blood and isolated a number of organs, including brain, brainstem, spinal cord, adrenal gland, liver, kidney, and testis, from two groups of three mice transduced with either AAV9 #1+#2 or AAV9-EGFP. After preparing genomic DNA from each organ, we examined the SaCas9/sgRNA-mediated editing efficiency using a T7 endonuclease I (T7E1) assay and found that in the experimental group, indels occurred at frequencies of 7% and 1% in the liver and adrenal gland (Figure 3D), respectively. We also examined HITI donor integration into the target site using PCR with two sets of primer pairs (arrows in Figure 3A) and confirmed the clear integration of the HITI donor through the PCR band size (Figure 3E) and Sanger sequencing results (Figure S4) in both the liver and adrenal gland. However, we could not detect indels or HITI donor integration in other organs (Figures S5A and S5B). As a negative control, we further delivered AAV9 #2 only, which contained the HITI donor, but not SaCas9 nucleases. In this case, we could not detect HITI donor integration in tested organs, including kidney and liver (Figure S5C).
We further performed quantitative real-time PCR to examine SaCas9 and ABCD1 mRNA expression in the brain, brainstem, spinal cord, adrenal gland, liver, kidney, and testis not only in the AAV9 #1+#2-injected group but also in control groups, including the PBS group, the AAV9-EGFP group, and the AAV9 #2-injected group. The SaCas9 expression levels were significantly increased in the AAV9 #1+#2-injected group, compared with the PBS group, the AAV9-EGFP group, or the AAV9 #2-injected group (Figure 4A), indicating that AAV9 vectors were delivered to most organs through intravenous injection as we expected. In addition, the ABCD1 expression levels were also significantly increased in the AAV9 #1+#2-injected group compared with the PBS group, the AAV9-EGFP group, or the AAV9 #2-injected group (Figure 4A). Moreover, we examined whether hABCD1 protein was expressed from the integrated donor in various organs, including brain, brainstem, spinal cord, liver, kidney, testis, and adrenal gland, using immunohistochemistry (IHC). Resultantly, we observed that cells were stained with hABCD1 in most tested regions (e.g., corpus callosum of the brain, brainstem, spinal cord, liver, kidney, testis, and adrenal cortex) in the AAV #1+#2-injected group, but not in the AAV9-GFP- or the AAV9 #2-injected groups. In particular, the hABCD1+ cells were more widely distributed in the liver and adrenal cortex of the AAV #1+#2-treated mice (Figure S6). These results strongly suggest that the hABCD1 cDNA was precisely integrated into the target site via HITI.
Figure 4.

The HITI method of mutant gene rescue induces hABCD1 gene expression and reduces VLCFA levels in Abcd1 KO mice
(A) SaCas9 and hABCD1 mRNA levels were significantly increased in organs from AAV9 #1+#2-treated mice (n = 3 for all organs) compared with organs from PBS- (n = 3 for all organs), AAV9-EGFP- (n = 3 for all organs), or AAV9 #2-treated mice (n = 3 for brain, brainstem, spinal cord, liver, testis, and kidney; n = 2 for adrenal gland). Error bars represent the mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, by one-way ANOVA with Bonferroni comparison. (B) The amounts of C24:0-LysoPC were significantly reduced in AAV9 #1+#2-treated mice compared with levels in WT mice, and the amounts of C26:0-LysoPC were significantly reduced in plasma from AAV9 #1+#2-treated mice compared with plasma from either WT or AAV9-EGFP-treated mice (n = 3 per group). Error bars represent the mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, by one-way ANOVA with Bonferroni comparison.
Therapeutic effect of in vivo HITI-mediated gene editing in the ALD mouse model
Finally, to test whether our HITI approach had a therapeutic effect, we compared the VLCFA levels in plasma from AAV9 #1+#2-injected mice with that in AAV9-EGFP-injected mice and normal wild-type (WT) mice, by analyzing the C24:0- and C26:0-LysoPC levels. The AAV9-EGFP Abcd1 KO group (C24:0-LysoPC, 90.30 ± 4.22; C26:0-LysoPC, 12.37 ± 1.10; n = 3) exhibited significantly higher C24:0-LysoPC and C26:0-LysoPC levels than the WT group (C24:0-LysoPC, 48.21 ± 4.21; C26:0-LysoPC, 2.26 ± 0.29; n = 3), an expected characteristic of Abcd1 KO mice. However, it is notable that AAV9 #1+#2-injected mice exhibited plasma C26:0-LysoPC levels that were significantly reduced, by 2.5-fold (4.99 ± 0.83; n = 3), compared with the AAV9-EGFP group (Figure 4B), implying that the in vivo gene editing had a therapeutic effect. Indeed, C26:0-LysoPC levels have diagnostic significance as a biochemical marker that reflects VLCFA levels in both ALD patients40,42,43 and ALD transgenic mice.40 These data indicate that a targeted integration of the ABCD1 cDNA at the Abcd1 locus can reduce VLCFA levels and ameliorate the effects of the Abcd1 deletion in ALD mice.
Discussion
In this study, we initially demonstrated both base editing and HITI-mediated mutant gene rescue strategies in ALD patient-derived fibroblasts. Although both strategies effectively rescue the ALD-associated point mutation, we found that HITI would be a more general method because it is effective regardless of the mutation type or locus. Despite the effectiveness of the HITI method, it was not easy to quantify the precise integration rates from bulk cells due to the large size of the HITI donor. Long-read sequencing technologies, such as nanopore sequencing44, 45, 46 or single-molecule real-time (SMRT) sequencing,47,48 will be necessary to address it.
Next, we applied the HITI method in vivo through the intravenous injection of two AAV9 vectors into ALD model mice. In contrast with the exogenous transferred gene that would be constitutively overexpressed regardless of cell-cycle stages, the integrated HITI donor genes (i.e., hABCD1) should be fully controlled by the endogenous Abcd1 promoter. Consequently, we observed clear integration of the HITI donor in the liver and adrenal gland, but not in other organs, at least under our experimental conditions. However, we observed hABCD1 mRNA expression in various organs, including brain, brainstem, spinal cord, adrenal gland, liver, kidney, and testis. For each cell of the HITI-treated mice, the number of integrated hABCD1 could be two in maximum, but the number of mRNAs transcribed from the integrated hABCD1 could be enriched by hundreds or thousands of times, which might be the reason why we detected the hABCD1 mRNA expression substantially compared with the hABCD1 integration. Additionally, we performed IHC in various organs to confirm the expression of hABCD1 protein from the integrated donor and found that some cells expressed hABCD1 proteins. As a result, we ultimately found that the plasma C26:0-LysoPC levels were significantly reduced in the HITI-treated group compared with the negative control group, indicating that our HITI strategy had a therapeutic effect. To our knowledge, this represents the first example of treating ALD via targeted integration of the normal gene. Although the exact amount of reduction in VLCFAs has not been known to be sufficient to ameliorate the disease progression, we showed a similar efficacy (∼2- to 3-fold reduction of VLCFAs in the Abcd1 KO mice), compared with previous reports demonstrating earlier trials using, for example, Lorenzo’s oil,49 bezafibrate,50 or chemical compounds that increase cAMP (forskolin, 8-bromo cAMP, and rolipram).51 Considering the 6-fold increase of C26:0-lysoPC in Abcd1 KO mice compared with the WT mice (Figure 4B), however, we should further improve the efficacy of HITI-mediated gene rescue strategy by enhancing HITI delivery efficiency or editing activity.
To date, many AAV serotypes, including AAV9, have been shown to bypass the blood-brain barrier when delivered through intravenous injection, which results in efficient targeting to the central nervous system,52, 53, 54 although all AAV serotypes can transduce liver most effectively following intravenous administration.55,56 A previous study showed that intravenous delivery of AAV9 led to widespread transduction of the brain and spinal cord.23 Similarly, we found that AAV9-mediated delivery of dual HITI vectors resulted in that the SaCas9 and the integrated hABCD1 were widely expressed in most organs, including the brain, brainstem, spinal cord, liver, testis, kidney, and adrenal gland, suggesting a potential for ameliorating the progression of all ALD phenotypes, such as the cerebral and AMN, as well as adrenal insufficiency.
In conclusion, we demonstrated both base editing and HITI-mediated mutant gene rescue strategies in ALD patient-derived cells. In addition, using an in vivo HITI method, we observed a significant increase of human ABCD1 expression and thereby a decrease of VLCFA level in the plasma in Abcd1 KO mice via AAV9-mediated ABCD1 gene integration through intravenous injection. Our results provide insight into the recovery from mutant or absent ABCD1, and a potential therapeutic strategy for future clinical trials using the CRISPR-Cas9 system.
Materials and methods
Subjects and approval
Human fibroblast samples were obtained from participants after their written informed consent was obtained. This consent procedure and the study were approved by the Institutional Review Board (IRB) of Severance Hospital, Yonsei University Health System (IRB No. 4-2012-0028).
Analysis of ALD-associated variations from three different databases
ALD-associated variations were collected from ClinVar (accessed GRCh37_20191202), gnomAD (v.2.1.1), and the ALD Mutation Database without duplication. The information about the mutated position and the mutation pattern was collected from each database, and 60 bp of peripheral sequence was extracted from the GRCh37 genome based on the mutated position. Effects on the encoded amino acids were determined using information about the ABCD1 CDS downloaded from NCBI. The analysis was divided into two parts: gene correction using (1) a base editor and (2) CRISPR-Pass. The number of possible base-editor-mediated gene corrections was calculated as a count of the mutations that could be repaired by CBEs or ABEs (mutations located in a 3- to 9-bp base editing activity window, from the 5′ end of sgRNA). Possible CRISPR-Pass-mediated gene corrections were counted when a mutated STOP codon could be converted to another codon. The analysis was programmed using Python 3.
Construction of sgRNA-expressing plasmids
The sgRNAs were designed using Cas-designer (http://www.rgenome.net/cas-designer/).57 Two complementary oligos for each target, purchased from Macrogen (South Korea), were annealed and cloned into pRG2 (#104174; Addgene), BPK2660 (#70709; Addgene), or pX601-AAV-CMV::NLS-SaCas9-NLS-3xHA-bGHpA;U6::BsaI-sgRNA (#61591; Addgene). The oligos used for sgRNA-expressing plasmids are listed in Table S1.
Construction of the HITI donor plasmid
To construct a plasmid containing a cABCD1-T2A-mCherry sequence, we linearized the pOTB7-cABCD1 plasmid using SpCas9 protein complexed with an in-vitro-transcribed sgRNA targeting the end of the ABCD1 CDS and assembled with a T2A-mCherry-encoding fragment using Gibson assembly (pOTB7-cABCD1-T2A-mCherry). For AAV production, the pX601-AAV-CMV::NLS-SaCas9-NLS-3xHA-bGHpA;U6::BsaI-sgRNA plasmid was used as a backbone. First, both the pX601-AAV-CMV::NLS-SaCas9-NLS-3xHA-bGHpA;U6::BsaI-sgRNA plasmid and the pOTB7-ABCD1-T2A-mCherry plasmid were digested with EcoRI and XhoI. pOTB7-cABCD1-T2A-mCherry plasmid digested with EcoRI and XhoI yielded three fragments, and the fragment containing cABCD1-T2A-mCherry was extracted and ligated with the linearized backbone plasmid. Then, the resulting plasmid containing the cABCD1-T2A-mCherry sequence was digested with XhoI and ligated with the rest of the ABCD1 gene fragment obtained from the pOTB7-cABCD1-T2A-mCherry plasmid. To insert target sequences at each end of the cABCD1-T2A-mCherry sequence, we linearized the plasmid using EcoRI and NotI and ligated with annealed target oligos. Then, the plasmid was linearized again using XbaI and ligated with annealed target oligos. All restriction enzymes were purchased from New England Biolabs, and the primers and oligos used for in vitro transcription are listed in Table S1.
Cell culture
293T cells (CRL-3216; ATCC), the patient-derived fibroblasts, and NIH 3T3 cells (CRL-165; ATCC) were grown in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum and 1% antibiotics (all components were purchased from Welgene).
Transfection
To measure sgRNA activity, we transfected sgRNA-expressing and WT Cas9-encoding plasmids into cells, after which we measured indel frequencies at the target site. To evaluate sgRNAs for human HITI targeting near the 5′ UTR of ABCD1, we seeded 100,000 293T cells into a 24-well plate 1 day before transfection and then transfected with 750 ng of SpCas9-encoding plasmid (#104171; Addgene) and 250 ng of sgRNA-expressing plasmid. To evaluate the activity of sgRNAs for mouse HITI targeting near the 5′ UTR of Abcd1, we transfected 50,000 NIH 3T3 cells with 750 ng of SaCas9-encoding plasmid (#64709; Addgene) and 250 ng of sgRNA-expressing plasmid using a Neon Transfection System with the following parameters: voltage, 1,400; width, 20 ms; number, 2. After selecting the sgRNA that was associated with the highest indel frequency, cells were transfected for HITI- or ABE-mediated mutant gene rescue. For ABE-mediated gene rescue, 150,000 patient-derived fibroblasts were transfected with 450 ng of ABE-encoding plasmid (#112095; Addgene) and 125 ng of sgRNA-expressing plasmid using a Neon Transfection System (Thermo Fisher Scientific) with the following parameters: voltage, 1,650; width, 10 ms; number, 3. For HITI-mediated gene rescue in the patient-derived fibroblasts, 150,000 cells were transfected with 375 ng of Cas9-encoding plasmid, 125 ng of sgRNA-expressing plasmid, and 375 ng of the HITI donor. To test HITI-mediated gene rescue in NIH 3T3 cells, we transfected 100,000 cells with 1 μg of pX601-AAV-CMV::NLS-SaCas9-NLS-3xHA-bGHpA;U6::BsaI-sgRNA plasmid expressing SaCas9 and the sgRNA for HITI and 1 μg of the HITI donor. The transfected cells were collected 3 days after transfection.
DNA isolation
Genomic DNA was isolated from cell pellets using NucleoSpin Tissue (Macherey-Nagel) according to the manufacturer’s protocol. Otherwise, cell pellets were resuspended in Proteinase K extraction buffer (40 mM Tris-HCl [pH 8.0; Sigma], 1% Tween 20 [Sigma], 0.2 mM EDTA [Sigma], 10 mg of Proteinase K, 0.2% Nonidet P-40 [VWR Life Science]) and then incubated at 60°C for 15 min and 98°C for 5 min. Genomic DNA was isolated from mouse tissues using NucleoSpin Tissue. 100 ng of isolated genomic DNA or 2–3 μL of Proteinase K extraction solution containing genomic DNA was amplified by PCR using SUN-PCR blend (SUN GENETICS) or KOD-Multi&Epi (KME-101; TOYOBO).
High-throughput sequencing
The amplified genomic DNA regions containing target sites were purified using Expin PCR SV mini (GeneAll) according to the manufacturer’s protocol and sequenced using a MiniSeq Sequencing System (Illumina). The results were analyzed using Cas-Analyzer (http://www.rgenome.net/cas-analyzer/)58 and BE-Analyzer (http://www.rgenome.net/be-analyzer/).59 The primers used for PCR are listed in Table S1.
Confirmation of HITI donor integration
The junction regions between endogenous ABCD1 (or Abcd1) and the HITI donor were amplified using relevant primer sets. For Sanger sequencing, genomic DNAs around the junction regions were subjected to two rounds of PCR and then purified using Expin PCR SV mini (GeneAll) or Expin Gel SV mini (GeneAll). The primers used for PCR are listed in Table S1.
T7E1 assay
The amplified genomic DNA regions containing target sites were subjected to denaturation and reannealing and were digested using 5 U of T7EI (M0302L; New England Biolabs) for 20 min at 37°C. The digested DNA was analyzed after electrophoresis on a 2.5% agarose gel using ImageJ. The intensities of the digested products were normalized to avoid bias caused by differences in the lengths of the digested and undigested products. The indel frequency was calculated with the following formula:
RNA extraction from the patient-derived fibroblasts and RT-PCR
RNA was extracted from the patient-derived fibroblasts using NucleoSpin RNA Plus (Macherey-Nagel), and cDNAs were synthesized using ReverTra Ace qPCR RT Master Mix (FSK-101; TOYOBO). Then, 1 μL of cDNA was used for RT-PCR. The following programs were used: 5 min at 95°C; 35 cycles (25 cycles for Glyceraldehyde-3-Phosphate Dehydrogenase [GAPDH]) of 30 s at 95°C, 30 s at 60°C, and 30 s at 72°C; and, finally, 3 min at 72°C.
Transgenic X-ALD mouse model
Chromosome X-linked Abcd1 gene KO mice (B6.129-Abcd1tm1Kan/J, stock no. 003716; Jackson Laboratories, Bar Harbor, ME, USA) and B6.129 WT mice were used for this study. All mice were housed in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) and provided food and water ad libitum with alternating 12-h light/dark cycles, according to animal protection regulations. The experimental procedures were approved by the Yonsei University Health System Institutional Animal Care and Use Committee (YUHS-IACUC approval number, 2016-0148; 2020-0047).
Genotyping
Genotyping of mice was performed based on a protocol from Jackson Laboratories. Genomic DNA (gDNA) was extracted from a 2-mm piece of each mouse tail using the standard procedure from a prepGEM Tissue Kit (ZyGEM, New Zealand). The tail tissue was incubated with 1 μL of prepGEM, 10 μL of Buffer Gold, and 89 μL of autoclaved deionized distilled water at 75°C for 15 min and 95°C for 5 min. The following primers were used for PCR: WT forward, 5′-CAC AGC CTC TCT CCT TAA GAC C-3′; WT reverse, 5′-CTC GTT GTC TAG GCA ACT GG-3′ (length of WT product, 217 bp); Mutant forward, 5′-CAC AGC CTC TCT CCT TAA GAC C-3′; Mutant reverse, 5′-CTT CTA TCG CCT TCT TGA CG-3′ (length of mutant product, 117 bp). Electrophoresis was performed after loading 10 μL of each PCR product on a 1% agarose gel.
AAV vector production
AAV vectors, including AAV9-EGFP (2.20 × 1013 gc/mL titer), AAV9-SaCas9-sgRNA (4.36 × 1013 gc/mL titer), and AAV9-HITI donor (3.80 × 1013 gc/mL titer), were produced by Vigene Biosciences (Vigene Biosciences, Shandong, China).
Intravenous injections
Six-week-old male Abcd1 KO mice (n = 6) were placed in a restrainer (Braintree Scientific, Braintree, MA, USA). Next, the tail was warmed in 40°C water for 30 s, after which the tail was wiped with 70% ethanol pads. A 100 μL volume of PBS containing 1.0 × 1012 gc AAV9-EGFP or AAV9-SaCas9-sgRNA and AAV9-HITI donor was slowly injected into a lateral tail vein, after which the injection site was gently clamped until it had stopped bleeding. Initially, mice were injected with AAV9-EGFP vector as a vector control; in these mice, no hABCD1 expression was detected; in subsequent experiments, mice received a PBS injection as a control. After the injection, mice were returned to their cages.
Tissue preparation
Three weeks after injection, mice were anesthetized with ketamine (100 mg/kg; Huons, Gyeonggi-do, Korea) with Rompun (10 mg/kg; Bayer, Gyeonggi-do, Korea) via intraperitoneal injection and perfused transcardially with PBS. Blood and a number of organs, including the brain, brainstem, spinal cord, adrenal gland, liver, kidney, and testis, were collected and stored at −80°C until further use.
RNA extraction from ALD model mouse tissues
Total RNA was extracted from mouse liver and adrenal gland using TRIzol (Invitrogen Life Technologies, Carlsbad, CA, USA) or using an isolation kit (RNeasy Mini Kit, QIAGEN, Stockholm, Sweden) according to the manufacturer’s instructions. After DNase digestion and clean-up procedures, the RNA samples were quantified, aliquoted, and stored at −80°C until further use. RNA purity was evaluated with the A260/A280 ratio and analyzed with an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA).
Quantitative real-time PCR
cDNAs were synthesized from sample RNAs with ReverTra Ace qPCR RT Master Mix with gDNA Remover (TOYOBO, Osaka, Japan). Then, 1 μL of cDNA in a total volume of 20 μL was used in the following reactions. Quantitative real-time PCR was performed in triplicate on a LightCycler 480 (Roche Applied Science, Mannheim, Germany) using LightCycler 480 SYBR Green Master Mix (Roche Applied Science). The thermocycler conditions were as follows: amplifications were performed starting with a 5-min template preincubation step at 95°C, followed by 40 cycles at 95°C for 20 s, 62°C for 20 s, and 72°C for 15 s. Melting curve analysis began at 95°C for 5 s, followed by 1 min at 60°C. The specificity of the amplification product was confirmed by melting curve analysis, which showed a distinct single sharp peak with the expected Tm for all samples. The Gapdh gene was used as an internal control. The expression level of each gene of interest was obtained using the 2−ΔΔCt method. The following reaction-specific primers were used for quantitative real-time PCR: hABCD1, forward 5′-GTG GCG AGA AGC AGA GAA TC-3′ and reverse 5′-ACC TTG AAG CGC ATG AAC TC-3′; CRISPR-SaCas9, forward 5′-ACC AGA TCC AAG ACC AGC GAC TAC-3′ and reverse 5′-TAC CAT TCT TTG ATG TCC TTC CAG-3′; mouse Gapdh, forward 5′-CAT CAC TGC CAC CCA GAA GAC TG-3′ and reverse 5′-ATG CCA GTG AGC TTC CCG TTC AG-3′.
Lipid analysis
To assess for the presence of free VLCFAs, we measured C24:0- and C26:0-LysoPC levels in mouse plasma from untreated B6.129 WT mice at 9 weeks of age and from Abcd1 KO mice at 3 weeks after intravenous gene delivery. 50 μL of plasma and 150 μL of internal standards (D4-Lignoceric, C24:0; D4-Hexacosanoic, C26:0; CDN isotope, Quebec, Canada) and VLCFA standards (Behenic [C22:0], Legnoceric [C24:0], hexacosanoic [C26:0]; Sigma, St. Louis, MO, USA) were extracted with methanol (Merck, Germany). After the extracts were dried at room temperature for 30 min, they were centrifuged at full speed (5 min, 4°C). The supernatants were filtered and transferred to liquid chromatography (LC) vials. 5-μL samples were analyzed by high-performance liquid chromatography (Agilent 1260 series; Agilent Technologies, CA, USA) connected to an MS/MS system (5500 QTRAP; AB SCIEX, USA). For quantification, LysoPCs were measured against internal standards using standard curves for the fatty acids in question.
IHC
Mice were euthanized and perfused with 0.9% normal saline and thereafter the same procedure with 4% paraformaldehyde (PFA) in PBS. The perfusion-fixed tissues, including brain, brainstem, spinal cord, liver, kidney, testis, and adrenal gland, were cryoprotected consecutively in 6% and 30% sucrose solution and allowed to be frozen in frozen section compound (Leica, Wetzlar, Germany) using isopentane. The tissues were serially cryosectioned at 16-μm thickness using cryomicrotome (Cryostat Leica 1860; Leica Biosystems, Buccinasco, Italy). The IHC staining was performed with mouse anti-human ABCD1 antibody (1:500; Origene, Rockville, MD, USA), as well as secondary antibodies such as Alexa Fluor 594 goat anti-mouse (1:400; Invitrogen). Finally, the sections were treated with Hoechst 33258 (1:1,000; Life Technologies, Grand Island, NY, USA) for staining cell nuclei and mounted on glass slides with a fluorescent mounting medium (Vectorshield; Vector, Burlingame, CA, USA). The images of stained sections were captured using a fluorescence microscopy (Zeiss Axio Imager.M2; Zeiss, Gottingen, Germany).
Statistical analysis
Statistical analysis was performed using Statistical Package for Social Sciences (SPSS) software (version 23.0; IBM Corporation, Armonk, NY, USA). Data are expressed as the mean ± standard error of the mean (SEM). The results from quantitative real-time PCR and VLCFA quantification were compared using one-way analysis of variance (ANOVA) with a post hoc Bonferroni comparison using SPSS software (version 25.0; IBM Corporation, Armonk, NY, USA). Statistical significance was accepted when p < 0.05.
Data availability
Sequencing data have been uploaded to the Sequence Read Archive under accession number BioProject: PRJNA673750. All other data are available from the authors upon reasonable request.
Acknowledgments
We thank Prof. Hyongbum (Henry) Kim for helpful comments during this study. This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (HI16C1012 to S.B. and S.-R.C.) and the Bio & Medical Technology Development Program of the National Research Foundation (2020M3A9I4036072 to S.B.).
Author contributions
S.B. and S.-R.C. conceived this project; S.-A.H. performed most of the in vitro study; J.H.S. performed the animal study and analyzed data; S.W. contributed to the animal study and analyzed data; J.H.Y. and A.B. managed the animals; G.-H.H. performed bioinformatics analyses; J.Y., S.P., and E.S.J. contributed to data analysis; and S.-A.H., J.H.S., S.B., and S.-R.C. wrote the manuscript with the approval of all other authors.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.05.022.
Contributor Information
Sangsu Bae, Email: sangsubae@hanyang.ac.kr.
Sung-Rae Cho, Email: srcho918@yuhs.ac.
Supplemental information
References
- 1.Mosser J., Douar A.M., Sarde C.O., Kioschis P., Feil R., Moser H., Poustka A.M., Mandel J.L., Aubourg P. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature. 1993;361:726–730. doi: 10.1038/361726a0. [DOI] [PubMed] [Google Scholar]
- 2.Kemp S., Huffnagel I.C., Linthorst G.E., Wanders R.J., Engelen M. Adrenoleukodystrophy - neuroendocrine pathogenesis and redefinition of natural history. Nat. Rev. Endocrinol. 2016;12:606–615. doi: 10.1038/nrendo.2016.90. [DOI] [PubMed] [Google Scholar]
- 3.Kemp S., Wanders R. Biochemical aspects of X-linked adrenoleukodystrophy. Brain Pathol. 2010;20:831–837. doi: 10.1111/j.1750-3639.2010.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Valianpour F., Selhorst J.J., van Lint L.E., van Gennip A.H., Wanders R.J., Kemp S. Analysis of very long-chain fatty acids using electrospray ionization mass spectrometry. Mol. Genet. Metab. 2003;79:189–196. doi: 10.1016/s1096-7192(03)00098-2. [DOI] [PubMed] [Google Scholar]
- 5.Vawter-Lee M.M., Hallinan B.E., Burrow T.A., Spaeth C.G., Arthur T.M. A Novel Catastrophic Presentation of X-Linked Adrenoleukodystrophy. JIMD Rep. 2015;24:97–102. doi: 10.1007/8904_2015_446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moser H.W., Mahmood A., Raymond G.V. X-linked adrenoleukodystrophy. Nat. Clin. Pract. Neurol. 2007;3:140–151. doi: 10.1038/ncpneuro0421. [DOI] [PubMed] [Google Scholar]
- 7.Engelen M., Kemp S., de Visser M., van Geel B.M., Wanders R.J., Aubourg P., Poll-The B.T. X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J. Rare Dis. 2012;7:51. doi: 10.1186/1750-1172-7-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turk B.R., Nemeth C.L., Marx J.S., Tiffany C., Jones R., Theisen B., Kambhampati S., Ramireddy R., Singh S., Rosen M., et al. Dendrimer-N-acetyl-L-cysteine modulates monophagocytic response in adrenoleukodystrophy. Ann. Neurol. 2018;84:452–462. doi: 10.1002/ana.25303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dubey P., Raymond G.V., Moser A.B., Kharkar S., Bezman L., Moser H.W. Adrenal insufficiency in asymptomatic adrenoleukodystrophy patients identified by very long-chain fatty acid screening. J. Pediatr. 2005;146:528–532. doi: 10.1016/j.jpeds.2004.10.067. [DOI] [PubMed] [Google Scholar]
- 10.van Geel B.M., Bezman L., Loes D.J., Moser H.W., Raymond G.V. Evolution of phenotypes in adult male patients with X-linked adrenoleukodystrophy. Ann. Neurol. 2001;49:186–194. doi: 10.1002/1531-8249(20010201)49:2<186::aid-ana38>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 11.Engelen M., Barbier M., Dijkstra I.M., Schür R., de Bie R.M., Verhamme C., Dijkgraaf M.G., Aubourg P.A., Wanders R.J., van Geel B.M., et al. X-linked adrenoleukodystrophy in women: a cross-sectional cohort study. Brain. 2014;137:693–706. doi: 10.1093/brain/awt361. [DOI] [PubMed] [Google Scholar]
- 12.Moser H.W., Borel J. Dietary management of X-linked adrenoleukodystrophy. Annu. Rev. Nutr. 1995;15:379–397. doi: 10.1146/annurev.nu.15.070195.002115. [DOI] [PubMed] [Google Scholar]
- 13.Moser H.W., Moser A.B., Hollandsworth K., Brereton N.H., Raymond G.V. “Lorenzo’s oil” therapy for X-linked adrenoleukodystrophy: rationale and current assessment of efficacy. J. Mol. Neurosci. 2007;33:105–113. doi: 10.1007/s12031-007-0041-4. [DOI] [PubMed] [Google Scholar]
- 14.Shapiro E., Krivit W., Lockman L., Jambaqué I., Peters C., Cowan M., Harris R., Blanche S., Bordigoni P., Loes D., et al. Long-term effect of bone-marrow transplantation for childhood-onset cerebral X-linked adrenoleukodystrophy. Lancet. 2000;356:713–718. doi: 10.1016/S0140-6736(00)02629-5. [DOI] [PubMed] [Google Scholar]
- 15.Mahmood A., Raymond G.V., Dubey P., Peters C., Moser H.W. Survival analysis of haematopoietic cell transplantation for childhood cerebral X-linked adrenoleukodystrophy: a comparison study. Lancet Neurol. 2007;6:687–692. doi: 10.1016/S1474-4422(07)70177-1. [DOI] [PubMed] [Google Scholar]
- 16.Greenfield A.L., Hauser S.L. Nucleic Acid-Based Therapeutics Relevant to Neuroimmune Conditions. Neurotherapeutics. 2019;16:314–318. doi: 10.1007/s13311-019-00740-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohashi T. Gene therapy for lysosomal storage diseases and peroxisomal diseases. J. Hum. Genet. 2019;64:139–143. doi: 10.1038/s10038-018-0537-5. [DOI] [PubMed] [Google Scholar]
- 18.Cartier N., Hacein-Bey-Abina S., Bartholomae C.C., Veres G., Schmidt M., Kutschera I., Vidaud M., Abel U., Dal-Cortivo L., Caccavelli L., et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 19.Eichler F., Duncan C., Musolino P.L., Orchard P.J., De Oliveira S., Thrasher A.J., Armant M., Dansereau C., Lund T.C., Miller W.P., et al. Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J. Med. 2017;377:1630–1638. doi: 10.1056/NEJMoa1700554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Copelan E.A. Hematopoietic stem-cell transplantation. N. Engl. J. Med. 2006;354:1813–1826. doi: 10.1056/NEJMra052638. [DOI] [PubMed] [Google Scholar]
- 21.Yong S.B., Chung J.Y., Song Y., Kim Y.H. Recent challenges and advances in genetically-engineered cell therapy. J. Pharm. Investig. 2018;48:199–208. doi: 10.1007/s40005-017-0381-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mingozzi F., High K.A. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat. Rev. Genet. 2011;12:341–355. doi: 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- 23.Gong Y., Mu D., Prabhakar S., Moser A., Musolino P., Ren J., Breakefield X.O., Maguire C.A., Eichler F.S. Adenoassociated virus serotype 9-mediated gene therapy for x-linked adrenoleukodystrophy. Mol. Ther. 2015;23:824–834. doi: 10.1038/mt.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jang H.K., Song B., Hwang G.H., Bae S. Current trends in gene recovery mediated by the CRISPR-Cas system. Exp. Mol. Med. 2020;52:1016–1027. doi: 10.1038/s12276-020-0466-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki K., Tsunekawa Y., Hernandez-Benitez R., Wu J., Zhu J., Kim E.J., Hatanaka F., Yamamoto M., Araoka T., Li Z., et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 2016;540:144–149. doi: 10.1038/nature20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suzuki K., Izpisua Belmonte J.C. In vivo genome editing via the HITI method as a tool for gene therapy. J. Hum. Genet. 2018;63:157–164. doi: 10.1038/s10038-017-0352-4. [DOI] [PubMed] [Google Scholar]
- 27.Spence E.F., Dube S., Uezu A., Locke M., Soderblom E.J., Soderling S.H. In vivo proximity proteomics of nascent synapses reveals a novel regulator of cytoskeleton-mediated synaptic maturation. Nat. Commun. 2019;10:386. doi: 10.1038/s41467-019-08288-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uezu A., Hisey E., Kobayashi Y., Gao Y., Bradshaw T.W., Devlin P., Rodriguiz R., Tata P.R., Soderling S. Essential role for InSyn1 in dystroglycan complex integrity and cognitive behaviors in mice. eLife. 2019;8:e50712. doi: 10.7554/eLife.50712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ou L., Przybilla M.J., Ahlat O., Kim S., Overn P., Jarnes J., O’Sullivan M.G., Whitley C.B. A Highly Efficacious PS Gene Editing System Corrects Metabolic and Neurological Complications of Mucopolysaccharidosis Type I. Mol. Ther. 2020;28:1442–1454. doi: 10.1016/j.ymthe.2020.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohmori T., Nagao Y., Mizukami H., Sakata A., Muramatsu S.I., Ozawa K., Tominaga S.I., Hanazono Y., Nishimura S., Nureki O., Sakata Y. CRISPR/Cas9-mediated genome editing via postnatal administration of AAV vector cures haemophilia B mice. Sci. Rep. 2017;7:4159. doi: 10.1038/s41598-017-04625-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chou S.J., Yang P., Ban Q., Yang Y.P., Wang M.L., Chien C.S., Chen S.J., Sun N., Zhu Y., Liu H., et al. Dual Supramolecular Nanoparticle Vectors Enable CRISPR/Cas9-Mediated Knockin of Retinoschisin 1 Gene-A Potential Nonviral Therapeutic Solution for X-Linked Juvenile Retinoschisis. Adv. Sci. (Weinh.) 2020;7:1903432. doi: 10.1002/advs.201903432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeong Y.K., Song B., Bae S. Current Status and Challenges of DNA Base Editing Tools. Mol. Ther. 2020;28:1938–1952. doi: 10.1016/j.ymthe.2020.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P., et al. Genome Aggregation Database Consortium The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee C., Hyun Jo D., Hwang G.H., Yu J., Kim J.H., Park S.E., Kim J.S., Kim J.H., Bae S. CRISPR-Pass: Gene Rescue of Nonsense Mutations Using Adenine Base Editors. Mol. Ther. 2019;27:1364–1371. doi: 10.1016/j.ymthe.2019.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 38.Thul P.J., Åkesson L., Wiking M., Mahdessian D., Geladaki A., Ait Blal H., Alm T., Asplund A., Björk L., Breckels L.M., et al. A subcellular map of the human proteome. Science. 2017;356:eaal3321. doi: 10.1126/science.aal3321. [DOI] [PubMed] [Google Scholar]
- 39.Forss-Petter S., Werner H., Berger J., Lassmann H., Molzer B., Schwab M.H., Bernheimer H., Zimmermann F., Nave K.A. Targeted inactivation of the X-linked adrenoleukodystrophy gene in mice. J. Neurosci. Res. 1997;50:829–843. doi: 10.1002/(SICI)1097-4547(19971201)50:5<829::AID-JNR19>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 40.van de Beek M.C., Dijkstra I.M., van Lenthe H., Ofman R., Goldhaber-Pasillas D., Schauer N., Schackmann M., Engelen-Lee J.Y., Vaz F.M., Kulik W., et al. C26:0-Carnitine Is a New Biomarker for X-Linked Adrenoleukodystrophy in Mice and Man. PLoS ONE. 2016;11:e0154597. doi: 10.1371/journal.pone.0154597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu J.F., Lawler A.M., Watkins P.A., Powers J.M., Moser A.B., Moser H.W., Smith K.D. A mouse model for X-linked adrenoleukodystrophy. Proc. Natl. Acad. Sci. USA. 1997;94:9366–9371. doi: 10.1073/pnas.94.17.9366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu C., Iwamoto T., Igarashi J., Miyajima T., Hossain M.A., Yanagisawa H., Akiyama K., Shintaku H., Eto Y. Application of a diagnostic methodology by quantification of 26:0 lysophosphatidylcholine in dried blood spots for Japanese newborn screening of X-linked adrenoleukodystrophy. Mol. Genet. Metab. Rep. 2017;12:115–118. doi: 10.1016/j.ymgmr.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hubbard W.C., Moser A.B., Liu A.C., Jones R.O., Steinberg S.J., Lorey F., Panny S.R., Vogt R.F., Jr., Macaya D., Turgeon C.T., et al. Newborn screening for X-linked adrenoleukodystrophy (X-ALD): validation of a combined liquid chromatography-tandem mass spectrometric (LC-MS/MS) method. Mol. Genet. Metab. 2009;97:212–220. doi: 10.1016/j.ymgme.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 44.Branton D., Deamer D.W., Marziali A., Bayley H., Benner S.A., Butler T., Di Ventra M., Garaj S., Hibbs A., Huang X., et al. The potential and challenges of nanopore sequencing. Nat. Biotechnol. 2008;26:1146–1153. doi: 10.1038/nbt.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deamer D., Akeson M., Branton D. Three decades of nanopore sequencing. Nat. Biotechnol. 2016;34:518–524. doi: 10.1038/nbt.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clarke J., Wu H.C., Jayasinghe L., Patel A., Reid S., Bayley H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 2009;4:265–270. doi: 10.1038/nnano.2009.12. [DOI] [PubMed] [Google Scholar]
- 47.Ardui S., Ameur A., Vermeesch J.R., Hestand M.S. Single molecule real-time (SMRT) sequencing comes of age: applications and utilities for medical diagnostics. Nucleic Acids Res. 2018;46:2159–2168. doi: 10.1093/nar/gky066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eid J., Fehr A., Gray J., Luong K., Lyle J., Otto G., Peluso P., Rank D., Baybayan P., Bettman B., et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 49.Morita M., Honda A., Kobayashi A., Watanabe Y., Watanabe S., Kawaguchi K., Takashima S., Shimozawa N., Imanaka T. Effect of Lorenzo’s Oil on Hepatic Gene Expression and the Serum Fatty Acid Level in abcd1-Deficient Mice. JIMD Rep. 2018;38:67–74. doi: 10.1007/8904_2017_32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Engelen M., Schackmann M.J., Ofman R., Sanders R.J., Dijkstra I.M., Houten S.M., Fourcade S., Pujol A., Poll-The B.T., Wanders R.J., Kemp S. Bezafibrate lowers very long-chain fatty acids in X-linked adrenoleukodystrophy fibroblasts by inhibiting fatty acid elongation. J. Inherit. Metab. Dis. 2012;35:1137–1145. doi: 10.1007/s10545-012-9471-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pahan K., Khan M., Singh I. Therapy for X-adrenoleukodystrophy: normalization of very long chain fatty acids and inhibition of induction of cytokines by cAMP. J. Lipid Res. 1998;39:1091–1100. [PubMed] [Google Scholar]
- 52.Manfredsson F.P., Rising A.C., Mandel R.J. AAV9: a potential blood-brain barrier buster. Mol. Ther. 2009;17:403–405. doi: 10.1038/mt.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Foust K.D., Nurre E., Montgomery C.L., Hernandez A., Chan C.M., Kaspar B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gray S.J., Matagne V., Bachaboina L., Yadav S., Ojeda S.R., Samulski R.J. Preclinical differences of intravascular AAV9 delivery to neurons and glia: a comparative study of adult mice and nonhuman primates. Mol. Ther. 2011;19:1058–1069. doi: 10.1038/mt.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Naso M.F., Tomkowicz B., Perry W.L., 3rd, Strohl W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs. 2017;31:317–334. doi: 10.1007/s40259-017-0234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang D., Tai P.W.L., Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019;18:358–378. doi: 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Park J., Bae S., Kim J.S. Cas-Designer: a web-based tool for choice of CRISPR-Cas9 target sites. Bioinformatics. 2015;31:4014–4016. doi: 10.1093/bioinformatics/btv537. [DOI] [PubMed] [Google Scholar]
- 58.Park J., Lim K., Kim J.S., Bae S. Cas-analyzer: an online tool for assessing genome editing results using NGS data. Bioinformatics. 2017;33:286–288. doi: 10.1093/bioinformatics/btw561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hwang G.H., Park J., Lim K., Kim S., Yu J., Yu E., Kim S.T., Eils R., Kim J.S., Bae S. Web-based design and analysis tools for CRISPR base editing. BMC Bioinformatics. 2018;19:542. doi: 10.1186/s12859-018-2585-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data have been uploaded to the Sequence Read Archive under accession number BioProject: PRJNA673750. All other data are available from the authors upon reasonable request.