

Abstract
A couple diagnosed as carriers for lamellar ichthyosis, an autosomal recessive rare disease, encountered two pregnancy losses. Their blood samples showed the same heterozygous c.607C>T mutation in the TGM1 gene. However, we found that about 98.4% of the sperm had mutations, suggesting possible de novo germline mutation. To explore the probability of correcting this mutation, we used two different adenine base editors (ABEs) combined with related truncated single guide RNA (sgRNA) to repair the pathogenic mutation in mutant zygotes. Our results showed that the editing efficiency was 73.8% for ABEmax-NG combined with 20-bp-length sgRNA and 78.7% for Sc-ABEmax combined with 19-bp-length sgRNA. The whole-genome sequencing (WGS) and deep sequencing analysis demonstrated precise DNA editing. This study reveals the possibility of correcting the genetic mutation in embryos with the ABE system.
Keywords: correction, pathogenic mutation, TGM1 gene, adenine base editing, mutant embryos
Graphical abstract
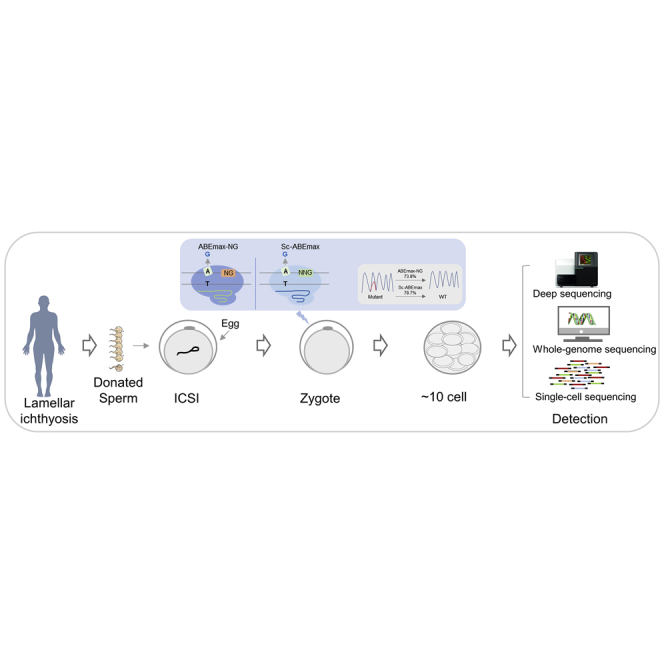
Dang et al. apply the adenine base editing system into the correction of one mutation in the TGM1 gene. Two different kinds of editors were used to correct it in human embryos. Whole-genome sequencing, deep sequencing, and RNA sequencing were used to detect the efficiency and safety.
Introduction
Lamellar ichthyosis (LI) (OMIM: 242300) is one kind of autosomal recessive congenital ichthyosis (ARCI), which is a group of rare cornification diseases with an estimated incidence of about 1 in 200,000.1 The affected newborn is usually encased with a collodion membrane at birth.2 Lamellar ichthyosis is a genetically heterogeneous disease, which has been linked to many genes, such as ABCA12, ALOXE3, and ALOX12B (https://omim.org/), and the most prevalent gene is the transglutaminase-1 gene (TGM1). Transglutaminase-1 protein functions in forming the cornified cell envelope, which is essential for the skin barrier.3 There are 170 kinds of mutations reported for the TGM1 gene, including 141 point mutations in the Human Gene Mutation Database (HGMD) (http://www.hgmd.cf.ac.uk/ac/index.php). Currently, the treatment for this disease is still a challenge. Gene therapy by providing the regular expression of TGM1 might provide curing treatment for this transglutaminase-1-deficient disease.4,5
CRISPR-Cas-related nucleases, base editors, transposases/recombinases, and prime editors provide powerful energy in genome editing.6 In particular, base editors show excellent efficiency for point mutation.7 Cytosine base editor (CBE) and adenine base editor (ABE) can efficiently convert C-to-T, or G-to-A, and A-to-G, or T-to-C without DNA double-strand break (DSB).8,9 Base editing systems have been applied in disease models,10 tumor treatment,11,12 and correcting genetic mutations.13,14 Our previous research showed the CBE could correct the pathogenic point mutation in human embryos, which would preclude the transmission of the pathogenic mutation.14 No research has been reported that corrected the pathogenic mutation using the ABE system in human embryos.
We fortuitously found a couple diagnosed with lamellar ichthyosis by symptoms and gene sequencing. Both had the heterozygous c.607C>T mutation in the TGM1 gene, which converted the glutamine to stop codon. This kind of mutation had been reported by other groups15,16 and recorded in HGMD. Previous research has shown the homozygous c.607C>T mutation would result in the disruption of TGM1 using RT-PCR and in situ hybridization.15 With the consent of the patients, we explored the probability of correcting this mutation in human embryos using the ABE system.
Results
Generation of the TGM1C607T cell model
After confirmation of the TGM1C607T mutation (Figure 1A), we explored to simulate and correct this mutation using CBE and ABE, respectively (Figure 1B). To efficiently get the homozygous specific mutant cell line, we designed two sgRNAs, which use different protospacer-adjacent motif (PAM) sequences (Figure 1C). We also chose four different CBEs: BEmax-AID,17 with wider window; BEmax-A3A-Y130F,18 harboring a broadened activity window and high efficiency; AncBE4max,19 with enhanced editing efficiency; and AncBEmax-NG,20 enabling relaxed PAM recognition. The dCas9 was used as control (Figure 1D). For mt-sg1, the BEmax-AID had about 57% editing efficiency at the C5 site, while BEmax-AID also showed the by-product editing for other C bases, which resulted in imprecise editing. The BEmax-A3A-Y130F editor had about 55% editing efficiency at the C5 site (Figure 1D), along with the lower efficiency for other C bases. The AncBE4max had about 45% editing efficiency at the C5 site and about 27% at C11. For mt-sg2, the efficiency of BEmax-A3A-Y130F was similar to mt-sg1, while for the non-target site it was higher (11% versus 25%) (Figure 1D). The editing efficiency for AncBE4max at the pathogenic point was about 65%, accompanied by increased editing at the non-target site (28% versus 36%) (Figure 1D; Figure S1). Finally, based on the editing efficiency of the target and non-target site, the BEmax-A3A-Y130F and mt-sg1 were selected to get the colony. After transfection, the GFP-positive cells were sorted one cell per well in 96-well plates. About 2 weeks later, the colonies were identified by sequencing, and the perfect cell line (100% editing in target site and 0% editing in non-target site) was stored (Figure 1E).
Figure 1.

Generation of TGM1C607T cell model
(A) The graphical representation shows the position of the pathogenic mutation. The target site is highlighted in red, and the related codon is underlined. (B) The procedure for simulating and correcting the pathogenic mutation in cell lines. For the first step, the mutant cell model is generated using the cytosine base editing (CBE) system combined with the mutation sgRNA (mt-sgRNA), and then the mutant cell model is corrected using the adenine base editing (ABE) system with the repair sgRNA (re-sgRNA). (C) sgRNAs used for generating mutant cell. The pathogenic site was at position 5 for mt-sg1 and position 6 for mt-sg2. (D) The editing efficiency of two sgRNAs combined with five different editors. Data are shown as mean ± SEM (n = 3 from three independent experiments). (E) The representative sequence chromatogram of mutant cell line harboring the homozygous mutation. The red star indicates the substituted or normal base.
Correction of the pathogenic point mutation in mutant cell line
Next, we explored the potential of correcting the pathogenic mutation using ABE. As there was no appropriate PAM sequence around the mutation, we explored two kinds of ABEs, ABEmax-NG (targeting NG PAM) and Sc-ABEmax (targeting NNG PAM).21,22 Previous studies showed truncation of sgRNA reduced off-target effects and narrowed the editing window for some target loci.23 The ABEmax-NG or Sc-ABEmax were co-transfected combined with truncated sgRNAs in the mutant cell line (Figures 2A and 2B). For ABEmax-NG editor, the editing efficiency at the pathogenic site for the four truncated sgRNAs (17 bp, 18 bp, 19 bp, and 20 bp) was 16.5%, 14.4%, 19.9%, and 20.9%, respectively (Figure 2C). Although previous research has shown the editing window for ABE7.10 was at positions 4 to 7,9 we did not find obvious editing (less than 0.01%) at the non-target site for the four sgRNAs (Figure 2C; Figure S2). For Sc-ABEmax editor, the sgRNA with the 19 bp sequence (re-Sc-sg19) showed the highest efficiency, with about 14.5%. For non-target sites, all were less than 0.01% (Figure 2D). Our results indicated the truncated sgRNAs had different editing patterns for this pathogenic mutation, and our data gave solid evidence the ABE system could efficiently correct the TGM1C607T mutation.
Figure 2.

Correction of the pathogenic mutation with truncated sgRNA
(A and B) sgRNAs used in ABEmax-NG group (A) or Sc-ABEmax group (B). The target sites are highlighted in red and with a numeric subscript. The non-target sites are highlighted in blue and with a numeric subscript. The PAM sequences are underlined. (C and D) The correction efficiency was calculated for the target and non-target sites in the ABEmax-NG group (C) or Sc-ABEmax group (D) with different truncated sgRNAs. Data are shown as mean ± SEM (n = 3 from three independent experiments).
Correction of the TGM1C607T mutation in human embryos
Next, we wondered about the effects of correcting the mutation in human embryos. Although the blood sample showed heterozygous c.607C>T, we found that 98.4% of the sperm was mutant genotype (Figure S3), which may be caused by the germline mutation.1 Using the discarded oocytes, we got the mature oocytes via an in vitro maturation (IVM) procedure (Figure 3A). These oocytes were then conducted using intracytoplasmic sperm injection (ICSI) with the patient sperm to get the mutant embryos. The embryos showed heterozygous mutants (Figure S4). Sixteen hours later, the in vitro transcribed mRNA of ABEmax-NG combined with re-NG-sg20 or the Sc-ABEmax combined with re-Sc-sg19 was microinjected into the cytoplasm.
Figure 3.

Correction of the pathogenic mutation in human embryos
(A) The graphical representation shows the procedure of correcting the pathogenic mutation in human embryos. The mutant embryos were generated using intracytoplasmic sperm injection (ICSI) with the mutant sperm. Then the mRNA of the ABE system was microinjected into the cytoplasm at the pronucleus stage. Two days later, the embryos were collected, and the genotype was analyzed using deep sequencing. (B) Morphological development of injected embryos. The embryos showed normal development after injection of the ABE system. (C) The representative sequencing chromatogram from the repaired embryos is shown. The red arrows indicate the corrected pathogenic site or normal base. The black arrows indicate non-target sites. (D) The percentage of the wild type (WT) and mutant genotypes at the pathogenic site. Seven embryos in the ABEmax-NG group and eight embryos in the Sc-ABEmax group were collected and analyzed using deep sequencing. (E) The editing efficiency was normalized and summarized for the target and non-target sites. The calculating formula is the average of (WT genotype percentage of each embryo − 50%)/0.5.
The development of the edited embryos was as normal as the control group (Figure 3B). Two days later, the edited embryos were collected and used for the following analysis. Some of the embryos showed the single wild genotype, indicating complete editing (Figure 3C; Figure S5). To further confirm the editing results, deep sequencing was performed to analyze the genotype (Figure 3D). Because the embryos contained no more than 10 cells, the minimum frequency for the mutant genotype (if any) is more than 5%, which means two embryos (embryo-5 and embryo-7) in the ABEmax-NG group and five embryos (embryo-8, embryo-9, embryo-11, embryo-13, and embryo-14) in the Sc-ABEmax group were completely corrected. We did not find obvious editing at the non-target site (Table S1). There was no significant difference between the ABEmax-NG group with 73.8% efficiency and the Sc-ABEmax with 78.7% efficiency after normalizing the editing efficiency (Figure 3E).
DNA off-target analysis by whole-genome sequencing (WGS) and deep sequencing
Previous studies have shown that ABE systems do not induce genome-wide DNA off-target effects.24 To further verify the precision of the corrected embryos, the off-target effects were investigated by WGS and amplicon-based deep sequencing. WGS was performed on Ctrl sgRNA, ABEmax-NG (embryo 7), and Sc-ABEmax (embryo 14)-injected embryos at a mean depth of 21×, 21×, and 22×, respectively (Figure 4D). A total of 1,709,219, 1,721,467, and 1,727,858 single-nucleotides polymorphisms (SNPs) were detected for the three groups (Figure 4A). There were 242,008 and 210,254 unique SNPs found in the ABEmax-NG- and Sc-ABEmax-injected embryos after filtering out the dbSNPs and control SNPs (Figure 4A). There was no significant increase in the number of SNPs and substitutions in the ABE-edited embryos compared with the control sgRNA group (Figures 4A and 4B). A total of 5,237 sites in the ABEmax-NG group were analyzed, and no off-target effects were detected (Figure 4C). For the Sc-ABEmax group, no off-target effect was found in 14,059 sites (Figure 4C). To further explore the potential off-target effects, deep sequencing was performed on all the edited embryos and 5 control embryos. The potential off-target sites were predicted with up to 5-base mismatches in the human genome,25 and the results showed no 0- or 1-base mismatches (Table S4). Seventeen top off-target sites on the list were selected and subjected to deep sequencing with an average of over 9 × 105 in depth. The results showed no obvious off-target at any of these sites (Figure 4E).
Figure 4.

DNA off-target analysis with WGS and deep sequencing on embryos
(A) Summary of SNP analysis by WGS. After filtering out naturally occurring variants in the human SNP database, 535,974 SNPs were obtained from the ABEmax-NG group and 559,247 SNPs in the Sc-ABEmax group. The number of A/T conversions is shown. dbSNPs, database of SNPs. (B) Heatmap of base substitution in control sg, ABEmax-NG, and Sc-ABEmax groups. (C) Summary of potential off-target site information. A total of 5,237 sites in ABEmax-NG-injected and 14,059 sites in the Sc-ABEmax group were analyzed. (D) A summary of WGS information. (E) Targeted deep sequencing was performed for 17 potential off-target sites. Seven embryos in the ABEmax-NG group, eight embryos in Sc-ABEmax, and five control sgRNA embryos were analyzed.
RNA off-target analysis in human embryos
Previous studies have indicated that ABE generated off-target RNA single nucleotide variations (SNVs).26,27 To further access the RNA off-target mutations in edited human embryos, RNA-seq analyses were performed. The control embryos occupied 123 SNVs, but we observed notably higher numbers of RNA SNVs in embryos injecting the ABE system, no matter with target sgRNA or with control sgRNA (Figure 5A). Nearly 90% of the RNA SNVs identified in ABE-injected embryos were mutations from A to I (Figures 5B and 5C). We also noticed the groups using target sgRNAs did not induce more off-target RNA mutations than control sgRNA groups, indicating the deaminase spontaneously induced the mutation. Recently, some improved editors have been raised, and these editors will be useful to reduce RNA off-target effects.28
Figure 5.

RNA off-target effects on corrected embryos
(A and B) RNA sequencing assay detecting RNA off-target SNVs at the human embryos. The bar graph displays the total number of SNVs (A) and the proportions of the relevant subsets (B). (C) The scatterplot displays the total SNV numbers in each injected group. Values in (A) and (B) are mean ± SEM from three biological replications as indicated by the dots.
Summary of mutations in TGM1 gene
Currently, about 170 different kinds of pathogenic mutations in the TGM1 gene were reported, of which 141 are point mutations (Figure 6A). Base editing systems show excellent efficiency and safety in simulating and correcting point mutations. For the reported mutations in the TGM1 gene, 51% were g-a and c-t mutations, and 14% were t-c and a-g mutations (Figure 6B). We analyzed the mutant sites and their PAM sequence and found 65 sites could be corrected with the ABE system, and the CBE system could be applied on 12 sites (Figure 6C). One noteworthy obstacle for the application of base editing systems is precision. For the ABE system, the percentage of point mutations that do not have the same base within ±1 bp is about 90%, and it drops to 58% within ±2 bp and 33% within ±3 bp (Figure 6C). The CBE systems occupy 35% within ±1 bp, 20% within ±2 bp, and 15% within ±3 bp, which means it is urgent to develop the new editors with narrow editing windows (Figure 6C).
Figure 6.

Summary of pathogenic mutations in the TGM1 gene
(A) Summary of the mutation type of TGM1 gene. All the mutations have been reported and recorded in the HGMD point mutations, including deletions, insertions, and indels. (B) The summaries of 12 kinds of point mutations. The g-a and c-t mutation occupy about 51%. The t-c and a-g mutation occupy about 14%. (C) Analysis of repairing effects of mutation sites by ABE/CBE. Potential repairing sgRNAs were analyzed based on mutant sites and PAM sequence. For mutations that could be corrected using ABE or CBE, the precision of correction was analyzed based on whether other bases could be modified. The three bases around the target site were considered.
Discussion
Approximately 130 million babies are born worldwide each year, of whom 7 million suffer from serious inherited genetic disorders.29 Up to November 9, 2020, there were reported 6,723 known molecular-based phenotypes associated with 4,318 different genes in the Online Mendelian Inheritance in Man. In addition, about 30–40 new genetic phenotypes are added every month. From the ClinVar database, there are approximately 844,269 unique variants, nearly half of which are point mutations.30 With the development of high-throughput sequencing, more and more genetic pathogenic mutations will be reported.31
Gene therapy provides an effective method to cure cancer and genetic diseases.32 It is reasonable and acceptable to edit the somatic mutations, and recently there have been many gene therapy clinical trials.33 For germline gene therapy (GGT), in September 2020, the International Commission on the Clinical Use of Human Germline Genome Editing reported that gene-editing methods were still far from mature enough and needed more research. There are still many kinds of diseases beyond our understanding. GGT provides more options for patients to make the best choice.34 The pressing issue is how to reinforce the existing regulations, not complete moratorium or bans.35 Gene editing based on DSBs induced by CRISPR-Cas9 may lead to off-target effects and frequent loss of the target chromosome in human embryos.36 Furthermore, when using CRISPR-Cas9 combined with the donor to corrected the mutant embryos, the edited embryos may contain multiple genotypes (mosaic) due to the predominance of non-homologous end joining.37 In this case, we cannot precisely predict the final genotype, and it is hard to guarantee safety. For base editing systems, especially the ABE system, we can precisely predict the final genotype.38 The main difference between the two methods is that the former method introduces new genotypes, while the base editing system does not. Base editing systems may be the better choice for GGT.
Lamellar ichthyosis is one kind of autosomal recessive congenital ichthyosis, and most of the pathogenic mutations are found in the TGM1 gene.24 About 170 different kinds of mutations have been recorded, and 141 of them are point mutations. We found a couple diagnosed as carriers for lamellar ichthyosis, and they encountered several pregnancy losses. Both had the heterozygous c.607C>T mutation. To our surprise, we found the sperm sample harbored 98.4% mutant genotype, which could explain the pregnancy losses. Previous research demonstrated germline mutations in TGM1 were identified in 55% of patients with ARCI.39 Base editing systems show an enormous advantage to simulate or correct point mutations.7 Our research used two different base editors to correct the c.607C>T mutation in human mutant embryos. For ABEmax-NG combined with re-sg20, seven embryos were collected, and two of them showed a completely wild genotype. The normalized editing efficiency was about 73.8%. For Sc-ABEmax combined with re-Sc-sg19, five embryos showed wild genotype among eight edited embryos, and the normalized editing efficiency was about 78.7%. The ABE8e,40 ABE8s,41 and prime editor30 may provide better editing efficiency, but more attention should be paid to the off-target editing, especially in embryos.
Recently, many engineering Cas enzymes that recognize different PAMs have been developed, especially the SpG and SpRY.6,42 The existing Cas enzymes nearly make it possible to find a PAM for any target site, and now the limitation for correcting the pathogenic mutation is the precision. Although several base editors have narrow editing windows, their efficiency and precision need enhancement.23
In summary, we achieved the first correction of a pathogenic mutation in human embryos using the ABE system. Our research provides the primary data to assess the probability of curing rare genetic diseases.
Materials and methods
Ethical statement
The study was approved by the Ethics Committee of the Third Affiliated Hospital of Guangzhou Medical University. All patients signed informed consent before sample collection. The patient with lamellar ichthyosis signed the informed consent for donating his semen samples for research. All the operations on the embryos were conducted at the Center for Reproductive Medicine.
Plasmid construction
The AncBE4max plasmid was obtained from Addgene (Addgene, 112094),19 with enhanced editing efficiency, especially under non-optimal conditions and previously moderately efficient gene sites. BEmax-A3A-Y130F was constructed using the DNA Assembly Cloning Kit (NEB, E5520S) by replacing the Apobec of AncBE4max with the A3A-Y130F fragment,18 which mediated efficient C-to-T base editing in methylated DNA regions and GpC dinucleotides. BEmax-AID was constructed by replacing the Apobec of AncBE4max using the AID fragment,17 achieving base editing with a broadened window. AncBEmax-NG was constructed by replacing the Cas9 of AncBE4max with Cas9-NG fragment.20 Sc-ABEmax plasmids were constructed using the DNA Assembly Cloning Kit by replacing the Cas9 of ABEmax (Addgene, 112095) with the ScCas9.21 AncBEmax-NG and Sc-ABEmax enable the relaxed PAM recognition, which increases targeting range.21,22 The sgRNA plasmids were constructed as previously described (Novoprotein, NR005-01B).14
Embryo culture
Immature oocytes were cultured in 25 μL G-1 PLUS (Vitrolife, 10128) medium covered with OVOIL (Vitrolife, 10029) in a 37°C tri-gas incubator (5% O2, 6% CO2, and 89% N2) until the first polar body appeared, which is a sign of oocyte maturation. Intracytoplasmic sperm injection with patient sperm was denoted as DAY0. Then, the injected fertilized eggs were cultured in 25 μL G-2-PLUS (Vitrolife, 10132) medium covered with OVOIL and cultured in a 37°C tri-gas incubator (5% O2, 6% CO2, and 89% N2) until DAY3.
Cell culture and transfection
HEK293T cells were cultured in DMEM (Hyclone, SH30243.01) supplemented with 10% fetal bovine serum (FBS) (v/v) (Gemini, 900-108) and 1% penicillin streptomycin (v/v) (Gibco, 15140122) and incubated at 37°C with 5% CO2 under humid conditions. Transfection was performed according to the manufacturer’s protocols (Thermo Fisher Scientific, 11668019). In brief, HEK293T cells were seeded on 24-well plates the day before transfection. Editor-expression vector (600 ng) was co-transfected with sgRNA-GFP plasmids (300 ng) using Lipofectamine 2000. Cells with the highest 20% of GFP signal were isolated by fluorescence-activated cell sorting 72 h later. The target fragment was amplified, and then the editing efficiency was analyzed by deep sequencing. sgRNAs used are listed in Table S2.
Flow cytometry
The sgRNA expression vectors were constructed by cloning annealed DNA oligos into BsaI-digested pGL3-U6-sgRNA-PGK-GFP (Addgene, 107721) plasmids, which expressed GFP fluorescence. Three days after transfection, HEK293T cells were separated by 0.25% trypsin, washed twice with cold PBS, and then resuspended in cell-sorting solution at a concentration of 106 cells/mL. Flow cytometry was then conducted with a BD FACSAria III Flow Cytometer (San Diego, CA, USA). Data were analyzed by FlowJo v.10 software (BD, San Diego, CA, USA). For target gene efficiency analysis, 20,000 GFP-positive cells were harvested for genomic DNA isolation. To obtain a mutation cell colony, a single cell was sorted and cultured in a 96-well plate and validated by sequencing.
In vitro transcription
In vitro transcription was performed as previously reported.43 In brief, the ABEmax-NG and Sc-ABEmax vectors were linearized by BbsI enzyme (NEB, R3539S) and the transcription was performed using the mMESSAGE mMACHINE T7 ULTRA Transcription Kit (AM1345) according to the manufacturer’s protocols. All the sgRNAs used for in vitro transcription were cloned into a pUC57-sgRNA expression vector with a T7 promoter. Then, the sgRNAs were amplified and transcribed in vitro using the MEGAshortscript T7 Transcription Kit (AM1354) and purified using the MEGAclear Transcription Clean-Up Kit (AM1908).
Intracytoplasmic sperm injection (ICSI) and microinjection
The procedure for ICSI and microinjection was the same as previously reported.14 Briefly, the immature oocytes were cultured in vitro to be mature oocytes. Then intracytoplasmic sperm injection was conducted using the patient’s sperm. After 16–18 h, the repair system containing the ABE editor with the sgRNA was microinjected into the embryos. Two days later, all the embryos were collected and used for the following process.
Genomic DNA extraction and amplification
Genomic DNA of cells was extracted using QuickExtract DNA Extraction Solution (Lucigen, QE09050) according to the manufacturer’s protocols. The genomic DNA of zygotes was amplified using the Discover-sc Single Cell Kit (Vazyme, N601-01). Primers synthesized by GenScript (Nanjing, China) are listed in Table S3.
Deep sequencing analysis
The on-target and potential off-target sites (Cas-OFFinder, http://www.rgenome.net/cas-offinder/)25 were amplified by touchdown PCR using Phanta Max Super-Fidelity DNA Polymerase (Vazyme, P505). The purified PCR products were sequenced using the Hiseq X-10 (2 × 150) platform. Primers synthesized by GenScript (Nanjing, China) used for deep sequencing are listed in Table S3.
WGS
WGS of human genomic DNA amplified from 3-day embryos was sequenced using Illumina HiSeq X Ten (2 × 150 paired-end [PE]) at Novogene Bioinformatics Institute (Beijing, China). All cleaned reads were mapped to the human reference genome (GRCh38/hg38) by BWA v.0.7.16 with default parameters. By removing duplicates with Sambamba v.0.6.7, sequence reads were realigned through Genome Analysis Toolkit (GATK v.3.7) IndelRealigner. The variants were identified by GATK HaplotypeCaller, and the criteria were: (1) sequencing depth between 1/3× and 3×, (2) variant confidence/quality by depth > 2, (3) root mean square (RMS) mapping quality (MQ) > 40.0, (4) Phred-scaled p value using Fisher’s exact test to detect strand bias < 60, (5) Z score from the Wilcoxon rank sum test of Alt versus Ref read MQs > −12.5, and (6) Z score from the Wilcoxon rank sum test of Alt versus Ref read position bias > −8. After filtering out variants in the SNP database and wild-type genome, potential off-target sites were predicted by CasOT-1.0, with up to 2-bp mismatch in seed region and 5-bp mismatch in nonseed region with NG/NNG PAM.
RNA off-target editing in embryos
For profiling of global RNA off-target editing, embryo RNA samples were sequenced using an Illumina HiSeq X Ten (2 × 150 PE) at the Novogene Bioinformatics Institute (Beijing, China), at a depth of ∼2 × 107 reads per sample. The reads were mapped to the human reference genome (GRCh38/hg38) by STAR software (v.2.5.1), annotated with GENCODE v.30. After removing duplication, GATK HaplotypeCaller (v.4.1.2) was used to identify variants and filter those under depth 2. All variants were quantified by bam readcount with parameters -q 20 -b 30. The depth for a given edit had to be at least 10×, and over 99% of the reads for these edits must support the reference allele. Only A-to-G edits in the transcribed strand were considered for downstream analysis.
Acknowledgment
This work was supported by grants from the National Natural Science Foundation of China (81170571), National Key Research and Development Project (2018YFC1003803), and Leading Talents of Guangdong Province Program (608285568031).
Author contributions
G.T., J.L., and G.L. conceived and designed the project. L.D., X. Zhou., X. Zhon., W.Y., S.H., H.L., Y.C., W.Z., L.Y., L.L., and G.L. performed the experiments. X.H., J.L., and G.T. supervised the project. L.D. and G.L. analyzed the data and wrote the paper.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.05.007.
Contributor Information
Guanglei Li, Email: ligl@shanghaitech.edu.cn.
Jianqiao Liu, Email: liujqssz@gzhmu.edu.cn.
Guoqing Tong, Email: drivftongguoqing@shutcm.edu.cn.
Supplemental information
Reference
- 1.Herman M.L., Farasat S., Steinbach P.J., Wei M.H., Toure O., Fleckman P., Blake P., Bale S.J., Toro J.R. Transglutaminase-1 gene mutations in autosomal recessive congenital ichthyosis: summary of mutations (including 23 novel) and modeling of TGase-1. Hum. Mutat. 2009;30:537–547. doi: 10.1002/humu.20952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Russell L.J., DiGiovanna J.J., Hashem N., Compton J.G., Bale S.J. Linkage of autosomal recessive lamellar ichthyosis to chromosome 14q. Am. J. Hum. Genet. 1994;55:1146–1152. [PMC free article] [PubMed] [Google Scholar]
- 3.Boeshans K.M., Mueser T.C., Ahvazi B. A three-dimensional model of the human transglutaminase 1: insights into the understanding of lamellar ichthyosis. J. Mol. Model. 2007;13:233–246. doi: 10.1007/s00894-006-0144-9. [DOI] [PubMed] [Google Scholar]
- 4.Choate K.A., Medalie D.A., Morgan J.R., Khavari P.A. Corrective gene transfer in the human skin disorder lamellar ichthyosis. Nat. Med. 1996;2:1263–1267. doi: 10.1038/nm1196-1263. [DOI] [PubMed] [Google Scholar]
- 5.Freedman J.C., Parry T.J., Zhang P., Majumdar A., Krishnan S., Regula L.K., O’Malley M., Coghlan S., Yogesha S.D., Ramasamy S., Agarwal P. Preclinical Evaluation of a Modified Herpes Simplex Virus Type 1 Vector Encoding Human TGM1 for the Treatment of Autosomal Recessive Congenital Ichthyosis. J. Invest. Dermatol. 2021;141:874–882.e6. doi: 10.1016/j.jid.2020.07.035. [DOI] [PubMed] [Google Scholar]
- 6.Anzalone A.V., Koblan L.W., Liu D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020;38:824–844. doi: 10.1038/s41587-020-0561-9. [DOI] [PubMed] [Google Scholar]
- 7.Rees H.A., Liu D.R. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018;19:770–788. doi: 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang F., Zhang W., Yang Q., Kang Y., Fan Y., Wei J., Liu Z., Dai S., Li H., Li Z., et al. Generation of a Hutchinson-Gilford progeria syndrome monkey model by base editing. Protein Cell. 2020;11:809–824. doi: 10.1007/s13238-020-00740-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X., Qian X., Wang B., Xia Y., Zheng Y., Du L., Xu D., Xing D., DePinho R.A., Lu Z. Programmable base editing of mutated TERT promoter inhibits brain tumour growth. Nat. Cell Biol. 2020;22:282–288. doi: 10.1038/s41556-020-0471-6. [DOI] [PubMed] [Google Scholar]
- 12.Crunkhorn S. Boost for base editing to treat inherited blood disorders. Nat. Rev. Drug Discov. 2020;19:307. doi: 10.1038/d41573-020-00051-x. [DOI] [PubMed] [Google Scholar]
- 13.Geurts M.H., de Poel E., Amatngalim G.D., Oka R., Meijers F.M., Kruisselbrink E., van Mourik P., Berkers G., de Winter-de Groot K.M., Michel S., et al. CRISPR-Based Adenine Editors Correct Nonsense Mutations in a Cystic Fibrosis Organoid Biobank. Cell Stem Cell. 2020;26:503–510.e7. doi: 10.1016/j.stem.2020.01.019. [DOI] [PubMed] [Google Scholar]
- 14.Zeng Y., Li J., Li G., Huang S., Yu W., Zhang Y., Chen D., Chen J., Liu J., Huang X. Correction of the Marfan Syndrome Pathogenic FBN1 Mutation by Base Editing in Human Cells and Heterozygous Embryos. Mol. Ther. 2018;26:2631–2637. doi: 10.1016/j.ymthe.2018.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao X., Lin Z., Yang H., Bu D., Tu P., Chen L., Wu H., Yang Y. New mutations in the transglutaminase 1 gene in three families with lamellar ichthyosis. Clin. Exp. Dermatol. 2009;34:904–909. doi: 10.1111/j.1365-2230.2009.03288.x. [DOI] [PubMed] [Google Scholar]
- 16.Liu J.J., Yuan Y.Y., Zhang X.Q., Li Z.M., Xu Y.S., Gao S.M., Cai J.F., Shao X.H., Lin X.H., Li B.X. Mutations of transglutaminase-1 in Chinese patients with autosomal recessive congenital ichthyosis: a case report with clinical and genetic analysis of Chinese cases reported in literature. Clin. Exp. Dermatol. 2015;40:56–62. doi: 10.1111/ced.12410. [DOI] [PubMed] [Google Scholar]
- 17.Jiang W., Feng S., Huang S., Yu W., Li G., Yang G., Liu Y., Zhang Y., Zhang L., Hou Y., et al. BE-PLUS: a new base editing tool with broadened editing window and enhanced fidelity. Cell Res. 2018;28:855–861. doi: 10.1038/s41422-018-0052-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X., Li J., Wang Y., Yang B., Wei J., Wu J., Wang R., Huang X., Chen J., Yang L. Efficient base editing in methylated regions with a human APOBEC3A-Cas9 fusion. Nat. Biotechnol. 2018;36:946–949. doi: 10.1038/nbt.4198. [DOI] [PubMed] [Google Scholar]
- 19.Koblan L.W., Doman J.L., Wilson C., Levy J.M., Tay T., Newby G.A., Maianti J.P., Raguram A., Liu D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018;36:843–846. doi: 10.1038/nbt.4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang S., Liao Z., Li X., Liu Z., Li G., Li J., Lu Z., Zhang Y., Li X., Ma X., et al. Developing ABEmax-NG with Precise Targeting and Expanded Editing Scope to Model Pathogenic Splice Site Mutations In Vivo. iScience. 2019;15:640–648. doi: 10.1016/j.isci.2019.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chatterjee P., Jakimo N., Jacobson J.M. Minimal PAM specificity of a highly similar SpCas9 ortholog. Sci. Adv. 2018;4:eaau0766. doi: 10.1126/sciadv.aau0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishimasu H., Shi X., Ishiguro S., Gao L., Hirano S., Okazaki S., Noda T., Abudayyeh O.O., Gootenberg J.S., Mori H., et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science. 2018;361:1259–1262. doi: 10.1126/science.aas9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim Y.B., Komor A.C., Levy J.M., Packer M.S., Zhao K.T., Liu D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017;35:371–376. doi: 10.1038/nbt.3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jin S., Zong Y., Gao Q., Zhu Z., Wang Y., Qin P., Liang C., Wang D., Qiu J.L., Zhang F., Gao C. Cytosine, but not adenine, base editors induce genome-wide off-target mutations in rice. Science. 2019;364:292–295. doi: 10.1126/science.aaw7166. [DOI] [PubMed] [Google Scholar]
- 25.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou C., Sun Y., Yan R., Liu Y., Zuo E., Gu C., Han L., Wei Y., Hu X., Zeng R., et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature. 2019;571:275–278. doi: 10.1038/s41586-019-1314-0. [DOI] [PubMed] [Google Scholar]
- 27.Grünewald J., Zhou R., Garcia S.P., Iyer S., Lareau C.A., Aryee M.J., Joung J.K. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature. 2019;569:433–437. doi: 10.1038/s41586-019-1161-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Y., Zhou C., Huang S., Dang L., Wei Y., He J., Zhou Y., Mao S., Tao W., Zhang Y., et al. A Cas-embedding strategy for minimizing off-target effects of DNA base editors. Nat. Commun. 2020;11:6073. doi: 10.1038/s41467-020-19690-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Church G. Compelling Reasons for Repairing Human Germlines. N. Engl. J. Med. 2017;377:1909–1911. doi: 10.1056/NEJMp1710370. [DOI] [PubMed] [Google Scholar]
- 30.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goldfeder R.L., Wall D.P., Khoury M.J., Ioannidis J.P.A., Ashley E.A. Human Genome Sequencing at the Population Scale: A Primer on High-Throughput DNA Sequencing and Analysis. Am. J. Epidemiol. 2017;186:1000–1009. doi: 10.1093/aje/kww224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dunbar C.E., High K.A., Joung J.K., Kohn D.B., Ozawa K., Sadelain M. Gene therapy comes of age. Science. 2018;359:eaan4672. doi: 10.1126/science.aan4672. [DOI] [PubMed] [Google Scholar]
- 33.Ginn S.L., Amaya A.K., Alexander I.E., Edelstein M., Abedi M.R. Gene therapy clinical trials worldwide to 2017: An update. J. Gene Med. 2018;20:e3015. doi: 10.1002/jgm.3015. [DOI] [PubMed] [Google Scholar]
- 34.Lea R.A., Niakan K.K. Human germline genome editing. Nat. Cell Biol. 2019;21:1479–1489. doi: 10.1038/s41556-019-0424-0. [DOI] [PubMed] [Google Scholar]
- 35.Wolf D.P., Mitalipov P.A., Mitalipov S.M. Principles of and strategies for germline gene therapy. Nat. Med. 2019;25:890–897. doi: 10.1038/s41591-019-0473-8. [DOI] [PubMed] [Google Scholar]
- 36.Zuccaro M.V., Xu J., Mitchell C., Marin D., Zimmerman R., Rana B., Weinstein E., King R.T., Palmerola K.L., Smith M.E., et al. Allele-Specific Chromosome Removal after Cas9 Cleavage in Human Embryos. Cell. 2020;183:1650–1664.e15. doi: 10.1016/j.cell.2020.10.025. [DOI] [PubMed] [Google Scholar]
- 37.Ma H., Marti-Gutierrez N., Park S.W., Wu J., Lee Y., Suzuki K., Koski A., Ji D., Hayama T., Ahmed R., et al. Correction of a pathogenic gene mutation in human embryos. Nature. 2017;548:413–419. doi: 10.1038/nature23305. [DOI] [PubMed] [Google Scholar]
- 38.Li G., Liu X., Huang S., Zeng Y., Yang G., Lu Z., Zhang Y., Ma X., Wang L., Huang X., Liu J. Efficient Generation of Pathogenic A-to-G Mutations in Human Tripronuclear Embryos via ABE-Mediated Base Editing. Mol. Ther. Nucleic Acids. 2019;17:289–296. doi: 10.1016/j.omtn.2019.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farasat S., Wei M.H., Herman M., Liewehr D.J., Steinberg S.M., Bale S.J., Fleckman P., Toro J.R. Novel transglutaminase-1 mutations and genotype-phenotype investigations of 104 patients with autosomal recessive congenital ichthyosis in the USA. J. Med. Genet. 2009;46:103–111. doi: 10.1136/jmg.2008.060905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richter M.F., Zhao K.T., Eton E., Lapinaite A., Newby G.A., Thuronyi B.W., Wilson C., Koblan L.W., Zeng J., Bauer D.E., et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020;38:883–891. doi: 10.1038/s41587-020-0453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaudelli N.M., Lam D.K., Rees H.A., Solá-Esteves N.M., Barrera L.A., Born D.A., Edwards A., Gehrke J.M., Lee S.J., Liquori A.J., et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020;38:892–900. doi: 10.1038/s41587-020-0491-6. [DOI] [PubMed] [Google Scholar]
- 42.Walton R.T., Christie K.A., Whittaker M.N., Kleinstiver B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science. 2020;368:290–296. doi: 10.1126/science.aba8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li G., Liu Y., Zeng Y., Li J., Wang L., Yang G., Chen D., Shang X., Chen J., Huang X., Liu J. Highly efficient and precise base editing in discarded human tripronuclear embryos. Protein Cell. 2017;8:776–779. doi: 10.1007/s13238-017-0458-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.