

Abstract
Transthyretin (TTR) amyloidosis is a hereditary life-threatening disease characterized by deposition of amyloid fibrils. The main causes of TTR amyloidosis are mutations in the TTR gene that lead to the production of misfolded TTR protein. Reducing the production of toxic protein in the liver is a validated strategy to treat TTR amyloidosis. In this study, we established a humanized mouse model that expresses mutant human TTR (hTTR; V30M) protein in the liver to model TTR amyloidosis. Then, we compared the efficiency of reducing the expression of mutant hTTR by dual adeno-associated virus 8 (AAV8)-mediated split SpCas9 with that by single AAV8-mediated Nme2Cas9 in this model. With two gRNAs targeting different exons, dual AAV-mediated split SpCas9 system achieved efficiencies of 37% and 34% reduction of hTTR mRNA and reporter GFP expression, respectively, in the liver. Surprisingly, single AAV-mediated Nme2Cas9 treatment resulted in 65% and 71% reduction of hTTR mRNA and reporter GFP, respectively. No significant editing was identified in predicted off-target sites in the mouse and human genomes after Nme2Cas9 targeting. Thus, we provide proof of principle for using single AAV-mediated CRISPR-Nme2Cas9 to effectively reduce mutant hTTR expression in vivo, which may translate into gene therapy for TTR amyloidosis.
Keywords: TTR amyloidosis, mouse model, gene therapy, AAV, Nme2Cas9
Graphical abstract
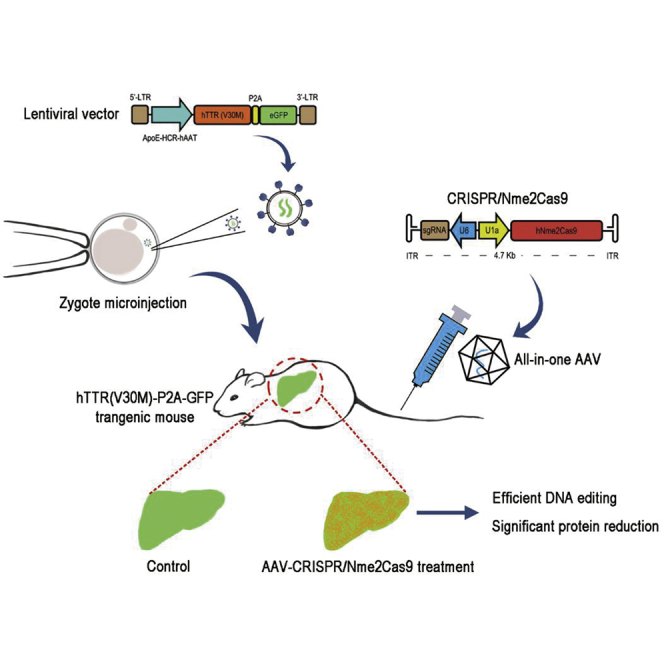
Huang and colleagues report that they generated a humanized mutant TTR transgene mouse model and compared the in vivo editing performance of the dual AAV-mediated split SpCas9 versus the all-in-one AAV-mediated Nme2Cas9 to disrupt human TTR expression. The Nme2Cas9 system showed robust efficiency and thus potential in treating TTR amyloidosis.
Introduction
Transthyretin (TTR) amyloidosis is an autosomal dominant hereditary disease caused by mutations in the TTR gene. TTR monomers are predominantly synthesized in the liver by hepatocytes and form tetrameric TTR complexes to transport thyroid hormones and vitamin A in plasma and cerebrospinal fluid. Changes in the single amino acid that result from point mutations of the TTR gene destabilize the tetramer to cause the aggregation of misfolded monomers into insoluble, extracellular amyloid fibril deposits. The accumulation of amyloid deposits in multiple organ systems causes progressive peripheral polyneuropathy, cardiomyopathy, nephropathy, and gastrointestinal dysfunction.1 Currently, it is estimated that approximately 50,000 people worldwide suffer from TTR amyloidosis.2 The average life expectancy of untreated patients is 3 to 15 years since symptom onset and clinical diagnosis. To date, more than 150 mutations have been identified as pathogenic. The TTR (V30M) mutation is the most common variant worldwide.3 In China, mainly in Han patients, as many as 20 different pathological TTR mutations have now been identified.4
The pathological mechanism of TTR amyloidosis has led to the notion that disease can be treated or prevented by approaches that reduce the production of misfolded monomers. The first strategy is to reduce the amount of mutant TTR by liver transplantation. The results in approximately 2,000 recipients who received a liver synthesizing only the wild-type (WT) protein showed that 80% of the patients survived for more than 10 years, and the arrest of the progression of the neuropathy was achieved in the majority.5 However, the shortage of donor organs greatly limits liver transplantation treatment. The second approach is to stabilize the mutant tetramers using small-molecule tafamidis to prevent amyloidogenic monomers from being released from circulating TTR tetramers.6,7 However, this stabilizer failed to prevent disease progression in many patients.8 Another available strategy for the treatment of the disease is to reduce the production of TTR monomers by targeting both WT and mutant TTR mRNAs using antisense oligonucleotides (ASOs) or small interfering RNAs (siRNAs). Recently, in 2018, Inotersen, an ASO drug, was approved by the European Medicines Agency (EMA) and later by the Food and Drug Administration (FDA) in the USA to treat hereditary TTR amyloidosis.9,10 In the same year, as the first approved siRNA drug, patisiran was also approved by the FDA.11 However, neither ASO nor siRNA can cure the disease. As long as mRNA is transcribed from the mutant TTR gene, patients need to receive repeated injections throughout their entire life, which causes a huge economic burden considering the expensive price of inotersen and patisiran treatments.
The emergence of engineered endonucleases such as zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas system enables researchers to manipulate mammalian genomic DNA in a site-specific manner. Since the first application in mammalian cells in 2013,12 CRISPR-Cas9 has been thought to have great potential in gene therapy because of its easy and versatile application.13 Successes have been achieved in treating hereditary diseases, especially by targeting the eye, muscle, and liver using nonviral or viral vectors to deliver the CRISPR-Cas9 system in preclinical animal models.14, 15, 16, 17, 18, 19 Progress in this rapidly evolving field has been highlighted by the recent FDA clinical trial approval for AGN-151587 (EDIT-101, Editas Medicine, Cambridge, MA, USA) to treat CEP290-associated Leber congenital amaurosis by delivering SaCas9-CEP290 guide RNA (gRNA) into photoreceptor cells via adeno-associated virus 5 (AAV5; clinicaltrials.gov ID: NCT03872479).20 Theoretically, targeting the TTR gene in the liver using Cas9 to knock out TTR expression is an ideal strategy to cure TTR amyloidosis. Based on this notion, Intellia Therapeutics developed a lipid nanoparticle (LNP) system to deliver SpCas9 mRNA and modified gRNA to mouse liver, and the treatment enabled significant editing of the mouse TTR gene in the liver, with a great reduction in the serum TTR protein level.21 By virtue of its efficient LNP system, the drug NTLA-2001 (Intellia Therapeutics, Cambridge, MA, USA) recently entered a phase 1 clinical trial as the first approved systemically delivered investigational CRISPR-Cas9 therapy to enter the clinic (clinicaltrials.gov ID: NCT04601051). However, LNP-based delivery systems still face critical barriers, such as the efficient packaging of CRISPR-Cas9 components into a single vector given the large size of the Cas9 mRNA, stability of the complex after administration into the blood, targeting specificity, and escape of the nanoparticles from the endosome after being consumed by targeting cells.22,23
AAV vectors are the leading carriers to deliver foreign genes in vitro or in vivo to treat a variety of diseases due to their low integration and low toxicity. Taking advantage of tropism to different organs of AAV serotypes, liver targeting efficiency can be increased by choosing AAV2, AAV5, AAV8, and AAVrh10.24,25 Preclinical and clinical successes in AAV-mediated gene therapies have helped AAV gain popularity as the ideal therapeutic vector.26 However, the most efficient CRISPR-SpCas9 system is too large to package into one AAV due to the limited capacity of the AAV vector. To settle this issue, split intein was used to develop the split SpCas9 system.27 Using a dual AAV-mediated split SpCas9 strategy, AAV-mediated CRISPR-Cas9, SpCas9-based epigenetic modifier, and base editing systems were successfully applied in animals to edit the genome in vivo.28, 29, 30 An alternative way to overcome the capacity limitation of AAV vectors is to explore and develop smaller Cas9 orthologs, such as SaCas9 and Nme2Cas9.31,32 Compared with SaCas9, Nme2Cas9 recognizes a simpler PAM (NNNNCC) and is therefore capable of targeting more loci in the genome.32 In addition, Nme2Cas9 and Nme2Cas9-based cytidine base editor (CBE) showed higher fidelity than SpCas9 and SpCas9-based CBE, respectively.32,33
However, whether dual or single AAV-mediated CRISPR-Cas9 system could efficiently knock out the mutant human TTR (hTTR) gene in the liver remains unknown. Here, we performed a proof-of-concept study to show that single AAV-mediated Nme2Cas9 more efficiently cleaved the hTTR gene and decreased hTTR expression in the liver of a hTTR (V30M) transgenic mouse model than dual AAV-mediated split SpCas9, showing that AAV-mediated CRISPR-Nme2Cas9-based gene therapy has better potential to treat TTR amyloidosis in humans.
Results
Generation of lentivirus-mediated hTTR (V30M) transgenic mice
TTR is a secretory protein mainly produced in the liver by hepatocytes, and the TTR (V30M) mutation is the most common pathological variant worldwide. To rapidly generate transgenic mice expressing mutant hTTR, we used lentivirus-mediated transgene in the study. First, we cloned the hTTR (V30M) CDS into a lentiviral plasmid harboring a potent artificial hepatocyte-specific promoter, ApoE-HCR-hAAT (Figure 1A), and verified hTTR (V30M) expression by transducing the mouse liver carcinoma cell line Hepa1-6. After transduction by the lentiviral vectors, Hepa1-6 cells showed a potent reporter GFP signal, and the hTTR (V30M) protein was detected by western blot assay (Figures 1B and 1C). Then, the concentrated lentiviral suspension was used for zygote injection to generate hTTR (V30M) transgenic mice (Figure 1D). To obtain mice with relatively consistent hTTR (V30M) expression, we used F2 generation transgenic mice bred from the same F1 father mating with WT mothers in the following experiments. Our previous study has showed that lentivirus-mediated transgenes were silenced by methylation modification and could be reactivated by 5-azacytidine (5-AzaC), a DNA methyltransferase inhibitor treatment administered in neonatal pups.34 Then, newborn F2 mice were injected with 5-AzaC to activate hTTR (V30M) expression. Five weeks after injection, the exogenous mutant hTTR protein was detected in the serum of 5-AzaC-treated mice but not the vehicle-treated mice, and results showed that serum hTTR concentration was consistent among the mice (Figure 1E). In addition, liver sections of 5-AzaC-treated mice showed intense reporter GFP expression in hepatocytes (Figure 1F). The above results indicated that the lentivirus-mediated transgenic mouse model with consistent hTTR (V30M) expression was qualified for assaying the efficiency of the CRISPR-Cas9 system in knocking down the pathological protein in vivo. Therefore, the 5-AzaC-treated F2 transgenic mice were used in the following treatment experiments.
Figure 1.

Generation of liver-specific expression hTTR (V30M) transgenic mouse model
(A) Schematic illustration of the lentiviral plasmid used for generating hTTR (V30M) transgenic mouse model. (B) Phase contrast and fluorescent images of Hepa1-6 cells after transduction by lentiviral vectors packaged using the plasmid in (A). Scale bar=200 μm. (C) Western blot showed that hTTR and hTTR (V30M) protein was detected in Hepa1-6 cells after transduction by lentiviral vectors harboring hTTR (LV-hTTR) and LV-hTTR (V30M). (D) Workflow of generating the lentivirus-mediated transgenic mice by micro-injection of lentiviral vectors into the zygotes and screening the F2 mice used in the treatment experiments. (E) Stable hTTR was detected in the serum of F2 hTTR (V30M) transgenic mice treated with 5-AzaC using a hTTR ELISA kit (n = 6). F2 mice treated with vehicle served as control (n = 4). (F) Fluorescent imaging of liver frozen sections showed that the reporter GFP was potently expressed in the liver of 5-AzaC-treated F2 hTTR (V30M) transgenic mice. F2 mice treated with vehicle served as control. Cell nuclei were shown by 4',6-diamidino-2-phenylindole (DAPI) staining. Scale bar=200 μm.
Dual AAV-mediated split CRISPR-SpCas9 treatment reduced hTTR (V30M) expression in the liver
Since SpCas9 was yet the most efficient Cas9 ortholog, we first tested the efficiency of the split SpCas9 system delivered by dual AAVs in editing the hTTR (V30M) transgene and reducing the protein expression. We designed 6 SpCas9-gRNAs targeting exon 1 or exon 2 of both the WT and mutant hTTR genes (Figure 2A) and validated the cleavage efficiency of the gRNAs by transfecting HEK293T cells. The T7E1 assay showed that the G1 and G4 gRNAs were the top two in editing the target sites (Figure 2B). Then, the G1 and G4 were cloned into the split SpCas9 vectors to generate the split CRISPR-SpCas9 system (Figure 2C). Cotransfection of the split SpCas9 vectors with gRNAs into HEK293T cells resulted in efficient cleavage in G1- and G4-targeted sites (Figure 2D). Thereafter, F2 transgenic mice were first treated with 5-AzaC to activate hTTR (V30M) expression and then a dose of 2 × 1012 viral genomes (VG) dual AAV8 (1 × 1012 VG for each terminal) carrying two terminals of split SpCas9, G1, and G4 gRNAs was administered to the 5-week-old transgenic mice by lateral tail vein injection. After treatment, the serum and liver were collected at the indicated times to evaluate the treatment effect (Figures 2E and 2F). Liver genomic DNA was isolated to test whether hTTR (V30M) was cleaved, and the T7E1 assay showed obvious cleavage in the targeted site in all treated mice (Figure 2G). The average indel efficiency of each mouse was 12.68% as revealed by targeted deep sequencing (Figure 2H). The qRT-PCR results showed that hTTR (V30M) mRNA expression in the liver of mice treated with AAV8-split SpCas9 with G1 and G4 gRNAs was reduced by 37% compared with those received AAV8-split SpCas9 with non-target gRNA (Figure 2I). However, we failed to detect any hTTR signal in liver lysates by western blot though hTTR was detected in cultured Hepa1-6 cells transduced with hTTR (V30M) (Figure S1). Since eGFP was linked after hTTR (V30M) by a self-cleaving P2A peptide,35 as long as Cas9 induced a frameshift indel on targeted site, GFP would no longer be expressed. Therefore, GFP expression could be used to represent the expression of the hTTR (V30M) protein in the liver. Then, GFP was detected by western blot, and the results showed that GFP expression was reduced by 34% (Figures 2J and 2K). Inferring from the reporter GFP expression, the hTTR (V30M) protein might be reduced to a similar level. However, serum hTTR level was not reduced after AAV8-split CRISPR-SpCas9 treatment (Figure S2A).
Figure 2.

Dual AAV-mediated split CRISPR-SpCas9 treatment reduced hTTR (V30M) expression
(A) Schematic illustration of hTTR gene and the targeted sites of SpCas9 gRNA candidates on the gene. (B) Cleavage efficiency of gRNA candidates (G1–G6) was tested by T7E1 assay using HEK293T cells. Non-targeted (NT) gRNA served as negative control. (C) Schematic illustration of the plasmids of N- and C-terminals of split CRISPR-SpCas9 system used for AAV packaging. (D) Validation of the split SpCas9 system with G1 or G4 gRNAs by transfecting HEK293T cells. Plasmid pX330 expressing full-length SpCas9 served as positive control. NT gRNA constructed into the split SpCas9 vectors served as negative control. (E) Schematic illustration of the hTTR (V30M)-P2A-eGFP transgene and the regions targeted by G1 and G4 gRNAs. (F) Timeline of the treatment experiment administering dual AAV8-split CRISPR-SpCas9 to the F2 hTTR (V30M) transgenic mice. (G) T7E1 assay showed the cleavage efficiency of dual AAV8-split SpCas9 with G1 and G4 targeting (hTTR G1+G4) in the liver genome of treated mice. Mice that received NT gRNA served as negative control. Each lowercase letter represented an individual mouse (NT gRNA, n = 1; hTTR G1+G4, n = 5). (H) The indel ratio of the targeted region in liver genome of mice received dual AAV8-split SpCas9 with G1 and G4 (hTTR G1+G4) or with NT gRNA treatment revealed by targeted deep sequencing (NT gRNA, n = 3; hTTR G1+G4, n = 5). (I) qRT-PCR result showed decreased hTTR (V30M) mRNA expression in the livers of mice received treatment of dual AAV8-split SpCas9 with G1 and G4 (hTTR G1+G4) compared with control mice that received AAV8-split SpCas9 with NT gRNA (NT gRNA, n = 5; hTTR G1+G4, n = 5). (J) Western blot assay showed decreased expression of the reporter GFP in the livers of mice that received treatment of dual AAV8-split SpCas9 with G1 and G4 (hTTR G1+G4) compared with control mice that received AAV8-split SpCas9 with NT gRNA. Each lane represented an individual mouse (NT gRNA, n = 4; hTTR G1+G4, n = 5). (K) Statistical graph of band intensity of the reporter GFP in (F).
Single AAV-mediated CRISPR-Nme2Cas9 treatment potently reduced hTTR (V30M) expression in the liver
Next, we tested the efficiency of a recently developed CRISPR-Nme2Cas9 system delivered by a single AAV vector in reducing hTTR (V30M) expression in the mouse model. Four Nme2Cas9 gRNAs were designed to target the exon 1 of the hTTR gene. Then, each gRNA was cloned into the Nme2Cas9_AAV plasmid to construct the all-in-one vector (Figures 3A and 3B). The gRNA cleavage efficiency was assayed by T7E1 after transfection into HEK293T cells, and the G3 gRNA showed the best performance (Figure 3C). Thereafter, all-in-one AAV8-Nme2Cas9-G3 was injected into 5-AzaC-treated F2 hTTR (V30M) transgenic mice to test the in vivo cleavage efficiency (Figure 3D). After treatment, no significant reduction in serum hTTR concentration was observed in mice treated with AAV8-Nme2Cas9-G3 compared with the mice treated with non-target gRNA (Figure S2B). But the T7E1 assay showed potent cleavage bands in the livers of all mice treated with the G3 gRNA (Figure 3E). And the indel ratio was up to 52%, as revealed by targeted deep sequencing (Figure 3F). The liver hTTR (V30M) mRNA level was decreased by 65% (Figure 3G) and the reporter GFP protein expression in the livers was significantly reduced by 71% in AAV8-Nme2Cas9-G3-treated mice (Figure 3H and 3I). The reduction of reporter GFP was further verified by liver sections showing potently decreased GFP-positive cells in the liver (Figure 3J). The above results indicated that single AAV-mediated CRISPR-Nme2Cas9 could greatly reduce the expression of hTTR (V30M) in the liver.
Figure 3.

Single AAV-mediated CRISPR-Nme2Cas9 treatment potently reduced hTTR (V30M) expression
(A) Schematic illustration of Nme2Cas9 gRNA candidates targeting the exon 1 of hTTR gene. (B) Schematic illustration of the all-in-one Nme2Cas9_AAV plasmid used for AAV packaging. (C) T7E1 assay showed the cleavage efficiency of Nme2Cas9 gRNA candidates using HEK293T cells. Duplicate experiments were represented. NT gRNA served as negative control. (D) Timeline of the treatment experiment administering single AAV8-mediated CRISPR-Nme2Cas9 to the F2 hTTR (V30M) transgenic mice. (E) T7E1 assay showed the cleavage efficiency of single AAV-mediated Nme2Cas9 with G3 gRNA (hTTR G3) in the liver genomes of transgenic mice. Mice that received NT gRNA served as negative control. Each lowercase letter represented an individual mouse (NT gRNA, n = 2; hTTR G3, n = 4). (F) The indel ratio of targeted sites in liver genome of mice received AAV8-Nme2Cas9-G3 (hTTR G3) treatment or NT gRNA treatment revealed by targeted deep sequencing (NT gRNA, n = 4; hTTR G3, n = 4). (G) qRT-PCR result showed decreased hTTR mRNA expression in the livers of mice that received treatment of AAV8-Nme2Cas9 with G3 gRNA (hTTR G3) compared with control mice that received NT gRNA (NT gRNA, n = 4; hTTR G3, n = 4). (H) Western blot assay showed decreased expression of the reporter GFP in the livers of mice that received treatment of AAV8-Nme2Cas9 with G3 gRNAs (hTTR G3) compared with control mice that received NT gRNA. Each lane represented an individual mouse (NT gRNA, n = 4; hTTR G3, n = 4). (I) Statistical graph of band intensity of the reporter GFP in (F). (J) Fluorescent images of liver frozen sections showed a significant reduction of the reporter GFP in the liver of mice that received treatment of AAV8-Nme2Cas9 with G3 gRNA (hTTR G3) compared with control mice that received NT gRNA. Cell nuclei were shown by DAPI staining. Scale bar=500 μm.
Off-target effects and targeted indel profile
Programmable nucleases bind and cleave genomic DNA in a targeted manner and can also cause off-target DNA cleavage at sites that share identical or highly homologous sequences. Off-target cleavage often results in indel mutations in off-target sites or chromosomal rearrangements such as deletions, inversions, and translocations.36,37 Therefore, off-target alterations of genome-editing tools have become a key concern in basic research and clinical medicine.38 Next, we determined the off-target editing events of the CRISPR-Nme2Cas9-G3 system in both mouse and human genomes based on deep sequencing of the top ten predicted off-target sites found by Cas-OFFinder.39 The results showed that no significantly increased indel reads were found in mouse livers and HEK293T cells receiving Nme2Cas9 hTTR-G3 treatment compared with control ones receiving non-target gRNA (Figure 4A). The indel profile of the targeted transgenic site in treated livers revealed that the majority of AAV-Nme2Cas9-G3-induced mutations were one base insertion and one or two base deletions resulting in frameshift mutations of the hTTR (V30M) gene. It’s worth noting that all of the top 5 indels in mouse genome introduced premature termination codons (PTCs; Figure S3). In contrast to the profile in the mouse genome, the main mutations were deletions in the human genome based on cultured HEK293T cells (Figure 4B).
Figure 4.

Off-target alteration and indel profile of Nme2Cas9 with hTTR G3 gRNA in mouse and human genomes
(A) The indel ratio of on-target and predicted off-target sites in the liver genomes of mouse livers that received AAV8-Nme2Cas9-G3 (hTTR G3) treatment or NT gRNA treatment (NT gRNA, n = 4; hTTR G3, n = 4), and HEK293T cells transfected with Nme2Cas9 with G3 gRNA or NT gRNA revealed by targeted deep sequencing. (B) The indel profile induced by Nme2Cas9 with hTTR G3 gRNA in the genomes of mouse livers and HEK293T cells. Frequencies of the top 5 were analyzed. +, insertion; –, deletion.
Discussion
TTR amyloidosis is a slowly progressive condition characterized by the formation of amyloid fibrils resulting from abnormal deposition of a mutant protein in the body’s organs and tissues. TTR is produced primarily in the liver, and a small amount of this protein is produced in the choroid plexus and in the retina. Reducing the production of mutant TTR by liver transplantation or gene therapies targeting both WT and mutant TTR mRNA was shown to be an effective way to slow down the progression of amyloidosis and alleviate clinical symptoms.5,40,41 In this study, we attempted to test the efficiency of AAV-mediated CRISPR-Cas9 systems in reducing the production of mutant pathological protein in the liver of a hTTR (V30M) transgenic mouse model.
SpCas9 is yet the most efficient ortholog in cleaving targeted DNA and the first one systematically administered to humans. First, we tried to test the effect of SpCas9 delivered by AAV in a humanized transgenic mouse model. The large size of SpCas9 presents a challenge in packaging it within a single AAV vector. Therefore, a split SpCas9 system was developed, bypassing the packaging limit using split inteins. In the system, each half of SpCas9 was fused to the corresponding split intein moiety and only upon coexpression does intein-mediated trans-splicing occur, and the full Cas9 protein is reconstituted to exert cleavage.27,42 In a previous study, we identified the Rma intein, and the N-573 split site was the most efficient combination after screening different inteins and split sites for the SpCas9 base editor.30 Hence, this combination was chosen for SpCas9 splicing and cloned into two individual AAV vectors. To boost the knockout efficiency, we delivered two gRNAs targeting exon 1 and exon 2 of both WT hTTR and mutant hTTR (V30M) genes together with each half of SpCas9. Referring to the dose (6 × 1013 VG/kg) used in the clinical trial for hemophilia (clinicaltrials.gov ID: NCT03520712), a total dose of 2 × 1012 VG (approximately corresponding to 8 × 1013/kg) AAV with 1 × 1012 VG for each half of SpCas9 was systematically administered to each mouse (weight of approximately 25 g).
Twelve weeks after treatment, we found that the liver hTTR (V30M) gene of the humanized mice was cleaved successfully by the dual AAV-mediated split CRISPR-SpCas9 system. We failed to directly detect hTTR protein in liver lysate using western blot assay (Figure S1). It has been reported that endogenous mouse TTR interacted with hTTR and formed stable heterotetramer in hTTR transgenic mouse.43 And complexed and non-complexed forms of TTR exhibited a difference in binding affinity of antibody,44 which might be the reason why hTTR protein could not be detected in liver lysate when it could be detected in cultured cell lysate using western blot. Since the reporter GFP was linked to hTTR (V30M) with a “self-cleaving” small peptide P2A,35 eGFP was cotranscribed with hTTR (V30M) so that these two proteins had a stoichiometric expression relationship. Indels leading to frameshift mutations in the hTTR (V30M) sequence would also disrupt GFP expression. By assaying GFP expression, we inferred that liver hTTR (V30M) expression was significantly reduced (Figure 2). In addition, there was no un-cleaved coupled hTTR and GFP was detected on the blots (data not shown). However, the circulating hTTR level did not change after AAV treatment (Figure S2A). Hereto, we speculated that the approximate 34% reduction in the liver was not sufficient to decrease the circulating hTTR. In the split SpCas9 system, only upon coentering two different AAVs into a single cell does intein-mediated trans-splicing occur, and the full Cas9 protein is reconstituted. Therefore, both the probability of coentering and the trans-splicing process could hamper the editing efficiency of this system in the liver.
Then, we turned to Nme2Cas9, a recently developed Cas9 ortholog that recognizes a widely accessible PAM (5′-NNNNCC-3′) similar to SpCas9 (5′-NGG-3′) but has a much smaller size and can be packaged with gRNA into a single AAV vector, bypassing the shortcomings of SpCas9.32 Next, a dose of 2 × 1012 VG AAV was given to the transgenic mouse to test the editing efficiency of single AAV-mediated CRISPR-Nme2Cas9 in the liver. Twelve weeks later, the treatment achieved a much more pronounced reduction in hTTR (V30M) expression inferred from the reporter GFP (up to 71%, Figure 3) in the liver than SpCas9. To our surprise, this pronounced reduction in hTTR (V30M) expression also failed to decrease the serum hTTR level (Figure S2). In contrast, Intellia Therapeutics posted that their LNP-mediated CRISPR-SpCas9 treatment achieved an editing efficiency of approximately 70% resulting a mean reduction of 97% in mouse serum TTR in CD1 mice.21 Zhao et al.45 found inconsistency between hepatic expression and serum concentration of hTTR in mice humanized at the TTR locus, which indicated that serum hTTR level could not precisely represent the expression level in the liver in transgenic mice with humanized TTR. Considering the stability of human/mouse TTR heterotetramer,43 we speculated that the very low production of hTTR (V30M) was sufficient to saturate the serum in our transgenic mice. Another possible reason was that mice were capable of clearing the exogenous protein efficiently, therefore keeping a stable low level of hTTR (V30M) in circulation. What’s more, data from clinical trials of two recently approved gene therapy drugs revealed a mean reduction of 81% with patisiran, whereas inotersen lowered the serum hTTR concentration by 71%.40,41,43 Therefore, although the serum hTTR (V30M) level of the transgenic mice failed to represent the therapeutic effects, the significant editing efficiency (52%) and reduction of protein production in the liver (71%) suggested that single AAV-mediated CRISPR-Nme2Cas9 has the potential to treat TTR amyloidosis in humans.
Since most of the patients are heterozygous, AAV-mediated Nme2Cas9 therapy in the study targets both WT and mutant alleles. Safety concerns on targeting both mutant and WT TTR alleles in patients were revealed by two clinical trials showing that no significantly adverse effect due to TTR reduction was observed.40,41 In addition, Episkopou et al.46 reported that TTR knockout mice appeared normal in the complete absence of mouse TTR in the serum. In addition, Nme2Cas9 has been reported as a naturally high-accuracy genome-editing platform in mammalian cells.32 Consistently, no indels could be detected at predicted off-target sites for G3 gRNA in either human or mouse genomes, while efficient on-target editing was readily detected. The indel profile in AAV-Nme2Cas9-hTTR G3-treated livers showed the top 5 indels were one base insertion and one or two base deletions. Theoretically, the small indels should not affect mRNA levels since they didn’t disrupt the primer binding sites on the transcripts, unless the transcripts became nonsense-mediated mRNA decay (NMD) targets. After analyzing the sequences with the most common indels listed in Figure 4, we exactly found that those indels introduced PTCs (Figure S3). The top 3 indels were 1 bp insertion and 2 bp deletions, which resulted in a PTC after the 18th and 17th amino acid, respectively. And the 4th and 5th most common indels were 1 bp deletion, which resulted in a PTC after the 31st amino acid (Figure S3). A PTC nearing the ATG starting codon could create a long 3′ UTR in the mRNA, making it become NMD targets that then led to decrease in mRNA level.47
Overall, we performed a proof-of-concept study to verify that single AAV-mediated CRISPR-Nme2Cas9 systems efficiently decreased mutant hTTR expression in the livers of a transgenic mouse model of TTR amyloidosis, showing that AAV-mediated CRISPR-Nme2Cas9-based gene therapy has the potential to treat TTR amyloidosis in humans. Furthermore, considering the high efficiency of the system, this strategy could also be applied to treat another hereditary liver disease, hypercholesterolemia, by targeting the PCSK9 or ANGPTL3 genes, to treat refractory infectious hepatitis B by targeting the viral genome and to treat liver carcinomas by targeting cancer cells.
Materials and methods
Mice and tissue collection
CD-1 mice were purchased from SPF Biotechnology (Beijing, China) and housed under standard conditions (22°C ± 1°C) in a specific pathogen-free animal facility with a 14/10-h light-dark cycle at Sun Yat-sen University. The Institutional Animal Care and Use Committee of Sun Yat-sen University, P.R. China approved all animal experiments. For tissue collection, the mice were sacrificed by an overdose of anesthesia using 2.5% Avertin prepared with 2, 2, 2-tribromoethanol (Sigma-Aldrich, T48402) and harvested liver tissue was stored at −80°C after being snap frozen by liquid nitrogen for DNA, RNA, and protein extraction or fixed by 4% paraformaldehyde (PFA) for frozen sectioning.
Plasmid construction
The full-length coding sequences of hTTR (V30M) were synthesized by IGE Biotechnology (Guangzhou, China) and tagged with eGFP reporter using a P2A peptide by overlap PCR. Then the DNA segment was cloned into a lentiviral plasmid pCCL-ApoE-HCR-hAAT-fLuc (Figure S4A), which contains the artificial hepatocyte-specific promoter ApoE-HCR-hAAT,48 to replace the fLuc cassette to generate pCCL-ApoE-HCR-hAAT-hTTR (V30M)-P2A-eGFP (Figure S4B). The gRNAs were designed by Cas-Designer, a web-based tool (http://www.rgenome.net/cas-designer/), and gRNA oligos of SpCas9 and Nme2Cas9 were cloned into pX330-U6-Chimeric_BB-CBh-hSpCas9 (pX330, Addgene, #42230) and Nme2Cas9_AAV (Addgene, #119924) to test the cleavage efficiency by transfecting HEK293T cells. To construct Split SpCas9 vectors, we amplified U6-driven gRNA expression cassette, Cbh driven N-terminal (1-573), and C-terminal (574-1368) of SpCas9 from pX330. The Rma inteins were synthesized by IGE Biotechnology and linked to terminals of SpCas9 by overlap PCR. Then the gRNA and SpCas9 expression cassettes were cloned into px601 AAV backbone (Addgene, #61591) to construct the split SpCas9 plasmids (Figure S4C).
Lentiviral vectors production
For lentiviral vectors production, HEK293T cells were plated onto 15-cm dishes at a density of 1 × 107 cells/dish. 24 h later, the cells were co-transfected with transfer plasmids and packaging plasmids using polyethylenimine (PEI, Polysciences, 23966-1). Briefly, for each 15-cm dish, 16 μg pCCL-ApoE-HCR-hAAT-hTTR (V30M)-P2A-eGFP, 12 μg psPAX2 (Addgene, #12260), and 16 μg pMD2.G (Addgene, #12259) were diluted into 1 mL Opti-MEM (GIBCO, 31985070). Then 96 μg PEI diluted in 1 mL Opti-MEM was mixed with the diluted plasmids. The mixture was left at room temperature for 15 min before adding to the culture cells. The lentivirus-containing culture medium was harvested at 48 and 72 h posttransfection and concentrated by ultracentrifugation with a 20% sucrose cushion using a Beckman Optima L-100XP Ultracentrifuge. Titer of viral suspension was determined by fluorescence-activated cell sorting (FACS). Briefly, 1 day before transduction, Hepa1-6 cells were seeded onto a 24-well plate at a density of 4 × 104 cells/well. Immediately before transduction, cells in counting wells were detached by trypsin and counted to obtain the cell number for transduction. Then cells were transduced by serial diluted viral vectors. 3 days after, the GFP-positive ratio of cells in each well was detected by FACS. The wells with GFP-positive ratios between 1% and 10% were used for calculating the titer. Titer = GFP-positive ratio × transduction cell number/transduction viral volume.
AAV production
For AAV production, HEK293T cells were plated onto 15-cm dishes at a density of 1 × 107 cells/dish. 24 h later, transfection was performed using PEI method. Briefly, for each 15-cm dish, 12 μg transfer plasmids and 20 μg pAAV2/8 (MiaoLing Plasmid Sharing Platform, P13271) were diluted into 1 mL Opti-MEM (GIBCO, 31985070). Then 96 μg PEI diluted in 1 mL Opti-MEM was mixed with the diluted plasmids. The mixture was left at room temperature for 15 min before adding to the culture cells. And collection of AAV was performed 96 h posttransfection according to the protocol provided by Addgene’s web site (https://www.addgene.org/protocols/aav-production-hek293-cells/). Briefly, cell cultured medium was collected and underwent polyethylene glycol (PEG) 8000 (Sigma-Aldrich, P5413) precipitation. And the detached cells were lysed by sonication to release AAV particles. Then the collected crude AAV solution was purified and concentrated by iodixanol gradient ultracentrifugation and ultrafiltration according to the protocol provided by Addgene’s web site (https://www.addgene.org/protocols/aav-purification-iodixanol-gradient-ultracentrifugation/). Titers of AAV stocks were determined by qRT-PCR using TB Green (Takara, RR430). Before releasing the viral DNA from the particles, all extra-viral DNA was removed by digestion with recombinant DNase I (Takara, 2270A). Then, the viral DNA was released by incubation with proteinase K (Takara, 9034) for 60 min at 50°C, followed by 10-min inactivation at 90°C. The primers targeting AAV2 ITR (forward primer [FP]: 5′-GGAACCCCTAGTGATGGAGTT-3′; reverse primer [RP]: 5′-CGGCCTCAGTGAGCGA-3′) were used.49 And commercialized AAV (BrainVTA, Wuhan, China), which was titrated with ATCC reference standard, was used as the reference standard.
Cell culture and transfection
HEK293T and Hepa1-6 cells were maintained in DMEM (Corning, 10-013-CV) supplemented with 10% FBS (Lonsera, S711-001S) and cultured at 37°C with 5% CO2. For gRNA targeting validation, HEK293T cells were seeded onto a 12-well plate at a density of 2 × 105 cells/well the day before transfection. Then 1.6 μg pX330 or Nme2Cas9_AAV plasmids with gRNA candidates were transfected into HEK293T cells using 4.8 μg PEI. Genomic DNA of transfected cell was isolated 48 h after transfection.
Genomic DNA extraction and T7E1 assay
Genomic DNA of cultured cells was extracted using AxyPrep Blood Genomic DNA Miniprep Kit (Axygen, AP-MN-BL-GDNA-250) and genomic DNA of mouse livers was isolated using E.Z.N.A. Tissue DNA Kit (Omega, D3396-02). The cleavage efficiency of each individual sgRNA was tested by the T7E1 assay (NEB, M0302) according to the manufacturer’s protocol. In brief, PCR product of target DNA was cleaned up using a QIAquick PCR Purification Kit (QIAGEN, 28106) and then 300 ng purified DNA was incubated with T7 Endonuclease I at 37°C for 40 min. Primers used in gRNA validation experiments were as follows: TTR-E1-FP, 5′-AAGCAGCCTAGCTCAGGAGA-3′ and TTR-E1-RP, 5′-AGCTCAGTAAGCTCAGTGGAA-3′ for hTTR exon 1; TTR-E2-FP, 5′-TGTCGACACTTACGTTCCTGAT-3′ and TTR-E2-RP, 5′-TGCTCAGGTTCCTGGTCACT-3′ for hTTR exon 2. Primers used in in vivo cleavage assay were FP 5′-GATCTTGCTACCAGTGGAACAGCC-3′ and RP 5′-GATGGCAGGACTGCCTCGG-3′.
Generation of hTTR (V30M) transgenic mice
Female mice (6–8 weeks old) were injected with 5 international units (IU) of pregnant mare serum gonadotropin (PMSG, Solarbio, P9970) and 7.5 IU human chorionic gonadotrophin (HCG, Sigma-Aldrich, 230734). After mating, the mice were sacrificed to harvest the zygotes. Micro-injection of lentiviral vectors into the perivitelline space of the zygote was modified from a Cold Spring Harbor protocol.50 In brief, between 50–100 pl of concentrated viral suspension with a titer of 5 × 105 to 5 × 106 TU/μL was injected beneath the zona pellucida of each zygote using a Femtojet microinjection manipulator (Eppendorf). After the injection, the zygotes were cultured for 4 h before transplantation into the oviducts of pseudopregnant females. Genotyping of newborn mice were performed using a KAPA HotStart Mouse Genotyping Kit (Kapa Biosystems, KK7352). Primers amplifying the integrated hTTR (V30M) segment were used for genotyping: FP 5′-GGCTCCTCTAGAGGTACCCGGG-3′ and RP 5′-GTTGCTTCCTTGGGATTGGTGACGACAGCCG-3′.
5′-AzaC treatment and AAV administration
The 5-AzaC powder (Selleck, S1782) was dissolved in 5% DMSO plus 30% PEG300 in sterile water at a concentration of 10 mg/mL and stored at −80°C. Before use, the drug was further diluted to 1 mg/mL. The treatment for the neonatal mice to activate silenced transgene expression was modified from previous reports.34,51 Briefly, each pup received two subcutaneous injections of 5-AzaC at doses of 25 and 50 μg at postnatal day 2 and day 9, respectively. The vehicle (0.5% DMSO+3% PEG300) was injected as a control. After the injections, the pups were returned to their mothers until weaning.
For AAV administration, mice at 5 weeks (20–25 g) of age received 2 × 1012 VG (8 × 1013–1 × 1014 VG/kg) AAV diluted in 200 μL PBS through lateral tail vein injection. For AAV-Split SpCas9 treatment, the dose of each terminal was 1 × 1012 VG per injection.
hTTR ELISA detection
Mouse serum was collected by retro-orbital bleed and was aliquoted and stored at −80°C before ELISA assay. The hTTR levels in mouse serum were determined using a Human Transthyretin/TTR ELISA Kit (Boster, EK1684). The ELISA plates were read on a BioTek microplate reader, and the hTTR concentrations were calculated by the BioTek Gen5 software using four-parameter logistic analysis.
Frozen section preparation
Harvested liver tissues were post-fixed in 4% PFA at 4°C overnight. Trimmed tissues were dehydrated with 30% sucrose in PBS overnight and then embedded in optimum cutting temperature compound (O.C.T., Sakura, 4583) before sectioning. Then, 8–10 μm sections were cut transversely using a cryostat (Leica). The slides were stained with 1 μg/mL DAPI (Sigma-Aldrich, D9542) for 10 min to show nuclei. Finally, the stained slides were mounted with anti-fade medium (Vector, H-1200) and imaged for GFP and DAPI using a fluorescence microscope (Zeiss).
qRT-PCR
Total RNA was extracted using RNAiso Plus (9108, Takara) and 1 μg of total RNA were used for cDNA synthesis using PrimeScript RT reagent Kit with gDNA Eraser (RR047A, Takara). qRT-PCR reactions were set up using the TB Green Premix Ex Taq II (RR820A, Takara). Relative hTTR expression values were normalized to GAPDH expression. The primers used are as follows: GAPDH, FP 5′-TCCCACTCTTCCACCTTCGATGC-3′, RP 5′-GGGTCTGGGATGGAAATTGTGAGG-3′; hTTR, FP 5′-AGTCCTGCCATCAATGTG-3′, RP 5′-CCAAGTGCCTTCCAGTAA-3′.
Western blot
Cultured cells or liver tissue were lysed in radioimmunoprecipitation assay (RIPA) buffer (GBCBIO, G3424) containing a protease inhibitor cocktail (Beyotime, P1005). The extracts were combined with 2 × Laemmli sample buffer (Bio-Rad, 1610737) and denatured by boiling for 10 min. Then, the samples were subjected to 10% SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (Bio-Rad, 1620177) by using a Trans-Blot Turbo Transfer System (Bio-Rad, USA). The blots were then blocked with 5% bovine serum albumin (Solarbio, A8010) for 1 h and incubated overnight at 4°C with the primary antibodies. Primary antibodies used in the study are as follows: rabbit anti-human TTR (1:1,000, Cell Signaling Technology, 29872), mouse anti-GAPDH (1:1,000, Cell Signaling Technology, 97166), mouse anti-GFP (1:400, Santa Cruz, sc-9996), and rabbit anti-β-actin (1:3,000, Abclonal, AC026). The blots were then washed with Tris-buffered saline containing 0.1% Tween-20 before incubation with IRDye 800CW goat anti-mouse immunoglobulin G (IgG; 1:4,000, LI-COR, 926-32210) or goat anti-rabbit IgG (1:4,000, LI-COR, 926-32211) secondary antibody for 2 h. The immunoreactive protein bands were detected and imaged by Odyssey system (LI-COR, USA), and the band intensity was measured by ImageJ software (NIH image, USA).
In silico off-target prediction
Cas-OFFinder39 was used to predict the off-target binding sites of Nme2Cas9 gRNA in the mouse (Mus musculus [mm10]) and human (Homo sapiens [GRCh38/hg38]) reference genomes with the following parameters: Pam type = Nme2Cas9 (5′-NNNNCC-3′), mismatch number = 4, DNA bulge size = 0, and RNA bulge size = 0. Primers were designed to amplify the loci flanking the top ten matches and listed in Table S1.
Target site deep sequencing
Amplification of target sites was conducted by PCR using barcoded primers and KOD FX (Toyobo, KFX-101). PCR product was cleaned up using a QIAquick PCR Purification Kit (QIAGEN, 28106) before deep sequencing. Deep sequencing was performed by Novogene (Beijing, China) using paired-end 150 bp Hiseq 2500 platform (Illumina). The sequencing depth for Nme2Cas9-G3 on-target site in mouse livers was 4 G, for off-target sites in mouse livers was 10 G, and for on-target and off-target sites in HEK293T cells was 2 G. Finally, the indels were determined using MATLAB script as previous report.52
Statistical analysis
The values of hTTR mRNA, GFP protein, and serum hTTR are presented as the mean ± SEM, and differences between two groups were analyzed using Student’s t test with the GraphPad Prism 9 (GraphPad Software, USA). The p values are indicated by ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. A p value of < 0.05 was considered statistically significant.
Acknowledgments
This study was funded by the National Key R&D Program of China (2017YFC1001901), the National Natural Science Foundation of China (31971365 and 32001063), the Guangdong Special Support Program (2019BT02Y276), the Guangdong Basic and Applied Basic Research Foundation (2019A1515010600 and 2020B1515120090), the Guangzhou Science and Technology Project (201803010020 and 201803010032), the Project funded by China Postdoctoral Science Foundation (2019M653205), and the Fundamental Research Funds for the Central Universities (19lgpy195).
Author contributions
J.H. and P.L. conceived and designed the project. J. Wen, J. Wu, and T.C. performed the experiments. Y.C., L.A., and P.L. provided technical support. J. Wen, S.Z., G.W., Y.H., P.Z., and J.Z. analyzed the data. J.H. and J. Wen wrote the manuscript. J.H. and J.Z. contributed to the final approval of the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.05.010.
Contributor Information
Jianxin Zhong, Email: 2269897342@qq.com.
Puping Liang, Email: liangpp5@mail.sysu.edu.cn.
Junjiu Huang, Email: hjunjiu@mail.sysu.edu.cn.
Supplemental information
References
- 1.Vieira M., Saraiva M.J. Transthyretin: a multifaceted protein. Biomol. Concepts. 2014;5:45–54. doi: 10.1515/bmc-2013-0038. [DOI] [PubMed] [Google Scholar]
- 2.Gertz M.A. Hereditary ATTR amyloidosis: burden of illness and diagnostic challenges. Am. J. Manag. Care. 2017;23(7, Suppl):S107–S112. [PubMed] [Google Scholar]
- 3.Plante-Bordeneuve V. Transthyretin familial amyloid polyneuropathy: an update. J. Neurol. 2018;265:976–983. doi: 10.1007/s00415-017-8708-4. [DOI] [PubMed] [Google Scholar]
- 4.Liu G., Ni W., Wang H., Li H., Zhang Y., Wang N., Wu Z. Clinical features of familial amyloid polyneuropathy carrying transthyretin mutations in four Chinese kindreds. J. Peripher. Nerv. Syst. 2017;22:19–26. doi: 10.1111/jns.12196. [DOI] [PubMed] [Google Scholar]
- 5.Ericzon B.G., Wilczek H.E., Larsson M., Wijayatunga P., Stangou A., Pena J.R., Furtado E., Barroso E., Daniel J., Samuel D., et al. Liver Transplantation for Hereditary Transthyretin Amyloidosis: After 20 Years Still the Best Therapeutic Alternative? Transplantation. 2015;99:1847–1854. doi: 10.1097/TP.0000000000000574. [DOI] [PubMed] [Google Scholar]
- 6.Tafamidis (Vyndaqel; Vyndamax) for Transthyretin Amyloid Cardiomyopathy. JAMA. 2020;324:505. doi: 10.1001/jama.2020.0753. [DOI] [PubMed] [Google Scholar]
- 7.Berk J.L., Suhr O.B., Obici L., Sekijima Y., Zeldenrust S.R., Yamashita T., Heneghan M.A., Gorevic P.D., Litchy W.J., Wiesman J.F., et al. Diflunisal Trial Consortium Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310:2658–2667. doi: 10.1001/jama.2013.283815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Planté-Bordeneuve V., Gorram F., Salhi H., Nordine T., Ayache S.S., Le Corvoisier P., Azoulay D., Feray C., Damy T., Lefaucheur J.P. Long-term treatment of transthyretin familial amyloid polyneuropathy with tafamidis: a clinical and neurophysiological study. J. Neurol. 2017;264:268–276. doi: 10.1007/s00415-016-8337-3. [DOI] [PubMed] [Google Scholar]
- 9.Keam S.J. Inotersen: First Global Approval. Drugs. 2018;78:1371–1376. doi: 10.1007/s40265-018-0968-5. [DOI] [PubMed] [Google Scholar]
- 10.Mahfouz M., Maruyama R., Yokota T. Inotersen for the Treatment of Hereditary Transthyretin Amyloidosis. Methods Mol. Biol. 2020;2176:87–98. doi: 10.1007/978-1-0716-0771-8_6. [DOI] [PubMed] [Google Scholar]
- 11.Wood H. FDA approves patisiran to treat hereditary transthyretin amyloidosis. Nat. Rev. Neurol. 2018;14:570. doi: 10.1038/s41582-018-0065-0. [DOI] [PubMed] [Google Scholar]
- 12.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fellmann C., Gowen B.G., Lin P.C., Doudna J.A., Corn J.E. Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat. Rev. Drug Discov. 2017;16:89–100. doi: 10.1038/nrd.2016.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu W., Wu Z. In Vivo Applications of CRISPR-Based Genome Editing in the Retina. Front. Cell Dev. Biol. 2018;6:53. doi: 10.3389/fcell.2018.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chemello F., Bassel-Duby R., Olson E.N. Correction of muscular dystrophies by CRISPR gene editing. J. Clin. Invest. 2020;130:2766–2776. doi: 10.1172/JCI136873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amoasii L., Hildyard J.C.W., Li H., Sanchez-Ortiz E., Mireault A., Caballero D., Harron R., Stathopoulou T.R., Massey C., Shelton J.M., et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362:86–91. doi: 10.1126/science.aau1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maeder M.L., Stefanidakis M., Wilson C.J., Baral R., Barrera L.A., Bounoutas G.S., Bumcrot D., Chao H., Ciulla D.M., DaSilva J.A., et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019;25:229–233. doi: 10.1038/s41591-018-0327-9. [DOI] [PubMed] [Google Scholar]
- 18.Richards D.Y., Winn S.R., Dudley S., Nygaard S., Mighell T.L., Grompe M., Harding C.O. AAV-Mediated CRISPR/Cas9 Gene Editing in Murine Phenylketonuria. Mol. Ther. Methods Clin. Dev. 2019;17:234–245. doi: 10.1016/j.omtm.2019.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zabaleta N., Barberia M., Martin-Higueras C., Zapata-Linares N., Betancor I., Rodriguez S., Martinez-Turrillas R., Torella L., Vales A., Olague C., et al. CRISPR/Cas9-mediated glycolate oxidase disruption is an efficacious and safe treatment for primary hyperoxaluria type I. Nat Commun. 2018;9:5454. doi: 10.1038/s41467-018-07827-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quinn J., Musa A., Kantor A., McClements M., Cehajic-Kapetanovic J., MacLaren R.E., et al. Genome editing strategies for treating human retinal degenerations. Hum. Gene Ther. 2020;32:247–259. doi: 10.1089/hum.2020.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finn J.D., Smith A.R., Patel M.C., Shaw L., Youniss M.R., van Heteren J., Dirstine T., Ciullo C., Lescarbeau R., Seitzer J., et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018;22:2227–2235. doi: 10.1016/j.celrep.2018.02.014. [DOI] [PubMed] [Google Scholar]
- 22.Chen F., Alphonse M., Liu Q. Strategies for nonviral nanoparticle-based delivery of CRISPR/Cas9 therapeutics. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020;12:e1609. doi: 10.1002/wnan.1609. [DOI] [PubMed] [Google Scholar]
- 23.Witzigmann D., Kulkarni J.A., Leung J., Chen S., Cullis P.R., van der Meel R. Lipid nanoparticle technology for therapeutic gene regulation in the liver. Adv. Drug Deliv. Rev. 2020;159:344–363. doi: 10.1016/j.addr.2020.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zincarelli C., Soltys S., Rengo G., Rabinowitz J.E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008;16:1073–1080. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 25.Wang L., Bell P., Somanathan S., Wang Q., He Z., Yu H., McMenamin D., Goode T., Calcedo R., Wilson J.M. Comparative Study of Liver Gene Transfer With AAV Vectors Based on Natural and Engineered AAV Capsids. Mol. Ther. 2015;23:1877–1887. doi: 10.1038/mt.2015.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang D., Tai P.W.L., Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019;18:358–378. doi: 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Truong D.J., Kühner K., Kühn R., Werfel S., Engelhardt S., Wurst W., Ortiz O. Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic Acids Res. 2015;43:6450–6458. doi: 10.1093/nar/gkv601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moretti A., Fonteyne L., Giesert F., Hoppmann P., Meier A.B., Bozoglu T., Baehr A., Schneider C.M., Sinnecker D., Klett K., et al. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat. Med. 2020;26:207–214. doi: 10.1038/s41591-019-0738-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moreno A.M., Fu X., Zhu J., Katrekar D., Shih Y.V., Marlett J., Cabotaje J., Tat J., Naughton J., Lisowski L., et al. In Situ Gene Therapy via AAV-CRISPR-Cas9-Mediated Targeted Gene Regulation. Mol. Ther. 2018;26:1818–1827. doi: 10.1016/j.ymthe.2018.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Y., Zhi S., Liu W., Wen J., Hu S., Cao T., et al. Development of Highly Efficient Dual-AAV Split Adenosine Base Editor for In Vivo Gene Therapy. Small Methods. 2020;4:2000309. [Google Scholar]
- 31.Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S., et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edraki A., Mir A., Ibraheim R., Gainetdinov I., Yoon Y., Song C.Q., Cao Y., Gallant J., Xue W., Rivera-Pérez J.A., Sontheimer E.J. A Compact, High-Accuracy Cas9 with a Dinucleotide PAM for In Vivo Genome Editing. Mol. Cell. 2019;73:714–726.e4. doi: 10.1016/j.molcel.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Z., Chen S., Jia Y., Shan H., Chen M., Song Y., Lai L., Li Z. Efficient and high-fidelity base editor with expanded PAM compatibility for cytidine dinucleotide. Sci. China Life Sci. 2021 doi: 10.1007/s11427-020-1775-2. Published January 6, 2020. [DOI] [PubMed] [Google Scholar]
- 34.Wen J., Wu J., Cao T., Zhi S., Chen Y., Aagaard L., Zhen P., Huang Y., Zhong J., Huang J. Methylation silencing and reactivation of exogenous genes in lentivirus-mediated transgenic mice. Transgenic Res. 2021;30:63–76. doi: 10.1007/s11248-020-00224-9. [DOI] [PubMed] [Google Scholar]
- 35.Kim J.H., Lee S.R., Li L.H., Park H.J., Park J.H., Lee K.Y., Kim M.K., Shin B.A., Choi S.Y. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS ONE. 2011;6:e18556. doi: 10.1371/journal.pone.0018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kosicki M., Tomberg K., Bradley A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018;36:765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang X.H., Tee L.Y., Wang X.G., Huang Q.S., Yang S.H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. Nucleic Acids. 2015;4:e264. doi: 10.1038/mtna.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adams D., Gonzalez-Duarte A., O’Riordan W.D., Yang C.C., Ueda M., Kristen A.V., Tournev I., Schmidt H.H., Coelho T., Berk J.L., et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018;379:11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 41.Benson M.D., Waddington-Cruz M., Berk J.L., Polydefkis M., Dyck P.J., Wang A.K., Planté-Bordeneuve V., Barroso F.A., Merlini G., Obici L., et al. Inotersen Treatment for Patients with Hereditary Transthyretin Amyloidosis. N. Engl. J. Med. 2018;379:22–31. doi: 10.1056/NEJMoa1716793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zetsche B., Volz S.E., Zhang F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 2015;33:139–142. doi: 10.1038/nbt.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tagoe C.E., Reixach N., Friske L., Mustra D., French D., Gallo G., Buxbaum J.N. In vivo stabilization of mutant human transthyretin in transgenic mice. Amyloid. 2007;14:227–236. doi: 10.1080/13506120701464396. [DOI] [PubMed] [Google Scholar]
- 44.Schweigert F.J., Gericke B., Wolfram W., Kaisers U., Dudenhausen J.W. Peptide and protein profiles in serum and follicular fluid of women undergoing IVF. Hum. Reprod. 2006;21:2960–2968. doi: 10.1093/humrep/del257. [DOI] [PubMed] [Google Scholar]
- 45.Zhao G., Li Z., Araki K., Haruna K., Yamaguchi K., Araki M., Takeya M., Ando Y., Yamamura K. Inconsistency between hepatic expression and serum concentration of transthyretin in mice humanized at the transthyretin locus. Genes Cells. 2008;13:1257–1268. doi: 10.1111/j.1365-2443.2008.01242.x. [DOI] [PubMed] [Google Scholar]
- 46.Episkopou V., Maeda S., Nishiguchi S., Shimada K., Gaitanaris G.A., Gottesman M.E., Robertson E.J. Disruption of the transthyretin gene results in mice with depressed levels of plasma retinol and thyroid hormone. Proc. Natl. Acad. Sci. USA. 1993;90:2375–2379. doi: 10.1073/pnas.90.6.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kishor A., Fritz S.E., Hogg J.R. Nonsense-mediated mRNA decay: The challenge of telling right from wrong in a complex transcriptome. Wiley Interdiscip. Rev. RNA. 2019;10:e1548. doi: 10.1002/wrna.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dalsgaard T., Cecchi C.R., Askou A.L., Bak R.O., Andersen P.O., Hougaard D., Jensen T.G., Dagnæs-Hansen F., Mikkelsen J.G., Corydon T.J., Aagaard L. Improved Lentiviral Gene Delivery to Mouse Liver by Hydrodynamic Vector Injection through Tail Vein. Mol. Ther. Nucleic Acids. 2018;12:672–683. doi: 10.1016/j.omtn.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aurnhammer C., Haase M., Muether N., Hausl M., Rauschhuber C., Huber I., Nitschko H., Busch U., Sing A., Ehrhardt A., Baiker A. Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum. Gene Ther. Methods. 2012;23:18–28. doi: 10.1089/hgtb.2011.034. [DOI] [PubMed] [Google Scholar]
- 50.Pease S. Microinjecting Lentivirus into Mouse Embryos. Cold Spring Harb. Protoc. 2018;2018 doi: 10.1101/pdb.prot093914. [DOI] [PubMed] [Google Scholar]
- 51.Jaenisch R., Schnieke A., Harbers K. Treatment of mice with 5-azacytidine efficiently activates silent retroviral genomes in different tissues. Proc. Natl. Acad. Sci. USA. 1985;82:1451–1455. doi: 10.1073/pnas.82.5.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.